

Abstract
Group II introns are catalytic RNAs that have been proposed to be the evolutionary precursors to the spliceosome. Most group II introns require accessory factors to splice efficiently in vivo, but few such factors have been identified. We have cloned the maize nuclear gene crs2, which is required for the splicing of nine group II introns in chloroplasts. CRS2 is related to peptidyl-tRNA hydrolase enzymes. However, CRS2 expression failed to rescue an Escherichia coli pthts mutant and CRS2 lacks several conserved amino acids that are important for the activity of the E.coli enzyme, indicating that it may lack peptidyl-tRNA hydrolase activity. CRS2 is localized to the chloroplast stroma, where it is found in a large salt-stable complex that contains RNA. CRS2 co-sediments with group II intron RNA during centrifugation of stroma through sucrose gradients, suggesting that CRS2 facilitates splicing via direct interaction with intron RNA. Sequence comparisons indicate how evolutionary tinkering may have allowed an enzyme that interacts with peptidyl-tRNAs to acquire a function in group II intron splicing.
Keywords: catalytic RNA/group II intron/plastid/RNA chaperone/splicing
Introduction
Group II introns, the largest of the known catalytic RNAs, are widely distributed in mitochondrial and chloroplast genomes, and are found less frequently in bacterial genomes (reviewed by Lambowitz et al., 1999). Some group II introns can self splice in vitro via two trans-esterification reactions that are identical to the chemical steps of spliceosomal splicing. However, this autocatalytic splicing activity is typically seen only in non-physiological, high salt conditions. It is not surprising, then, that most or all group II introns require accessory factors to splice efficiently in vivo. Some depend on a related group of proteins known as maturases, which are en coded in an open reading frame within the intron itself. However, most group II introns do not encode maturases. Although genetic data have implicated ‘non-maturase’ accessory proteins in the splicing of many group II introns (Seraphin et al., 1989; Goldschmidt-Clermont et al., 1990; Wiesenberger et al., 1992; Jenkins et al., 1997; Perron et al., 1999), little is known about how these facilitate the splicing reactions.
Land plant chloroplasts provide a rich source of group II intron targets for potential splicing factors. In contrast to fungal mitochondria and Chlamydomonas reinhardtii chloroplasts, which contain few group II introns, land plant chloroplasts contain ∼17 group II introns (the precise number varies between species). Previously, we described a nuclear gene in maize, crs2, which is required for the splicing of numerous group II introns in the chloroplast (Jenkins et al., 1997). Group II introns have been divided into two subgroups, IIA and IIB, based on several structural differences (Michel et al., 1989). crs2 has an intriguing specificity in that it is required for the splicing of nine of the ten chloroplast introns in subgroup IIB, but for none of the seven introns in subgroup IIA (Jenkins et al., 1997; Vogel et al., 1999). To elucidate how the crs2 gene product facilitates group IIB splicing, we have now cloned crs2. We found that crs2 encodes a protein with homology to a class of enzymes called peptidyl-tRNA hydrolases (PTHs) (Menninger, 1976). However, CRS2 does not complement an Escherichia coli pthts mutant and lacks several amino acid residues that are important for the activity of the E.coli enzyme, suggesting that it may not have PTH activity. CRS2 is localized to the stromal compartment of the chloroplast, where it is found in a large complex containing RNA and multiple polypeptides. This complex co-sediments in sucrose gradients with group IIB introns, suggesting that CRS2 participates directly in splicing by interacting with intron RNA.
Results
Molecular cloning of the crs2 gene
Two crs2 alleles had been recovered from maize lines with active Mutator (Mu) transposons: crs2-1 exhibits severe group IIB splicing defects, which lead to a loss of chloroplast ribosomes and a severe loss of leaf chlorophyll; crs2-2 has a weaker phenotype in that some splicing of crs2-dependent introns can be detected and mutant leaf accumulates some chlorophyll (Jenkins et al., 1997). To identify Mu insertions that are genetically linked to crs2-1, DNAs from distantly related homozygous mutant seedlings were analyzed by Southern hybridization using probes corresponding to each member of the Mu family. A Mu1 probe hybridized to a 3.8 kb BglII fragment present in all mutant seedlings but absent in all closely related homozygous wild-type plants (data not shown). This fragment was cloned from a size-selected genomic library generated from a BglII digest of crs2-1 mutant DNA. A map of the cloned fragment and the nucleotide sequence surrounding the Mu insertion site are shown in Figure 1. Southern and PCR analyses of the analogous genomic region in mutants carrying the crs2-2 allele (data not shown) revealed a Mu8 insertion in close proximity to the Mu1 insertion in crs2-1 (Figure 1).

Fig. 1. Organization of the cloned portion of the crs2 gene. (A) Map of the genomic BglII fragment containing Mu insertions that disrupt crs2. Dashed lines represent introns and heavy solid lines are exons. Transcription continues downstream of this region. The Mu8 insertion was accompanied by a deletion of 97 bp, as indicated. Primers 1 and 2 were used in PCRs with a Mu end primer to identify and map the Mu8 insertion site in crs2-2. (B) Nucleotide sequence of the crs2 gene in the region flanking the Mu insertion sites. The sequence shown begins 247 nt upstream of the start codon and ends within the first exon. The location of the Mu1 insertion in crs2-1 is shown by a box marking the 9 bp duplicated upon insertion. The insertion site of the Mu8 element in crs2-2 is indicated, with brackets showing the 97 bp that are deleted in this allele. The 5′ end of the 5′ RACE product is indicated (5′ end cDNA) and the predicted start codon is printed in bold. The complete nucleotide sequence of the crs2 cDNA has been deposited in DDBJ/EMBL/GenBank, accession No. AAF225708.
A probe containing sequences flanking the cloned Mu1 insertion detected a 1.5 kb mRNA on RNA gel blots (data not shown). A full-length cDNA (1544 bp) corresponding to this genomic region contains an open reading frame of 256 codons, a 5′ untranslated region (UTR) of 103 bp and a 3′ UTR of 700 bp. The Mu1 insertion in crs2-1 lies 9 bp upstream of the putative start codon (Figure 1); the Mu8 insertion in crs2-2 maps 29 bp upstream of the putative start codon and is flanked by a 97 bp deletion (Figure 1). The accumulation of the corresponding mRNA is severely reduced in both crs2-1 and crs2-2 mutants (Figure 2A). Antiserum raised against a recombinant antigen generated from the cDNA detected a leaf protein that was found at reduced levels in crs2-2 mutants and was undetectable in crs2-1 mutants (Figure 2B). The fact that two independent mutant alleles have Mu insertions disrupting the cloned gene and that the abundance of gene product correlates with the severity of the mutant phenotype indicates that the cloned cDNA represents the crs2 mRNA.

Fig. 2. Expression of the crs2 gene is disrupted in crs2-1 and crs2-2 mutants. (A) RNase-protection assay of crs2 mRNA. The probe for the crs2 mRNA was derived by in vitro transcription of the crs2 genomic sequence to the ‘right’ of the Mu1 insertion in Figure 1A. A probe complementary to a portion of the maize actin mRNA was included as an internal control. Thirty micrograms of total leaf RNA from wild-type, crs2-1, crs2-2 or w1 seedlings, or an equivalent amount of tRNA were analyzed. w1, an ivory maize mutant with a global and tight defect in chloroplast gene expression, was included to control for any effects on crs2 RNA accumulation that might be associated with defects in chloroplast development. Because the crs2 probe included intron sequence, the 253 nt fragment protected by wild-type mRNA is considerably smaller than the probe. The undigested probes (lanes 2 and 3) correspond to 1/200th as much probe as was included in each reaction. (B) Immunoblot showing CRS2 protein levels in crs2 mutants. Twenty micrograms of total leaf protein (or the indicated dilutions) from wild-type, crs2-1, crs2-2 or w1 seedlings were fractionated by SDS–PAGE. Upper panel: immunoblot probed with a CRS2 antibody. Lower panel: the same filter stained with Ponceau S to illustrate equal loading of the protein.
CRS2 is localized to the chloroplast stroma
The phenotype of crs2 mutants and the fact that the CRS2 N-terminus has features of a chloroplast-targeting sequence (overall basic charge and rich in hydroxylated amino acids) suggested that CRS2 is a chloroplast protein. Indeed, the crs2 gene product can be imported into isolated pea chloroplasts (Figure 3A); its size after import is consistent with the removal of a transit peptide of ∼5 kDa. Furthermore, immunoblots probed with CRS2 antiserum demonstrated that CRS2 is enriched in isolated chloro-plasts with respect to its concentration in total leaf extracts (Figure 3B). Immunoblots of chloroplast subfractions (Figure 3C) showed that CRS2 is found predominantly in the chloroplast stroma. A small proportion may also be bound to the thylakoid membrane.

Fig. 3. CRS2 is localized to the chloroplast stroma. (A) Import of the crs2 gene product into isolated pea chloroplasts. Radiolabeled CRS2 was generated by in vitro translation of the full-length crs2 cDNA in the presence of [35S]methionine (TP-translation product). After a 30 min incubation with intact pea chloroplasts, aliquots were further incubated without (–) or with (+) thermolysin to degrade proteins external to the chloroplast. Intact chloroplasts were then repurified and the proteins were fractionated by SDS–PAGE and visualized by autoradiography. (B) CRS2 is enriched in the chloroplast fraction of leaf tissue. Ten micrograms of total leaf protein or 10 µg of protein from purified intact maize chloroplasts (CPS) were fractionated by SDS–PAGE and analyzed on an immunoblot. The blot was sequentially probed with antibodies to CRS2, the chloroplast protein CRP1 (Fisk et al., 1999), the mitochondrial protein malate dehydrogenase (MDH) and the cytosolic protein phosphoenolpyruvate carboxylase (PEPCase). (C) CRS2 is localized to the chloroplast stroma. Purified chloroplasts from wild-type leaves (CPS) were fractionated into stroma, thylakoid (thy) and inner envelope (IE) fractions. The same proportion of each fraction was separated by SDS–PAGE and analyzed on an immunoblot. The blot was probed sequentially with antibodies to CRS2, the stromal protein CRP1 (Fisk et al., 1999), the thylakoid lumen protein OE33 and the inner envelope protein IM35.
CRS2 is closely related to PTHs but may lack PTH activity
The predicted crs2 gene product is closely related to a class of enzymes called PTHs (Figure 4). PTHs cleave the ester bond linking the tRNA and nascent peptide when peptidyl-tRNAs are released prematurely from the ribosome (Menninger, 1976). A loss of PTH function is lethal in E.coli because it leads to the depletion of certain free tRNAs (Hergue-Harnard et al., 1996). A predicted Arabidopsis protein exhibits a high degree of identity with CRS2 and likely represents the CRS2 ortholog (Figure 4). These two plant proteins have an N-terminal extension when aligned with bacterial PTHs. These extensions are predicted by the TargetP algorithm (Emanuelsson et al., 1999) to be chloroplast transit peptides, and their size is consistent with that of the cleaved CRS2 transit peptide after import into isolated chloroplasts (Figure 3A).

Fig. 4. Multiple sequence alignment of CRS2 with related Arabidopsis and bacterial proteins. The alignment includes sequences of the putative CRS2 ortholog in Arabidopsis (DDBJ/EMBL/GenBank accession No. BAB09443) and three bacterial PTHs (E.coli, accession No. P23932; Bacillus subtilis, accession No. P37470; Synechocystis PCC6803, accession No. Q59989). Solid circles indicate residues shown to be essential for the catalytic activity of the E.coli PTH (Schmitt et al., 1997; Fromant et al., 1999). Open squares indicate residues important for the binding of substrate by the E.coli enzyme (Fromant et al., 1999). The gray bar highlights a putative RNA-binding region that is found in predicted CRS2 proteins in plants, but not in their PTH ancestor. The lower box shows an alignment of this region of predicted plant CRS2s with a region of the yeast protein Cbp2 that has been implicated in binding group I introns and in facilitating group I intron splicing (Tirupati et al., 1999). The C-terminal sequences of the presumed CRS2 orthologs in iceplant (accession No. AI026363) and aspen (accession No. AI162403) were derived from EST sequences. Sequence alignments were calculated using ClustalW 1.8 (Thompson et al., 1994) and Boxshade (Bioinformatics group, ISREC, Lausanne, Switzerland), with default parameters.
All amino acid residues that are known to be required for the catalytic activity of the E.coli PTH (Schmitt et al., 1997; Fromant et al., 1999) are conserved in CRS2 (Figure 4, filled circles), suggesting that CRS2 may have PTH activity. If CRS2 retained PTH activity, it seemed likely that CRS2 expression would complement an E.coli pthts mutant, since the E.coli enzyme functions as a monomer (Koessel, 1969) and tRNA structures are highly conserved between plastids and E.coli. To test this possibility, a CRS2 expression construct encoding the mature form of CRS2 (i.e. lacking its chloroplast-targeting sequence), or an analogous E.coli PTH expression construct, was introduced into E.coli pthts cells. The E.coli PTH expression construct restored growth of the pthts cells at the non-permissive temperature as expected (Figure 5A, filled squares). However, CRS2 expression (Figure 5B) failed to complement the mutation (Figure 5A, filled diamonds), suggesting that CRS2 lacks PTH activity. In fact, two amino acid residues that are essential for substrate binding by the E.coli PTH (Fromant et al., 1999) and are highly conserved among bacterial PTHs have not been maintained in the plant CRS2s (open squares in Figure 4). When these residues were mutated to alanine in the E.coli enzyme, its Km increased dramatically and PTH activity was reduced 100-fold (Fromant et al., 1999). Even a conservative R134H mutation reduced E.coli PTH activity 10-fold (Garcia-Villegas et al., 1991). Since CRS2, its Arabidopsis ortholog and all other available plant CRS2 sequences have non-conservative changes at both positions, they may be unable to bind peptidyl-tRNA.
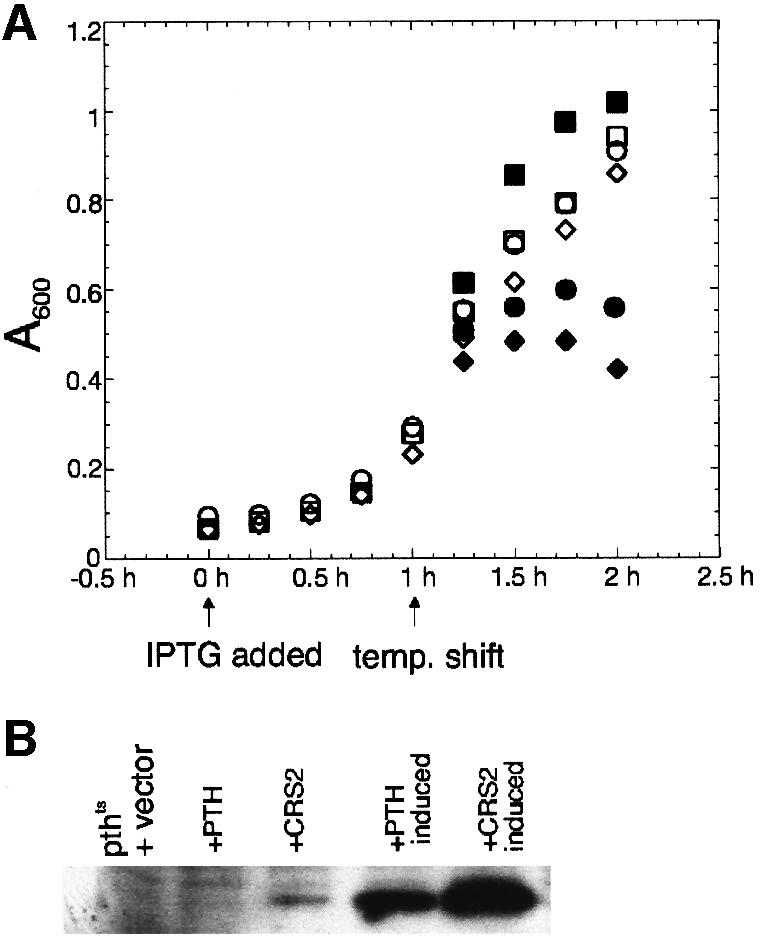
Fig. 5. CRS2 fails to complement an E.coli pthts mutant. (A) The pthts strain was transformed with either a clone expressing E.coli PTH (squares), the mature form of CRS2 (diamonds) or empty vector (circles), and grown at the permissive temperature of 30°C (open symbols). After reaching A600 = 0.1, 8 µM IPTG was added to induce protein expression from the plasmids (t = 0). After an additional 1 h of growth at 30°C, one half of each culture was shifted to the non-permissive temperature of 42°C (filled symbols). Cells were grown for one additional hour at both temperatures and A600 was measured every 15 min. Growth of the uninduced strains containing the PTH or CRS2 expression plasmid was monitored under the same temperature shift regime. The growth curves were superimposable with those for the corresponding induced cultures, indicating that even very low level expression of the E.coli PTH was sufficient for rescue, and that low level expression of CRS2 did not permit rescue (not shown). (B) Immunoblot showing CRS2 and PTH levels in the transformed E.coli pthts strain. Cells were harvested at t = 1 h from cultures treated with IPTG or from uninduced control cultures. Total proteins were prepared by resuspending pelleted cells from 1 ml of culture in 100 µl of SDS–PAGE sample buffer. Samples were heated to 90°C for 5 min and 10 µl of each were analyzed on an immunoblot by probing with the CRS2 antibody. The CRS2 antibody cross-reacts with the PTH protein. +PTH, cultures containing the PTH expression plasmid; +CRS2, cultures containing the CRS2 expression plasmid.
CRS2 is found in a salt-stable ribonucleoprotein complex in the chloroplast stroma
CRS2 did not promote the splicing of its target introns when they were co-expressed in E.coli (data not shown), suggesting that CRS2 is not sufficient to activate splicing. To assess whether other factors associate with CRS2 in vivo, the size of native CRS2 in stromal extracts was determined. Following sucrose gradient sedimentation of crude stroma, essentially all of the CRS2 was found in a peak that sediments more rapidly than Rubisco (550 kDa) and more slowly than ribosomes (Figure 6A, C and D). The size of the CRS2 complex (or complexes) was estimated to be 500–800 kDa by size exclusion chroma-tography (Figure 7A). The complex is stable in KOAc concentrations between 60 and 400 mM (data not shown), supporting the notion that it is physiologically relevant. Treatment of stroma with micrococcal nuclease (MNase) reduced the size of the CRS2 complex to ∼70 kDa (Figures 6B and 7C), whereas treatment with DNase had no detectable effect (Figure 7B). Therefore, CRS2 is found in a salt-stable complex that contains RNA.

Fig. 6. CRS2 is in a high molecular weight complex that contains nucleic acid. Stromal extracts were sedimented through sucrose gradients after treatment with micrococcal nuclease [+MNase (B)] or after incubation without MNase using identical conditions (A). The gradients were divided into 35 fractions. An immunoblot containing 13% of each fraction was probed with the CRS2 antibody. Twenty micrograms of stromal protein (str) were included on the immunoblots as a reference. CRS2 was undetectable in fractions 23–35 (not shown). After MNase treatment, a small proportion of CRS2 co-sediments with ribosomes, possibly due to non-specific binding. (C) Results of re-probing the blot shown in (B) with an antibody that detects the large subunit of Rubisco (RbcL); reprobing of the blot in (A) gave the same profile (not shown). (D) A260 profile of the +MNase gradient illustrating that ribosomes peak in fractions 18–21. The A260 profile of the –MNase gradient was similar, except that there was substantially less absorbance in fractions 1–5 (data not shown).

Fig. 7. Size exclusion chromatography of the native, DNase-treated and MNase-treated CRS2 complex. One microgram of stromal protein (A), stromal protein that had been treated with DNase I (B) or stromal protein that had been treated with MNase (C) was fractionated on a Superdex 200 gel filtration column. Column fractions were assayed for CRS2 by probing immunoblots with the CRS2 antibody. Twenty micrograms of stroma (str) were included for reference. The sizes of proteins in each fraction were calculated by comparison with molecular weight standards, and are indicated above each lane (in kDa). In (A) and (B), CRS2 was undetectable in fractions containing proteins <220 kDa. In (C), fractions 1–29, containing proteins >150 kDa, did not contain significant amounts of CRS2.
The native CRS2 complex co-sediments with intron RNA
Because CRS2 facilitates group II intron splicing, it seemed plausible that the RNA component of the CRS2 complex includes group II intron sequences. To address this possibility, the sedimentation of intron RNA was compared with that of CRS2 during centrifugation of stroma through sucrose gradients (Figure 8). CRS2 co-sediments with intron RNAs from two genes (petB and ndhB) whose splicing is crs2 dependent. In contrast, intron RNA from the atpF gene, whose splicing is crs2 independent, sediments more rapidly. The gel mobility of these intron RNAs indicates that they correspond in size to excised intron. However, it is plausible that they are derived from the partial degradation of pre-mRNAs down to nuclease-resistant intron ‘cores’ during preparation of the stromal extracts. In either case, these results support the notion that CRS2 binds with specificity to its target introns.
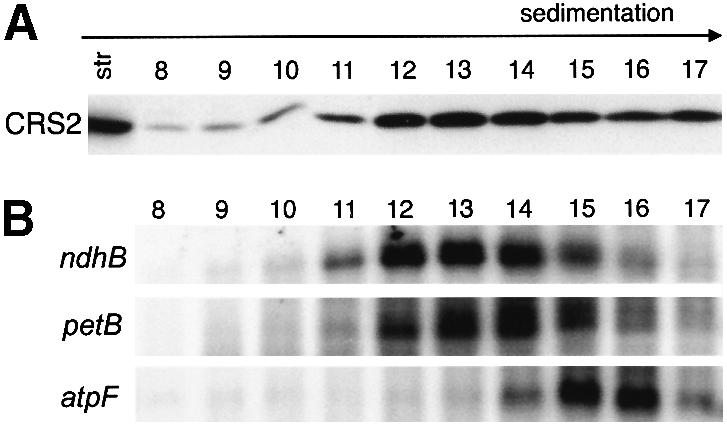
Fig. 8. CRS2 co-sediments with group II introns during sucrose gradient centrifugation of chloroplast stroma. (A) Immunoblot showing the distribution of CRS2 in sucrose gradient fractions. Stromal extract (2 mg protein) was sedimented through a sucrose gradient. Protein from each gradient fraction was analyzed on immunoblots by probing with the CRS2 antibody. Twenty micrograms of stromal protein (str) were included for reference. (B) RNA gel blots showing distribution of intron RNA in sucrose gradient fractions. RNA gel blots of RNA purified from the same gradient fractions analyzed in (A) were hybridized with radiolabeled probes specific for ndhB, petB or atpF intron. The remaining fractions (1–7 and 18–30) did not contain significant amounts of CRS2 or intron RNA (not shown). The intron RNAs detected migrated with RNA markers of 700–800 nt, the size of the excised introns. Although unspliced pre-mRNAs accumulate to high levels in chloroplasts (Jenkins et al., 1997), no other transcripts were detected with these probes in any gradient fraction (data not shown), presumably due to partial degradation of unspliced mRNA precursors during this experimental protocol.
Discussion
Our results show that CRS2, a nucleus-encoded protein required for the splicing of group II introns in maize chloroplasts, is a PTH homolog. CRS2 is bound to an RNA-containing complex in the chloroplast stroma that likely contains its target introns. These findings, together with the genetic data showing that CRS2 is required specifically for subgroup IIB splicing (Jenkins et al., 1997; Vogel et al., 1999), suggest that CRS2 facilitates splicing directly, via interaction with intron RNA.
Other than the intron-encoded maturase protein family, just two proteins that function specifically in the splicing of group II introns were described previously: MRS2 in yeast and MAA2 in C.reinhardtii. Mutations in the yeast MRS2 gene disrupt group II intron splicing in mitochondria (Wiesenberger et al., 1992), but recent data suggest that the splicing defects are indirect, resulting from altered Mg2+ homeostasis in the mutant mitochondria (Bui et al., 1999). Mutations in the C.reinhardtii nuclear gene Maa2 disrupt the trans-splicing of a split group II intron in the chloroplast psaA gene (Goldschmidt-Clermont et al., 1990; Perron et al., 1999). How MAA2 participates in splicing has not been established.
CRS2 joins the list of factors that were recruited from enzymes that interact with tRNA to facilitate the splicing of self-splicing introns. Neurospora CYT18 is a mitochondrial tyrosyl-tRNA synthetase that facilitates the splicing of group I introns (Lambowitz et al., 1999), and MAA2 in C.reinhardtii chloroplasts has homology to pseudouridine synthases with tRNA substrates (Perron et al., 1999). Mutagenesis of conserved active site residues in MAA2 did not disrupt its ability to promote splicing, suggesting that it may not require pseudouridine synthase activity for its trans-splicing function (Perron et al., 1999). Similarly, our results suggest that the ancestral PTH activity of CRS2 may not be required for its splicing activity and, indeed, that this activity may have been lost.
Comparison of plant CRS2s with bacterial PTH sequences may reveal changes that allowed CRS2 to acquire a new function as a facilitator of group IIB intron splicing. A suggestive feature is the C-terminal extension found in plant CRS2s (gray bar, Figure 4), which is composed of alternating basic and aromatic residues. Among the four plant species for which CRS2 C-terminal sequence is available, there is some variation within this extension, but all variants maintain the pattern of basic and aromatic residues (box, Figure 4). This extension resembles a six-amino-acid motif (RYRYKF) in the group I intron splicing factor Cbp2, which contacts intron RNA and is essential for Cbp2 to promote splicing (Tirupati et al., 1999) (box, Figure 4). We hypothesize that the acquisition of this C-terminal extension was an essential step in the evolution of CRS2 from its PTH ancestor. This may be analogous to the acquisition of an N-terminal domain in CYT18, which is essential for its group I splicing activity and is not found in bacterial or yeast tyrosyl-tRNA synthetases (reviewed in Lambowitz et al., 1999). It should be noted that two types of PTH homolog can be identified from maize and Arabidopsis genome sequences. The putative CRS2 orthologs are more similar to one another than they are to the second PTH gene in each genome, and include the C-terminal extension discussed above. The second type of PTH in each species lacks the C-terminal extension and may function as a bona fide PTH in one or more cellular compartment.
Complexity of the CRS2 particle in the chloroplast stroma
During size exclusion chromatography of MNase-treated stroma, CRS2 elutes with proteins of ∼70 kDa (Figure 7C), despite the fact that the CRS2 monomer is only 23 kDa. It is possible that the 70 kDa CRS2 complex contains an RNA fragment bound to a CRS2 monomer. Alternatively, CRS2 may be found in a proteinaceous homo- or hetero-multimer. The native CRS2 complex (>600 kDa) is significantly larger than the sum of the masses of excised intron (∼260 kDa) and the MNase-resistant CRS2 complex (∼70 kDa). Therefore, other proteins (or RNAs) are likely to be bound to the complex in such a way that their association with CRS2 is not maintained after MNase treatment. Among these may be factors that are involved in splicing, recycling splicing components by dissociating the complex, or intron degradation.
These biochemical findings are in accord with genetic data suggesting that chloroplast group II introns may generally require interactions with multiple proteins to splice efficiently in vivo. Splicing of the group IIA intron in the maize chloroplast atpF gene requires, at a minimum, the nuclear gene product CRS1 and also a chloroplast gene product, likely the putative maturase MatK (Jenkins et al., 1997). Analogously, the products of multiple nuclear genes participate in the trans-splicing of each of the two group II introns in the C.reinhardtii chloroplast psaA gene (Choquet et al., 1988; Goldschmidt-Clermont et al., 1990). Evidence presented here suggests that CRS2 may also interact with other proteins while carrying out its splicing function. It is possible that multisubunit particles akin to a mini-spliceosome may typically be involved in group II intron splicing, in contrast to the relatively simple pictures drawn from studies of the less intricately structured introns in group I.
Role of CRS2 in group II intron splicing
The mechanism by which proteins increase the activity of self-splicing introns has been studied for only three proteins: CYT18 and Cbp2, which promote group I intron splicing, and the bacterial LtrA maturase, which promotes the splicing of the group II intron in which it is encoded (reviewed in Weeks, 1997; Lambowitz et al., 1999). In all three cases, the purified recombinant protein is sufficient to promote splicing and binds with high affinity and specificity to intron RNA. The broad picture that has emerged is that these proteins facilitate splicing by aiding intron folding, either by capturing and stabilizing a folding intermediate or by stabilizing interactions within the folded intron. It is possible that CRS2 likewise facilitates the folding of group IIB introns. It is also plausible, however, that other proteins in the CRS2 complex promote intron folding and that CRS2 participates more directly in splicing chemistry. Unlike the CYT18-, Cbp2- and LtrB-dependent introns, there is no evidence that CRS2-dependent introns can self-splice, even in the presence of high salt. The conservation in CRS2 of PTH active site residues (most notably H20) is interesting in this regard, and raises the intriguing possibility that the protein and RNA cooperate during catalysis, with each providing functional groups that promote different steps of the reaction.
Materials and methods
Plant growth and genetic analyses
The crs2-2 allele was previously called crs* and the crs2-1 allele was previously called crs2 (Jenkins et al., 1997). crs2-2 was tested for its ability to complement crs2-1 by intercrossing heterozygous plants. Each of 10 crosses involving different pairs of heterozygous plants yielded ∼1/4 chlorophyll-deficient mutants with defects in the splicing of crs2-dependent introns (data not shown). Therefore, the two mutations fail to complement. Seedlings used for protein, RNA and DNA extraction were grown in a growth chamber (16 h days at 28°C and 400 µE/m2/s; 8 h nights at 25°C) and harvested at 10–12 days after planting, when seedlings had between two and three leaves.
Nucleic acid extraction and analysis
Genomic DNA was extracted from seedlings and analyzed on Southern blots by probing with digoxigenin-labeled Mu probes, as described previously (Voelker et al., 1997). RNA was extracted from seedling leaf with TRIzol reagent (Gibco-BRL) according to the manufacturer’s instructions. RNase protection analyses were performed as previously described (Barkan et al., 1994).
Molecular cloning methods
To clone the genomic 3.8 kb BglII fragment containing a Mu1 insertion linked to crs2, 28 µg of DNA from a homozygous crs2-1 mutant were digested with 70 U of BglII and fractionated in a 0.8% agarose gel. A gel slice containing DNA fragments of 3.6–3.9 kb was excised, and DNA was extracted using the Qiaex II gel extraction kit, following the manufacturer’s instructions (Qiagen). One hundred nanograms of the purified DNA were ligated into 1 µg of Zap Express BamHI CIAP-treated phage arms (Stratagene), packaged and used to infect XL1-Blue MRF′ cells. Plaques (4 × 105) were screened by probing lifts with a radioactive Mu1 probe. This led to the identification of the crs2 genomic clone shown in Figure 1A.
Genomic Southern blots of crs2-2 mutant DNA with a crs2 probe revealed an insertion within the same BglII fragment. The site and identity of this insertion in the crs2-2 allele were determined by PCR amplification of crs2-2 DNA with a Mu primer that reads outward from the terminal inverted repeat of all Mu elements (AGAGAAGCCAACGCCAWCGCCTCYATTTCGTC) in conjunction with crs2-specific primers (primer 1, CACAATCGAATATTGACGGTAACA; or primer 2, TACCAATTCCCAGAAGCGAC; see Figure 1). The Mu primer and primer 1 generated a fragment of 211 bp. The Mu primer and primer 2 generated a fragment of 541 bp. These were cloned into Bluescript SK+ (Stratagene) and their DNA sequence determined. Polymorphisms within the amplified portion of the Mu terminal inverted repeat identified the insertion in crs2-2 as a Mu8 element.
A maize seedling leaf cDNA library (inbred line B73, Pioneer Hi-Bred) (Fisk et al., 1999) was screened with a radiolabeled probe generated from the genomic clone (Figure 1A), yielding a 1378 bp cDNA. Additional 5′ sequence was obtained by 5′ RACE as described in Fisk et al. (1999), using primer 2 (Figure 1) for first strand cDNA synthesis. The 167 bp 5′ RACE product was cloned in-frame with the cDNA obtained from the library to reconstruct the full-length cDNA. DNA sequence analysis was performed by Yanling Wang in the Institute of Molecular Biology DNA Sequencing Facility. The nucleotide sequence of the full-length crs2 cDNA has been deposited in DDBJ/EMBL/GenBank (accession No. AAF225708).
Preparation and analysis of proteins
Methods for the extraction of total leaf protein, SDS–PAGE and immunoblot analysis are described in Barkan (1998). Chloroplasts were purified from the leaves of 10-day-old seedlings (Barkan, 1998) and fractionated into stromal, thylakoid and envelope fractions as described in Fisk et al. (1999). Chloroplast import assays were performed as in Fisk et al. (1999). Protein for import reactions was generated from a clone containing the entire crs2 open reading frame ligated into the pGEM4Z vector (Promega). Radiolabeled CRS2 was synthesized in vitro with the Novagen STP3 coupled transcription–translation kit, and 2 × 105 c.p.m. were used per import reaction.
To prepare stroma, intact chloroplasts were burst by osmotic lysis in Ex buffer [30 mM HEPES–KOH pH 8, 60 mM KOAc, 10 mM Mg(OAc)2, 2 mM dithiothreitol (DTT)] containing a protease inhibitor cocktail (2 µg/ml aprotinin, 2 µg/ml leupeptin, 1 µg/ml pepstatin, 1 mM phenylmethylsulfonyl fluoride) with repeated vortexing and incubation on ice for 30 min. Membranes were pelleted from lysed plastids by centrifugation at 29 000 r.p.m. for 30 min in a TLA 100.2 rotor in a Beckman table-top ultracentrifuge. The supernatant (the stromal fraction) was collected and either used immediately or stored at –80°C.
Sucrose gradient sedimentation analysis
To test the effect of MNase on the CRS2 complex, 500 µl of Ex buffer containing 2 mg of stromal protein were adjusted to 10 mM DTT and 5 mM CaCl2. Forty units of MNase (Sigma) were added, the sample was incubated for 10 min at 22°C, and the reaction was stopped by the addition of 10 mM EGTA. The untreated stroma analyzed in Figure 6 was treated identically except MNase was not added. The KOAc concentration of both samples was then adjusted to 200 mM. Two hundred and fifty microliter samples were layered on sucrose gradients (10–40% sucrose prepared in Ex buffer containing 200 mM KOAc) and centrifuged in a Beckman SW55.1 rotor at 48 000 r.p.m. for 80 min at 4°C. Fractions of 150 µl were collected.
Stroma analyzed in Figure 8 was prepared as described above except that 0.4 U/µl rRNasin (Promega) was added to the Ex buffer during chloroplast lysis. Stroma (2 mg protein in 250 µl) was layered on 10–40% linear sucrose gradients prepared in Ex buffer containing 200 mM KOAc. Gradients were centrifuged in a Beckman SW55.1 rotor at 48 000 r.p.m. for 3 h and 10 min at 4°C. RNA was extracted from 130 µl of each fraction with 0.75 ml of TRIzol Reagent (Gibco-BRL), following the manufacturer’s instructions. One quarter of each RNA sample was analyzed by RNA gel blot hybridization. Intron-specific probes were generated by PCR amplification of leaf DNA using primers corresponding to the intron termini. Probes were radiolabeled by random hexamer priming in the presence of [32P]dCTP.
Size exclusion chromatography
A Pharmacia Superdex 200 HR 10/30 column was equilibrated at 4°C in at least three column volumes of Kex buffer [30 mM HEPES–KOH pH 8, 200 mM KOAc, 10 mM Mg(OAc)2, 2 mM DTT]. The KOAc concentration in the stromal sample (1 mg protein in 300 µl) was adjusted to 200 mM. The sample was centrifuged in a microfuge at 15 000 r.p.m. for 15 min at 4°C to pellet insoluble material, and was filtered (S & S Uniflo-13, 0.2 CA). Two hundred and fifty microliters of the filtrate were loaded onto the column and fractions were eluted in Kex buffer at a rate of 0.4 ml/min; 200 µl fractions were collected. The molecular weight range of each fraction was calculated by comparison with elution profiles of size standards included in Pharmacia’s high and low molecular weight gel filtration calibration kits. Protein in each fraction was precipitated by the addition of 1 ml of ice-cold ethanol, overnight incubation at –20°C, and centrifugation at 3000 g for 15 min. Pellets were resuspended in 1× SDS sample buffer prior to SDS–PAGE and immunoblot analysis with CRS2 antibody.
To test whether the CRS2 complex contains DNA (Figure 7), 1 mg of stromal protein in 300 µl of Ex buffer was incubated with 2600 U of DNase I (Gibco-BRL) at 22°C for 15 min. The KOAc concentration was then adjusted to 200 mM and the sample applied to the gel filtration column. To assay the size of the CRS2 complex after MNase treatment, 1 mg of stromal protein (250 µl), nuclease treated as described above, was analyzed by chromatography on a Superdex 200 column.
Antisera
A recombinant CRS2 antigen was generated by ligating a crs2 cDNA fragment encoding amino acids 33–256 (Figure 4) into the Pet 28b+ vector (Novagen), yielding an in-frame translation product with an N-terminal His6 tag. The fusion protein was purified on a nickel column and injected into rabbits for the production of polyclonal antisera. Antisera were generated by the University of Oregon antibody facility.
Antibodies to CRP1, OE33 and AtpB were described previously (Fisk et al., 1999; McCormac and Barkan, 1999). The following antibodies were kindly donated: the MDH antibody by Kathy Newton (University of Missouri), the PEP carboxylase antibody by William Taylor (CSIRO, Canberra) and the IM35 antibody by Danny Schnell (Rutgers University).
Test for complementation of an E.coli pthts mutant
A CRS2 expression cassette was generated by PCR amplification of a segment of the crs2 cDNA clone, using primers containing a 5′ NcoI site (CRS2N, GGCCCATGGAATACACGCCCT) and a 3′ BamHI site (CRS2B, CCGGATCCTCAAACATTCAAC). This insert was cloned into the NcoI and BamHI sites of pQE 60 (Qiagen). The expressed CRS2 was designed to mimic E.coli PTH at its N-terminus, containing the initiating methionine followed by residues 57–256 of CRS2 (Figure 4); no residues were added from translation of vector sequence. An E.coli PTH expression cassette was generated by PCR of E.coli DNA using primers containing a 5′ NspI site (PTHN, CAAAAAAACATGTCGATTAAATTG) and a 3′ BamHI site (PTHB, CCGGATCCGCAGACAACGACTTA). The product was cloned into the SphI and BamHI sites of pQE 70 (Qiagen). The expressed E.coli protein included the entire PTH sequence with no additional residues. The pthts strain MF100 was generously provided by John Menninger (University of Iowa). To minimize expression of CRS2 and PTH in the uninduced cells, MF100 was initially transformed with the pREP4 plasmid (Qiagen), which expresses the lac repressor. The pthts strain was then transformed with either the CRS2 expression plasmid, PTH expression plasmid or pQE 70 without insert.
Acknowledgments
Acknowledgements
The authors would like to thank Roz Carrier and Macie Walker for expert technical assistance; Dianna Fisk for providing chloroplast samples and for assistance with chromatography; Gerry Ostheimer and members of the Barkan laboratory for helpful discussions; and Roz Carrier, Brad Till, Gerry Ostheimer, John Davenport, Kenny Watkins and Bea Darimont for providing helpful comments on the manuscript. This work was supported by NSF grant MCB-9904666 to A.B. and NIH Training Grant GM 07413 (B.D.J).
REFERENCES
- Barkan A. (1998) Approaches to investigating nuclear genes that function in chloroplast biogenesis in land plants. Methods Enzymol., 297, 38–57. [Google Scholar]
- Barkan A., Walker,M., Nolasco,M. and Johnson,D. (1994) A nuclear mutation in maize blocks the processing and translation of several chloroplast mRNAs and provides evidence for the differential translation of alternative mRNA forms. EMBO J., 13, 3170–3181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bui D., Gregan,J., Jarosch,E., Ragnini,A. and Schweyen,R. (1999) The bacterial magnesium transporter CorA can functionally substitute for its putative homologue Mrs2p in the yeast inner mitochondrial membrane. J. Biol. Chem., 274, 20438–20443. [DOI] [PubMed] [Google Scholar]
- Choquet Y., Goldschmidt-Clermont,M., Girard-Bascou,J., Kueck,U., Bennoun,P. and Rochaix,J.-D. (1988) Mutant phenotypes support a trans-splicing mechanism for the expression of the tripartite psaA gene in the C. reinhardtii chloroplast. Cell, 52, 903–913. [DOI] [PubMed] [Google Scholar]
- Emanuelsson O., Nielsen,H. and von Heijne,G. (1999) ChloroP, a neural network-based method for predicting chloroplast transit peptides and their cleavage sites. Protein Sci., 8, 978–984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fisk D.G., Walker,M.B. and Barkan,A. (1999) Molecular cloning of the maize gene crp1 reveals similarity between regulators of mitochondrial and chloroplast gene expression. EMBO J., 18, 2621–2630. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fromant M., Plateau,P., Schmitt,E., Mechulam,Y. and Blanquet,S. (1999) Receptor site for the 5′-phosphate of elongator tRNAs governs substrate selection by peptidyl-tRNA hydrolase. Biochemistry, 38, 4982–4987. [DOI] [PubMed] [Google Scholar]
- Garcia-Villegas M., Vega,F.D.L., Galindo,J., Segura,M., Buckingham,R. and Guarneros,G. (1991) Peptidyl-tRNA hydrolase is involved in lambda inhibition of host protein synthesis. EMBO J., 10, 3549–3555. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goldschmidt-Clermont M., Girard-Bascou,J., Choquet,Y. and Rochaix,J.-D. (1990) Trans-splicing mutants of Chlamydomonas reinhardtii. Mol. Gen. Genet., 223, 417–425. [DOI] [PubMed] [Google Scholar]
- Hergue-Harnard V., Mora,L., Guarneros,G. and Buckingham,R. (1996) The growth defect in Escherichia coli deficient in peptidyl-tRNA hydrolase is due to starvation for Lys-tRNAlys. EMBO J., 15, 2826–2833. [PMC free article] [PubMed] [Google Scholar]
- Jenkins B.D., Kulhanek,D.J. and Barkan,A. (1997) Nuclear mutations that block group II RNA splicing in maize chloroplasts reveal several intron classes with distinct requirements for splicing factors. Plant Cell, 9, 283–296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koessel H. (1970) Purification and properties of peptidyl-tRNA hydrolase from Escherichia coli. Biochim. Biophys. Acta, 204, 191–202. [DOI] [PubMed] [Google Scholar]
- Lambowitz A., Caprara,M., Zimmerly,S. and Perlman,P. (1999) Group I and group II ribozymes as RNPs: Clues to the past and guides to the future. In Gesteland,R., Cech,T. and Atkins,J. (eds), The RNA World. Cold Spring Harbor Laboratory Press, Cold Spring Harbor, NY, pp. 451–485.
- McCormac D. and Barkan,A. (1999) A nuclear gene in maize required for the translation of the chloroplast atpB/E mRNA. Plant Cell, 11, 1709–1716. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Menninger J. (1976) Peptidyl-transfer RNA dissociates during protein synthesis from ribosomes of Escherichia coli.J. Biol. Chem., 251, 3392–3398. [PubMed] [Google Scholar]
- Michel F., Umesono,K. and Ozeki,H. (1989) Comparative and functional anatomy of group II catalytic introns—a review. Gene, 82, 5–30. [DOI] [PubMed] [Google Scholar]
- Perron K., Goldschmidt-Clermont,M. and Rochaix,J.-D. (1999) A factor related to pseudouridine synthases is required for chloroplast group II intron trans-splicing in Chlamydomonas reinhardtii. EMBO J., 18, 6481–6490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmitt E., Mechulam,Y., Fromant,M., Plateau,P. and Blanquet,S. (1997) Crystal structure at 1.2 Å resolution and active site mapping of Escherichia coli peptidyl-tRNA hydrolase. EMBO J., 16, 4760–4769. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seraphin B., Simon,M., Boulet,A. and Faye,G. (1989) Mitochondrial splicing requires a protein from a novel helicase family. Nature, 337, 84–87. [DOI] [PubMed] [Google Scholar]
- Thompson J., Higgins,D. and Gibson,T. (1994) Clustal W: improving the sensitivity of progressive multiple sequence alignment through sequence weighting, position-specific gap penalties and weight matrix choice. Nucleic Acids Res., 22, 4673–4680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tirupati H., Shaw,L. and Lewin,A. (1999) An RNA binding motif in the Cbp2 protein required for protein-stimulated RNA catalysis. J. Biol. Chem., 274, 30393–30401. [DOI] [PubMed] [Google Scholar]
- Voelker R., Mendel-Hartvig,J. and Barkan,A. (1997) Transposon-disruption of a maize nuclear gene, tha1, encoding a chloroplast SecA homolog: in vivo role of cp-SecA in thylakoid protein targeting. Genetics, 145, 467–478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vogel J., Boerner,T. and Hess,W. (1999) Comparative analysis of splicing of the complete set of chloroplast group II introns in three higher plant mutants. Nucleic Acids Res., 27, 3866–3874. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weeks K. (1997) Protein-facilitated RNA folding. Curr. Opin. Struct. Biol., 7, 336–342. [DOI] [PubMed] [Google Scholar]
- Wiesenberger G., Waldherr,M. and Schweyen,R. (1992) The nuclear gene MRS2 is essential for the excision of group II introns from yeast mitochondrial transcripts in vivo. J. Biol. Chem., 267, 6963–6969. [PubMed] [Google Scholar]