

Abstract
We have studied the ubiquitination and degradation patterns of the human securin/PTTG protein. We show that, in contrast to budding yeast pds1, securin degradation is catalyzed by both fzy (fizzy/cdc20) and fzr (fizzy-related/cdh1/hct1). Both fzy and fzr also induce the APC/C to ubiquitinate securin in vitro. Securin degradation is mediated by an RXXL destruction box and a KEN box, and is inhibited only when both sequences are mutated. Interestingly, the non-degradable securin mutant is also partially ubiquitinated by fzy and fzr in vitro. Expressing the non-degradable securin mutant in cells frequently resulted in incomplete chromatid separation and gave rise to daughter cells connected by a thin chromatin fiber, presumably of chromosomes that failed to split completely. Strikingly, the mutant securin did not prevent the majority of sister chromatids from separating completely, nor did it prevent mitotic cyclin degradation and cytokinesis. This phenotype, reminiscent of the fission yeast cut (cells untimely torn) phenotype, is reported here for the first time in mammals.
Keywords: APC/cyclosome/fizzy/PTTG/securin
Introduction
Sister chromatid separation in mitosis is rigorously controlled to ensure equal chromosome segregation. The century-old mystery of how chromosomes dissociate at anaphase has been solved elegantly both in yeast (Uhlmann et al., 1999) and in mammalian cells (Waizenegger et al., 2000). The two sister chromatids are glued together by a group of proteins collectively termed cohesins (Michaelis et al., 1997). One of these cohesins, Scc1p, is cleaved at the metaphase–anaphase transition by the evolutionarily conserved separin protein. Premature activation of separin is prevented by binding to securin, i.e. pds1 (precocious dissociation of sister chromatids) in budding yeast (Cohen-Fix et al., 1996; Yamamoto et al., 1996; Ciosk et al., 1998) and cut2 (cells untimely torn) in fission yeast (Funabiki et al., 1996). Homologs of pds1/cut2 were identified recently as PTTG (pituitary tumor-transforming gene) in vertebrates (Pei and Melmed, 1997; Zou et al., 1999), and as Pim1 in Drosophila (Leismann et al., 2000). All securins, i.e. pds1, cut2, PTTG and Pim1, bind separin but share no sequence homology except for the RXXL destruction box (d-box), which targets them for degradation.
The degradation of cyclin B and sister chromatid separation are independent events which rely on the same pathway of ubiquitin-mediated proteolysis (Holloway et al., 1993). The study of this pathway has gained considerable momentum since the discovery of the cyclosome (Sudakin et al., 1995), also called the APC (anaphase-promoting complex) (King et al., 1995). The APC/cyclosome (APC/C) is a ubiquitin ligase (E3) complex containing about a dozen different subunits which ubiquitinates mitotic cyclins (Sudakin et al., 1995), securin (Cohen-Fix et al., 1996; Funabiki et al., 1996; Zou et al., 1999; Leismann et al., 2000) and other cell cycle proteins (Morgan, 1999; Zachariae and Nasmyth, 1999). The APC/C is activated by WD repeat proteins, which are not permanent components of the complex but interact with it transiently in a cell cycle-specific manner. These proteins have been identified in all eukaryotes and fall into two distinct groups: (i) fizzy (Drosophila) (Sigrist et al., 1995), p55CDC (rat and human) (Weinstein et al., 1994), cdc20 (budding yeast) (Visintin et al., 1997) and slp1 (fission yeast) (Matsumoto, 1997); and (ii) fizzy-related (Drosophila, Xenopus and mammals) (Sigrist and Lehner, 1997; Fang et al., 1998b; Kramer et al., 1998; Lorca et al., 1998), hct1/cdh1 (budding yeast) (Schwab et al., 1997) and ste9 (fission yeast) (Kitamura et al., 1998). The activation pattern of the APC/C is remarkably conserved from yeast to man. It is activated at metaphase and its activity persists until the G1–S transition (Amon et al., 1994; Brandeis and Hunt, 1996). The APC/C initially is activated by fizzy (fzy) and this activation requires phosphorylation of APC/C subunits by cdk1 (Shteinberg et al., 1999; Kramer et al., 2000; Rudner and Murray, 2000). The APC/Cfzy is negatively regulated by the metaphase checkpoint mechanism (Fang et al., 1998a; Hwang et al., 1998; Kallio et al., 1998) preventing its premature activation.
Fzr activates the APC/C in anaphase and in G1, and this activation leads to the degradation of fzy (Shirayama et al., 1998; Pfleger and Kirschner, 2000). Fzr binds both phosphorylated, mitotic, and unphosphorylated, interphase APC/C. Fzr is phosphorylated by cdk1, and in mammals also by cdk2. This phosphorylation inhibits its binding to the APC/C and prevents APC/Cfzr activation from S phase to anaphase (Zachariae et al., 1998; Lukas et al., 1999; Listovsky et al., 2000). According to the prevailing model (Morgan, 1999; Zachariae and Nasmyth, 1999), APC/Cfzy activity is required for the degradation of securin while APC/Cfzr activity, which appears later in mitosis, is essential for the degradation of mitotic cyclins, fzy and other substrates, leading up to cytokinesis. Fzr is further responsible for the G1-specific APC/C activity (Sigrist and Lehner, 1997), as well as for its activity in arrested and differentiated cells (Gieffers et al., 1999).
We show here that human securin is ubiquitinated and degraded by fzy and fzr. Securin ubiquitination and degradation depend on a conserved RXXL d-box and on a KEN box (single-letter amino acid code). A non-degradable double mutant securin was also ubiquitinated, albeit to a lesser degree than the wild-type protein. We also show that the expression of non-degradable securin in mammalian cells does not prevent the bulk of the chromatids from separating and allows for the completion of mitosis and cytokinesis. It nevertheless partially interferes with sister chromatid separation, giving rise to daughter cells connected by a thin thread of chromatin.
Results
The APC/C is associated with fzr during G1
We examined the interaction of fzr with the APC/C during the different phases of the mammalian cell cycle in relation to cyclin B1 degradation. As cyclin B1 levels are determined both by transcription and by degradation, we used a constitutively transcribed cyclin B1–CAT (chloramphenicol acetyl transferase) reporter. Cyclin B1–CAT codes for the N-terminal 106 residues of cyclin B1, including its d-box, and is degraded like the endogenous cyclin (Brandeis and Hunt, 1996). A d-box mutant of B1–CAT is as stable as wild-type CAT protein. NIH 3T3 cells stably expressing the cyclin B1–CAT reporter were synchronized with nocodazole for 16 h and prometaphase cells were collected by shake-off. The cells were washed, replated in fresh medium and harvested at intervals for an entire cell cycle of 18 h. The APC/C was immunoprecipitated with an antibody directed against one of its subunits, cdc27 (King et al., 1995). Figure 1A shows that the levels of cdc27 remained constant throughout the cell cycle. Fzr, in contrast, co-immunoprecipitated with the APC/C only during G1. This closely correlates with the APC/C activity, as demonstrated by the B1–CAT activity (Figure 1B). Fzr dissociation from the APC/C and its inactivation thus take place at roughly the same time.
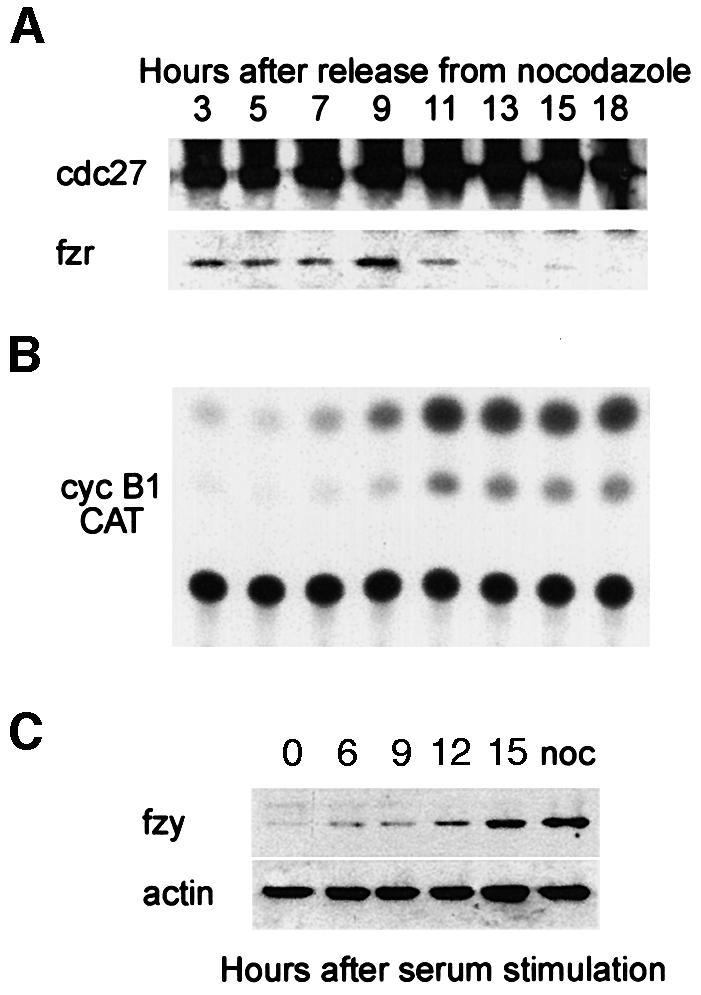
Fig. 1. Fzr binding to the APC/C is associated with its G1-specific activity. (A) NIH 3T3 cells stably expressing cyclin B1–CAT were synchronized at prometaphase with nocodazole and subsequent shake-off of the rounded cells. They were washed and released into fresh medium and harvested at the indicated time points. Cells synchronized by this method pass the G1–S transition at ∼7–9 h. Cell extracts were immunoprecipitated with anti-cdc27 beads. The immunoprecipitates were analyzed by immunoblotting with fzr and cdc27 antibodies. (B) The cells were analyzed further for CAT activity, an indication of the stability of the cyclin B1–CAT and thus of APC/C activity or inactivity. (C) Serum-starved, G0-arrested fibroblasts were stimulated with serum to re-enter the cell cycle and were harvested at the indicated time points. They were immunoblotted with fzy antibodies and with α-actin antibodies which served as a loading control. Entry into S phase takes place at 12–15 h after stimulation. An extract of nocodazole-arrested cells, at a time when fzy levels peak in the cell, was used as a positive control.
We have shown previously that fzy is absent during G1 (Inbal et al., 1999). To test that the same is also true for G0, we arrested cells by serum starvation and immunoblotted them at different time points after stimulation with serum. Figure 1C shows that fzy is absent during G0 and appears only when cells reach S phase, 12–15 h after stimulation. Taken together, these results suggest that all APC/C activity observed in G1 and G0 is due to the APC/Cfzr and not the APC/Cfzy.
Securin degradation is mediated by the APC/Cfzy
The activity of the APC/Cfzy is limited to a narrow time window during mitosis. To obtain cells with active APC/Cfzy, we transfected them with non-degradable cyclin B1 to arrest them in telophase with separated and condensed chromatids (Gallant and Nigg, 1992; Wheatley et al., 1997). These cells maintain an active APC/Cfzy and the high cdk1 activity in them prevents fzr from binding to the APC/C (Listovsky et al., 2000). We transfected cells with an indestructible green fluorescent protein (GFP)–cyclin B1 expression vector and either treated them with nocodazole or enabled them to proceed and arrest in telophase. Both prometaphase- and telophase-arrested cells were harvested by mitotic shake-off and analyzed by immunoblotting. Figure 2 shows that both securin and cyclin B1 are stable in prometaphase and are degraded in telophase, presumably by the APC/Cfzy.
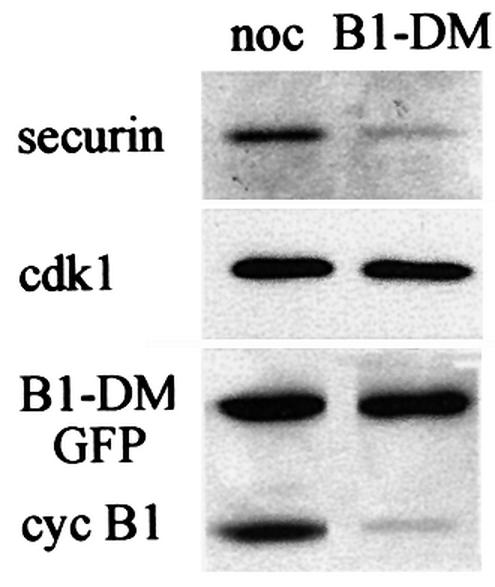
Fig. 2. APC/Cfzy-mediated degradation of securin. Cells were transfected with non-degradable GFP–cyclin B1 and either arrested in prometaphase with nocodazole or allowed to proceed and arrest in telophase. Cells were obtained by mitotic shake-off, and securin and cyclin B1 levels were analyzed by immunoblotting. Cdk1, which is stable in G1, served as a loading control.
The degradation patterns of securin and cyclin B1 are identical
To study securin degradation in mammalian cells, we fused the N-terminal 87 residues of the human securin gene (a kind gift of Drs M.W.Kirschner and H.Zou) to the CAT gene to make a securin–CAT reporter. We used only the N-terminal part of securin comprising its d-box (RKALGTVNR) to minimize the chance that securin–CAT will bind separin and interfere with the cell cycle. The securin–CAT reporter was stably transfected into NIH 3T3 cells, as described for the cyclin B1–CAT reporter, and its fate was followed along the cell cycle. Figure 3A shows the securin–CAT activity pattern to be similar to that of cyclin B1–CAT shown in Figure 1B. Figure 3B shows that the constitutively expressed securin–CAT reporter is also degraded in G0 when fzy is undetectable (Figure 1C) and all the APC/C activity is due to the APC/Cfzr.
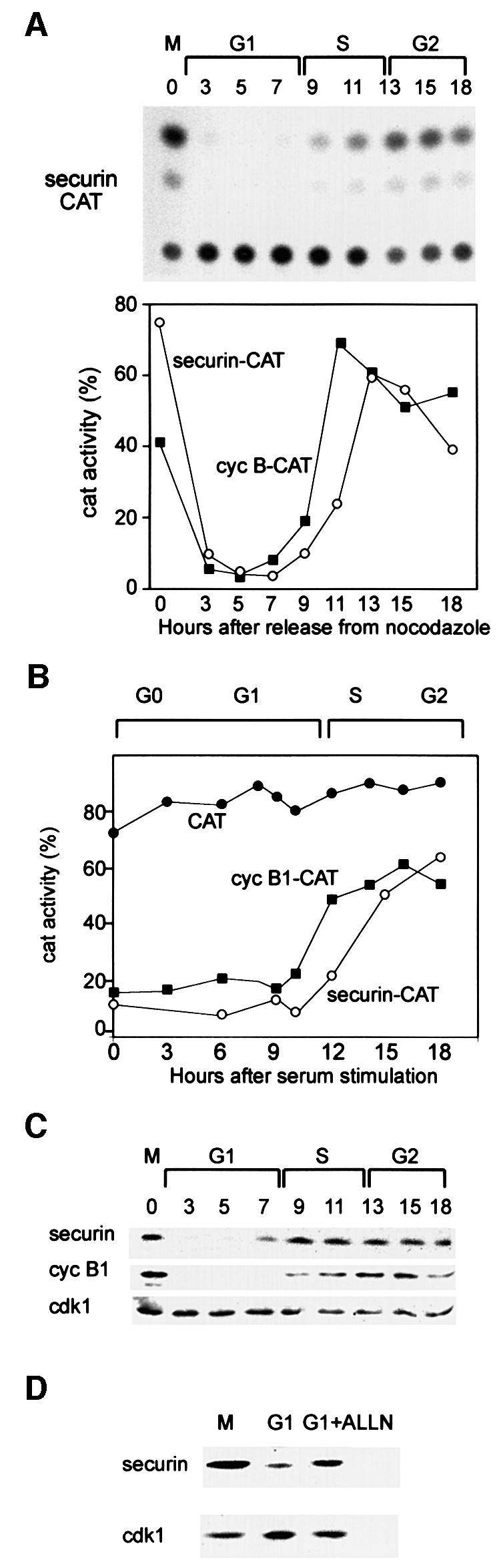
Fig. 3. Securin and cyclin B1 have a similar degradation pattern in G1. (A) Cells were stably transfected with the securin–CAT expression vector which constitutively transcribes a reporter fusion protein between the N-terminal 87 residues of human securin and a bacterial CAT protein. Cells were synchronized as described and assayed for CAT activity. The CAT activity was calculated as the ratio of the amount of diacetylated [14C]chloramphenicol to that of total [14C]chloramphenicol (diacetylated + non-acetylated) as quantified by a phosphoimager. The changes in the activities of securin–CAT (open circles) and cyclin B1–CAT (filled squares) (shown in Figure 1) were plotted as a function of the cell cycle phases when released from a nocodazole block. (B) Cells stably expressing CAT (filled circles), cyclin B1–CAT (filled squares) and securin–CAT (open circles) were arrested by serum deprivation in G0 and released by serum stimulation. Cells were harvested and analyzed for CAT activity at the indicated time points. (C) Cells were synchronized at prometaphase by nocodazole arrest and mitotic shake-off, released into fresh medium and harvested at the indicated time points for immunoblotting with securin, cyclin B1 and cdk1 antibodies. (D) Prometaphase-arrested cells were washed and released into fresh medium for 2 h, and then either harvested or grown for an additional 2 h with or without ALLN. Cell extracts were immunoblotted with securin or cdk1 antibodies. Cdk1 is constant in G1 and served as a loading control.
We further examined the expression pattern of the endogenous securin (Figure 3C) and found that it was identical to that of cyclin B1 as well as to that of fzy (Inbal et al., 1999), which is an APC/Cfzr substrate (Pfleger and Kirschner, 2000). Cyclin B1 is not transcribed in G1; however, it is synthesized from the mRNA left in the G1 cells (Brandeis and Hunt, 1996). We treated G1 cells with the proteasome inhibitor ALLN to test whether the absence of securin is a result of its ongoing degradation. Figure 3D shows that ALLN-treated G1 cells accumulated securin, suggesting that securin is not only a potential substrate of G1-specific proteolysis but is indeed both actively synthesized and degraded in G1.
Fzr overexpression and cdk1 inhibition induce securin destruction
The experiments described so far show that securin is degraded in the cell when the APC/Cfzy or the APC/Cfzr are active. This holds both for endogenous securin and for the constitutively expressed securin–CAT fusion protein. We next attempted to translate this ‘guilt by association’ to a more causal relationship. Fzr overexpressed in prometaphase-arrested cells is capable of premature activation of the APC/C to degrade cyclin B1 (Listovsky et al., 2000). Figure 4A shows that fzr overexpression also leads to efficient degradation of the securin–CAT reporter in prometaphase.
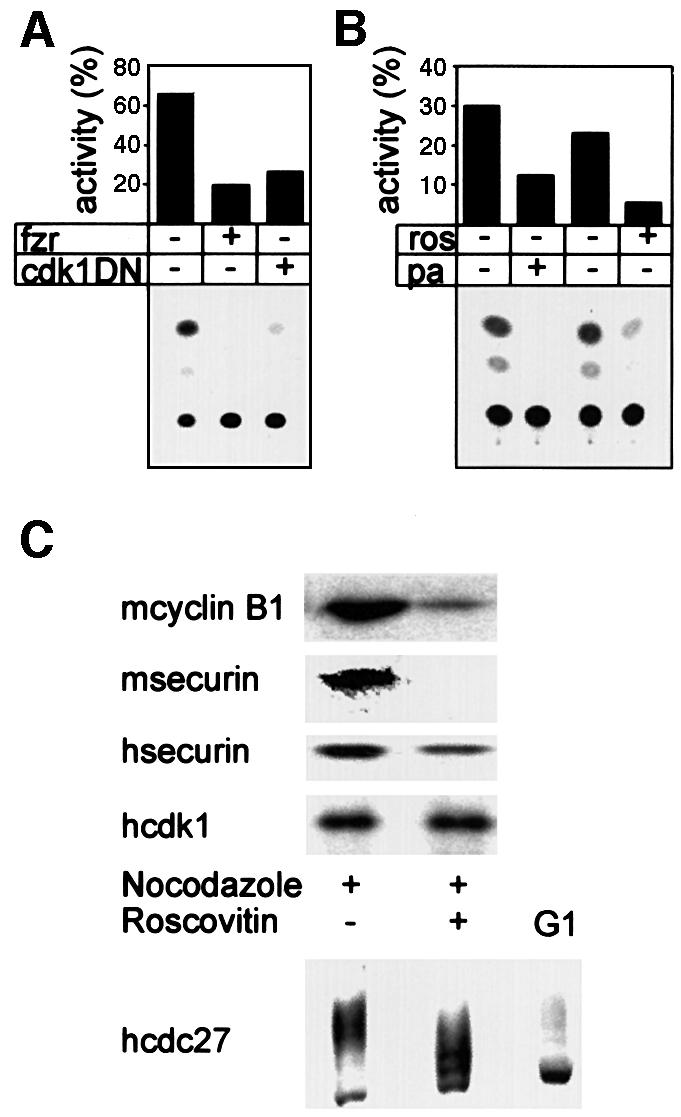
Fig. 4. Fzr overexpression and cdk1 inactivation lead to securin degradation in prometaphase. (A) Cells were co-transfected with securin–CAT with a fzr expression vector, a cdk1DN expression vector or an empty vector. They were arrested with nocodazole, harvested by shake-off and analyzed for CAT activity. Both fzr overexpression and cdk1 inhibition strongly activated the APC/C-specific degradation of securin–CAT in prometaphase. (B) Cells expressing securin–CAT were arrested in prometaphase by nocodazole and subsequently treated for 3 h with the cdk inhibitors roscovitin and purvalanol A, in the presence of nocodazole. They were then assayed for CAT activity. (C) Mouse (m) NIH 3T3 fibroblasts or human (h) HeLa cells were treated as in (B) and immunoblotted with securin, cyclin B1, cdk1 and cdc27 antibodies. The last antibody was used to show that roscovitin indeed leads to cdk1 inhibition in vivo as seen by the decrease in the phosphorylation of cdc27, a known cdk1 phosphorylation substrate.
The phosphorylation of fzr by cdk2–cyclin A in S phase and G2, and by cdk1–cyclin B in early M, inhibits fzr premature binding to the APC/C. Cdk can be inactivated in prometaphase, either by a cdk1 dominant-negative expression vector or by the cdk1- and cdk2-specific inhibitor roscovitin. This inactivation leads to fzr-specific binding and activation of the APC/C, and to premature cyclin B1 degradation (Listovsky et al., 2000). Figure 4 shows that cdk inactivation in prometaphase also leads to securin degradation. This was achieved either by co-transfection of dominant-negative cdk1 (Figure 4A) or by treatment with the cdk inhibitors roscovitin or purvalanol A (Figure 4B). The treatment of mouse or human prometaphase cells with roscovitin also induced the degradation of endogenous securin and cyclin B1 (Figure 4C). Roscovitin activity is reversible and its in vivo activity cannot be tested by in vitro assay of cdk kinase activity in extracts of treated cells. We therefore investigated the phosphorylation of cdc27, which is a known cdk1 substrate (Shteinberg et al., 1999; Kramer et al., 2000; Rudner and Murray, 2000). Figure 4C shows that cdc27 is significantly less phosphorylated upon roscovitin treatment. We are aware, however, that this is only a very indirect method by which to assess the inhibition of cdk1 by roscovitin. These results stress the importance of cdk1, and presumably also of cdk2, for preventing premature securin degradation before the onset of metaphase.
Both fzy and fzr activate the APC/C to ubiquitinate securin in vitro
To support our observations that securin degradation is mediated in vivo by the APC/Cfzy as well as the APC/Cfzr, we used an in vitro ubiquitination assay. The substrate for this assay was a full-length 35S-labeled securin synthesized in reticulocyte lysate. The reaction mix contained a partially purified APC/C from mitotic HeLa cells, E1, E2 (UbcH10), an energy-regenerating system, ubiquitin and ubiquitin aldehyde. The assay was performed in the absence or presence of fzy, fzr, or both fzy and fzr. Figure 5A shows that securin was not modified by the APC/C alone (lane 1), but when incubated with fzy (lane 2) or fzr (lane 3) it was converted to much more slowly migrating forms, presumably by ubiquitination. Fzy and fzr had a roughly equal ubiquitination activity and we did not observe any synergism when both of them were added (lane 4), suggesting that each of them is capable of maximal activation of this reaction.
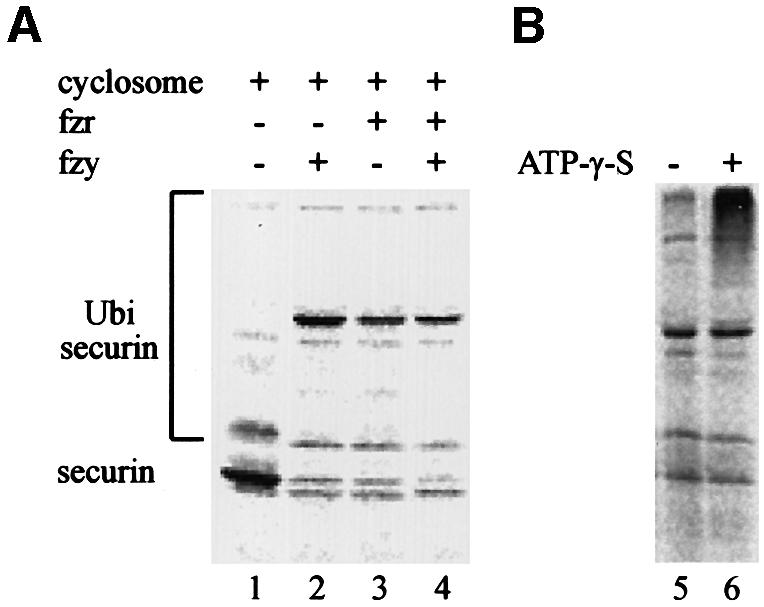
Fig. 5. Both fzy and fzr can activate the APC/C to ubiquitinate securin. (A) An in vitro ubiquitination assay was performed on 35S-labeled securin expressed in reticulocyte lysate (lane 1) with a partially purified APC/C from mitotic HeLa cells in the presence of bacterially expressed E1, E2-C, an energy-regenerating system and ubiquitin aldehyde, which inhibits de-ubiquitination. This reaction was supplemented with either fzy (lane 2), fzr (lane 3) or both (lane 4). (B) Some of the ubiquitinated securin must have been degraded by the proteasome in the reticulocyte lysate. We therefore repeated the ubiquitination reaction in the presence (lane 6) or absence (lane 5) of ATP-γ-S, which enables ubiquitination but not proteasome-specific degradation.
Most of the securin in the reaction was degraded, probably by the proteasome present in the reticulocyte lysate. To show that this degradation is proteasome dependent, we replaced the ATP and the energy-regenerating system in the reaction with ATP-γ-S. This ATP analog is capable of supporting ubiquitination but not proteasome-specific degradation. Figure 5B shows that when ATP was substituted with ATP-γ-S, the substrate was no longer degraded but accumulated as more slowly migrating forms of presumably highly ubiquitinated securin.
Degradation of securin can be signaled by two conserved motifs
The human securin gene has a conserved d-box RKALGTVNR. We mutated this d-box to AKAAGTVNR in the securin–CAT reporter (securin-DM–CAT), and to our surprise this protein was only partially stabilized and was still degraded in the same distinct cell cycle-specific manner as the wild-type reporter during G1 (Figure 6) and G0 (data not shown). The analogous mutation in cyclin B1–CAT completely stabilized the fusion protein (Brandeis and Hunt, 1996). We examined the sequence of securin and detected a putative KEN box 10 residues from its N-terminus. The KEN box recently has been discovered to be an APC/Cfzr-specific degradation signal which drives the degradation of vertebrate fzy as well and other proteins (Pfleger and Kirschner, 2000). We mutated the KEN sequence to KAA, preserving the lysine residue which might be a ubiquitination target. This mutation had little effect on the degradation of the securin–CAT reporter. The introduction of both a KAA and d-box mutation into securin–CAT, however, completely abolished its cell cycle-specific oscillations (Figure 6) and its degradation in G0 (data not shown).
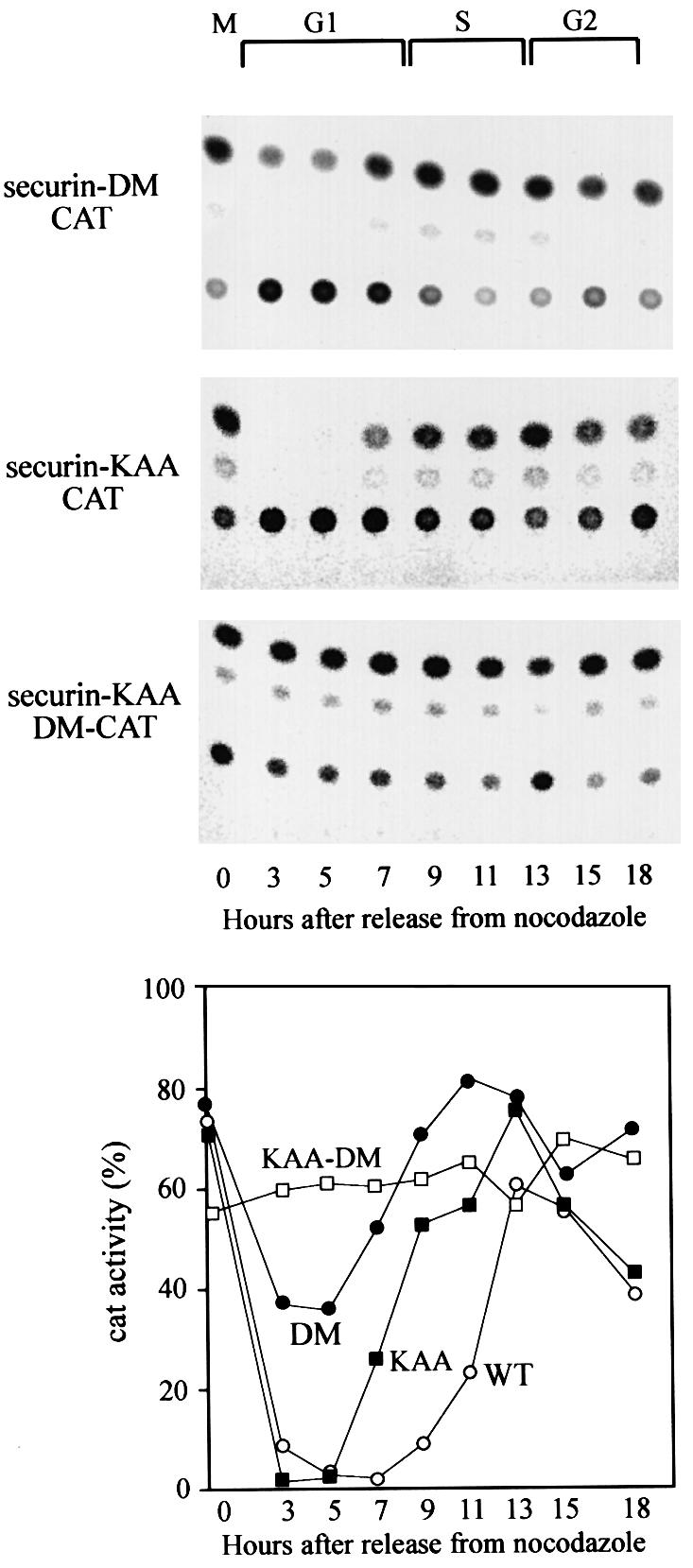
Fig. 6. Securin has two conserved sequences that signal its APC/C-specific degradation. Cells stably expressing securin-DM–CAT (filled circles), which carries a mutated d-box, securin-KAA–CAT (filled squares) with a mutated KEN box and securin-KAA-DM–CAT (open squares) with a double mutation were generated. These cells were released from a nocodazole block, harvested at the indicated time points and the CAT assays are shown in the top panels. The quantified CAT activity of the above mutants, as well as of wild-type securin–CAT (open circles), shown in Figure 3, is plotted in the bottom panel.
Full-length securin-DM and securin KAA were ubiquitinated in vitro by both the APC/Cfzy and the APC/Cfzr. Surprisingly the non-degradable double mutant was also ubiquitinated to some extent (Figure 7A). We thus expected to find an additional ubiquitination/degradation signal at the C-terminal half of securin. This part is present in the full-length securin used as a substrate of the in vitro ubiquitination reaction but is absent in securin–CAT. We prepared two new securin–CAT constructs, one full-length with mutated KEN and d-boxes (fl-sec-KAA-DM–CAT) and a second with only the C-terminal part of securin (sec-C-term–CAT). Figure 7B shows that both constructs were completely stable at the M–G1 transition, indicating that no additional degradation signals are present in securin.

Fig. 7. Both degradable and non-degradable securin mutants are ubiquitinated by the APC/C. (A) Full-length securin–CAT, securin-DM–CAT, securin-KAA–CAT and securin-KAA-DM–CAT were expressed in reticulocyte lysate and tested for ubiquitination as described in the legend to Figure 5, in the presence of ATP-γ-S. (B) Plasmids expressing either full-length non-degradable securin–CAT (fl-sec-KAA-DM–CAT) or the C-terminal half of securin fused to CAT (sec-C-term–CAT) were transfected into cells. Cells were arrested in prometaphase and were either harvested immediately for CAT assay or first released into G1 for 3 h.
Non-degradable securin interferes with complete sister chromatid separation
To check the in vivo significance of securin degradation, we transfected cells with expression vectors of a wild-type securin and a non-degradable double KAA-DM securin mutant. To identify the transfected cells, we co-transfected them with a vector expressing GFP-tagged histone H2A. GFP–H2A is incorporated into the chromatin and has no effect on the cells, as even lines stably expressing it can be propagated over many generations (A.Laronne and M.Brandeis, unpublished). When Zou et al. (1999) added a DM mutant of securin to Xenopus embryos their chromosomes failed to segregate. Surprisingly, the transfected NIH 3T3 cells did not arrest in metaphase, or in any other stage of mitosis. We observed many pairs of adjacent cells with green nuclei that had divided recently. However, many of the daughter cells that expressed the non-degradable, but not the wild-type, securin were connected by a thin thread of green chromatin (Figure 8A). Staining of these cells with the DNA-specific dye Hoechst 33258 confirmed that the connecting thread included DNA. These DNA connections between the chromatin of the divided cells show that chromatid separation was incomplete. Many hundreds of cells were visualized in several different experiments and ∼1/3 of them were connected by chromatin threads. Figure 8A, panel 4 shows an example of a case where the daughter nuclei failed to separate. Panel 6 shows an example where the size of the two daughter nuclei was not identical. Some HeLa cells transfected with non-degradable securin showed the same failure to separate their chromatids (panel 7), but the proportion of incomplete separation was <5% compared with ∼30% in NIH 3T3 cells.

Fig. 8. Expression of indestructible securin interferes with chromatid separation. (A) NIH 3T3 cells were transfected with vectors expressing non-degradable (panels 1, 2, 4, 5 and 6) and wild-type (panel 3) securin together with a GFP–histone H2A expression vector (green panels). Cells were stained further in vivo with Hoechst 33258 (blue panels). About 1/3 of the cells expressing the mutant securin failed to separate all their chromatids prior to cytokinesis and remained connected by a thin chromatin string (arrow), or even failed to complete nuclear division altogether (panel 4). The same phenotype was observed in some HeLa cells expressing non-degradable securin (panel 7). (B) Cells transfected with an empty vector or with non-degradable securin were arrested in prometaphase and either harvested immediately or released first into G1 for 3 h. Cell extracts subsequently were immunoblotted with securin antibodies. (C) Cells were transfected with an expression vector for myc-tagged wild-type and non-degradable securin. Extracts prepared from transfected and untransfected cells were immunoprecipitated with myc tag (9E10) antibodies and immunoblotted with separin and securin antibodies. (D) Cells were co-transfected with expression vectors for securin (wild-type or non-degradable), non-degradable cyclin B1 and GFP–H2A. Cells were arrested in mitosis and were harvested by shake-off. Chromosome spreads were prepared as described in Materials and methods. Cells transfected with wild-type, and the majority of those transfected with non-degradable securin, completely dissociated chromatids (panel 1). However, many of those transfected with non-degradable securin still had a few (panel 2; arrow shows a separated chromosome), most (panel 3; arrow shows an unseparated chromosome) or all (panel 4) chromatids paired.
We used immunoblotting to estimate the extent of overexpression of the non-degradable securin in the transfected cells. In the experiment shown in Figure 8B, cells were transfected with the full-length securin-KAA-DM expression vector, arrested with nocodazole in prometaphase and released into G1. The level of securin in the extract of transfected cells was ∼10 times higher than in the extract of mock-transfected cells. Not more than 1/5 of the cells used for the extract took up the expression vector and they must, therefore, have expressed about two orders of magnitude more non-degradable than endogenous securin. Nevertheless, these cells were capable of completing mitosis and entering G1 without degrading this securin and apparently without considerable cell loss (Figure 8B).
Securin binds to separin and prevents it from cleaving the cohesins that glue sister chromatids together. The non-degradable securin carries only four point mutations. It could not be ruled out, however, that metaphase arrest is not observed because these mutations prevent securin from binding separin. We therefore expressed wild-type and KAA-DM myc-tagged securin in cells and immunoprecipitated them with antibodies against the tag. Figure 8C shows an immunoblot of these immunoprecipitates with separin and securin antibodies. Both wild-type and mutant securins precipitated equal amounts of separin, showing that the mutations did not affect the capacity of securin to bind separin. Cells transfected with the mutant myc-tagged securin showed the same phenotype of incomplete chromatid separation demonstrated by the untagged version (data not shown).
The failure of cyclin B to degrade causes cells to arrest in telophase with separated and condensed chromatids (Gallant and Nigg, 1992; Wheatley et al., 1997). Non-degradable securin expression did not cause such an arrest, as already observed in frog embryos (Zou et al., 1999). Once the APC/C has been activated upon the release of the metaphase checkpoint, cyclin B seems to be degraded regardless of securin degradation. Cyclin B degradation led to chromosome decondensation and prevented the direct study of the chromosomes in cells expressing the non-degradable securin. We co-transfected cells with expression vectors for securin (wild-type or non-degradable), non-degradable cyclin B1 and GFP–histone H2A. The non-degradable securin construct was used in 3-fold excess over the cyclin construct to ensure securin expression. The transfected cells were arrested in mitosis with condensed chromatids due to the persisting cyclin B activity. We prepared chromosome spreads from these cells and observed the GFP-positive chromosomes. Chromatids were separated in almost all cells transfected with wild-type securin (Figure 8D, panel 1). In contrast, chromatids in many of the cells transfected with non-degradable securin were either fully (panel 4) or partially (panels 2 and 3) paired.
Discussion
We have studied the degradation of mammalian securin and have described four main observations which will be discussed below. First, securin degradation is mediated by both fizzy and fizzy-related. Secondly, cell cycle-specific ubiquitination, and degradation, of securin are mediated by two signals: an RXXL d-box and a KEN box. Thirdly, even the non-degradable securin is partially ubiquitinated by the APC/C. Finally, securin degradation is dispensable for complete separation of most chromatids and is not essential for completion of mitosis or for cytokinesis.
The pattern of mammalian securin degradation by the APC/C pathway of ubiquitin-mediated proteolysis is very similar to the degradation of cyclin B1 both in mitosis and during G1 and G0. The APC/C is activated initially by fzy upon the release of the spindle checkpoint at the metaphase transition. Fzy is degraded later in mitosis and is replaced by the fzr APC/C activator. Fzr activates the APC/C in G1 and G0 (Figure 1A) and, during these phases, fzy is unstable and completely absent (Inbal et al., 1999; and Figure 1C). A real-time observation of APC/Cfzy-specific degradation is difficult and the precise timing of switching between fzy and fzr during mitosis is not known. Cells arrested with non-degradable cyclin B1, however, arrest in telophase with an active APC/Cfzy. Both cyclin B1 and securin are degraded in these cells (Figure 2). The observation that securin degradation can, and does, also take place during G1 (Figure 3) strongly suggested that it is, like cyclin B, also an APC/Cfzr substrate. To support this point, we showed that securin can be degraded prematurely by the overexpression of fzr in prometaphase (Figure 4). cdk1 and cdk2 phosphorylate fzr and prevent it from activating the APC/C. Inhibition of cdk in prometaphase prematurely activated the APC/Cfzr to degrade both cyclin B and securin at the wrong time. This stresses the importance of cdk2 and cdk1 activity for the preservation of securin until its scheduled degradation. Finally, we showed that both fzy and fzr are capable of specifically inducing the APC/C to ubiquitinate securin in vitro (Figure 5).
These findings are different from securin-pds1 degradation in budding yeast, where it is degraded only by the fzy homolog cdc20 and precedes the proteolysis of clb2 by 20 min, a fair part of their rapid cell cycle. The observation that indestructible cyclin B arrests mammalian cells (Gallant and Nigg, 1992; Wheatley et al., 1997) as well as Xenopus embryos (Holloway et al., 1993) in telophase suggested that metazoan securin and cyclin B are also degraded by separate mechanisms. Recent real-time observations, however, showed that mammalian cyclin B starts to be degraded during metaphase (Clute and Pines, 1999). The role of fzy and fzr in the activation of the APC/C are unknown and there is little evidence in metazoa that they confer any substrate specificity. The only known example is that fzy cannot be ubiquitinated by itself but only by fzr (Pfleger and Kirschner, 2000). This might be an exception governed by a specific mechanism which prevents fzy from self-destruction.
The observation that a human securin with a mutated d-box is degraded in a cell cycle-specific manner is surprising. A closer look at the sequence of human securin reveals a putative KEN box. This sequence was shown recently to be the signal targeting fzy and other proteins for APC/C-specific degradation (Pfleger and Kirschner, 2000). This box is not evolutionarily conserved in securin, even in closely related species (Zou et al., 1999), and its mutagenesis to KAA had no effect on securin stability. However, when this mutation was introduced into the unstable d-box mutant, it was completely stabilized (Figures 6 and 8B). The existence of two different degradation signals is not unprecedented. The fission yeast securin cut2 (Funabiki et al., 1996) and the Xenopus cyclin B1 (Pfleger and Kirschner, 2000) have two d-boxes, and cdc6 has a d-box and a KEN box (Petersen et al., 2000).
We show that securin is ubiquitinated specifically by the fzy- and fzr-activated APC/C in vitro. The d-box, KEN box and even the non-degradable double mutant were also partially ubiquitinated. A related observation was made previously by Klotzbücher et al. (1996) who replaced the d-box of cyclin B1 with that of cyclin A. This chimeric protein was no longer degraded, but was still partially ubiquitinated. Most of the ubiquitinated non-degradable securin (Figure 7A) did not accumulate as the high molecular weight form, which could be the form that is targeted for degradation.
Cells transfected with non-degradable securin underwent mitosis and cytokinesis. In many cases, however, they failed to separate all their chromatids and remained connected by a thin thread of chromatin. Chromosome spreads (Figure 8D) suggest that this chromatin thread represents one or more non-separated chromosomes. This phenotype is reminiscent of the cut phenotype of fission yeast but was never reported for mammalian cells. In view of the high levels of non-degradable securin persisting in the cells (Figure 8B), it is astonishing that cells were capable of separating most of their chromatids. This is in contrast to observations in other organisms (Cohen-Fix et al., 1996; Funabiki et al., 1996; Zou et al., 1999; Leismann et al., 2000) and to the prevailing model that securin prevents premature chromatid separation. In vertebrates, the loss of most of the cohesins happens before metaphase (Losada et al., 1998) and the release of cohesins from the chromatids is both cleavage- and separin-dependent, and non-cleavage- and separin-independent (Waizenegger et al., 2000). Our observations suggest that mammalian chromatids lose enough of their cohesins without separin activity to enable most of them to separate even in the presence of non-degradable securin.
Cells transiently expressing dominant-negative UbcH10, the ubiquitin carrier (E2) of the APC/C, arrest at metaphase (Townsley et al., 1997). A plausible explanation for this arrest was that cells could not degrade securin and mitotic cyclins. However, when we co-expressed non-degradable mutants of both securin and cyclin B1, cells arrested in telophase. This indicates that APC/C substrates other then securin and cyclin B must be degraded for the metaphase–anaphase transition to occur. A likely candidate could be the recently discovered Xkid frog chromokinesin. Xkid is degraded by the APC/C and is essential for the positioning of the chromosomes on the metaphase plate and for the maintenance of this plate (Antonio et al., 2000; Funabiki and Murray, 2000). If mammalian kid has similar properties, its stabilization could be involved in the metaphase arrest caused by dominant-negative UbcH10.
It is also of great interest that cells are capable of completing mitosis and cytokinesis without separating all their chromosomes. We observed that securin and cyclin B degradation can proceed in parallel and are not interdependent. Similar observations were made in Xenopus embryos (Zou et al., 1999). This is probably the most fundamental difference between mammalian securin and its budding yeast homolog pds1, which is part of the checkpoint mechanism and prevents fzr activation and completion of mitosis (Cohen-Fix and Koshland, 1999; Shirayama et al., 1999; Tinker-Kulberg and Morgan, 1999). We nevertheless find it striking that no checkpoint mechanism prevents cells with incompletely separated chromatids from undergoing cytokinesis.
Fission yeast cut8 is required for proteasome localization to the nuclear periphery and securin-cut2 degradation is delayed in cut8 mutants (Tatebe and Yanagida, 2000). Interestingly, nematode embryos that are depleted of the nuclear lamina protein lamin divide but daughter nuclei remain connected by chromatin (Liu et al., 2000), similarly to the phenotype caused by non-degradable securin. We currently are investigating the dependence of securin degradation on the integrity of the nuclear lamina.
The identification of human securin as the PTTG oncogene was unexpected. Our results here suggest a possible explanation for the tumor-promoting activity of securin/PTTG. The non-degradable variant of securin leads to a high degree of genetic instability and unequal separation of chromatin to daughter cells. The incomplete separation could also lead to chromosome breakage due to the pulling force of the spindle and of the dividing cells. It is easy to imagine that overexpression of the wild-type variant could have a similar effect, even though at a lower rate.
Materials and methods
Plasmids
Securin–CAT was made by subcloning the HindIII fragment of pCS2-securin (a gift of H.Zou and M.Kirschner), which includes the first 263 nucleotides of human securin, into the HindIII site of pSV-CAT (Brandeis and Hunt, 1996) upstream of the CAT reporter gene. This expression vector codes for a fusion protein of the N-terminal 87 amino acids with the CAT protein driven by an SV40 promoter. Cyclin B1–CAT is a similar vector which expresses a fusion between the N-terminal 106 amino acids of mouse cyclin B1 and CAT (Brandeis and Hunt, 1996). The various mutants, securin-DM–CAT, securin-KAA–CAT and securin-KAA-DM–CAT, were made by the quick-change site-directed mutagenesis method (Stratagene). Securin-KS was made by cloning the securin BamHI–SalI fragment of pCS2-securin into Bluescript KS downstream of a T7 promoter. This plasmid was used to synthesize full-length securin in a coupled transcription–translation reticulocyte lysate reaction (Promega) in the presence of [35S]methionine. The mutants securin-DM-KS, securin-KAA-KS and securin-KAA-DM-KS were made by site-directed mutagenesis or by subcloning. The mammalian expression vectors pEF-securin and pEF-securin-KAA-DM were made by subcloning the genes into a pEF-plink2 expression vector. pCS2-myc-securin-KAA-DM was made by subcloning the mutant securin into pCS2-myc-securin (Zou et al., 1999). The fzr expression vectors pEF-fzr (Listovsky et al., 2000) and cdk1dn (van den Heuvel and Harlow, 1993) have been described previously. All inserts were sequenced to ensure that they contained the correct mutations and did not harbor any additional ones.
Cell culture, transfections and synchronization
Cells were maintained in Dulbecco’s modified Eagle’s medium (DMEM) supplemented with glutamine, penicillin/streptomycin and 10% fetal calf serum (FCS; Beit Haemek) and transfected by the CaPO4 co-precipitation method (Ausubel et al., 1994). The various CAT fusion expression vectors were co-transfected with a G418 resistance vector and resistant colonies were pooled. For transient transfection experiments of mitotic cells, the cells were co-transfected with the various plasmids as described in the Results. They were incubated overnight with the precipitate, washed the following morning, allowed to recover for several hours and were treated with nocodazole for ∼16 h. Prometaphase-arrested cells were obtained by shake-off and were processed for CAT assay or immunoblotting. Stable transfectants were synchronized either by nocodazole and shake-off, or by serum deprivation in 0.5% FCS for 48 h. The former were washed and released into fresh medium without nocodazole and the latter were released with 10% FCS. The following drugs were dissolved in dimethylsulfoxide (DMSO) and used at the indicated concentrations: nocodazole (Sigma) 2 µM; ALLN (Sigma) 150 µg/ml; roscovitin (Biomol) 28 µM; and purvalanol A (Alexis) 30 µM.
Antibodies
The following antibodies were used: rabbit securin and separin antibodies were a gift of H.Zou and M.Kirschner; mouse monoclonal antibodies against cdc27 (AF3), fzr (AR38), cdk1 (A17) and cyclin B1 (152) were a gift of J.Gannon and T.Hunt; rabbit fzr antibodies were a gift of C.Lehner; and goat p55cdc (fzy) and α-actin were purchased from SCB. AF3 antibodies were covalently coupled to protein A–Affiprep beads (Bio-Rad) as described (Harlow and Lane, 1987). CAT assays were performed by standard methods using [14C]chloramphenicol (Amersham) and acetyl-CoA (Roche).
In vitro ubiquitination assays
The in vitro ubiquitination assays were performed as previously described (Yudkovsky et al., 2000). The substrates of ubiquitination were prepared by in vitro transcription and translation of securin-KS and its mutant derivatives in a coupled reticulocyte lysate system (Promega) in the presence of [35S]methionine. Each reaction contained partially purified APC/C, E1, E2-C, an energy-regenerating system and fzy or fzr made in a reticulocyte system with non-radioactive methionine.
Green fluorescent chromosome spreads
Cells were co-transfected with expression vectors for cyclin B1-DM, securin (wild-type and non-degradable) and GFP–histone H2A. Cells were arrested as round mitotic cells and were obtained by shake-off. They were treated for 10 min with 0.5% KCl, applied to polylysine-coated coverslips and spun for 2 min at 1315 g (the coverslips were placed into inverted caps of 50 ml nunc tubes taped to capped 50 ml tubes). The cells were then fixed for 5 min in 3.5% formaldehyde and washed several times with phosphate-buffered saline (PBS) with 0.1% NP-40, and mounted with moviol. The slides were visualized on an inverted IX70 Olympus microscope and captured with a DVC-1300 digital camera. The images were analyzed with Northern Eclipse 5.0 (empix) and Photoshop 5 (Adobe) programs.
Acknowledgments
Acknowledgements
We are deeply indebted to Avram Hershko and Yana Yudkovsky for generous help with the in vitro assays and for many fruitful discussions. We would like to thank Hui Zou and Marc Kirschner for kindly providing human securin cDNA and antibodies against securin and separin, Christian Lehner for anti-fzr antibodies, and Julian Gannon and Tim Hunt for many of the other antibodies used for this study. We thank Chaya Miller and Tamar Listovsky for help throughout this project, and Mira Korner for dedicated sequencing. We also thank Yosef Gruenbaum for critical reading of the manuscript. This work was funded by grants from the Israeli Science Foundation (ISF, 124/98) and from the Israel–US Binational science fund (BSF, 9800123).
REFERENCES
- Amon A., Irniger,S. and Nasmyth,K. (1994) Closing the cell cycle circle in yeast: G2 cyclin proteolysis initiated at mitosis persists until the activation of G1 cyclins in the next cycle. Cell, 77, 1037–1050. [DOI] [PubMed] [Google Scholar]
- Antonio C., Ferby,I., Wilhelm,H., Jones,M., Karsenti,E., Nebreda,A. and Vernos,I. (2000) Xkid, a chromokinesin required for chromosome alignment on the metaphase plate. Cell, 102, 425–435. [DOI] [PubMed] [Google Scholar]
- Ausubel F.M., Brent,R., Kingston,R.E., Moore,D.D., Seidman,J.G., Smith,J.A. and Struhl,K. (1994) Current Protocols in Molecular Biology. John Wiley & Sons Inc., New York, NY.
- Brandeis M. and Hunt,T. (1996) The proteolysis of mitotic cyclins in mammalian cells persists from the end of mitosis until the onset of S phase. EMBO J., 15, 5280–5289. [PMC free article] [PubMed] [Google Scholar]
- Ciosk R., Zachariae,W., Michaelis,C., Shevchenko,A., Mann,M. and Nasmyth,K. (1998) An ESP1/PDS1 complex regulates loss of sister chromatid cohesion at the metaphase to anaphase transition in yeast. Cell, 93, 1067–1076. [DOI] [PubMed] [Google Scholar]
- Clute P. and Pines,J. (1999) Temporal and spatial control of cyclin B1 destruction in metaphase. Nature Cell Biol., 1, 82–87. [DOI] [PubMed] [Google Scholar]
- Cohen-Fix O. and Koshland,D. (1999) Pds1p of budding yeast has dual roles: inhibition of anaphase initiation and regulation of mitotic exit. Genes Dev., 13, 1950–1959. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cohen-Fix O., Peters,J.M., Kirschner,M.W. and Koshland,D. (1996) Anaphase initiation in Saccharomyces cerevisiae is controlled by the APC-dependent degradation of the anaphase inhibitor Pds1p. Genes Dev., 10, 3081–3093. [DOI] [PubMed] [Google Scholar]
- Fang G., Yu,H. and Kirschner,M.W. (1998a) The checkpoint protein MAD2 and the mitotic regulator CDC20 form a ternary complex with the anaphase-promoting complex to control anaphase initiation. Genes Dev., 12, 1871–1883. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fang G., Yu,H. and Kirschner,M.W. (1998b) Direct binding of CDC20 protein family members activates the anaphase-promoting complex in mitosis and G1. Mol. Cell, 2, 163–171. [DOI] [PubMed] [Google Scholar]
- Funabiki H. and Murray,A. (2000) The Xenopus chromokinesin Xkid is essential for metaphase chromosome alignment and must be degraded to allow anaphase chromosome movement. Cell, 102, 411–424. [DOI] [PubMed] [Google Scholar]
- Funabiki H., Yamano,H., Kumada,K., Nagao,K., Hunt,T. and Yanagida,M. (1996) Cut2 proteolysis required for sister-chromatid separation in fission yeast. Nature, 381, 438–441. [DOI] [PubMed] [Google Scholar]
- Gallant P. and Nigg,E.A. (1992) Cyclin B2 undergoes cell cycle-dependent nuclear translocation and, when expressed as a non-destructible mutant, causes mitotic arrest in HeLa cells. J. Cell Biol., 117, 213–224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gieffers C., Peters,B.H., Kramer,E.R., Dotti,C.G. and Peters,J.M. (1999) Expression of the CDH1-associated form of the anaphase-promoting complex in postmitotic neurons. Proc. Natl Acad. Sci. USA, 96, 11317–11322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harlow E. and Lane,D. (1987) Antibodies: A Laboratory Manual. Cold Spring Harbor Laboratory Press, Cold Spring Harbor, NY.
- Holloway S.L., Glotzer,M., King,R.W. and Murray,A.W. (1993) Anaphase is initiated by proteolysis rather than by the inactivation of maturation-promoting factor. Cell, 73, 1393–1402. [DOI] [PubMed] [Google Scholar]
- Hwang L.H., Lau,L.F., Smith,D.L., Mistrot,C.A., Hardwick,K.G., Hwang,E.S., Amon,A. and Murray,A.W. (1998) Budding yeast Cdc20: a target of the spindle checkpoint. Science, 279, 1041–1044. [DOI] [PubMed] [Google Scholar]
- Inbal N., Listovsky,T. and Brandeis,M. (1999) The mammalian Fizzy and Fizzy-related genes are regulated at the transcriptional and post-transcriptional levels. FEBS Lett., 463, 350–354. [DOI] [PubMed] [Google Scholar]
- Kallio M., Weinstein,J., Daum,J., Burke,D. and Gorbsky,G. (1998) Mammalian p55CDC mediates association of the spindle checkpoint protein Mad2 with the cyclosome/anaphase-promoting complex and is involved in regulating anaphase onset and late mitotic events. J. Cell Biol., 141, 1393–1406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- King R.W., Peters,J.M., Tugendreich,S., Rolfe,M., Hieter,P. and Kirschner,M.W. (1995) A 20S complex containing CDC27 and CDC16 catalyzes the mitosis-specific conjugation of ubiquitin to cyclin B. Cell, 81, 279–288. [DOI] [PubMed] [Google Scholar]
- Kitamura K., Maekawa,H. and Shimoda,C. (1998) Fission yeast Ste9, a homolog of Hct1/Cdh1 and Fizzy-related, is a novel negative regulator of cell cycle progression during G1-phase. Mol. Biol. Cell, 9, 1065–1080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klotzbücher A., Stewart,E., Harrison,D. and Hunt,T. (1996) The ‘destruction box’ of cyclin A allows B-type cyclins to be ubiquitinated, but not efficiently destroyed. EMBO J., 15, 3053–3064. [PMC free article] [PubMed] [Google Scholar]
- Kramer E.R., Gieffers,C., Holzl,G., Hengstschlager,M. and Peters,J.M. (1998) Activation of the human anaphase-promoting complex by proteins of the CDC20/Fizzy family. Curr. Biol., 8, 1207–1210. [DOI] [PubMed] [Google Scholar]
- Kramer E.R., Scheuringer,N., Podtelejnikov,A.V., Mann,M. and Peters,J.M. (2000) Mitotic regulation of the APC activator proteins CDC20 and CDH1. Mol. Biol. Cell, 11, 1555–1569. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leismann O., Herzig,A., Heidmann,S. and Lehner,C. (2000) Degradation of Drosophila PIM regulates sister chromatid separation during mitosis. Genes Dev., 14, 2192–2205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Listovsky T., Zor,A., Laronne,A. and Brandeis,M. (2000) Cdk1 is essential for mammalian cyclosome/APC regulation. Exp. Cell Res., 255, 184–191. [DOI] [PubMed] [Google Scholar]
- Liu J., Ben-Shahar,T.R., Riemer,D., Treinin,M., Spann,P., Weber,K., Fire,A. and Gruenbaum,Y. (2000) Essential roles for Caenorhabditis elegans lamin gene in nuclear organization, cell cycle progression and spatial organization of nuclear pore complexes. Mol. Biol. Cell, 11, 3937–3947. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lorca T., Castro,A., Martinez,A., Vigneron,S., Morin,N., Sigrist,S., Lehner,C., Doree,M. and Labbe,J. (1998) Fizzy is required for activation of the APC/cyclosome in Xenopus egg extracts. EMBO J., 17, 3565–3575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Losada A., Hirano,M. and Hirano,T. (1998) Identification of Xenopus SMC protein complexes required for sister chromatid cohesion. Genes Dev., 12, 1986–1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lukas C., Sorensen,C.S., Kramer,E., Santoni-Rugiu,E., Lindeneg,C., Peters,J.M., Bartek,J. and Lukas,J. (1999) Accumulation of cyclin B1 requires E2F and cyclin-A-dependent rearrangement of the anaphase-promoting complex. Nature, 401, 815–818. [DOI] [PubMed] [Google Scholar]
- Matsumoto T. (1997) A fission yeast homolog of CDC20/p55CDC/Fizzy is required for recovery from DNA damage and genetically interacts with p34cdc2. Mol. Cell. Biol., 17, 742–750. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Michaelis C., Ciosk,R. and Nasmyth,K. (1997) Cohesins: chromosomal proteins that prevent premature separation of sister chromatids. Cell, 91, 35–45. [DOI] [PubMed] [Google Scholar]
- Morgan D. (1999) Regulation of the APC and the exit from mitosis. Nature Cell Biol., 1, E47–E53. [DOI] [PubMed] [Google Scholar]
- Pei L. and Melmed,S. (1997) Isolation and characterization of a pituitary tumor-transforming gene (PTTG). Mol. Endocrinol., 11, 433–441. [DOI] [PubMed] [Google Scholar]
- Petersen B.O. et al. (2000) Cell cycle- and cell growth-regulated proteolysis of mammalian CDC6 is dependent on APC-CDH1. Genes Dev., 14, 2330–2343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pfleger C.M. and Kirschner,M.W. (2000) The KEN box: an APC recognition signal distinct from the D box targeted by Cdh1. Genes Dev., 14, 655–665. [PMC free article] [PubMed] [Google Scholar]
- Rudner A.D. and Murray,A.W. (2000) Phosphorylation by Cdc28 activates the Cdc20-dependent activity of the anaphase-promoting complex. J. Cell Biol., 149, 1377–1390. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schwab M., Lutum,A. and Seufert,W. (1997) Yeast Hct1 is a regulator of Clb2 cyclin proteolysis. Cell, 90, 683–693. [DOI] [PubMed] [Google Scholar]
- Shirayama M., Zachariae,W., Ciosk,R. and Nasmyth,K. (1998) The Polo-like kinase Cdc5p and the WD-repeat protein Cdc20p/fizzy are regulators and substrates of the anaphase promoting complex in Saccharomyces cerevisiae. EMBO J., 17, 1336–1349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shirayama M., Toth,A., Galova,M. and Nasmyth,K. (1999) APC(Cdc20) promotes exit from mitosis by destroying the anaphase inhibitor Pds1 and cyclin Clb5. Nature, 402, 203–207. [DOI] [PubMed] [Google Scholar]
- Shteinberg M., Protopopov,Y., Ganoth,D., Listovsky,T., Brandeis,M. and Hershko,A. (1999) Phosphorylation of the cyclosome is required for its stimulation by Fizzy/Cdc20. Biochem. Biophys. Res. Commun., 260, 193–198. [DOI] [PubMed] [Google Scholar]
- Sigrist S.J. and Lehner,C.F. (1997) Drosophila fizzy-related down-regulates mitotic cyclins and is required for cell proliferation arrest and entry into endocycles. Cell, 90, 671–681. [DOI] [PubMed] [Google Scholar]
- Sigrist S.J., Jacobs,H., Stratmann,R. and Lehner,C.F. (1995) Exit from mitosis is regulated by Drosophila fizzy and the sequential destruction of cyclins A, B and B3. EMBO J., 14, 4827–4838. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sudakin V., Ganoth,D., Dahan,A., Heller,H., Hershko,J., Luca,F.C., Ruderman,J.V. and Hershko,A. (1995) The cyclosome, a large complex containing cyclin-selective ubiquitin ligase activity, targets cyclins for destruction at the end of mitosis. Mol. Biol. Cell, 6, 185–197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tatebe H. and Yanagida,M. (2000) Cut8, essential for anaphase, controls localization of 26S proteasome, facilitating destruction of cyclin and cut2. Curr. Biol., 10, 1329–1338. [DOI] [PubMed] [Google Scholar]
- Tinker-Kulberg R.L. and Morgan,D.O. (1999) Pds1 and Esp1 control both anaphase and mitotic exit in normal cells and after DNA damage. Genes Dev., 13, 1936–1949. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Townsley F., Aristarkhov,A., Beck,S., Hershko,A. and Ruderman,J. (1997) Dominant-negative cyclin-selective ubiquitin carrier protein E2-C/UbcH10 blocks cells in metaphase. Proc. Natl Acad. Sci. USA, 94, 2362–2367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Uhlmann F., Lottspeich,F. and Nasmyth,K. (1999) Sister-chromatid separation at anaphase onset is promoted by cleavage of the cohesin subunit Scc1. Nature, 400, 37–42. [DOI] [PubMed] [Google Scholar]
- van den Heuvel S. and Harlow,E. (1993) Distinct roles for cyclin-dependent kinases in cell cycle control. Science, 262, 2050–2054. [DOI] [PubMed] [Google Scholar]
- Visintin R., Prinz,S. and Amon,A. (1997) CDC20 and CDH1: a family of substrate-specific activators of APC-dependent proteolysis. Science, 278, 460–463. [DOI] [PubMed] [Google Scholar]
- Waizenegger I., Hauf,S., Meinke,A. and Peters,J. (2000) Two distinct pathways remove mammalian cohesin from chromosome arms in prophase and from centromeres in anaphase. Cell, 103, 399–410. [DOI] [PubMed] [Google Scholar]
- Weinstein J., Jacobsen,F.W., Hsu-Chen,J., Wu,T. and Baum,L.G. (1994) A novel mammalian protein, p55CDC, present in dividing cells is associated with protein kinase activity and has homology to the Saccharomyces cerevisiae cell division cycle proteins Cdc20 and Cdc4. Mol. Cell. Biol., 14, 3350–3363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wheatley S.P., Hinchcliffe,E.H., Glotzer,M., Hyman,A.A., Sluder,G. and Wang,Y. (1997) CDK1 inactivation regulates anaphase spindle dynamics and cytokinesis in vivo. J. Cell Biol., 138, 385–393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamamoto A., Guacci,V. and Koshland,D. (1996) Pds1p is required for faithful execution of anaphase in the yeast, Saccharomyces cerevisiae. J. Cell Biol., 133, 85–97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yudkovsky Y., Shteinberg,M., Listovsky,T., Brandeis,M. and Hershko,A. (2000) Phosphorylation of Cdc20/fizzy negatively regulates the mammalian cyclosome/APC in the mitotic checkpoint. Biochem. Biophys. Res. Commun., 271, 299–304. [DOI] [PubMed] [Google Scholar]
- Zachariae W. and Nasmyth,K. (1999) Whose end is destruction: cell division and the anaphase-promoting complex. Genes Dev., 13, 2039–2058. [DOI] [PubMed] [Google Scholar]
- Zachariae W., Schwab,M., Nasmyth,K. and Seufert,W. (1998) Control of cyclin ubiquitination by CDK-regulated binding of Hct1 to the anaphase promoting complex. Science, 282, 1721–1724. [DOI] [PubMed] [Google Scholar]
- Zou H., McGarry,T.J., Bernal,T. and Kirschner,M.W. (1999) Identification of a vertebrate sister-chromatid separation inhibitor involved in transformation and tumorigenesis. Science, 285, 418–422. [DOI] [PubMed] [Google Scholar]