

Abstract
Transformation by oncogenic Ras requires the function of the Rho family GTPases. We find that Ras-transformed cells have elevated levels of RhoA-GTP, which functions to inhibit the expression of the cell cycle inhibitor p21/Waf1. These high levels of Rho-GTP are not a direct consequence of Ras signalling but are selected for in response to sustained ERK–MAP kinase signalling. While the elevated levels of Rho-GTP control the level of p21/Waf, they no longer regulate the formation of actin stress fibres in transformed cells. We show that the sustained ERK–MAP kinase signalling resulting from transformation by oncogenic Ras down-regulates ROCK1 and Rho-kinase, two Rho effectors required for actin stress fibre formation. The repression of Rho- dependent stress fibre formation by ERK–MAP kinase signalling contributes to the increased motility of Ras-transformed fibroblasts. Overexpression of the ROCK target LIM kinase restores actin stress fibres and inhibits the motility of Ras-transformed fibroblasts. We propose a model in which Ras and Rho signalling pathways cross-talk to promote signalling pathways favouring transformation.
Keywords: LIM kinase/MAP kinase/p21Waf1/Ras/Rho/ROCK
Introduction
Small GTPases of the Ras superfamily control a wide range of cellular processes by switching between inactive GDP- and active GTP-bound states. When bound to GTP, these proteins regulate cell behaviour by binding to effector molecules and altering their localization, protein–protein interactions and activity. Small GTPase proteins can be divided into subfamilies: members of the Ras subfamily regulate cell proliferation and differentiation, whereas members of the Rho subfamily were first identified as regulators of the actin cytoskeleton but also affect gene expression and proliferation. The importance of Ras subfamily genes in control of cell proliferation is demonstrated by the high frequency of mutations that activate N-Ras and K-Ras in tumours (Bos, 1989). Several of the Ras effectors that contribute to oncogenesis have been identified: these include Raf, which in turn activates the extracellular signal-regulated kinase (ERK)–mitogen-activated protein (MAP) kinase pathway, phosphatidylinositol 3-kinase (PI-3-kinase) and the RalGDS family of activators of Ral GTPases (White et al., 1995, 1996; Rodriguez-Viciana et al., 1997).
RhoA, Rac1 and Cdc42 are the best characterized members of the Rho family. RhoA regulates the formation of actin stress fibres and focal adhesions by binding the effectors mDia and the closely related kinases ROCK1/Rho-kinase (Amano et al., 1997; Watanabe et al., 1999; Tominaga, 2000). ROCK/Rho-kinase activates LIM kinase, which subsequently phosphorylates and inhibits the actin cable-severing protein cofilin (Maekawa et al., 1999; Ohashi et al., 2000). Rac and Cdc42 regulate the formation of filopodia and lamellipodia, respectively, and are involved in cell migration (Ridley et al., 1992; Nobes and Hall, 1995, 1999). In addition to regulating the cytoskeleton, Rho family proteins can promote cell cycle entry (Olson et al., 1995) and are required for oncogenic Ras to induce transformation of mouse fibroblasts (Qiu et al., 1995a,b, 1997). Intriguingly, transformed cells generally lack the actin stress fibres characteristic of Rho activity, leading to the interpretation that Rho activity is reduced in these cells (Izawa et al., 1998).
While Rho family GTPases are required for transformation by oncogenic Ras, the contribution that they make to the transformed phenotype and the underlying mechanisms has not been uncovered. It is not clear whether signalling from Rho family GTPases is required for the altered control of cell proliferation characteristic of transformed cells or whether it contributes to other aspects of the transformed phenotype such as altered morphology. One mechanism by which RhoA may function in transformation is to suppress the inhibitory high levels of the cell cycle regulator p21/Waf1 induced by sustained activation of the ERK–MAP kinase pathway by oncogenic Ras (Olson et al., 1998). However, suppression of inhibitory levels of p21/Waf1 is not the only Rho signalling pathway involved in Ras transformation since ROCK and Rho-kinase have been shown to be required for Ras transformation in some systems but are not responsible for regulating p21/Waf1 levels (Sahai et al., 1999). Another potentially significant role for Rho family proteins in tumorigenesis is in cell migration and invasion; RhoC has been found to be highly expressed in metastatic variants (Clark et al., 2000), and RhoA, Rac and Cdc42 have been shown to be required for cell migration in model systems (Itoh et al., 1999; Nobes and Hall, 1999; Banyard et al., 2000). Members of the Rho family have been found overexpressed in a variety of human tumours and, in some cases, this correlates with poor prognosis (Suwa et al., 1998; Fritz et al., 1999).
While Rho family GTPase signalling is required for transformation, few studies have investigated the activity of Rho family GTPases in transformed cells. Mira et al. (2000) found increased Rac3 activity in transformed breast epithelial cells, whereas Zondag et al. (2000) reported that Rac activity is reduced and Rho activity increased in Ras-transformed MDCK cells. Furthermore, it is unclear whether elevated activity of Rho family proteins is required for transformation by Ras or whether only their basal activity is required.
In order to investigate the interactions between small GTPases, we have measured the activity of Rho, Rac and Cdc42 in Swiss-3T3 fibroblasts and Ras-transformed derivatives. RhoA-GTP levels are elevated in Ras-transformed cells whereas Rac-GTP and Cdc42-GTP levels are reduced. However, the activity of all three proteins is required for proliferation. The elevated levels of RhoA-GTP do not occur as a direct consequence of acute Ras signalling but are selected for in cells with sustained ERK–MAP kinase activity. While active RhoA is required for regulation of p21/Waf1 levels in transformed fibroblasts and some human colon carcinoma cell lines, it is uncoupled from regulation of the actin cytoskeleton. This uncoupling arises through ERK–MAP kinase signalling leading to down-regulation of ROCK1 and Rho-kinase, loss of stress fibres and increased motility in the transformed cells. Overexpression of LIM kinase, a ROCK/Rho-kinase substrate, restores actin stress fibres but not growth control to the transformed cells. Thus, in transformed cells, ERK–MAP kinase selectively uncouples RhoA-GTP from one of its effector pathways but it remains coupled to down-regulating the high levels of p21/Waf1 induced by the ERK–MAP kinase pathway.
Results
RhoA, Rac and Cdc42 are required for proliferation of Ras-transformed cells
Microinjection experiments in Swiss-3T3 cells have revealed that activated Ras protein will only drive proliferation when RhoA is in its active GTP-bound state (Olson et al., 1998). Swiss-3T3 cells, like early passage mouse embryo fibroblasts, but unlike other established lines of fibroblasts, have very low levels of Rho signalling in the absence of serum growth factors (E.Sahai, M.F.Olson and C.J.Marshall, unpublished data). Thus, unlike other non-transformed cell lines with high basal activity of RhoA, these cells provide a useful system for studying regulation of RhoA signalling. We found that Swiss-3T3 cells stably transformed by Ras, but not Swiss-3T3 cells microinjected with activated Ras protein or parental Swiss-3T3 cells, proliferated in serum-free conditions (Figure 1 and data not shown). This observation suggests that the transformed cells either have lost the requirement for Rho-GTP or have elevated Rho-GTP.

Fig. 1. Rho, Rac and Cdc42 function are required for Ras-dependent proliferation. Swiss-3T3 cells or a Ras-transformed Swiss-3T3 cell line were seeded at 100 000 cells/3.5 cm dish and serum starved 24 h later. Cells were microinjected with expression plasmids or treated with 10 µM Y-27632 16 h after starvation. BrdU was added 24 h after starvation and cells were fixed and stained for BrdU incorporation 16 h later (average of at least three experiments ± standard error).
To determine whether Rho signalling was required for the proliferation of Ras-transformed cells, we microinjected an expression construct for C3 toxin, which ribosylates and inactivates RhoA. Inhibition of Rho with C3 blocked the proliferation of the Ras-transformed cells in serum-free conditions, while a control green fluorescent protein (GFP) expression construct had no effect; similar results were obtained using a cell-permeable derivative of C3 (data not shown). While these experiments demonstrated the requirement for Rho signalling, inhibition of the Rho effector ROCK/Rho-kinase using the pharmacological inhibitor Y-27632 did not affect the proliferation of the Ras-transformed cells. These data demonstrate that RhoA but not ROCK/Rho-kinase function is required for the Ras-dependent proliferation.
To test whether other Rho family members are required for the proliferation of Ras-transformed cells, we microinjected inhibitory T>N17 mutants of Rac1 and Cdc42 into parental and Ras-transformed cells and monitored cell cycle progression. Inhibition of either Rac or Cdc42 dramatically reduced but did not completely block the proliferation of Ras-transformed cells (Figure 1). Similar results were obtained by expressing the Rac/Cdc42-binding domain of Pak, which titrates Rac and Cdc42 into non-functional complexes (data not shown). These data show that Rac and Cdc42, like Rho, are required for the serum-independent proliferation of Ras-transformed cells.
Regulation of RhoA activity by Ras transformation and confluence
The requirement for Rho family proteins for the proliferation of Ras-transformed cells may indicate that cells transformed by Ras have increased activity of these proteins; alternatively, the basal activity of these proteins may be required for proliferation. To discriminate between these possibilities, we measured the activity of endogenous RhoA, Rac1 and Cdc42 in Swiss-3T3 cells and Ras-transformed derivatives using pull-down assays that only capture the active GTP-bound form of the GTPases. There was little difference between Rho-GTP levels in subconfluent parental Swiss-3T3 cells and a Ras-transformed cell line (Figure 2A). However, at confluence, Rho-GTP levels remained high in the Ras-transformed cell line but were significantly reduced in parental Swiss-3T3 cells. The increase in RhoA-GTP levels in the Ras-transformed cells occurred without a change in the total amount of RhoA. All the experiments analysing proliferation and protein levels described here were performed at the higher cell density. The elevated levels of RhoA-GTP in the Ras-transformed cells could be increased further by serum stimulation; in both untransformed and Ras-transformed cells, RhoA-GTP levels peaked at 1 min after serum stimulation (Figure 2B). These data demonstrate that a higher proportion of RhoA is in the active GTP-bound state in Ras-transformed Swiss-3T3 cells.

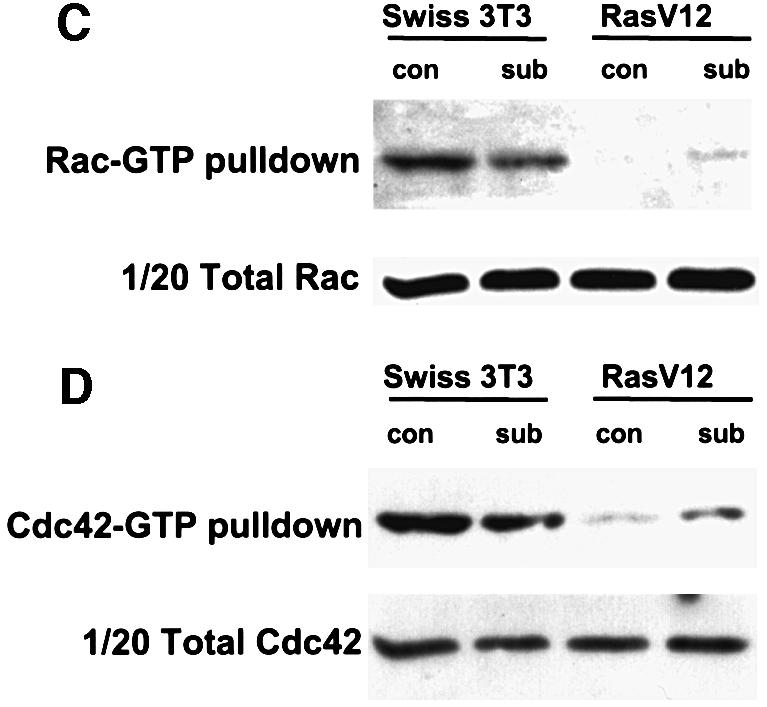
Fig. 2. Changes in RhoA, Rac and Cdc42 activity in Ras-transformed cells. (A) Swiss-3T3 cells or a Ras-transformed Swiss-3T3 cell line were seeded at either 125 000 (subconfluent) or 500 000 (confluent) cells/10 cm dish and serum starved 48 h later. At 24 h after serum starvation, RhoA-GTP levels were analysed (representative of one of at least three experiments). (B) Swiss-3T3 cells or a pool of Ras-transformed derivatives were seeded at 500 000 cells/10 cm dish and serum starved 48 h later. At 24 h after serum starvation, 15% donor calf serum was added for the number of minutes indicated and RhoA-GTP levels were analysed (representative of one of three experiments). (C) Swiss-3T3 cells or a Ras-transformed Swiss-3T3 cell line were seeded at either 125 000 (subconfluent) or 500 000 (confluent) cells/10 cm dish and serum starved 48 h later. At 24 h after serum starvation, Rac-GTP levels were analysed (representative of one of three experiments, one of which was performed with a pool of Ras-transformed cells). (D) As for (C) except that lysates and pull-down were blotted for Cdc42.
Rac and Cdc42 activity are reduced in Ras-transformed cells
We measured the activity of endogenous Rac1 and Cdc42 in the parental and Ras-transformed cells. Even though both Rac and Cdc42 are required for Ras-dependent proliferation, the levels of active Rac1 and Cdc42 were lower in Ras-transformed cells than in parental cells. Similar results were obtained comparing pools of control and Ras-transformed Swiss-3T3 cells. In contrast to RhoA activity, cell density had little effect on Rac1-GTP and Cdc42-GTP levels in non-transformed cells (Figure 2C and D). These observations demonstrate that there is no requirement for elevated levels of Rac1 and Cdc42 activity for the proliferation of Ras-transformed cells.
Rho-GTP regulates p21/Waf1 levels in Ras-transformed cells
Olson et al. (1998) previously showed that unless RhoA is active, microinjection of cells with oncogenic Ras fails to induce DNA synthesis because high levels of the cell cycle regulator p21/Waf1 are induced via the ERK–MAP kinase pathway. The data presented above demonstrate that Rho is required for cell cycle progression and that the levels of Rho-GTP are elevated in Ras-transformed cells. Therefore, we tested whether Rho-GTP regulates p21/Waf1 levels in Ras-transformed cells. Parental Swiss-3T3 and Ras-transformed cells were serum starved and treated with cell-permeable C3 to inactivate RhoA or with 10 µM Y-27632 to inactivate ROCK/Rho-kinase, and levels of the cell cycle regulators p21/Waf1 and p27/Kip1 were measured. Parental cells had very low levels of p21/Waf1 whereas Ras-transformed cells had slightly higher levels that were increased greatly upon treatment with C3 (Figure 3A, lower panel). Treatment of cells with ROCK/Rho-kinase inhibitor did not affect p21/Waf1 levels. Levels of p27/Kip1 were not greatly affected by treatment with C3 or Y-27632 (Figure 3A upper panel). We also investigated the effect of C3 and Y-27632 treatment on the levels of p21/Waf1 and p27/Kip1 in Raf-transformed Swiss-3T3 cells, and in BE and HCT15 human colon carcinoma cell lines that contain oncogenic K-ras mutations (S.Hooper and C.J.Marshall, unpublished data). As with the Ras-transformed fibroblasts, inhibition of Rho, but not ROCK/Rho-kinase, led to elevated levels of p21/Waf1 in these cell lines (Figure 3A and B). The level of p27/Kip1 was increased slightly by both C3 and Y-27632 treatment of BE cells but completely unaffected in HCT15 cells (Figure 3B). Thus, in Swiss-3T3, BE and HCT15 cells, Rho appears to play little role in regulation of the levels of p27/Kip1; this contrasts with studies in IIC9 fibroblasts and FRTL-5 cells (Hirai et al., 1997; Weber et al., 1997). These results demonstrate that in transformed cells containing oncogenic Ras, just as in the microinjection experiments, Rho-GTP reduces p21/Waf1 to levels that enable growth.

Fig. 3. Rho regulates p21/Waf1 levels in Ras-transformed cells and colon carcinoma cell lines. (A) Swiss-3T3 cells, a pool of Ras-transformed Swiss-3T3 cells or a pool of Raf:ER-transformed cells (cultured permanently in 100 nM tamoxifen) were seeded at 100 000 cells/3.5 cm dish and serum starved 24 h later. At the time of serum starvation, either recombinant 500 nM TAT-C3 or 10 µM Y-27632 was added. Cells were harvested after a further 24 h and analysed by western blotting (one of two experiments shown). (B) BE and HCT15 human colon carcinoma cells were treated as above.
Elevated Rho-GTP levels in transformed cells are a consequence of selection not direct signalling
The increased Rho-GTP levels we observe in Ras-transformed cells may be a direct result of Ras-regulated signalling pathways or an indirect result of selection during the establishment of the transformed cell lines. To determine whether the elevated Rho-GTP levels in Ras-transformed cells are a direct consequence of either Raf/MEK/ERK or PI-3-kinase signalling, we inhibited these pathways with pharmacological agents. Neither inhibition of MEK with U0126 nor inhibition of PI-3-kinase with LY294002 reduced the RhoA-GTP levels in pools of confluent Ras-transformed cells (Figure 4A); in one Ras-transformed clone, inhibition of PI-3-kinase reduced RhoA-GTP levels but they were still much higher than in parental Swiss-3T3 cells (data not shown). The efficacy of MEK inhibition was confirmed by monitoring levels of phosphorylated active ERK–MAP kinase (data not shown). These results demonstrate that Raf/MEK/ERK and PI-3-kinase signalling are not required for the elevated RhoA-GTP levels in Ras-transformed cells. Olson et al. (1995) previously demonstrated that activated MEK, like activated Ras, was only mitogenic in the presence of active Rho signalling; we therefore used cells containing an inducible activated version of Raf-1 to test further whether elevated Rho-GTP was a consequence of selection rather than direct signalling. Swiss-3T3 cells were infected with a retrovirus containing a tamoxifen-regulatable version of Raf-1, Raf:ER (Woods et al., 1997). As previously described, induction of ERK–MAP kinase activity by high level tamoxifen treatment initially led to growth arrest (Woods et al., 1997 and data not shown) but, after 2 weeks treatment, proliferating cells had grown out. These cells had elevated ERK–MAP kinase activity, grew to high density and had a slightly more refractile appearance (Figure 4B and data not shown). RhoA-GTP levels were elevated in these transformed cells compared with untreated cells and cells in which ERK–MAP kinase had been activated by tamoxifen treatment for 6 h (Figure 4B). Both the removal of tamoxifen and treatment with U0126 resulted in reduced ERK–MAP kinase activity and a flattened morphology (Figure 4B and data not shown). In contrast to ERK–MAP kinase activity, Rho-GTP levels remained high even after the removal of tamoxifen (Figure 4B). The persistence of high Rho-GTP levels in Raf:ER-transformed cells even when Raf has been deactivated, together with the lack of effect of MEK inhibitor on Rho-GTP levels in Ras-transformed cells suggest that the elevated Rho-GTP levels observed in these transformed cells are not a direct result of MAP kinase signalling but are selected for in response to high levels of MAP kinase signalling.

Fig. 4. Isolation of cells transformed by Raf leads to selection for high Rho-GTP levels. (A) Swiss-3T3 cells or a pool of Ras-transformed derivatives were seeded at 500 000 cells/10 cm dish and serum starved 48 h later, and either vehicle, 10 µM U0126 or 20 µM LY294002 added. At 24 h after serum starvation, RhoA-GTP levels were analysed (representative of one of at least three experiments). (B) Upper panels: a pool of Raf:ER-expressing Swiss-3T3 derivatives was cultured either without tamoxifen (naïve –), with 100 nM tamoxifen for 6 h (naïve +), with 100 nM tamoxifen for at least 4 weeks (transformed +) or with 100 nM tamoxifen for at least 4 weeks and then the tamoxifen withdrawn 72 h prior to harvesting [transformed (–)]. Cells were seeded at 500 000 cells/10 cm dish and serum starved 48 h later. At 24 h after serum starvation, RhoA-GTP levels were analysed (representative of one of three experiments). Lower panel: Raf:ER cells were treated as above and transformed cells were also treated with U0126 and lysates western blotted for active phospho-ERK.
MAP kinase signalling uncouples Rho-GTP from stress fibre formation
Activation of Rho by serum stimulation promoted the formation of actin stress fibres in untransformed fibroblasts; however, in Ras-transformed fibroblasts, the high levels of Rho-GTP induced by serum did not lead to stress fibre formation (Figure 5A, see also Figure 2B). These data suggest that the increased levels of active Rho-GTP in Ras-transformed cells are not coupled to the formation of actin stress fibres. To test this hypothesis, we microinjected RhoAV14, a constitutively active RhoA mutant. In untransformed cells but not in Ras-transformed cells, RhoAV14 led to increased numbers of stress fibres; interestingly, in Ras-transformed cells, RhoAV14 led to a slight increase in diffuse F-actin staining, suggesting that while Rho-GTP is uncoupled from stress fibre formation, it is still able to regulate some aspects of the actin cytoskeleton in these cells. The observation that in Ras-transformed cells Rho-GTP is uncoupled from actin stress fibre formation suggests that signalling pathways activated by Ras may inhibit signalling downstream of RhoA. We tested whether either the Raf/MEK/ERK or PI-3-kinase pathways were responsible for uncoupling Rho-GTP from actin stress fibre formation. Overnight treatment of Ras-transformed cells with the PI-3-kinase inhibitor LY294002 had little effect on stress fibre formation; however, treatment with the MEK inhibitor U0126 restored actin stress fibres (Figure 5A and data not shown). Inhibition of MEK in parental Swiss-3T3 cells slightly increased the numbers of stress fibres observed in serum-starved cells. To eliminate the possibility that U0126 may restore actin stress fibres by acting on targets other than MEK1/2, we used an unrelated MEK inhibitor PD098095 and stable expression of dominant-negative MEK constructs; consistent with the effects being MEK dependent, both had similar effects to those of U0126 (data not shown). To examine whether activation of the ERK–MAP kinase pathway could uncouple active Rho from stress fibre formation, we made use of Swiss-3T3 cells containing tamoxifen-regulatable Raf:ER. Activation of the Raf/MEK/ERK pathway following treatment with tamoxifen inhibited serum-induced stress fibre formation even though RhoA-GTP levels were stimulated to the same degree; consistent with this, RhoAV14 microinjection did not lead to stress fibre formation though there was a slight increase in F-actin staining and some cell rounding (Figure 5B and data not shown). These results argue that prolonged signalling through ERK–MAP kinase suppresses the ability of active Rho to promote the formation of stress fibres. This uncoupling of Rho from the regulation of the actin cytoskeleton is selective as it remains coupled to regulation of p21/Waf1.


Fig. 5. ERK–MAP kinase signalling uncouples Rho-GTP from stress fibre formation. (A) Swiss-3T3 cells or a Ras-transformed Swiss-3T3 cell line were seeded at 100 000 cells/3.5 cm dish, serum starved 24 h later and treated with 10 µM U0126 where indicated. Cells were fixed and phalloidin stained 24 h after starvation; microinjection was performed 6 h before harvesting and stimulation with donor calf serum 30 min before harvesting. (B) A pool of Raf:ER-expressing Swiss-3T3 derivatives was seeded at 100 000 cells/3.5 cm dish, serum starved 24 h later and treated with 100 nM tamoxifen where indicated. Cells were fixed and phalloidin stained 24 h after starvation; microinjection was performed 6 h before harvesting and stimulation with donor calf serum 30 min before harvesting. Panels are 120 µm × 120 µm.
ROCK/Rho-kinase signalling is compromised in Ras-transformed fibroblasts
To determine how Rho signalling is uncoupled from stress fibre formation, we tested which signalling molecules downstream of Rho were able to regulate the actin cytoskeleton in Ras-transformed cells. The effects on the cytoskeleton of microinjection of ROCK1, mDia2 and LIM kinase expression constructs into parental and Ras-transformed cells were compared. ROCK1 produced thick actin fibres in Swiss-3T3 cells but had no effect in Ras-transformed cells (Figure 6A); however, full-length ROCK1 was expressed poorly in Ras-transformed cells (data not shown). A constitutively active truncation mutant of ROCK1, which was expressed comparably in both parental and transformed cells, promoted more profound actin fibre assembly in Swiss-3T3 cells than the full-length ROCK1; in contrast, it promoted cell contraction and membrane blebbing in Ras-transformed cells. This suggests that exogenously expressed ROCK1 can regulate actin architecture in Ras-transformed cells. Elsewhere, we have investigated further the links between ROCK1 and membrane blebbing (Coleman et al., 2001). LIM kinase, which acts downstream of ROCK/Rho-kinase to cause actin re-modelling through inactivating the actin-destabilizing protein cofilin, had similar effects in both parental and Ras-transformed cells and led to the appearance of F-actin patches and a few actin fibres. In both parental and Ras-transformed cells, mDia2 increased F-actin staining and actin-rich protrusions, often with small ruffles. These results demonstrate that while mDia and LIM kinase can function in Ras-transformed cells in the same way as in non-transformed cells, ROCK1 function is disrupted in Ras-transformed cells. Thus, signalling from RhoA through ROCK/Rho-kinase is inhibited by the sustained activation of the ERK–MAP kinase pathway resulting from transformation by Ras. The difference in phenotype observed after truncated ROCK1 is expressed in parental and Ras-transformed cells indicates that abrogation of ROCK/Rho-kinase signalling is not the sole mechanism by which MAP kinase signalling alters cell morphology.


Fig. 6. ROCK/Rho-kinase signalling is compromised in Ras-transformed cells. (A) Swiss-3T3 cells or a Ras-transformed Swiss-3T3 cell line were seeded at 100 000 cells/3.5 cm dish and serum starved after 24 h. Cells were fixed and phalloidin stained 24 h after starvation; microinjection was performed 6 h before harvesting. Panels are 80 µm × 80 µm. (B) Swiss-3T3 cells or a Ras-transformed Swiss-3T3 cell line were seeded at 500 000 cells/10 cm dish, serum starved 48 h later and treated with 10 µM U0126 where indicated. Cell lysates were harvested 24 h after starvation and analysed by western blot. (C) Swiss-3T3 cells or a Ras-transformed Swiss-3T3 cell line were seeded at 500 000 cells/10 cm dish and serum starved 48 h later. Where indicated, cells were stimulated with donor calf serum (+DCS) for 20 min prior to preparation of Triton X-100-soluble and -insoluble fractions.
Since full-length ROCK1 appeared to be expressed poorly in Ras-transformed cells, we examined the expression levels of ROCK1 and Rho-kinase in parental and Ras-transformed cells. Western blotting showed that both ROCK1 and Rho-kinase expression were reduced in Ras-transformed cells, although the reduction in Rho-kinase levels was only slight (Figure 6B). As we have shown that treatment of Ras-transformed cells with U0126 to block activation of ERK–MAP kinase restores actin stress fibres, we examined the effect of U0126 treatment on ROCK1 and Rho-kinase levels. Figure 6B shows that U0126 treatment increased the level of ROCK1 and Rho-kinase expression to levels similar to those in non-transformed parental Swiss-3T3 cells. Changes in the levels of tropo myosin have been implicated in the altered morphology of Ras-transformed cells (Janssen et al., 1998); however, we found that tropomyosin levels were not changed in the Ras-transformed cells (Figure 6B). To compare ROCK1/Rho-kinase activity between the parental and Ras-transformed cells, we carried out immune complex kinase assays; although we could immunoprecipitate ROCK1 from parental cells, we were unable to do so from the Ras-transformed cells (data not shown). This failure to immunoprecipitate ROCK1 from Ras-transformed cells is due to ROCK1 shifting to a Triton X-100-insoluble fraction. Analysis of Triton X-100-soluble and -insoluble fractions showed that both ROCK1 and Rho-kinase were translocated from the soluble fraction to the insoluble fraction in both serum-starved and stimulated Ras-transformed cells (Figure 6C); this was partially reverted by U0126 (data not shown). In contrast, the distribution of tropomyosin between the soluble and insoluble fractions was only slightly altered in the Ras-transformed cells, and RhoA was found exclusively in the soluble fraction in both parental and Ras-transformed cells (Figure 6C and data not shown). Thus, in the Ras-transformed cells, ROCK1 and Rho-kinase are largely translocated away from the cell fraction in which RhoA is found, providing an additional mechanism for the uncoupling of RhoA-GTP from ROCK/Rho-kinase-dependent stress fibre formation.
Overexpression of LIM kinase restores stress fibres and reduces motility but does not affect growth control in Ras-transformed cells
To determine whether the inhibition of ROCK/Rho-kinase function and altered morphology observed in Ras-transformed cells is important for their deregulated growth, we sought to revert their morphology by more prolonged expression of either ROCK/Rho-kinase or LIM kinase. Prolonged expression of active ROCK1 or Rho-kinase constructs, either by microinjection or retroviral infection, could not be tolerated by the Ras-transformed cells (E.Sahai and M.F.Olson, unpublished data). Prolonged expression of LIM kinase following microinjection (24 h) or retroviral infection did not cause the formation of F-actin patches observed in the short-term experiments (6 h) but led to flatter less refractile cells and restored the formation of actin stress fibres (contrast Figures 6A and 7A). Actin-rich ruffles were sometimes observed in addition to stress fibres in cells microinjected with LIM kinase (Figure 7A). The growth properties of these LIM kinase-overexpressing Ras-transformed cells were measured by monitoring bromodeoxyuridine (BrdU) incorporation into cells maintained in serum-free media. As shown previously, Ras-transformed cells proliferate in the absence of serum whereas untransformed cells do not (Figure 7B). Two Ras-transformed clones stably infected with a LIM kinase retrovirus proliferated to a similar degree to cells stably infected with a control retrovirus or uninfected cells. These results demonstrate that the ERK–MAP kinase-dependent down-regulation of ROCK/ Rho-kinase and loss of actin stress fibres are not required for the deregulated proliferation of Ras-transformed cells.

Fig. 7. Prolonged LIM kinase overexpression reverts the morphology and motility of Ras-transformed cells but not their deregulated proliferation. (A) Swiss-3T3 cells, a Ras-transformed Swiss-3T3 cell line and a Ras-transformed cell line stably infected with a LIM kinase retrovirus were seeded at 100 000 cells/3.5 cm dish and serum starved 24 h later. Cells were fixed and phalloidin stained 24 h after starvation; where indicated, stimulation with donor calf serum was performed 30 min before harvesting. Panels are 120 µm × 120 µm. (B) Swiss-3T3 cells, a Ras-transformed Swiss-3T3 cell line or Ras-transformed cell lines stably infected with control or LIM kinase retroviruses were seeded at 100 000 cells/3.5 cm dish and serum starved 24 h later. BrdU was added 24 h after starvation and cells were fixed and stained for BrdU incorporation 16 h later (average of two experiments ± range). (C) Cells were starved for 24 h and, where indicated, treated with 10 µM U0126 for 16 h prior to video-microscopy. Data plotted are the average distance moved by a cell in µm/h ± SD based on two independent experiments monitoring ∼20 cells per experiment. Asterisks indicate that data sets differ from Ras-transformed cells with a P-value <0.001 (two-tailed Student’s t-test).
In addition to deregulated proliferation, oncogenic Ras leads to increased motility of fibroblasts (Trahey et al., 1987). To determine whether the down-regulation of ROCK–LIM kinase signalling by the ERK–MAP kinase pathway affected motility, we performed time lapse microscopy. The movement of cells in serum-free medium over a 3 h period was traced and subsequently measured. Ras-transformed cells were considerably more motile than the parental Swiss-3T3 cells; their increased mobility was greatly reduced by treatment with the MEK inhibitor U0126 (Figure 7C). Overexpression of LIM kinase in three independent clones reduced the motility of the Ras-transformed cells to a similar degree to U0126 treatment (Figure 7C). Treatment of parental Swiss-3T3 cells with Y-27632 did not increase their motility (data not shown); therefore, the increased motility of Ras-transformed cells is not solely a consequence of down-regulating ROCK– LIM kinase signalling. Taken together, these data suggest that ERK–MAP kinase-dependent down-regulation of ROCK–LIM kinase signalling causes both the loss of actin stress fibres and increased motility of Ras-transformed cells.
Discussion
We have shown that signalling through the Rho family GTPases RhoA, Rac and Cdc42 is required for cells transformed by oncogenic Ras to proliferate. Interestingly, their activities are not affected in the same way; Rho activity is increased in Ras-transformed cells yet Rac and Cdc42 activity is reduced. The elevated level of Rho activity is required to down-regulate the cell cycle regulator p21/Waf1 and permit cell proliferation. However, while RhoA is activated in Ras-transformed cells, the elevated level of MAP kinase signalling selectively uncouples Rho-GTP from inducing actin stress fibres. This uncoupling of high RhoA activity from stress fibre formation plays a role in the increased cell motility of Ras-transformed cells (see Figure 8).

Fig. 8. Model of Ras–Rho cross-talk during the process of transformation.
In Ras-transformed cells, the activation state of RhoA no longer responds to signals generated by high cell densities, with the consequence that confluent Ras-transformed cells have levels of RhoA-GTP as high as subconfluent cells. This disregulation of Rho activation does not seem to be the direct result of signalling pathways activated by Ras, but rather it is an event that is selected for in the isolation of transformed cell lines (see below). We have also observed elevated RhoA-GTP levels in Ras-transformed NIH-3T3 cells (M.Rosario and C.J.Marshall, unpublished observations); this contrasts strikingly with short-term experiments performed by Zondag et al. (2000), which showed that Ras decreased Rho activity. These results further support the model that Rho activity in transformed cells is not a consequence of direct signalling. Previous studies have shown that there is a complex relationship between the activation state of the various Rho family GTPases. In short-term microinjection assays, activation of Cdc42 leads to Rac activation, which in turn activates RhoA (Nobes and Hall, 1995). In transient transfection studies, oncogenic Ras and platelet-derived growth factor (PDGF) stimulation led to increased Rac-GTP but decreased Rho-GTP levels (Zondag et al., 2000). Thus, the changes in activity of Rho family GTPases observed in Ras-transformed cells do not follow previously described signalling hierarchies (Nobes and Hall, 1995). These discrepancies may reflect the differences in analysing connections between signalling pathways in transient assays compared with the consequences of selection for transformation. It is also apparent that there are cell type-specific influences on the levels of activation of Rho family GTPases in transformed cells. Analysis of Rho-GTP and Rac-GTP levels in epithelial cell models of Ras transformation reveals dramatic differences between cell types: Ras-transformed MDCK cells have high Rho and low Rac activity (Zondag et al., 2000), while we found the opposite in Ras-transformed HB4A human mammary epithelial cells and unchanged Rho and Rac activity in Ras-transformed HA1 kidney cells (E.Sahai and C.J.Marshall, unpublished observations). These disparate observations strengthen the view that selective pressures rather than direct signalling events determine the level of activity of Rho family GTPases in transformed cells. These selective pressures will vary depending on cellular and genetic backgrounds.
While Rac and Cdc42 are required for proliferation of Ras-transformed Swiss-3T3 cells, their activities are reduced in Ras-transformed cells. High levels of ERK– MAP kinase signalling can block proliferation (Woods et al., 1997), and it is possible that an analogous situation occurs with Rac and Cdc42 activity. Perhaps moderate activity of these cytoskeletal regulators is needed to enable the formation of signalling complexes, such as at focal adhesions. If the activity is too low, these complexes cannot be formed and, if the activity is too high, aberrant cytoskeletal organization may prevent proliferation.
Expression of p21/Waf1 following mitogenic stimulation is dependent on the ERK–MAP kinase pathway (Balmanno and Cook, 1999; Bottazzi et al., 1999). In the control of cell cycle progression, p21/Waf1 has a dual role, it serves as an assembly factor for active complexes of D-type cyclins and their cyclin-dependent kinases (CDKs) and is an inhibitor of CDK2 (Cheng et al., 1999). In previous studies employing transient assays of Ras-driven proliferation, the absence of signalling through RhoA resulted in the Ras-driven ERK–MAP kinase pathway inducing levels of p21/Waf1 that are inhibitory to cell cycle progression (Olson et al., 1998). We now show that signalling through RhoA is required to suppress growth inhibitory levels of p21/Waf1 in Ras-transformed Swiss-3T3 cells and in two human colorectal cancer cell lines. In these human cell lines, inhibition of the Raf/MEK/ERK and PI-3-kinase Ras effector pathways does not affect Rho-GTP levels, supporting the model that Rho activity is determined by selection; however, the levels of Rho-GTP in these cells compared with untransformed cells could not be determined due to the lack of genotypically matched controls (E.Sahai, unpublished observation). We propose that cells with high levels of Rho activity are selected for because they counteract the high levels of p21/Waf1 induced by oncogenic Ras and proliferate, while cells with low Rho activity remain growth arrested by high p21/Waf1 levels. Interestingly, the Ras-transformed Swiss-3T3 cells have higher levels of p21/Waf1 than parental non-transformed cells. However, these cells proliferate presumably because the levels of p21/Waf1 are such that they enable the assembly of active cyclin D–CDK complexes rather than inhibit CDK2. We propose that the elevated levels of Rho-GTP in the transformed cells set a threshold level of p21/Waf1 that is compatible with proliferation.
Potentially, several different Rho effector pathways are important for transformation and, depending upon cell type and the oncogenic lesions, the pathways involved may vary. The regulation of p21/Waf1 levels by RhoA is not mediated by ROCK/Rho-kinase signalling as treatment of Ras-transformed Swiss-3T3 cells with the ROCK/Rho-kinase inhibitor Y-27632 does not increase p21/Waf1 levels or prevent deregulated proliferation. However, a distinct pathway involving phosphorylation of ERM proteins by ROCK/Rho-kinase is required for the transformed phenotype of Ras-, Dbl- and mNET-transformed NIH-3T3 fibroblasts and some colon carcinoma cells (Sahai et al., 1999; Tran Quang et al., 2000). In cells that require ROCK/Rho-kinase function for transformation, it is possible that a moderate threshold level of ROCK/Rho-kinase activity is required; indeed, we find that elevated Rho-kinase activity prevents proliferation of NIH-3T3 cells (M.F.Olson, unpublished data). This may explain why the most active truncation mutant of ROCK1 does not co-operate with Raf to transform NIH-3T3 cells while a less active truncation does (Ishizaki et al., 1997; Sahai et al., 1999).
While Rho-GTP is elevated in Swiss-3T3 cells transformed by Ras or Raf:ER, it is no longer coupled to stress fibre formation. This uncoupling is a consequence of sustained activation of the ERK–MAP kinase pathway since transient activation of Rho and ERK–MAP kinase by serum stimulation leads to the formation of stress fibres. Khosravi-Far et al. (2001) found that NIH-3T3 cells transformed with Ras effector mutants unable to activate ERK–MAP kinase had stress fibres similar to RhoAL63-transformed cells; this is probably due to the failure of these mutants to induce ERK–MAP kinase signalling and uncouple Rho from stress fibre formation. The molecular basis for the uncoupling of RhoA-GTP is at least in part at the level of ROCK/Rho-kinase function since we found that Ras-transformed cells could respond to mDia and to LIM kinase but not to ROCK1. Stable expression of LIM kinase leads to morphological reversion of the Ras-transformed cells with restoration of the actin cytoskeleton. This is likely to be a direct result of LIM kinase regulating the activity of cytoskeletal proteins not an indirect result of activating transcription of cytoskeletal genes via SRF as expression of a constitutively active SRF mutant failed to restore actin stress fibres (Sotiropoulos et al., 1999 and data not shown).
The inability of RhoA-GTP to activate ROCK/Rho-kinase appears to be a consequence of both reduced protein levels and the translocation of ROCK/Rho-kinase away from RhoA into a Triton X-100-insoluble fraction. It is possible that ROCK/Rho-kinase may be activated in the insoluble fraction by Rho-independent mechanisms and may phosphorylate a different subset of targets. The reduction in protein levels was a consequence of ERK– MAP kinase signalling since inhibition of the ERK–MAP kinase pathway restored the levels of ROCK1 and Rho-kinase. Zuber et al. (2000) reported that expression of mRNA for Rho-kinase was dramatically down-regulated in Ras-transformed fibroblasts, suggesting that a com ponent of this effect is transcriptional. However, we found reduced expression of full-length ROCK1 from an exogenous construct, suggesting that there is also a post-transcriptional component. While the effects of the ERK–MAP kinase pathway on Rho signalling to the cytoskeleton are most pronounced, parental Swiss-3T3 cells treated with U0126 have increased numbers of stress fibres in the absence of serum. This suggests that in non-transformed cells as well as transformed cells, ERK–MAP kinase signalling modulates the ability of active Rho to promote actin rearrangements. In transformed cells with sustained activation of ERK–MAP kinase, signalling downstream of RhoA is uncoupled from regulation of stress fibre formation.
Ras-transformed cells are highly motile (Trahey et al., 1987), and we have shown that the uncoupling of Rho-GTP from stress fibre formation contributes to this increased motility. Treatment of Ras-transformed fibroblasts with MEK inhibitor or LIM kinase overexpression recouples Rho-GTP to stress fibre formation and reduces motility. While the uncoupling of RhoA-GTP from actin stress fibres is important for increased motility, inhibition of ROCK/Rho-kinase is not sufficient to cause a motile phenotype; this suggests that changes in other cytoskeletal regulators are required for the increased motility of Ras-transformed cells. These data would suggest that high numbers of stress fibres inhibit cell motility in fibroblasts. The relationship between Rho–ROCK signalling and cell motility is complex, with seemingly contradictory results from different systems (Itoh et al., 1999; Nobes and Hall, 1999; Clark et al., 2000). We speculate that, as with cell proliferation, certain threshold levels of Rho–ROCK signalling are required for cell motility; both excessive activation and inhibition of Rho can prevent cell invasion (Banyard et al., 2000). If Rho–ROCK activity is too high, cells may be unable to remodel focal adhesions and become immobilized as a consequence. If ROCK/Rho-kinase activity is too low, cells may not be able to generate the contractile force needed to pull the rest of the cell behind the leading edge.
These studies show that there is reciprocal cross-talk between the Ras and Rho signalling pathways in transformation. Ras signalling through the ERK–MAP kinase pathway has growth stimulatory effects via the induction of cyclin D1 (Albanese et al., 1995). However, when Rho activity is low, signalling through the ERK–MAP kinase pathway leads to growth inhibition because inhibitory high levels of p21/Waf1 result. Therefore, during transformation, there is selection for cells with high levels of Rho-GTP to oppose inhibitory levels of p21/Waf1. While Rho signalling is opposing the growth inhibitory effects of the ERK–MAP kinase pathway, ERK–MAP kinase signalling opposes RhoA signalling to the cytoskeleton, thereby promoting motility (Figure 8). Since RhoA can still signal to suppress p21/Waf1 levels, this inhibition of RhoA signalling by ERK–MAP kinase must only operate on some of the downstream targets of Rho. Thus, during the process of transformation, there is selective cross-talk between Ras and Rho signalling pathways to favour effector pathways that promote proliferation and motility.
Materials and methods
Microinjections and immunofluorescence
Needles were pulled with a Narishige vertical needle puller. All plasmids were injected in Tris-buffered saline (TBS) at a concentration of 100 ng/µl, except EF-GFP, which was at 25 ng/µl. For cell cycle assays, BrdU was added 8 h post-injection and cells were harvested 24 h post injection; for cytoskeletal assays, cells were harvested 6 h post-injection. To harvest, cells were washed with phosphate-buffered saline (PBS), fixed with 4% formaldehyde in PBS and permeablized with 0.2% Triton X-100 in PBS. After several PBS washes, cells were stained either for the actin cytoskeleton with Texas red–phalloidin (Molecular Probes) or for BrdU incorporation by treatment with 2 U/µl DNase I (Boehringer Mannheim) in PBS and 2.5 mM MgCl2 at 37°C for 1 h before addition of 1/2 volume of anti-BrdU rat monoclonal antibody (Institute of Cancer Research, Sutton) for 1 h; after several PBS washes, Texas red-conjugated anti-rat secondary antibody (Jackson Stratech) was added for 1 h, cells were mounted after several further PBS washes and viewed using a Bio-Rad MRC1024 confocal microscope. Microinjected cells were identified by GFP fluorescence.
Small G-protein pull-down assays
The Rho-GTP pull-down assay is modified from Ren et al. (1999) and described in detail in Coleman et al. (2001): for experiments done at confluence, 500 000 cells were plated in 10 cm dishes and serum starved after 48 h; for subconfluent experiments, 125 000 cells were plated in 10 cm dishes. Cells were lysed 24 h after serum starvation. The Rac-GTP and Cdc42-GTP pull-down assay was as described for Rho-GTP except that cell lysis and washes were done with 50 mM Tris pH 7.2, 100 mM NaCl, 5 mM MgCl2, 1 mM dithiothreitol (DTT), 10% glycerol, 1% NP-40 + protease inhibitors. Glutathione S-transferase (GST)–PAK (rat PAKα amino acids 1–252) was used in place of rhotekin.
Cell motility assays
After serum starvation for 24 h, the motility of cells (∼30% confluent) was determined by recording phase contrast images once a minute with a video recorder for several hours; to determine the scale of the image, a slide with known graduations was recorded. The recordings were played back and the movement of the cells over a 3 h period was traced manually. The lengths of the tracings were measured with a ruler, scaled appropriately and divided by three to give the distance moved per hour.
Plasmids, antibodies, TAT-C3 and miscellaneous
EF-C3, EF-RacN17, EF-Cdc42N17, EF-RhoAV14, pGEX-rhotekin and pGEX-PAK-Nterm were gifts from R.Treisman; pcDNA3-LIM kinase 1 was a gift from P.Aspenström; pBabepuroLIMK was made by blunt subcloning LIM kinase into the SnaBI site; pBabepuroRaf: ER-YY was a gift from M.McMahon; EF-mDia2ΔGBD was a gift from A.Alberts; pCAGGS-ROCK1Δ3 and pCAGGS-ROCK1 were gifts from S.Narumiya; EGFP-C1 was from Clontech; and pLXSN and pLXSN-H-rasV12 were gifts from J.Downward. Antibodies: ROCK1 (Transduction labs #R81520), Rho-kinase (Transduction labs #R54520), RhoA (Santa Cruz #sc418), Rac1 (Upstate Biotech #05-389), Cdc42 (Santa Cruz #sc8401), phosphoERK (Sigma #M8159), p21 (Santa Cruz #sc397), p27 (Transduction labs #K25020) and tropomyosin (Sigma #T2780). U0126 (Promega) was used at 10 µM, LY294002 (Biomol #ST-420) was used at 20 µM, and Y-27632 (Welfide) was used at 10 µM. For western blot analysis, cell were lysed in the same lysis buffer as used for the Rho-GTP pull-downs; protein levels were determined using the Bio-Rad Protein Assay system and subsequently equalized. Western blots were stripped in 0.15 M glycine pH 2.5, 0.4% SDS for 15 min. Cytoskeletal fractionation was performed as described (Algrain et al., 1993). TAT-C3 was made as described in Coleman et al. (2001) and used at 500 nM in cell culture media.
Cell lines
Retroviral constructs were transfected using Lipofectamine (Gibco-BRL) in BOSC 293 packaging cells; retrovirus was harvested from the medium after 40 h. Exponentially growing Swiss-3T3 cells were infected with virus mixed with 4 µg/ml polybrene (Sigma H-9268) and selected with 2.5 µg/ml puromycin or 1 mg/ml geneticin sulfate. HCT15 and BE cell lines were gifts from L.Kelland (Institute of Cancer Research, Sutton).
Acknowledgments
Acknowledgements
We thank members of the laboratory for help and advice throughout this study, Jurgen Riedl for help with TAT-C3, Hugh Paterson for microscopy advice, Marta Rosario for comments on the manuscript, Welfide for providing Y-27632, and numerous laboratories for plasmids (laboratories listed in Materials and methods). E.S. and C.J.M. are funded by the Cancer Research Campaign, M.F.O. is funded by the Royal Society.
REFERENCES
- Albanese C., Johnson,J., Watanabe,G., Eklund,N., Vu,D., Arnold,A. and Pestell,R.G. (1995) Transforming p21ras mutants and c-Ets-2 activate the cyclin D1 promoter through distinguishable regions. J. Biol. Chem., 270, 23589–23597. [DOI] [PubMed] [Google Scholar]
- Algrain M., Turunen,O., Vaheri,A., Louvard,D. and Arpin,M. (1993) Ezrin contains cytoskeleton and membrane binding domains accounting for its proposed role as a membrane–cytoskeletal linker. J. Cell Biol., 120, 129–139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Amano M., Chihara,K., Kimura,K., Fukata,Y., Nakamura,N., Matsuura,Y. and Kaibuchi,K. (1997) Formation of actin stress fibers and focal adhesions enhanced by Rho-kinase. Science, 275, 1308–1311. [DOI] [PubMed] [Google Scholar]
- Balmanno K. and Cook,S.J. (1999) Sustained MAP kinase activation is required for the expression of cyclin D1, p21Cip1 and a subset of AP-1 proteins in CCL39 cells. Oncogene, 18, 3085–3097. [DOI] [PubMed] [Google Scholar]
- Banyard J., Anand-Apte,B., Symons,M. and Zetter,B.R. (2000) Motility and invasion are differentially modulated by Rho family GTPases. Oncogene, 19, 580–591. [DOI] [PubMed] [Google Scholar]
- Bos J.L. (1989) ras oncogenes in human cancer: a review. Cancer Res., 49, 4682–4689. [PubMed] [Google Scholar]
- Bottazzi M.E., Zhu,X., Bohmer,R.M. and Assoian,R.K. (1999) Regulation of p21cip1 expression by growth factors and the extracellular matrix reveals a role for transient ERK activity in G1 phase. J. Cell Biol., 146, 1255–1264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheng M., Olivier,P., Diehl,J.A., Fero,M., Roussel,M.F., Roberts,J.M. and Sherr,C.J. (1999) The p21Cip1 and p27Kip1 CDK ‘inhibitors’ are essential activators of cyclin D-dependent kinases in murine fibroblasts. EMBO J., 18, 1571–1583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clark E.A., Golub,T.R., Lander,E.S. and Hynes,R.O. (2000) Genomic analysis of metastasis reveals an essential role for RhoC. Nature, 406, 532–535. [DOI] [PubMed] [Google Scholar]
- Coleman M.L., Sahai,E.A., Yeo,M., Bosch,M., Dewar,A. and Olson,M. (2001) Membrane blebbing during apoptosis results from caspase-mediated activation of ROCK I. Nature Cell Biol., in press. [DOI] [PubMed] [Google Scholar]
- Fritz G., Just,I. and Kaina,B. (1999) Rho GTPases are over-expressed in human tumors. Int. J. Cancer, 81, 682–687. [DOI] [PubMed] [Google Scholar]
- Hirai A. et al. (1997) Geranylgeranylated rho small GTPase(s) are essential for the degradation of p27Kip1 and facilitate the progression from G1 to S phase in growth-stimulated rat FRTL-5 cells. J. Biol. Chem., 272, 13–16. [PubMed] [Google Scholar]
- Ishizaki T., Naito,M., Fujisawa,K., Maekawa,M., Watanabe,N., Saito,Y. and Narumiya,S. (1997) p160ROCK, a Rho-associated coiled-coil forming protein kinase, works downstream of Rho and induces focal adhesions. FEBS Lett., 404, 118–124. [DOI] [PubMed] [Google Scholar]
- Itoh K., Yoshioka,K., Akedo,H., Uehata,M., Ishizaki,T. and Narumiya,S. (1999) An essential part for Rho-associated kinase in the transcellular invasion of tumor cells. Nature Med., 5, 221–225. [DOI] [PubMed] [Google Scholar]
- Izawa I., Amano,M., Chihara,K., Yamamoto,T. and Kaibuchi,K. (1998) Possible involvement of the inactivation of the Rho–Rho-kinase pathway in oncogenic Ras-induced transformation. Oncogene, 17, 2863–2871. [DOI] [PubMed] [Google Scholar]
- Janssen R.A., Veenstra,K.G., Jonasch,P., Jonasch,E. and Mier,J.W. (1998) Ras- and Raf-induced down-modulation of non-muscle tropomyosin are MEK-independent. J. Biol. Chem., 273, 32182–32186. [DOI] [PubMed] [Google Scholar]
- Khosravi-Far R., White,M.A., Westwick,J.R., Solski,P.A., Chrzanowska- Wodnicka,M., Van Aelst,L., Wigler,M.H. and Der,C.J. (2001) Oncogenic Ras activation of Raf/mitogen-activated protein kinase-independent pathways is sufficient to cause tumorigenic transformation. Mol. Cell. Biol., 16, 3923–3933. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maekawa M. et al. (1999) Signaling from Rho to the actin cytoskeleton through protein kinases ROCK and LIM-kinase. Science, 285, 895–898. [DOI] [PubMed] [Google Scholar]
- Mira J.P., Benard,V., Groffen,J., Sanders,L.C. and Knaus,U.G. (2000) Endogenous, hyperactive Rac3 controls proliferation of breast cancer cells by a p21-activated kinase-dependent pathway. Proc. Natl Acad. Sci. USA, 97, 185–189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nobes C.D. and Hall,A. (1995) Rho, rac and cdc42 GTPases regulate the assembly of multimolecular focal complexes associated with actin stress fibers, lamellipodia and filopodia. Cell, 81, 53–62. [DOI] [PubMed] [Google Scholar]
- Nobes C.D. and Hall,A. (1999) Rho GTPases control polarity, protrusion and adhesion during cell movement. J. Cell Biol., 144, 1235–1244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ohashi K., Nagata,K., Maekawa,M., Ishizaki,T., Narumiya,S. and Mizuno,K. (2000) Rho-associated kinase ROCK activates LIM-kinase 1 by phosphorylation at threonine 508 within the activation loop. J. Biol. Chem., 275, 3577–3582. [DOI] [PubMed] [Google Scholar]
- Olson M.F., Ashworth,A. and Hall,A. (1995) An essential role for Rho, Rac and Cdc42 GTPases in cell cycle progression through G1. Science, 269, 1270–1272. [DOI] [PubMed] [Google Scholar]
- Olson M.F., Paterson,H.F. and Marshall,C.J. (1998) Signals from Ras and Rho GTPases interact to regulate expression of p21Waf1/Cip1. Nature, 394, 295–299. [DOI] [PubMed] [Google Scholar]
- Qiu R.G., Chen,J., Kirn,D., McCormick,F. and Symons,M. (1995a) An essential role for Rac in Ras transformation. Nature, 374, 457–459. [DOI] [PubMed] [Google Scholar]
- Qiu R.G., Chen,J., McCormick,F. and Symons,M. (1995b) A role for Rho in Ras transformation. Proc. Natl Acad. Sci. USA, 92, 11781–11785. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qiu R.G., Abo,A., McCormick,F. and Symons,M. (1997) Cdc42 regulates anchorage-independent growth and is necessary for Ras transformation. Mol. Cell. Biol., 17, 3449–3458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ren X.D., Kiosses,W.B. and Schwartz,M.A. (1999) Regulation of the small GTP-binding protein Rho by cell adhesion and the cytoskeleton. EMBO J., 18, 578–585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ridley A.J., Paterson,H.F., Johnston,C.L., Diekmann,D. and Hall,A. (1992) The small GTP-binding protein rac regulates growth factor-induced membrane ruffling. Cell, 70, 401–410. [DOI] [PubMed] [Google Scholar]
- Rodriguez-Viciana P., Warne,P.H., Khwaja,A., Marte,B.M., Pappin,D., Das,P., Waterfield,M.D., Ridley,A. and Downward,J. (1997) Role of phosphoinositide 3-OH kinase in cell transformation and control of the actin cytoskeleton by Ras. Cell, 89, 457–467. [DOI] [PubMed] [Google Scholar]
- Sahai E., Ishizaki,T., Narumiya,S. and Treisman,R. (1999) Transformation mediated by RhoA requires activity of ROCK kinases. Curr. Biol., 9, 136–145. [DOI] [PubMed] [Google Scholar]
- Sotiropoulos A., Gineitis,D., Copeland,J. and Treisman,R. (1999) Signal-regulated activation of serum response factor is mediated by changes in actin dynamics. Cell, 98, 159–169. [DOI] [PubMed] [Google Scholar]
- Suwa H., Ohshio,G., Imamura,T., Watanabe,G., Arii,S., Imamura,M., Narumiya,S., Hiai,H. and Fukumoto,M. (1998) Overexpression of the rhoC gene correlates with progression of ductal adenocarcinoma of the pancreas. Br. J. Cancer, 77, 147–152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tominaga T., Sahai,E., Chardin,P., McCormick,F., Courtneidge,S.A. and Alberts,A.S. (2000) Diaphanous-related formins bridge Rho GTPase and Src tyrosine kinase signaling. Mol. Cell, 5, 13–25. [DOI] [PubMed] [Google Scholar]
- Trahey M., Milley,R.J., Cole,G.E., Innis,M., Paterson,H., Marshall,C.J., Hall,A. and McCormick,F. (1987) Biochemical and biological properties of the human N-ras p21 protein. Mol. Cell. Biol., 7, 541–544. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tran Quang C., Gautreau,A., Arpin,M. and Treisman,R. (2000) Ezrin function is required for ROCK-mediated fibroblast transformation by the Net and Dbl oncogenes EMBO J., 19, 4565–4576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Watanabe N., Kato,T., Fujita,A., Ishizaki,T. and Narumiya,S. (1999) Cooperation between mDia1 and ROCK in Rho-induced actin reorganization. Nature Cell Biol., 1, 136–143. [DOI] [PubMed] [Google Scholar]
- Weber J.D., Hu,W., Jefcoat,S.C.,Jr, Raben,D.M. and Baldassare,J.J. (1997) Ras-stimulated extracellular signal-related kinase 1 and RhoA activities coordinate platelet-derived growth factor-induced G1 progression through the independent regulation of cyclin D1 and p27. J. Biol. Chem., 272, 32966–32971. [DOI] [PubMed] [Google Scholar]
- White M.A., Nicolette,C., Minden,A., Polverino,A., Van Aelst,L., Karin,M. and Wigler,M.H. (1995) Multiple Ras functions can contribute to mammalian cell transformation. Cell, 80, 533–541. [DOI] [PubMed] [Google Scholar]
- White M.A., Vale,T., Camonis,J.H., Schaefer,E. and Wigler,M.H. (1996) A role for the Ral guanine nucleotide dissociation stimulator in mediating Ras-induced transformation. J. Biol. Chem., 271, 16439–16442. [DOI] [PubMed] [Google Scholar]
- Woods D., Parry,D., Cherwinski,H., Bosch,E., Lees,E. and McMahon,M. (1997) Raf-induced proliferation or cell cycle arrest is determined by the level of Raf activity with arrest mediated by p21Cip1. Mol. Cell. Biol., 17, 5598–5611. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zondag G.C., Evers,E.E., ten Klooster,J.P., Janssen,L., van der Kammen,R.A. and Collard,J.G. (2000) Oncogenic Ras down regulates Rac activity, which leads to increased Rho activity and epithelial–mesenchymal transition. J. Cell Biol., 149, 775–782. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zuber J., Tchernitsa,O.I., Hinzmann,B., Schmitz,A.C., Grips,M., Hellriegel,M., Sers,C., Rosenthal,A. and Schafer,R. (2000) A genome-wide survey of RAS transformation targets. Nature Genet., 24, 144–152. [DOI] [PubMed] [Google Scholar]