

Abstract
Werner syndrome (WS) is marked by early onset of features resembling aging, and is caused by loss of the RecQ family DNA helicase WRN. Precisely how loss of WRN leads to the phenotypes of WS is unknown. Cultured WS fibroblasts shorten their telomeres at an increased rate per population doubling and the premature senescence this loss induces can be bypassed by telomerase. Here we show that WRN co-localizes with telomeric factors in telomerase-independent immortalized human cells, and further that the budding yeast RecQ family helicase Sgs1p influences telomere metabolism in yeast cells lacking telomerase. Telomerase-deficient sgs1 mutants show increased rates of growth arrest in the G2/M phase of the cell cycle as telomeres shorten. In addition, telomerase-deficient sgs1 mutants have a defect in their ability to generate survivors of senescence that amplify telomeric TG1–3 repeats, and SGS1 functions in parallel with the recombination gene RAD51 to generate survivors. Our findings indicate that Sgs1p and WRN function in telomere maintenance, and suggest that telomere defects contribute to the pathogenesis of WS and perhaps other RecQ helicase diseases.
Keywords: ALT/replicative senescence/SGS1/telomeres/Werner syndrome
Introduction
Three human diseases are caused by loss of RecQ family DNA helicases: Werner syndrome (WS), Rothmund– Thompson syndrome (RTS) and Bloom syndrome (BS) (Ellis et al., 1995; Yu et al., 1996; Kitao et al., 1999). WS, RTS and, to a small degree, BS are marked by the premature onset of features resembling aging (Epstein et al., 1966; German, 1995; Lindor et al., 2000), arguing that this class of proteins has roles in preventing the accumulation of genomic lesions that contribute to phenotypes of aging. Individuals with WS are essentially normal as children, but as young adults develop premature graying and loss of hair, thinning of the dermis and loss of subcutaneous fat, bilateral ocular cataracts, and forms of osteoporosis and arteriosclerosis (Epstein et al., 1966). They also have an increased incidence of cancer, in particular sarcomas, a feature shared with RTS (Lindor et al., 2000). Several abnormalities in WS cells have been described, including elevated levels of DNA deletions and chromosome translocations, and sensitivity to 4-nitroquinoline-1-oxide and the topoisomerase I poison camptothecin (reviewed by Shen and Loeb, 2000). Which, if any, of these defects contributes to the gross pathology of WS is unknown.
Replicative senescence refers to the finite replicative potential of many cultured human cell types (Hayflick, 1980). Most somatic human cells, including primary fibroblasts, lack telomerase activity and shorten their telomeres with cell division (Harley et al., 1990). The shortening of telomeres appears to be a cause of replicative senescence because transgenic expression of telomerase can bypass senescence in several cell types (Bodnar et al., 1998; Vaziri and Benchimol, 1998; Yang et al., 1999). Cultured WS fibroblasts senesce prematurely, and shorten their telomeres at a faster rate per population doubling (Schulz et al., 1996); studies of replicative senescence in RTS and BS have not been reported. As in wild-type (wt) fibroblasts, the senescence of WS and BS fibroblasts can be bypassed by expression of telomerase (Ouellette et al., 2000; Wyllie et al., 2000; our unpublished observations), arguing that telomeric events, rather than other cellular defects, lead to senescence in these mutant cells.
Similar to primary human fibroblasts, yeast cells in which telomerase has been genetically inactivated lose telomeric repeats and senesce with cell division (Lundblad and Szostak, 1989). For example, Saccharomyces cerevisiae cells lacking the telomerase RNA component TLC1 shorten telomeres and lose viability, resulting in the death of most cells in the population (Singer and Gottschling, 1994). The terminal phenotype of senescing yeast cells has not been characterized. Rare cells escape senescence and these survivors maintain their telomeres without telomerase (Lundblad and Blackburn, 1993). This maintenance depends on the recombination gene RAD52 and either RAD50 or RAD51 (Lundblad and Blackburn, 1993; Le et al., 1999; Teng and Zakian, 1999), and so appears to involve recombination among telomeric DNA repeats (reviewed by Kass-Eisler and Greider, 2000). Two different types of survivors arise: type I have a repeat consisting of subtelomeric Y′ elements and short telomeric TG1–3 repeats amplified at their chromosome termini, while type II survivors have little amplification of Y′ elements, but instead have long TG1–3 repeats extending up to several kilobase pairs (kbp) (Lundblad and Blackburn, 1993; Teng and Zakian, 1999).
Telomerase-independent telomere maintenance mechanisms also occur in human cells. Primary human cells may be induced to bypass senescence by oncoproteins, such as SV40 large T antigen. Such cells shorten their telomeres to a point referred to as crisis, where the shortened telomeres lead to genome instability and apoptosis. Rare cells survive crisis and maintain their telomeres either by expression of telomerase or by a telomerase-independent mechanism (or mechanisms) termed ALT (alternative lengthening of telomeres; Bryan et al., 1997). ALT cells are immortalized and are characterized by telomeres of heterogeneous size that extend beyond 50 kbp in length (reviewed in Colgin and Reddel, 1999). ALT cells are also distinguished by nuclear structures, referred to as ALT-associated promyelocytic leukemia (AA-PML) bodies, which contain telomeric repeat DNA, the telomere repeat binding proteins TRF1 and TRF2, and the PML protein (Yeager et al., 1999). ALT is believed to involve recombination among telomeric DNA repeats, based largely on analogy with yeast survivors of senescence. Observations supporting simi-larities between yeast survivor pathways and human ALT include: (i) the long and heterogeneous telomeric repeats of type II survivors and of human ALT cells; and (ii) the requirements for RAD50, RAD51 and RAD52 in yeast survivor pathways, and the presence of human RAD50, RAD51 and RAD52 proteins in AA-PML bodies (Yeager et al., 1999; Zhu et al., 2000). Approximately 5% of human tumors use an ALT mechanism to maintain telomeres (Bryan et al., 1997; Colgin and Reddel, 1999).
Saccharomyces cerevisiae has a single RecQ family member, SGS1 (Gangloff et al., 1994). Sgs1 mutants display elevated levels of recombination, particularly among repeated DNA sequences including the ribosomal DNA and subtelomeric Y′ elements (Gangloff et al., 1994; Watt et al., 1996). When combined with mutation of another helicase, SRS2, sgs1 mutation causes extremely poor growth. The poor growth can be bypassed by rad51 mutation, indicating that SGS1 is important for preventing deleterious recombination events (Gangloff et al., 2000). WRN and BLM can each complement certain sgs1 recombination phenotypes, demonstrating conservation of function among the family of helicases (Yamagata et al., 1998).
Here we demonstrate localization of WRN with telomeric factors in ALT cells, a finding that prompted us to study telomeric functions of SGS1. In cells lacking telomerase, sgs1 mutants senesce prematurely and shorten their telomeres rapidly. We demonstrate for the first time that senescing yeast cells arrest at a specific phase in the cell cycle, G2/M. Senescing sgs1 mutant cells show higher rates of growth arrest at any given telomere length, a phenotype which is not observed in telomerase-positive sgs1 cells. We also show that SGS1 functions in parallel with RAD51 in the generation of survivors of senescence. The findings support the hypothesis that telomere defects contribute to the pathogenesis of WS and perhaps RTS and BS.
Results
Localization of WRN with telomeric factors in human ALT cells
WRN localizes predominantly in the nucleolus in many cell types (Marciniak et al., 1998). However, indirect immunofluorescence of WRN in immortalized human cells that lack telomerase, ALT cells (Bryan et al., 1997), revealed that in addition to nucleolar localization, WRN co-localizes in nuclear foci with the telomere repeat binding factors TRF2 (Figure 1) and TRF1 (data not shown). ALT cells are characterized by long and heterogeneous telomeres and by nuclear structures, dubbed AA-PML, containing telomeric repeat DNA, telomere repeat binding factors and several proteins involved in DNA recombination; these bodies are typically present in 5% of cycling interphase cells (Yeager et al., 1999). WRN localizes to such bodies, as assessed by co-staining known AA-PML body proteins (Yeager et al., 1999; Zhu et al., 2000), including PML, hRAD52 and NBS1 (Figure 1 and data not shown). WRN co-localization with TRF1 and TRF2 was observed in all six ALT cell lines examined. These included both SV40 T-antigen-transformed cell lines (WI38 75.1, VA13, LM216J and AG07066) and spontaneously occurring tumor cell lines (Saos2 and U2OS). We did not observe co-localization of WRN with PML or telomeric factors in primary fibroblast cell lines (e.g. BJ, and WI38) or in telomerase-positive cell lines (e.g. HeLa, and hTERT-immortalized BJ fibroblasts). Our inability to detect WRN at telomeres in primary or telomerase-positive cells may reflect a specific association between WRN and ALT-maintained telomeres. Alternatively, these findings may be a consequence of the limited sensitivity of immunofluorescence, because many proteins present at telomeres in both ALT and primary cells are detected more readily in ALT cells (e.g. NBS1; Zhu et al., 2000). Nonetheless, the co-localization of WRN with telomere components under at least some conditions suggests that WRN could have direct effects at telomeres.

Fig. 1. Co-localization of WRN with telomeric factors in human ALT fibroblasts. Cultured WI38 75.1 fibroblasts were examined by indirect immunofluorescence and confocal microscopy with antibodies to WRN, TRF2 and PML, as indicated. The cell in the center of the field showing co-localization of WRN with TRF2 and PML is representative of the 5% of cells in a WI38 75.1 culture that possess AA-PML bodies.
Premature senescence of yeast sgs1 mutants
The above findings prompted us to investigate a possible role for the yeast WRN homolog SGS1 in telomere metabolism. SGS1 has been reported to have no effect on steady-state telomere length, although sgs1 mutants do have elevated levels of recombination among subtelomeric Y′ elements (Watt et al., 1996). Yeast have constitutive telomerase activity, but genetic inactivation of components of the yeast telomerase complex causes telomeres to shorten with each cell division and results in the eventual senescence of the clone, similar to the telomere shortening and senescence of cultured human primary fibroblasts (Lundblad and Szostak, 1989; Singer and Gottschling, 1994).
To investigate functions of SGS1 in senescing yeast cells, we sporulated diploids heterozygous for deletion mutations in SGS1 and the telomerase RNA TLC1 (Singer and Gottschling, 1994). The sgs1 tlc1 double mutant segregants senesced more rapidly than tlc1 mutants in liquid culture (Figure 2A; see Materials and methods) and in serial streaks on plates (not shown). Telomere length in the senescing cultures was assessed by probing XhoI-digested DNA with a fragment of the subtelomeric Y′ elements present on most chromosome ends (e.g. Figure 2B). As reported (Watt et al., 1996), sgs1 mutants had telomere lengths similar to wt (Figure 2B, lanes 1 and 2 and data not shown), although analysis of 13 pairs of SGS1 and sgs1 segregants did indicate a slightly greater length in the sgs1 mutants (mean lengths and standard deviations of Y′-containing terminal restriction fragments: 1301 ± 35 versus 1324 ± 30 bp, P <0.1). To measure initial telomere shortening rates, isolated segregants were grown in liquid culture until sufficient cells were obtained for Southern analysis of Y′-containing telomere lengths, and the number of population doublings was calculated (see Materials and methods). Comparing senescing segregants to the wt segregant within each tetrad yielded mean rates and standard deviations of 5.7 ± 1.0 and 7.2 ± 1.7 bp per population doubling for the tlc1 single and sgs1 tlc1 double mutants, respectively (N = 13 pairs, P <0.01). Comparing tlc1 to wt and sgs1 tlc1 to sgs1 segregants within each tetrad yielded shortening rates of 5.6 ± 1.0 and 8.0 ± 1.2 bp per population doubling (N = 12 pairs, P <0.001). Thus, like senescing cultured WS fibroblasts, sgs tlc1 mutants shorten telomeres at an increased rate per population doubling.


Fig. 2. Senescence of tlc1 and sgs1 tlc1 cells. (A) Senescence in liquid culture. Values are from three independent pairs of tlc1 (open squares) and sgs1 tlc1 (closed diamonds) mutant cultures for the first 13 days of growth in liquid. Control wt and sgs1 cultures grown in parallel gave mean levels of 1.9 × 108 and 1.5 × 108 cells/ml. Similar results were obtained for the first 3 days of liquid culture comparing 16 other spore pairs from an independent cross. (B) Y′-containing telomeres were visualized by digesting genomic DNA with XhoI and probing with Y′ sequences distal to the cut site. Lanes 1 and 2 are wt and sgs1 mutant cells, and lanes 3–6 and 7–10 are from senescing tlc1 and sgs1 tlc1 mutants, respectively. Lanes 3 and 6 are from reference patches of segregants (day 0) and other lanes are from liquid culture. Days of growth in liquid, population doubling levels and size markers (M, in kbp) are indicated.
Sgs1 mutants have a hypersensitive arrest phenotype in the absence of telomerase
The faster telomere shortening in sgs1 tlc1 cultures could result from effects of Sgs1p on the rate of telomere shortening in dividing cells or from an increased tendency of sgs1 mutants to cease cell division as telomeres shorten. In the latter case, viable cells must undergo additional divisions to compensate for arrested cells, thereby further shortening their telomeres and clonal lifespan. We thus examined the phenotypes of senescing cells, using flow cytometry and microscopy. tlc1 mutants accumulated in the G2/M phase of the cell cycle as they approached senescence (Figure 3A). The accumulation of cells in the G2/M compartment is accompanied by an increase in cells with <1N (and perhaps >2N) DNA content. These changes might reflect debris from dying cells or perhaps chromosome fragmentation or aneuploidy in the senescing cells. The progressive G2/M accumulation also occurred in the sgs1 tlc1 mutants, but was more dramatic and occurred at earlier generations and longer telomere lengths. For example, the cell cycle pattern for generation 20 sgs1 tlc1 cells is not seen until generation 75 for tlc1 cells, yet the telomeres of generation 30 sgs1 tlc1 cells are clearly longer than those of generation 75 tlc1 cells (Figure 2B, lanes 5 and 7). Fluorescence microscopy of cells stained with 4′,6-diamidino-2-phenylindole (DAPI) and anti-tubulin antibodies to visualize DNA and the nuclear spindle revealed an accumulation of large budded cells, many with nuclei wedged in bud necks and with short spindles (Figure 3B and data not shown), characteristic of G2/M arrest (Pinto and Winston, 2000). The frequency of such cells paralleled the changes observed by flow cytometry, and the sgs1 tlc1 mutants again arrested with this morphology more frequently at longer telomere lengths (Figure 3C). Cells with the G2/M phenotype might have permanently arrested or might have only paused in their cell cycle progression. To distinguish between these possibilities, we tested cells from senescing liquid cultures for their ability to divide after growth for 36 h on YPD plates. Cells and microcolonies were observed by microscopy, and isolated large single- or double-budded cells were considered to have arrested permanently. After the first day of growth in liquid media, 28% of sgs1 tlc1 cells failed to divide, compared with only 1, 5 and 22% of the tlc1 cells on the first, fourth and seventh days, respectively. Under these conditions, >98% of control wt and sgs1 cells formed colonies. In conclusion, a greater fraction of sgs1 tlc1 mutant cells permanently exit the cell cycle (and presumably die) at all telomere lengths as the culture senesces. This G2/M arrest occurs in only a small fraction of sgs1 cells (Figure 3C), indicating that the G2/M arrest in tlc1 and sgs1 tlc1 cells is due to a telomere-related defect. This might reflect a role for Sgs1p helicase activity in avoiding or resolving deleterious recombination events at telomeres.



Fig. 3. Arrest phenotypes of senescing tlc1 and sgs1 tlc1 cells. (A) The first cell cycle profile on each graph is from wt or sgs1 cells, which differed in the fraction of cells in G2/M (44 and 55%, respectively). The later profiles are senescing tlc1 or sgs1 tlc1 cells from one spore pair, taken from segregant colonies or on the indicated day and population doubling (PD) of liquid culture (same cultures as used for Figure 2B). Similar results were obtained for two other spore pairs. Note that the day 8 sgs1 tlc1 culture contains ALT survivors. (B) Two examples of typical large-budded sgs1 tlc1 cells at day 5 of liquid culture, apparently arrested in G2/M, stained with DAPI and anti-TUB2 tubulin antibodies. Arrested tlc1 cells had the same phenotype as sgs1 tlc1 cells (not shown). (C) Quantitation of the fraction of cells with large buds, short nuclear spindles and nuclei near or in the bud neck. Mean and standard deviations for three cultures of each type are shown; at least 100 cells were counted for each sample. Genotypes and population doublings and days in liquid culture are indicated. The mean telomere lengths and standard deviations for sgs1 tlc1 on day 0, and for tlc1 on day 4 and 6 were 1139 ± 44, 1142 ± 15 and 1096 ± 14 bp, respectively.
Role of SGS1 in the type II survivor pathway
Although most yeast cells lacking telomerase cease dividing because of shortening telomeres, a small fraction of cells escape senescence and maintain their telomeres by recombination-dependent mechanisms (Lundblad and Blackburn, 1993; Teng and Zakian, 1999). The most frequent type of survivor (type I) maintains telomeres by amplifying a unit consisting of a subtelomeric Y′ element and a short stretch of telomeric TG1–3 DNA, and has a variable growth rate. With continued cell division, type I cells may convert to faster growing type II survivors, which maintain telomeres via amplification of long and heterogeneous terminal TG1–3 repeats. Conversion from type I to type II is seen most readily in liquid culture where the more rapidly growing cells overtake the population (Teng and Zakian, 1999). sgs1 tlc1 mutants generated survivors with efficiency similar to tlc1 mutants, although the emergence of survivors was preceded by a more prolonged period of poor growth in the double mutants (Figure 2A and data not shown). To test whether this period of poor growth of sgs1 tlc1 cells might be associated with a qualitative defect in the generation of survivors, we examined the telomeres of survivors by Southern analysis. Genomic DNA was digested with XhoI, or a combination of endonucleases with four-base recognition sequences that do not cut TG1–3 repeats, and probed with poly(dG)–(dT); type I cells yield discrete bands corresponding to amplified Y′ DNA, and type II cells yield more heterogeneous patterns (Teng and Zakian, 1999). Twelve tlc1 and nine sgs1 tlc1 survivor colonies each generated from independent spore segregants were grown for 120 generations in liquid culture and monitored for the emergence of type II survivors. Ten of the tlc1 cultures adopted type II character by 10 generations of growth, one by 20 generations, and one maintained a mixed type I/II phenotype for 120 generations (Figure 4A, lanes 1–3 and 7–9 and data not shown). In contrast, all of the tlc1 sgs1 cultures maintained a clear type I character for at least 120 generations (Figure 4A, lanes 4–6 and 10–12 and data not shown), indicating that SGS1 is important for the transition from type I to II. When SGS1 was deleted from established type II survivors, the double mutant survivors grew much more poorly than sgs1 cells or tlc1 survivors (Figure 4B). The poor growth of sgs1 tlc1 type II survivors might be due to difficulties in the replication or stabilization of TG1–3 repeats, and may explain why these cells do not accumulate in liquid culture.

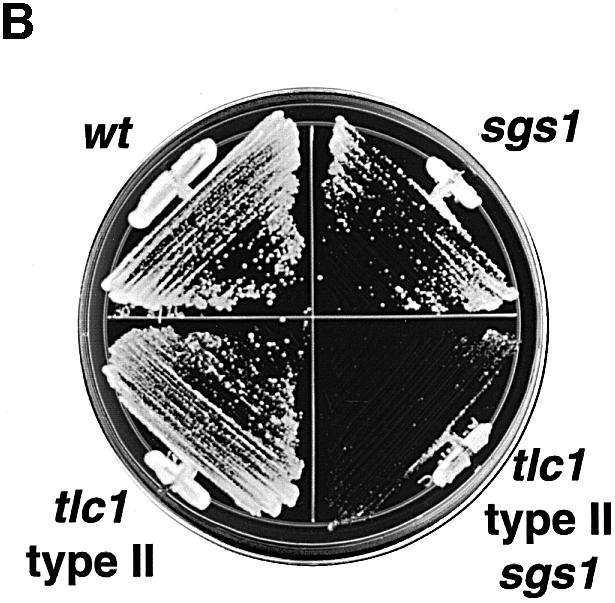
Fig. 4. Dependence of senescence survivor phenotypes on SGS1. (A) Examples of telomere structures of tlc1 survivors, three tlc1 (lanes 1–3 and 7–9) and three sgs1 tlc1 (lanes 4–6 and 10–12). Newly emerged and independent survivor colonies from senescence on plates were grown for an additional 120 generations in liquid culture. Genomic DNA was digested with XhoI, which enables visualization of tandemly amplified Y′ elements (type I pattern), or with AluI, HaeIII, HinfI and MspI, which digest genomic DNA into small fragments but leave amplified TG1–3 sequences intact (type II pattern) (Teng and Zakian, 1999). Blots of digested DNA were probed with poly(dG)–(dT) to detect all telomeric tracts, as well as Y′ sequences, which have short interspersed dG–dT DNA (Lundblad and Szostak, 1989; Le et al., 1999). Note that the two Y′ bands correspond to the two different size classes of Y′ elements: Y′S and Y′L. (B) Growth after 42 h on rich media of wt, sgs1, a type II survivor and the same type II survivor from which SGS1 has been deleted.
SGS1 is important for the RAD51-independent survivor pathway
There are at least two recombination pathways by which tlc1 survivors can arise, involving either RAD50 or RAD51 (Le et al., 1999). We addressed whether Sgs1p was a component of either pathway defined by these RAD genes. Diploids triply heterozygous for tlc1, sgs1 and rad50 or rad51 deletions were sporulated and the segregants were carried through senescence in liquid (Figure 5A) or on plates (Figure 5B and data not shown). rad50 rad51 tlc1 mutants were also generated as controls. As expected, each of 13 tlc1 and nine sgs1 tlc1 mutants generated survivors on plates (Figure 5B and data not shown) just as they had in liquid culture (Figure 2A). sgs1 rad50 tlc1 cells, although slow growing, also generated survivors efficiently (Figure 5B and data not shown). In contrast, sgs1 rad51 tlc1 mutants, like rad50 rad51 tlc1 mutants, grew well initially but failed to generate survivors in liquid culture (Figure 5A). In 17 of 18 trials on plates, no sgs1 rad51 tlc1 survivors were observed (Figure 5B and data not shown). In the 18th trial, as well as in nine of 17 trials for rad50 rad51 tlc1 mutants, survivors appeared. In this same rad50 rad51 tlc1 strain, survivors were not reported previously (Le et al., 1999). The larger number of cells used for each passage in our plate assays (>107), compared with 2 × 106 for liquid assays, may have allowed for the selection of rare bypass suppressor mutants. We conclude that SGS1 functions in parallel with RAD51 in the generation of survivors, and plays a role at least as important as RAD50 in the generation of survivors that emerge in the absence of RAD51.


Fig. 5. SGS1 dependence of the emergence of survivors in tlc1 cells lacking RAD50 or RAD51. (A) Senescence of rad51 tlc1, rad50 rad51 tlc1 and sgs1 rad51 tlc1 mutants in liquid culture, using three, two and five independent spores, respectively. The mean results for days 1–10 in liquid culture are shown. (B) Examples of senescing cells during growth on solid medium. For comparison, the upper left plate shows growth of TLC1 cells of the indicated genotypes after 42 h of growth. Clockwise from the top, the genotypes are wt, sgs1, rad50, rad51, sgs1 rad50 and sgs1 rad51. The remaining plates illustrate serial restreaks of senescing cells of the indicated genotypes (restreak number is indicated; 1 is the first restreak from the reference patch made from spore colonies). Cultures were first carried through senescence on separate plates and the time course was then reconstructed on the plates shown. The genotypes are tlc1, tlc1 rad50, tlc1 rad51, tlc1 sgs1, tlc1 rad50 rad51, tlc1 sgs1 rad50 and tlc1 sgs1 rad51, moving top to bottom and left to right. Plates were incubated for 54 h, except for tlc1 and tlc1 rad51, which were incubated for 42 h, and tlc1 sgs1 rad50, which was incubated for 67 h to adjust for different growth rates. sgs1 rad51 double mutants cultured in parallel did not senesce (not shown).
Discussion
We have observed defects in sgs1 mutant cells lacking telomerase, both during the senescence of the cells and in survivors of senescence. The central defect in senescing sgs1 cells is a greater tendency to arrest growth as telomeres shorten. There are three related, and non-exclusive, interpretations of this defect. One possibility is that loss of telomerase or telomere shortening causes structural changes that induce recombination or the formation of G-quadruplex complexes (see below) between telomeres. If such events cannot be resolved efficiently in the absence of the Sgs1p helicase, G2/M arrest could result. Secondly, if SGS1 plays a role in an intra-S checkpoint (Frei and Gasser, 2000), in sgs1 mutants telomere lesions may persist through S phase and later trigger a G2/M arrest. A third possibility is that SGS1 is normally involved in a telomerase-independent repair pathway, perhaps mechanistically related to its role in the survivor pathway (see below). In this model, a telomere suddenly and critically shortened by damage at a point in the cell cycle when telomerase is not active [telomerase appears only to lengthen telomeres in late S phase (Marcand et al., 2000)] might be repaired by an SGS1-dependent pathway. Our data indicate that the increased rate of telomere shortening in senescing sgs1 tlc1 populations is most simply explained by a more rapidly diminishing fraction of dividing cells, although an additional direct effect on the rate of shortening of telomeres in viable cells cannot be excluded. Primary fibroblasts from individuals with WS have also been observed to exit the cell cycle more frequently than passage-matched control cells (Faragher et al., 1993). Regardless of whether SGS1 has direct effects on telomere shortening or only on the propensity of cells to arrest as telomeres shorten, our data indicate that SGS1 has important roles in senescing cells that depend on loss of telomerase because TLC1 sgs1 mutant cells do not show significant cell cycle arrest.
The localization of WRN with telomere factors, at least in ALT cells, and the functional experiments with telomerase-deficient sgs1 mutants, together with previous reports of telomere abnormalities in Werner cells (Schulz et al., 1996; Tahara et al., 1997), support a role for telomere abnormalities in the genesis of the WS phenotype. Also supporting this idea are the recent demonstration that WRN interacts with the Ku protein (Cooper et al., 2000; Li and Comai, 2000), which is part of telomeric chromatin in human cells (Hsu et al., 1999), and also the demonstration that WRN, assisted by RPA, can unwind complexes formed of telomeric DNA in vitro (Ohsugi et al., 2000). If telomerase masks a role for WRN at telomeres, as it does for SGS1, then WS phenotypes would be expected to occur in slowly dividing tissues that lack telomerase, rather than rapidly proliferating tissues that contain progenitor cells with active telomerase. This prediction holds well for much of WS pathology, e.g. in the dermis, vascular endothelium, adipose tissue and bone (Epstein et al., 1966; Shen and Loeb, 2000). It is also consistent with the absence of pathology related to cell loss in the epidermis, bone marrow and gastrointestinal tract that would otherwise be expected if telomerase-positive progenitors could not replenish these tissues. Some features of WS, like premature hair loss, are not readily explained by this hypothesis, but may be a consequence of other WRN functions or may be secondary to defects in other tissues. Our results also suggest that the relative unimportance of WRN in mice (Lombard et al., 2000) could be explained by the dramatic differences in mouse and human telomere biology (reviewed by Sherr and DePinho, 2000; Wright and Shay, 2000). Mouse chromosomes have telomeres that are about five times longer than human, and mouse tissues have more abundant telomerase activity (Chadeneau et al., 1995; Prowse and Greider, 1995). Moreover, mice are relatively unaffected by the loss of telomerase: cultured mouse fibroblasts and intact mice lacking the mTR telomerase RNA show no evidence of senescence induced by telomere shortening and do not appear to incur pathologies until the point where telomeres become short enough to induce crisis (Blasco et al., 1997; Lee et al., 1998; Rudolph et al., 1999). It will be informative to test whether mice lacking both mTR and WRN develop synthetic defects.
SGS1 functions in parallel with RAD51 in the generation of survivors of senescence, placing SGS1 in the RAD50-dependent survivor pathway (Le et al., 1999). SGS1 is also required for the emergence of type II survivors. Even though SGS1 can be disrupted in preformed type II survivors, the resulting cells have a very poor growth rate, which could explain the inability of sgs1 tlc1 mutants to generate type II survivors. These observations are consistent with the recent demonstration that RAD50 is also required for the emergence of type II survivors, while RAD51 appears to be required for the emergence of type I survivors (Teng et al., 2000).
The function of SGS1 in a RAD51-independent survivor pathway raises the possibility that SGS1 might participate in a break-induced-replication (BIR) mechanism at telomeres. In BIR, a chromosome end invades a recipient chromosome and copies the recipient by replication to the end of the chromosome (Haber, 1999). BIR is RAD51 independent in yeast (Malkova et al., 1996) and has been suggested as a mechanism of telomere maintenance in survivors (Le et al., 1999). After invasion, the BIR intermediate resembles structures believed to reinitiate replication from stalled replication forks (Haber, 1999), and there is evidence that both Sgs1p and WRN localize to and function at stalled replication forks (Constantinou et al., 2000; Frei and Gasser, 2000). The physical interaction of WRN with the replication proteins proliferating cell nuclear antigen, polymerase δ and RPA (Brosh et al., 1999; Huang et al., 2000; Kamath-Loeb et al., 2000) is also consistent with this notion. In addition, Sgs1p and WRN are potent G-quadruplex DNA helicases in vitro (Fry and Loeb, 1999; Sun et al., 1999). Perhaps during a BIR-like event, the long TG1–3 repeats of type II survivors are prone to forming G-quadruplexes, or related secondary structures, which require processing by Sgs1p. The poor growth of type II survivors that lack SGS1 supports this idea.
Might WRN be involved in human ALT? It is intriguing that WS and RTS patients are particularly prone to sarcomas (Lindor et al., 2000), and that sarcomas appear to be more susceptible to ALT pathways than are carcinomas (Colgin and Reddel, 1999), the latter being typically positive for telomerase. Telomere maintenance by subtelomeric repeat amplification can occur in mouse cells, suggesting that a mammalian equivalent of the type I survivor pathway may exist (Niida et al., 2000). Moreover, we have identified an unusual ALT WS fibroblast line that has very short telomeric repeats but amplified telomere-associated sequences (our unpublished data), also reminiscent of yeast type I survivor telomeres. In yeast, the type I pathway is associated with unstable growth, possibly reflecting genome instability. If loss of WRN or RTS biases ALT cells toward a type I pathway, this might contribute to genome instability and the occurrence of sarcomas in WS and RTS. It will be informative to examine the telomeres of tumor cells from individuals with these disorders.
In summary, our findings indicate that the Sgs1p and WRN helicases play important roles in telomere biology, suggesting that a telomere defect is relevant to the pathophysiology of WS and perhaps other RecQ helicase diseases.
Materials and methods
Yeast strains and methods
Yeast were cultured at 30°C with standard media. All strains were isogenic and derived from JKM111 and the rad51 and rad50 derivatives JKM114 and JKM116 (Le et al., 1999). SGS1 was deleted using the sgs1Δ::hisG-URA3 contruct pPP69 (Park et al., 1999). TLC1 was deleted using PCR-mediated replacement of transcript bases 141–1212 with a kanMX G418-selectable marker (Guldener et al., 1996). Double and triple mutants were constructed by deleting one copy of TLC1 in diploids heterozygous for sgs1, or sgs1 and rad50 or rad51 followed by sporulation. All genotypes were confirmed by auxotrophy/G418 resistance and by PCR amplification of fragments that crossed mutation breakpoints and were thus different sizes for wt and mutant alleles. Senescence experiments were performed starting with spore products from reference plates (estimated population doubling of 30). Liquid senescence experiments were as described (Le et al., 1999), with rounds of inoculation of 2 × 106 cells into 5 ml of YPD followed by growth for 23 h, counting of cells with a Coulter counter, and re-inoculation. Plate senescence experiments were performed using a mixture of five colonies (10–20 × 106 cells) and growth for 3 days per restreak. Genotypes of survivors were confirmed as for starting spore products. Continued passage of survivors in liquid was performed by diluting saturated cultures 1000-fold serially every 1.5 (tlc1) or 3 days (sgs1 tlc1), each passage counted as 10 doublings. Viability of cells senescing in liquid culture was assessed by plating 1000 cells, from each of the six cultures analyzed in Figure 2A, and observing cells/colonies using microscopy after 36 h of growth. At least 100 cells/colonies were counted per culture, and isolated large single- and double-budded cells were considered arrested.
Human cells
WI38 75.1, a clone of WI38 fetal lung fibroblasts immortalized with the SV40 large T antigen, were obtained from the Coriell cell repository, and were grown as described (Marciniak et al., 1998). The ALT phenotype of WI38 75.1 was determined using standard telomeric TRF blots and TRAP assays for telomerase (Counter et al., 1998). Hela S3, WI38, VA13, AG07066, U2OS, Saos2 and BJ cells were obtained from the American Type Culture Collection or the Coriell cell repositories. LM216J and hTERT-immortalized BJ fibroblasts were as described (Murnane, 1986; Hahn et al., 1999).
Telomere analysis
Southern analysis was performed essentially as described (Le et al., 1999). Briefly, digested yeast genomic DNA was separated on 1% agarose gels, transferred to Hybond-XL (Amersham) and hybridized to random-primed poly(dG)–(dT) or a 784 bp PCR-generated Y′ fragment (GACTGCAGGG…GAAAGCAGGG) lying between the terminal XhoI site to the start of the terminal TG1–3 repeats. Washed blots were visualized using a Molecular Dynamics PhosphorImager. To measure initial telomere shortening rates, entire spore colonies were dissected from plates, resuspended in 5 ml of liquid YPD and grown to an OD of 1–3. Cell number was determined with a Coulter counter and population doublings from the original spore calculated as log2(final cell number). Data files generated from telomere blots probed with the 784 bp Y′ fragment (above) were analyzed for mean TRF length using NIH Image and Microsoft Excel software. The weighted average was used to determine mean telomere length: Σ(ODi × lengthi)/Σ(ODi). P values for differences between telomere lengths and rates of shortening were calculated using a two-tailed, unpaired two-sample t-test.
Flow cytometry
Log-phase cells were prepared as described (Mills et al., 1999), except that they were sonicated gently prior to fixation, all centrifugations were at 2000 g and digestions were at 50°C. Propidium iodide-stained cells were analyzed on a Beckman-Dickson FACScan using CellQuest and ModFit software; 30 000 cells were aquired per sample.
Immunofluorescence
For yeast, log-phase cells were fixed in formaldehyde and spheroplasted with zymolyase as described (Mills et al., 1999). DNA was stained with DAPI and microtubules were visualized using the mouse anti-TUB2 antibody BIB2 and an anti-mouse Cy3 conjugate (Amersham) diluted 1:200 in phosphate-buffered saline/1% bovine serum albumin/0.1% Triton X-100. Digital images were obtained using a SPOT RT CCD camera and software. Human fibroblasts were grown on multi-welled slides, fixed and stained for WRN using an affinity-purified rabbit anti-WRN antibody as described (Marciniak et al., 1998). Mouse monoclonal anti-TRF-2 antibodies (Imgenex) and anti-PML antibodies (Santa Cruz Biotechnology) were used at dilutions of 1:800 and 1:200, respectively. Secondary antibodies were a goat anti-rabbit fluorescein isothiocyanate (Vector Labs) at 1:100 and goat anti-mouse Cy3 conjugate (Amersham) at 1:300. Microscopy was performed at the WM Keck Foundation Biological Imaging Facility at the Whitehead Institute on a Zeiss laser scanning confocal microscope, sampling 0.8 µm optical sections.
Acknowledgments
Acknowledgements
We thank J.Haber for yeast strains, F.Solomon for the TUB2 antibody, R.Weinberg for support and advice, and D.McNabb, S.Imai, P.Park, C.Armstrong, H.Vaziri, B.Brown, M.Kaeberlein, P.Defossez and the other Guarente laboratory members for advice and reagents. L.G. is supported by grants from NIH, the Ellison Medical Foundation, the Seaver Foundation, and the Howard and Linda Stern Fund. F.B.J. and R.A.M. are supported by NIA K-08 awards. W.C.H. is, and F.B.J. was, supported by a Howard Hughes Medical Institute postdoctoral fellowship. S.A.S. is supported by an American Cancer Society Fellowship. W.C.H. and S.A.S. are Herman and Margaret Sokol postdoctoral fellows.
REFERENCES
- Blasco M.A., Lee,H.W., Hande,M.P., Samper,E., Lansdorp,P.M., DePinho,R.A. and Greider,C.W. (1997) Telomere shortening and tumor formation by mouse cells lacking telomerase RNA. Cell, 91, 25–34. [DOI] [PubMed] [Google Scholar]
- Bodnar A.G. et al. (1998) Extension of life-span by introduction of telomerase into normal human cells. Science, 279, 349–352. [DOI] [PubMed] [Google Scholar]
- Brosh R.M. Jr, Orren,D.K., Nehlin,J.O., Ravn,P.H., Kenny,M.K., Machwe,A. and Bohr,V.A. (1999) Functional and physical interaction between WRN helicase and human replication protein A. J. Biol. Chem., 274, 18341–18350. [DOI] [PubMed] [Google Scholar]
- Bryan T.M., Englezou,A., Dalla-Pozza,L., Dunham,M.A. and Reddel,R.R. (1997) Evidence for an alternative mechanism for maintaining telomere length in human tumors and tumor-derived cell lines. Nature Med., 3, 1271–1274. [DOI] [PubMed] [Google Scholar]
- Chadeneau C., Siegel,P., Harley,C.B., Muller,W.J. and Bacchetti,S. (1995) Telomerase activity in normal and malignant murine tissues. Oncogene, 11, 893–898. [PubMed] [Google Scholar]
- Colgin L.M. and Reddel,R.R. (1999) Telomere maintenance mechanisms and cellular immortalization. Curr. Opin. Genet. Dev., 9, 97–103. [DOI] [PubMed] [Google Scholar]
- Constantinou A., Tarsounas,M., Karow,J.K., Brosh,R.M., Bohr,V.A., Hickson,I.D. and West,S.C. (2000) Werner’s syndrome protein (WRN) migrates Holliday junctions and co-localizes with RPA upon replication arrest. EMBO rep., 1, 80–84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cooper M.P., Machwe,A., Orren,D.K., Brosh,R.M., Ramsden,D. and Bohr,V.A. (2000) Ku complex interacts with and stimulates the Werner protein. Genes Dev., 14, 907–912. [PMC free article] [PubMed] [Google Scholar]
- Counter C.M., Hahn,W.C., Wei,W., Caddle,S.D., Beijersbergen,R.L., Lansdorp,P.M., Sedivy,J.M. and Weinberg,R.A. (1998) Dissociation among in vitro telomerase activity, telomere maintenance and cellular immortalization. Proc. Natl Acad. Sci. USA, 95, 14723–14728. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ellis N.A., Groden,J., Ye,T.Z., Straughen,J., Lennon,D.J., Ciocci,S., Proytcheva,M. and German,J. (1995) The Bloom’s syndrome gene product is homologous to RecQ helicases. Cell, 83, 655–666. [DOI] [PubMed] [Google Scholar]
- Epstein C.J., Martin,G.M., Schultz,A.L. and Motulsky,A.G. (1966) Werner’s syndrome: a review of its symptomatology, natural history, pathologic features, genetics and relationship to the natural aging process. Medicine (Baltimore), 45, 177–221. [DOI] [PubMed] [Google Scholar]
- Faragher R.G., Kill,I.R., Hunter,J.A., Pope,F.M., Tannock,C. and Shall,S. (1993) The gene responsible for Werner syndrome may be a cell division ‘counting’ gene. Proc. Natl Acad. Sci. USA, 90, 12030–12034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frei C. and Gasser,S.M. (2000) The yeast Sgs1p helicase acts upstream of Rad53p in the DNA replication checkpoint and colocalizes with Rad53p in S-phase-specific foci. Genes Dev., 14, 81–96. [PMC free article] [PubMed] [Google Scholar]
- Fry M. and Loeb,L.A. (1999) Human Werner syndrome DNA helicase unwinds tetrahelical structures of the fragile X syndrome repeat sequence d(CGG)n. J. Biol. Chem., 274, 12797–12802. [DOI] [PubMed] [Google Scholar]
- Gangloff S., McDonald,J.P., Bendixen,C., Arthur,L. and Rothstein,R. (1994) The yeast type I topoisomerase Top3 interacts with Sgs1, a DNA helicase homolog: a potential eukaryotic reverse gyrase. Mol. Cell. Biol., 14, 8391–8398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gangloff S., Soustelle,C. and Fabre,F. (2000) Homologous recombination is responsible for cell death in the absence of the Sgs1 and Srs2 helicases. Nature Genet., 25, 192–194. [DOI] [PubMed] [Google Scholar]
- German J. (1995) Bloom’s syndrome. Dermatol. Clin., 13, 7–18. [PubMed] [Google Scholar]
- Guldener U., Heck,S., Fiedler,T., Beinhauer,J. and Hegemann,J.H. (1996) A new efficient gene disruption cassette for repeated use in budding yeast. Nucleic Acids Res., 24, 2519–2524. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haber J.E. (1999) DNA recombination: the replication connection. Trends Biochem. Sci., 24, 271–275. [DOI] [PubMed] [Google Scholar]
- Hahn W.C., Counter,C.M., Lundberg,A.S., Beijersbergen,R.L., Brooks,M.W. and Weinberg,R.A. (1999) Creation of human tumour cells with defined genetic elements. Nature, 400, 464–468. [DOI] [PubMed] [Google Scholar]
- Harley C.B., Futcher,A.B. and Greider,C.W. (1990) Telomeres shorten during ageing of human fibroblasts. Nature, 345, 458–460. [DOI] [PubMed] [Google Scholar]
- Hayflick L. (1980) Cell aging. Annu. Rev. Gerontol. Geriatr., 1, 26–67. [Google Scholar]
- Hsu H.L., Gilley,D., Blackburn,E.H. and Chen,D.J. (1999) Ku is associated with the telomere in mammals. Proc. Natl Acad. Sci. USA, 96, 12454–12458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang S., Beresten,S., Li,B., Oshima,J., Ellis,N.A. and Campisi,J. (2000) Characterization of the human and mouse WRN 3′→5′ exonuclease. Nucleic Acids Res., 28, 2396–2405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kamath-Loeb A.S., Johansson,E., Burgers,P.M. and Loeb,L.A. (2000) Functional interaction between the Werner Syndrome protein and DNA polymerase δ. Proc. Natl Acad. Sci. USA, 97, 4603–4608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kass-Eisler A. and Greider,C.W. (2000) Recombination in telomere-length maintenance. Trends Biochem. Sci., 25, 200–204. [DOI] [PubMed] [Google Scholar]
- Kitao S., Shimamoto,A., Goto,M., Miller,R.W., Smithson,W.A., Lindor,N.M. and Furuichi,Y. (1999) Mutations in RECQL4 cause a subset of cases of Rothmund–Thomson syndrome. Nature Genet., 22, 82–84. [DOI] [PubMed] [Google Scholar]
- Le S., Moore,J.K., Haber,J.E. and Greider,C.W. (1999) RAD50 and RAD51 define two pathways that collaborate to maintain telomeres in the absence of telomerase. Genetics, 152, 143–152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee H.W., Blasco,M.A., Gottlieb,G.J., Horner,J.W.,2nd, Greider,C.W. and DePinho,R.A. (1998) Essential role of mouse telomerase in highly proliferative organs. Nature, 392, 569–574. [DOI] [PubMed] [Google Scholar]
- Li B. and Comai,L. (2000) Functional interaction between Ku and the Werner syndrome protein in DNA end processing. J. Biol. Chem., 275, 28349–28352. [DOI] [PubMed] [Google Scholar]
- Lindor N.M., Furuichi,Y., Kitao,S., Shimamoto,A., Arndt,C. and Jalal,S. (2000) Rothmund–Thomson syndrome due to RECQ4 helicase mutations: report and clinical and molecular comparisons with Bloom syndrome and Werner syndrome. Am. J. Med. Genet., 90, 223–228. [DOI] [PubMed] [Google Scholar]
- Lombard D.B. et al. (2000) Mutations in the WRN gene in mice accelerate mortality in a p53-null background. Mol. Cell. Biol., 20, 3286–3291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lundblad V. and Blackburn,E.H. (1993) An alternative pathway for yeast telomere maintenance rescues est1- senescence. Cell, 73, 347–360. [DOI] [PubMed] [Google Scholar]
- Lundblad V. and Szostak,J.W. (1989) A mutant with a defect in telomere elongation leads to senescence in yeast. Cell, 57, 633–643. [DOI] [PubMed] [Google Scholar]
- Malkova A., Ivanov,E.L. and Haber,J.E. (1996) Double-strand break repair in the absence of RAD51 in yeast: a possible role for break-induced DNA replication. Proc. Natl Acad. Sci. USA, 93, 7131–7136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marcand S., Brevet,V., Mann,C. and Gilson,E. (2000) Cell cycle restriction of telomere elongation. Curr. Biol., 10, 487–490. [DOI] [PubMed] [Google Scholar]
- Marciniak R.A., Lombard,D.B., Johnson,F.B. and Guarente,L. (1998) Nucleolar localization of the Werner syndrome protein in human cells. Proc. Natl Acad. Sci. USA, 95, 6887–6892. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mills K.D., Sinclair,D.A. and Guarente,L. (1999) MEC1-dependent redistribution of the Sir3 silencing protein from telomeres to DNA double-strand breaks. Cell, 97, 609–620. [DOI] [PubMed] [Google Scholar]
- Murnane J.P. (1986) Inducible gene expression by DNA rearrangements in human cells. Mol. Cell. Biol., 6, 549–558. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Niida H., Shinkai,Y., Hande,M.P., Matsumoto,T., Takehara,S., Tachibana,M., Oshimura,M., Lansdorp,P.M. and Furuichi,Y. (2000) Telomere maintenance in telomerase-deficient mouse embryonic stem cells: characterization of an amplified telomeric DNA. Mol. Cell. Biol., 20, 4115–4127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ohsugi I., Tokutake,Y., Suzuki,N., Ide,T., Sugimoto,M. and Furuichi,Y. (2000) Telomere repeat DNA forms a large non-covalent complex with unique cohesive properties which is dissociated by Werner syndrome DNA helicase in the presence of replication protein A. Nucleic Acids Res., 28, 3642–3648. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ouellette M.M., McDaniel,L.D., Wright,W.E., Shay,J.W. and Schultz,R.A. (2000) The establishment of telomerase-immortalized cell lines representing human chromosome instability syndromes. Hum. Mol. Genet., 9, 403–411. [DOI] [PubMed] [Google Scholar]
- Park P.U., Defossez,P.A. and Guarente,L. (1999) Effects of mutations in DNA repair genes on formation of ribosomal DNA circles and life span in Saccharomyces cerevisiae. Mol. Cell. Biol., 19, 3848–3856. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pinto I. and Winston,F. (2000) Histone H2A is required for normal centromere function in Saccharomyces cerevisiae. EMBO J., 19, 1598–1612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prowse K.R. and Greider,C.W. (1995) Developmental and tissue-specific regulation of mouse telomerase and telomere length. Proc. Natl Acad. Sci. USA, 92, 4818–4822. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rudolph K.L., Chang,S., Lee,H.W., Blasco,M., Gottlieb,G.J., Greider,C. and DePinho,R.A. (1999) Longevity, stress response and cancer in aging telomerase-deficient mice. Cell, 96, 701–712. [DOI] [PubMed] [Google Scholar]
- Schulz V.P., Zakian,V.A., Ogburn,C.E., McKay,J., Jarzebowicz,A.A., Edland,S.D. and Martin,G.M. (1996) Accelerated loss of telomeric repeats may not explain accelerated replicative decline of Werner syndrome cells. Hum. Genet., 97, 750–754. [DOI] [PubMed] [Google Scholar]
- Shen J.C. and Loeb,L.A. (2000) The Werner syndrome gene: the molecular basis of RecQ helicase-deficiency diseases. Trends Genet., 16, 213–220. [DOI] [PubMed] [Google Scholar]
- Sherr C.J. and DePinho,R.A. (2000) Cellular senescence: mitotic clock or culture shock? Cell, 102, 407–410. [DOI] [PubMed] [Google Scholar]
- Singer M.S. and Gottschling,D.E. (1994) TLC1: template RNA component of Saccharomyces cerevisiae telomerase. Science, 266, 404–409. [DOI] [PubMed] [Google Scholar]
- Sun H., Bennett,R.J. and Maizels,N. (1999) The Saccharomyces cerevisiae Sgs1 helicase efficiently unwinds G–G paired DNAs. Nucleic Acids Res., 27, 1978–1984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tahara H. et al. (1997) Abnormal telomere dynamics of B-lymphoblastoid cell strains from Werner’s syndrome patients transformed by Epstein–Barr virus. Oncogene, 15, 1911–1920. [DOI] [PubMed] [Google Scholar]
- Teng S.C. and Zakian,V.A. (1999) Telomere–telomere recombination is an efficient bypass pathway for telomere maintenance in Saccharomyces cerevisiae. Mol. Cell. Biol., 19, 8083–8093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Teng S., Chang,J., McCowan,B. and Zakian,V.A. (2000) Telomerase-independent lengthening of yeast telomeres occurs by an abrupt Rad50p-dependent, Rif-inhibited recombinational process. Mol. Cell, 6, 947–952. [DOI] [PubMed] [Google Scholar]
- Vaziri H. and Benchimol,S. (1998) Reconstitution of telomerase activity in normal human cells leads to elongation of telomeres and extended replicative life span. Curr. Biol., 8, 279–282. [DOI] [PubMed] [Google Scholar]
- Watt P.M., Hickson,I.D., Borts,R.H. and Louis,E.J. (1996) SGS1, a homologue of the Bloom’s and Werner’s syndrome genes, is required for maintenance of genome stability in Saccharomyces cerevisiae. Genetics, 144, 935–945. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wright W.E. and Shay,J.W. (2000) Telomere dynamics in cancer progression and prevention: fundamental differences in human and mouse telomere biology. Nature Med., 6, 849–851. [DOI] [PubMed] [Google Scholar]
- Wyllie F.S., Jones,C.J., Skinner,J.W., Haughton,M.F., Wallis,C., Wynford-Thomas,D., Faragher,R.G. and Kipling,D. (2000) Telomerase prevents the accelerated cell ageing of Werner syndrome fibroblasts. Nature Genet., 24, 16–17. [DOI] [PubMed] [Google Scholar]
- Yamagata K., Kato,J., Shimamoto,A., Goto,M., Furuichi,Y. and Ikeda,H. (1998) Bloom’s and Werner’s syndrome genes suppress hyperrecombination in yeast sgs1 mutant: implication for genomic instability in human diseases. Proc. Natl Acad. Sci. USA, 95, 8733–8738. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang J., Chang,E., Cherry,A.M., Bangs,C.D., Oei,Y., Bodnar,A., Bronstein,A., Chiu,C.P. and Herron,G.S. (1999) Human endothelial cell life extension by telomerase expression. J. Biol. Chem., 274, 26141–26148. [DOI] [PubMed] [Google Scholar]
- Yeager T.R., Neumann,A.A., Englezou,A., Huschtscha,L.I., Noble,J.R. and Reddel,R.R. (1999) Telomerase-negative immortalized human cells contain a novel type of promyelocytic leukemia (PML) body. Cancer Res., 59, 4175–4179. [PubMed] [Google Scholar]
- Yu C.E. et al. (1996) Positional cloning of the Werner’s syndrome gene. Science, 272, 258–262. [DOI] [PubMed] [Google Scholar]
- Zhu X.-D., Kuster,B., Mann,M., Petrini,J.H. and de Lange,T. (2000) Cell-cycle-regulated association of RAD50/MRE11/NBS1 with human telomeres. Nature Genet., 25, 347–352. [DOI] [PubMed] [Google Scholar]