

Abstract
Wild-type p53 protein can markedly stimulate base excision repair (BER) in vitro, either reconstituted with purified components or in extracts of cells. In contrast, p53 with missense mutations either at hot-spots in the core domain or within the N-terminal transactivation domain is defective in this function. Stimulation of BER by p53 is correlated with its ability to interact directly both with the AP endonuclease (APE) and with DNA polymerase β (pol β). Furthermore, p53 stabilizes the interaction between DNA pol β and abasic DNA. Evidence that this function of p53 is physiologically relevant is supported by the facts that BER activity in human and murine cell extracts closely parallels their levels of endogenous p53, and that BER activity is much reduced in cell extracts immunodepleted of p53. These data suggest a novel role for p53 in DNA repair, which could contribute to its function as a key tumor suppressor.
Keywords: AP endonuclease/base excision repair/DNA polymerase β/p53
Introduction
The tumor suppressor p53 has been proposed to be involved in maintaining stability of the genome through either cell cycle arrest or apoptosis (reviewed by Ko and Prives, 1996; Giaccia and Kastan, 1998; Prives and Hall, 1999). It is believed that stress signals like DNA damage induce expression of p53, which in turn arrests the cell cycle, presumably to allow DNA repair (Lane, 1992), or drives cells to apoptosis if the damage is too extensive (Liebermann et al., 1995). Consistent with a role for p53 in protecting genome integrity, p53-deficient murine fibroblasts demonstrate chromosomal abnormalities, which appear at early passage in homozygous null cells and at later passage in heterozygous cells (Harvey et al., 1993). p53-deficient cells also exhibit a higher tolerance to genetic abnormalities arising from radiation as well as spontaneously (Lee et al., 1994). Evidence suggesting a role of p53 in DNA repair in vivo is supported by observations that (i) p53 increases global but not transcription-coupled nucleotide excision repair (Hwang et al., 1999 and references therein); (ii) p53 reduces point mutation frequency of a shuttle vector modified by several chemical mutagens (Courtemanche and Anderson, 1999); and (iii) p53 inactivation by HPV16 results in increased mutagenesis in human cells (Yu et al., 1997). So far, most observations of a role of p53 in DNA repair seem to involve nucleotide excision repair (NER) (Ford et al., 1998; Lloyd and Hanawalt, 2000) and it is believed that p53 plays an indirect role in NER, probably by transactivating the p48 gene (and perhaps some others), which is essential for efficient repair of certain DNA lesions (Hwang et al., 1999; Tang et al., 2000).
DNA base damage generated by ionizing radiation, simple alkylating agents, as well as endogenous hydrolytic and oxidative processes is corrected by the base excision repair (BER) pathway (reviewed by Seeberg et al., 1995). Thus, BER is critically important for regulating spontaneous mutation. Partly because deamination of cytosine to uracil in DNA genomes is one of the most common spontaneous premutagenic events and occurs hundreds of times a day in every single cell (Lindahl, 1993; Friedberg et al., 1995), uracil-containing DNA has been an excellent model substrate for studying BER. A number of models for eukaryotic BER of uracil-containing DNA have been proposed (Matsumoto and Bogenhagen, 1991; Dianov and Lindahl, 1994; Friedberg et al., 1995; Lindahl et al., 1995). Human BER has been reconstituted with purified human proteins: uracil-DNA glycosylase (UDG), AP endonuclease (HAP1), DNA polymerase β (pol β) and either DNA ligase III or DNA ligase I (Kubota et al., 1996; Nicholl et al., 1997). The scaffold protein XRCC1, which interacts with both DNA pol β and DNA ligase III (Caldecott et al., 1994, 1995), is not absolutely required for the reaction but suppresses strand displacement by DNA pol β, allowing for more efficient ligation after filling of a single-nucleotide patch (Kubota et al., 1996).
In human cells, the major 5′ AP endonuclease is termed APE or HAP1 (Chen et al., 1991; Demple et al., 1991). However, the same gene product was identified as an activator of DNA-binding activity of several transcription factors, including AP-1 (Xanthoudakis and Curran, 1992; Xanthoudakis et al., 1992, 1994; Walker et al., 1993) and p53 (Jayaraman et al., 1997), and as such is also known as Ref-1. For the purpose of simplicity in this study we refer to APE/HAP1/Ref-1 as APE. The APE product belongs to a large family of nucleases related to exonuclease III of Escherichia coli (Demple and Harrison, 1994; Barzilay and Hickson, 1995), and human APE protein can be separated into two functionally distinct regions, with the N-terminal domain possessing the AP-1 stimulatory activity (Walker et al., 1993; Xanthoudakis et al., 1994) and the C-terminal domain containing the AP endonuclease activity. In mammalian cells, most BER synthesis is carried out by DNA pol β (Singhal et al., 1995; Sobol et al., 1996). It is believed that DNA pol β not only functions as the polymerase, but also catalyzes the excision of the deoxyribose phosphate (5′-dRp) by the β-elimination (AP lyase) activity of its N-terminal domain (Matsumoto and Kim, 1995; Piersen et al., 1996). APE and DNA pol β function in a coordinated manner during BER. APE interacts with DNA pol β, loads it onto DNA abasic sites and stimulates its AP lyase activity (Bennett et al., 1997).
Our interest in a possible role for p53 in BER was primarily spurred by the observation that APE is a potent activator of p53 in vitro and in vivo (Jayaraman et al., 1997; Gaiddon et al., 1999) and that APE can interact directly with p53 (Gaiddon et al., 1999). Additionally, Offer et al. (1999) reported that BER activity in cell extracts correlates with levels of wild-type p53. These observations prompted us to test the possibility that p53 might directly influence the process of BER. In this paper, we report that purified p53 protein can markedly stimulate BER in vitro and that BER activity in vivo correlates with the level of p53. The ability of p53 to stimulate BER correlates with its ability to interact with DNA pol β. Hot-spot tumor-derived p53 mutants do not significantly enhance BER, supporting the possibility that the stimulatory effect of wild-type p53 may contribute to its ability to suppress tumorigenesis.
Results
p53 stimulates DNA BER in vitro
Eukaryotic BER of uracil-containing DNA has been reconstituted with several purified proteins: UDG, APE, DNA pol β and either DNA ligase III or DNA ligase I (Kubota et al., 1996; Nicholl et al., 1997). Our first goal was to examine whether p53 affects BER of a reconstituted system. Using a 21 bp uracil-containing DNA, we found that purified baculovirus-expressed p53 stimulated reconstituted BER activity dramatically (up to >9-fold) (Figure 1A). Consistent with a previous report (Bogenhagen and Pinz, 1998), T4 DNA ligase could also ligate the single-nucleotide filled-in repair product, and the presence of T4 DNA ligase did not affect the stimulation of BER by p53. That no repair activity was detected with a control DNA sample, which was not pre-treated with UDG, excludes the possibility that the incorporation of dCMP was due to causes other than BER. When we used a longer template (55 bp uracil-containing DNA) in the reconstituted reaction, the same result was observed (data not shown). Moreover, when the 55 bp uracil-containing DNA was incubated with whole-cell extracts made from a p53-null human cell line H1299, BER was again strongly stimulated by adding purified p53 (Figure 1B).

Fig. 1. p53 stimulates DNA BER in vitro. BER experiments with AP-DNA were performed as described in Materials and methods. Baculovirus-expressed purified p53 protein (200, 400, 800 ng) was added to BER reaction mixtures containing either purified APE and DNA pol β (A) or whole-cell extracts made from H1299 cells (B). As controls, the 21 bp U-DNA that was not treated with UDG [lane 1, (A)] and a 55 bp DNA that has a cytosine at the position of the uracil [lane 1, (B)] were used as templates. (C) Three hundred or 600 ng of bacterially expressed His-tagged p53 and p53 depletion mutants [p53 core domain (100–300), p53ΔC30 (1–363), p53ΔN96 (97–393) and the p53 C-terminus (300–393)] were added into reconstituted BER reaction mixtures. (D) Five hundred nanograms of each protein were run on an SDS gel, which was silver stained: full-length, lane 1; ΔN96, lane 2; ΔC30, lane 3; the core domain, lane 4; the C-terminus, lane 5. The numbers on the left show migration positions of the indicated molecular weight markers (in kDa) run in lane M.
To understand which domain of p53 is responsible for the observed stimulation of BER, the abilities of a series of purified E.coli-expressed wild-type and deleted p53 proteins to regulate BER were examined (Figure 1C). Both full-length p53 (1–393) and p53ΔC30 (1–363) could stimulate BER, while neither p53ΔN96 (97–393) nor the central core domain (100–300) affected this process. This suggests that the N-terminal transactivation domain and the central specific DNA-binding domain of p53 are together required to stimulate BER. It has been reported that the C-terminus of p53 binds tightly to various forms of DNA damage such as DNA ends, strand breaks and deletion/insertion mismatches (Bakalkin et al., 1995; Lee et al., 1995; Reed et al., 1995). The inhibition of BER by the p53 C-terminus (300–393) might be explained by its high affinity for DNA containing single-nucleotide gaps, which are intermediate products of BER (J.Ahn, unpublished results).
Attenuated stimulation of BER by tumor-derived p53 mutants
Mutations of the p53 gene occur in ∼50% of tumors derived from the major forms of human cancer (reviewed by Nigro et al., 1989; Hollstein et al., 1991, 1994). Nearly all of the thousands of tumor-derived mutations identified to date are located within the central DNA-binding domain, and all six hot-spots of mutation are in this region. Because our results suggest that the central domain of p53 is involved in the stimulation of BER, we examined the abilities of two tumor-derived hot-spot p53 mutants, R248W and R175H, to affect BER. To this end, we compared purified baculovirus-expressed wild-type p53 with purified baculovirus-expressed mutant p53 both in the reconstituted BER system and in H1299 whole-cell extracts (Figure 2). When the same amounts of proteins were added to the reconstituted BER reactions, the two mutant p53 proteins showed much reduced stimulation (Figure 2B). Strikingly, when tested in H1299 whole-cell extracts, the two p53 mutants failed to stimulate BER only at high protein concentrations (Figure 2C). One possible explanation for this is that mutant p53 at high concentrations tends to aggregate, and some unknown factor(s) in the cell may facilitate this aggregation.
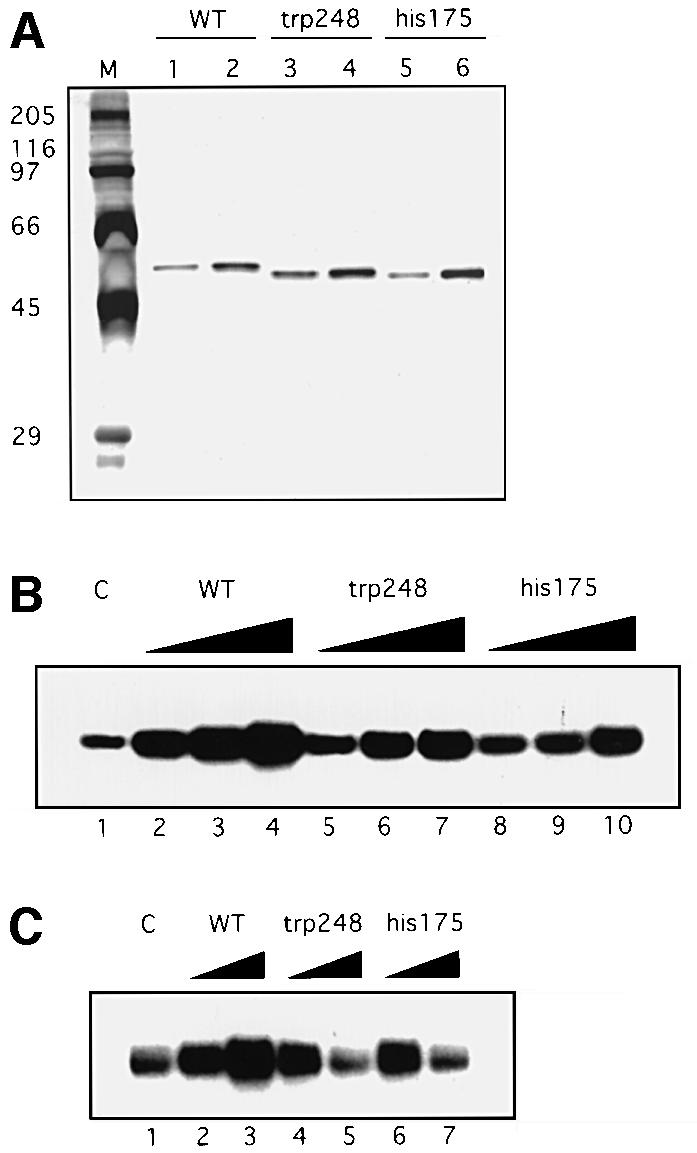
Fig. 2. Tumor-derived p53 mutants have reduced stimulation of BER. p53 mutant proteins, R248W and R175H, and flu-tagged wild-type p53 protein were immunopurified as described in Materials and methods. (A) Silver-stained SDS gel with proteins (300 and 600 ng) as indicated. Lane M contains molecular weight marker proteins with sizes indicated on the left. (B) Wild-type, R248W or R175H p53 proteins (200, 400, 600 ng) were added to reconstituted BER reaction mixtures as in Figure 1A. (C) Three hundred or 600 ng of the indicated proteins were added to BER reaction mixtures containing H1299 whole-cell extracts as in Figure 1B.
p53 stabilizes the interaction between DNA pol β and abasic DNA
To understand the mechanism underlying the stimulation of BER by p53, we studied the effect of p53 on several aspects of the BER process. Bennett et al. (1997) have shown that APE interacts physically with DNA pol β, facilitates the loading of DNA pol β onto DNA abasic sites, and accelerates excision of the 5′-dRp by DNA pol β. To study whether p53 may regulate the loading function of APE, we used an end-labeled 21 bp AP-DNA oligonucleotide as the probe in an electrophoretic mobility shift assay (EMSA). As shown in Figure 3A, p53 stimulated APE-facilitated loading of DNA pol β onto DNA AP sites (compare lanes 12 and 15). The reason that we used two different buffer conditions for the EMSA was to illustrate the migration positions of APE–AP-DNA and DNA pol β–AP-DNA. APE bound to AP-DNA only in the absence of Mg2+ as previously described (Bennett et al., 1997). When Mg2+ was added to support the cleavage activity of APE (Wilson et al., 1997), APE no longer bound to the incised AP-DNA.

Fig. 3. p53 stabilizes the interaction between DNA pol β and AP-DNA. (A) p53 stimulates APE-facilitated loading of DNA pol β onto AP-DNA. EMSA with end-labeled 21 bp AP-DNA was performed as described. APE (20 ng), DNA pol β (20, 60 or 120 ng) and baculovirus-expressed p53 (80 ng) were added to EMSA reaction mixtures as indicated. The migrations of APE, DNA pol β and p53 are indicated by the black, gray and white arrowheads, respectively. (B) p53 slows the dissociation of DNA pol β from AP-DNA. EMSA reaction mixtures (in Mg2+ buffer), containing APE, DNA pol β and end-labeled AP-DNA, lacked (lanes 1–4) or contained (lanes 5–8) p53. Excess unlabeled competitor AP-DNA was added to each reaction mixture at time zero. After the indicated time, mixtures were loaded onto a running 8% polyacrylamide gel. (C) A graphic representation of the PhosphorImager-quantified result of the competition/dissociation assay.
We then asked whether p53 stabilizes the interaction between DNA pol β and AP-DNA using a competition/dissociation assay. Excessive unlabeled AP-DNA competitor was added to EMSA reactions containing APE and DNA pol β, and the dissociation of the DNA pol β–AP-DNA complex was compared in the absence and presence of p53. p53 indeed significantly stabilized the interaction between DNA pol β and AP-DNA (Figure 3B and C). APE was required for the stabilization of the DNA pol β–AP-DNA complex by p53. Without APE, no matter whether AP-DNA or incised AP-DNA was used, we could not detect a stabilization of the DNA pol β–AP-DNA complex by p53 (data not shown).
p53 directly interacts with DNA pol β
When we used the 55 bp AP-DNA as the probe in the EMSA experiment, we found that p53 and DNA pol β could together ‘supershift’ the 55 bp AP-DNA probe (Figure 4A). The p53 antibody PAb421, which binds to the C-terminus of p53, is known to inhibit its binding to non-specific DNA (Wang et al., 1993). Because the p53–DNA pol β–AP-DNA complex may be formed through separate protein–DNA interactions, PAb421 was added to the EMSA reactions with the reasoning that if p53 binds to AP-DNA through its C-terminus the binding will be inhibited by this antibody. As shown in Figure 4A, the p53–DNA pol β–AP-DNA complex was not inhibited but rather was further shifted by PAb421, suggesting that a ternary complex was formed through protein–protein interaction between p53 and DNA pol β.

Fig. 4. p53 interacts with DNA pol β. (A) p53 and DNA pol β together supershift the 55 bp AP-DNA probe. EMSA with end-labeled 55 bp AP-DNA was performed as described. APE (20 ng), DNA pol β (20 ng), p53 (80 ng) and PAb421 (100 or 200 ng) were added to reaction mixtures (in Mg2+ buffer) as indicated. The migrations of p53 alone and the p53–DNA pol β complex are indicated by the black and white arrowheads, respectively. (B) Far-western blotting was performed as described. Full-length HDM2 and a truncated HDM2 (dl166), which does not bind to p53, were used as the positive and negative controls, respectively. Left panel shows Ponceau S-stained image of the nitrocellulose filter; right panel shows the result of incubation of filter with p53 followed by probing with PAb421. (C) Co-immunoprecipitation was performed as described in Materials and methods. Precipitations of PAb421-cross-linked beads were resolved by SDS–PAGE, followed by detection with PAb421 and antiserum against DNA pol β. (D) Protein cross-linking was performed as described in Materials and methods. Proteins were separated in a step-gradient SDS–PAGE gel. A silver-stained image of the gel is shown.
To confirm a direct interaction between p53 and DNA pol β, three different methods were used: far-western blotting (Figure 4B), co-immunoprecipitation (Figure 4C) and protein cross-linking (Figure 4D). Full-length HDM2 and a truncated HDM2 (dl166), which does not bind to p53, were used in far-western blotting as the positive and negative control, respectively. As shown in Figure 4, all three lines of evidence showed that p53 physically interacts with DNA pol β.
BER activity in cell extracts closely parallels their levels of endogenous p53
We wished to determine whether the stimulatory effect of p53 on BER in vitro could be replicated in a more physiological context. To this end, BER activity was examined in LNCaP cell extracts before and after their endogenous wild-type p53 was induced by γ-irradiation. As previously observed (Shieh et al., 1997), p53 levels in LNCaP cells increased, reaching a peak within 1 h, and then subsided (Figure 5A). When BER activities were tested using the uracil-containing 55 bp DNA as the probe, the extent of BER activity correlated strikingly with the levels of endogenous p53 protein (Figure 5A, left panel). Because a factor(s) other than p53 may be induced by ionizing radiation and be responsible for the change in BER activity, we used tetracycline-inducible H1299 cells in which p53 expression can be induced by removing tetracycline from the medium. Consistent with previous findings (Chen et al., 1996), p53 levels in the induced cells did not increase in response to the same dosage of γ-irradiation (Figure 5B), and, whether p53 was induced or not, BER activity did not change after γ-irradiation (Figure 5B and C). Note, however, that H1299 cells in which p53 was expressed had significantly higher BER activity in vitro (averaging 3.2-fold) than in the same cells without p53 (compare Figure 5B and C), further supporting the likelihood that p53 levels are directly linked to BER activity.

Fig. 5. BER activity correlates with p53 levels in cells. (A–C) Cell cultures at 70% confluence were exposed to 7 Gy γ-irradiation. At 0, 0.5, 1, 2, 3 and 4 h after irradiation, cells were collected and whole-cell extracts were prepared. Then BER was assayed using the 55 bp U-DNA. p53 protein in aliquots of extracts was detected by western blotting using PAb421. To monitor cell protein concentrations, actin was detected using an anti-actin antibody (Sigma). (D) Depletion of p53 reduces BER activity. Cell extracts (3 µg) made from LNCaP cells 2 h after γ-irradiation were pre-incubated with PAb421-coated magnetic Dynabeads (lanes 3–5) or beads alone (lane 2) and assayed for BER activity. Lane 1, untreated cell extract; lanes 4 and 5, 300 or 600 ng of baculovirus-expressed p53 were added into depleted extracts, respectively. (E) p53 protein levels in the cell extracts (15 µg) were detected by western blotting. Lane 1, untreated cell extract; lane 2, depleted with control beads; lane 3, extract depleted with PAb421-coated beads; lanes 4 and 5, 60 and 120 ng of p53 proteins were added to PAb421-depleted extracts, respectively.
It was reported by Offer et al. (1999) that extracts of cells depleted of p53 show low BER activities. We confirmed and extended this observation in an experiment in which p53 was immunodepleted from cell extracts made from γ-irradiated LNCaP cells using PAb421-coated magnetic beads. When BER activities of these cell extracts were compared with those of extracts depleted with control beads, ∼1/4 of the control BER activity was left after p53 was removed. When purified p53 was added to the p53-depleted cell extracts, BER activity was restored to its former high level (Figure 5D). The result of the immunodepletion experiment may be partly explained by the physical interaction between p53 and DNA pol β, but our result that adding back purified p53 restores the BER activity of the depleted cell extracts supports the likelihood that p53 plays a direct role in BER of the cells.
Transactivation-defective N-terminal mutant forms of p53 do not interact with DNA pol β and cannot stimulate BER
The reduced stimulation of BER by tumor-derived core domain missense mutant forms of p53 (Figure 2) supports the likelihood that the central region of p53 is required for its stimulation of BER. However, our result with deleted forms of p53 (Figure 1C) suggested that the N-terminus of p53 is required as well. To study further a possible role for the p53 N-terminus in enhancing BER, we examined a doubly mutated form of human p53, L22Q/W23S, previously shown to be defective in both sequence-specific transactivation of p53 target genes and interaction with a number of proteins including Mdm2, adenovirus E1B (Lin et al., 1994, 1995), TAFs (Lu and Levine, 1995) and p300 (Gu et al., 1997). When added either to reconstituted BER reaction mixtures or nuclear extracts, there was a dramatic difference in the stimulatory abilities of wild-type and L22Q/W23S p53 proteins, with the latter being virtually inert in affecting the repair reaction (Figure 6B). Since the N-terminus of p53 is involved in protein–protein interactions in which the above mutant is faulty, it was of interest to determine whether the ability of p53 to bind to DNA pol β (as documented in Figure 4) was affected by mutation of residues 22 and 23. Indeed, while wild-type and two core-domain mutants bound similarly to DNA pol β, the 22/23 mutant p53 was dramatically impaired in this interaction (Figure 6C).

Fig. 6. A mutant p53 (L22Q/W23S) does not interact with DNA pol β and loses the ability to stimulate BER. Baculovirus-expressed wild-type and mutant p53 proteins were immunopurified as described in Materials and methods. (A) Silver-stained SDS gel with proteins (300 and 600 ng) as indicated. Lane M contains molecular marker proteins with sizes indicated on the left. (B) Wild-type and L22Q/W23S p53 proteins (300 and 600 ng) were added to reconstituted BER reaction mixtures (lanes 2–5) and BER reaction mixtures containing H1299 whole-cell extracts (lanes 7–10). (C) The co-immunoprecipitation assay was performed as described in Materials and methods. Wild-type and mutant p53 proteins incubated with DNA pol β and Dynabeads coated with 18S monoclonal antibody against DNA pol β were resolved by SDS–PAGE, transferred to nitrocellulose and probed with PAb421 and 18S. Fifty percent of the inputs of p53 proteins are shown on the left.
Murine embryo fibroblasts (MEFs) expressing either wild-type or mutant p53 (L25Q/W26S), which mimics human mutant L22Q/W23S p53, allowed us to compare BER in extracts of murine cells. p53-null MEFs or MEFs expressing wild-type p53 or 25/26 mutant p53 were γ-irradiated prior to lysis and either subjected to SDS–PAGE–western analysis or assessed for BER activity as performed above (e.g. Figure 5). Cells expressing wild-type p53 had low, but detectable levels of p53 that increased after irradiation, while cells containing mutant p53 had overall higher levels of p53 that did not increase after irradiation, consistent with what was observed previously (Jimenez et al., 2000). Extracts of p53-null or 25/26 p53 showed essentially comparable levels of BER (Figure 7B). In contrast, extracts of cells expressing wild-type p53 had dramatically increased levels of BER, which were greatly reduced if extracts were first pre-depleted with anti-p53 antibody (Figure 7B and C). Here, the levels of BER in wild-type p53 expressing MEFs did not increase after γ-irradiation, suggesting that even the relatively small amount of detectable p53 in unirradiated cells was sufficient for maximal augmentation of BER activity. Taken together, our data indicate that the p53 N-terminus interaction with DNA pol β is required for p53 stimulation of BER.

Fig. 7. Extracts of cells with wild-type p53 have more BER activity than p53–/– or mutant p53 (L25Q/W26S) cell extracts. Cultures of MEF cells as indicated were γ-irradiated and extracts were prepared as described in Figure 5. (A) p53 protein in aliquots of extracts was detected by western blotting using a mixture of antibodies, including PAb421, PAb240 and PAb246. DNA pol β protein was detected using the 18S monoclonal antibody. In (B) and (C), BER activity was assayed using the 55 bp U-DNA fragment as in Figure 1B. Either aliquots of extracts prepared at the indicated times after γ-irradiation of MEFs were directly assayed for BER activity (B) or aliquots of the wild-type p53-containing samples above were either untreated (lanes 1–6) or incubated with PAb 421-coated Dynabeads (lanes 7–12) or Dynabeads alone (lanes 13–18). The BER activities in either the untreated samples or the different Dynabead supernatants were assayed (C).
Discussion
In this study we provide evidence for a direct role of p53 in the process of BER. Support for this assertion is derived from our demonstration that p53 markedly stimulates a reconstituted BER system in vitro, and that BER activity in cell extracts correlates closely with the cellular levels of p53. Physical interactions among p53, APE and DNA pol β are likely to be involved in the stimulation of BER by p53.
p53 transmits signals from multiple cellular stresses to numerous cellular outputs, which together serve to protect cells from accruing the consequences of genotoxic damage. Although there is strong support for the roles of DNA-damage checkpoint factors such as ATM (Banin et al., 1998; Canman et al., 1998; Khanna et al., 1998), Chk1 and Chk2 (Chehab et al., 2000; Shieh et al., 2000) in signaling to p53, there is less clarity as to how such mediator and effector kinases find p53 protein in stressed cells. It has been demonstrated, however, that p53 itself has the ability to interact with several forms of DNA in addition to double-stranded B form DNA, such as DNA with deletions or insertions, Holliday junctions, as well as double- and single-stranded DNA or DNA ends (reviewed by Jayaraman and Prives, 1999). We have discovered, in fact, that p53 binds with a higher affinity to DNA with single- or 4-nucleotide gaps than it does to double-stranded consensus site DNA (J.Ahn, J.Zhou and C.Prives, unpublished). Thus, we might envisage that p53 directly receives signals at sites of DNA damage, including sites undergoing BER. Based on the observations described in this paper, it is entirely possible that p53 is not only a passive responder to such damage, but may play an active role in helping to resolve the lesions. One would hope to eventually identify conditions where complexes could be visualized containing p53 and APE or pol β (or both) at sites of BER. If APE and p53 do function as a complex, different events may happen at p53 target promoters (where APE serves as a transcriptional co-activator) and on AP-DNA sites. It is attractive to speculate further that, upon interaction with the DNA and proteins involved in this process, p53 becomes modified and is then liberated to serve as a transcriptional activator of genes involved in cell arrest or, depending on the context, cell death.
p53 is a modular protein with a number of structurally and functionally characterized functional domains. Both the central core and the N-terminal region of the protein are required for p53 to stimulate BER. The core region, which contains the sequence-specific binding activity, sustains the great majority of missense mutations in human cancers, and it is thus significant that tumor-derived mutants were defective in their ability to augment BER. p53 interacts with DNA pol β through its N-terminal region. The N-terminus contains at least two functional domains: a bi-partite transactivation region extending to approximately residue 60, and a PXXP domain (amino acids 60–90), which has a role in p53-mediated apoptosis. Our results with mutant forms of p53 bearing substitutions in N-terminal residues suggest that the integrity of the transactivation region is required for both the interaction between p53 and DNA pol β, and the stimulation of BER by p53. It is not known at present whether the PXXP domain is also involved in the ability of p53 to stimulate BER. It was unexpected that the C-terminus is not directly involved in BER, since this region of p53 is responsible for most of its non-specific interactions with DNA, including gapped DNA. This strongly suggests that the major mode by which p53 functions in BER is through protein–protein interactions and, as speculated above, that the C-terminus may help localize p53 to sites of BER but is not actually functionally involved in this process. It is well documented that p53 can be activated by various types of DNA damage, which trigger a series of phosphorylation, de-phosphorylation and acetylation events on the p53 protein (reviewed by Giaccia and Kastan, 1998; Prives, 1998). Such modifications occur at several sites within both the N- and C-termini of p53, and it will be interesting to examine whether any of these are involved in regulating the ability of p53 to serve as a BER stimulatory factor.
Based on our results, p53 stabilization of the DNA pol β–AP-DNA complex is likely to be the mechanism underlying the stimulation of BER by p53. DNA pol β functions in two steps during the process of BER: the re-synthesis catalyzed by its polymerase activity, and the excision of the 5′-dRp catalyzed by its AP lyase activity (Matsumoto and Kim, 1995; Piersen et al., 1996). The fact that excision of the 5′-dRp is the rate-limiting step of BER (Srivastava et al., 1998) makes it reasonable that p53 stimulates BER through stabilizing the interaction of DNA pol β and AP-DNA. Stabilization of DNA pol β–AP-DNA by p53 requires APE, because it is only observed in the presence of APE (data not shown). APE and DNA pol β work in a closely coordinated way during the process of BER. We hypothesize that APE, DNA pol β and p53 may form a trimeric complex when binding to AP-DNA, and APE leaves after catalyzing the incision at the 5′-side of the AP site.
Another type of BER is long-patch repair, which involves gap-filling of 4–7 nucleotides and requires DNase IV (FEN1) and PCNA (Frosina et al., 1996; Klungland and Lindahl, 1997). If DNA pol β is also the major repair polymerase for long-patch BER, it is reasonable to speculate that p53 can also stimulate long-patch BER. By stabilizing the interaction of DNA pol β and AP-DNA, p53 may upregulate the fidelity of DNA pol β by reducing the frame-shift infidelity caused by strand slippage during the dissociation–reassociation phase of a polymerization reaction (Kunkel, 1985; Osheroff et al., 1999). The 3′-to-5′ exonuclease activity of p53 (Mummenbrauer et al., 1996) may also provide a proofreading function for DNA pol β, since it has been shown that p53 increases the fidelity of DNA synthesis catalyzed by DNA pol α (Huang, 1998). In recent years, additional roles of DNA pol β have been suggested, most involving synthesis to fill relatively short-gapped intermediates in recombination and replication (Yoo et al., 1994; Plug et al., 1997).
It is a surprise that DNA pol β is not induced by γ-irradiation, since it is known that ionizing radiation causes various forms of base modification in addition to DNA strand breaks. It is known that alkylating agents such as MMS induce upregulation of DNA pol β (Fornace et al., 1989). So far, it is not clear whether MMS-induced upregulation of DNA pol β is p53 dependent. Our experimental results showing that DNA pol β is not trans-regulated by p53 in MEF cells after γ-irradiation as well as in tetracycline-inducible H1299 cells (data not shown) suggest that DNA pol β is not transcriptionally regulated by p53.
In unstressed cells, BER may be viewed as a ‘housekeeping’ function in which oxidative damage to DNA arising from normal metabolic processes is repaired by DNA glycosylases and other components of the BER machinery. We hypothesize that p53, normally held in check by Mdm2, is not involved in this process. However, agents of stress such as those that induce p53 itself, e.g. alkylating agents or γ-rays, also initiate base damage and ensuing BER (Seeberg et al., 1995). It is attractive to speculate that such induction allows p53 to function in two ways: as a regulator of transcription and as a facilitator of BER. Since both activities are compromised in either tumor-derived p53 mutants or in the N-terminal mutants used in our studies, they may each be key components of the ability of p53 to serve as the major tumor suppressor of the mammalian cell.
Materials and methods
Cells, proteins and oligonucleotide substrates
Human H1299-p53-14 and LNCaP cell lines were grown in RPMI medium containing 10% fetal bovine serum (FBS) (HyClone). MEF cell lines were grown in Dulbecco’s modified Eagle’s medium (DMEM) containing 15% FBS, 0.1 g/l sodium pyruvate, 100 µM 2-mercaptoethanol, 2 mM l-glutamine, 10 µg/ml ciprofloxacin hydrochloride (Bayer). To maintain tetracycline-inducible H1299-p53-14 cells, tetracycline (4.5 µg/ml; Gibco-BRL) was present in the culture medium; p53 expression was induced in H1299-p53-14 cells, grown to 60–70% confluence, by washing three times in medium lacking tetracycline and then incubation in tetracycline-free medium overnight. Baculovirus-expressed p53 protein (Jayaraman and Prives, 1995), and bacterially expressed His-tagged APE (Xanthoudakis et al., 1992) and murine DNA pol β (Date et al., 1988), were purified as described. Constructs expressing His-tagged p53 and p53 deletion mutant proteins were generated as described (Ko et al., 1997). Proteins were expressed in E.coli BL21(DE3)(pLysS) (Novagen) growing at 19°C, and then chromatographically purified over Ni-NTA–agarose (Qiagen), Hi-trap Heparin (Pharmacia) and Superose 6 (Pharmacia) columns. Single-stranded oligonucleotide, GAAATCAAGTUTGCAGGACTT (21mer) or AGCTACCAGTACTGCACGAAU(C)TAAGCAATCCTGTAATTGTGGTCATAGCTGATCC (55mer), was annealed to its complementary strand with a G residue opposite U. Annealed double-stranded oligonucleotides were gel purified.
In vitro BER assay
Whole-cell extracts were prepared as described (Wood, 1988). For the reconstituted BER reaction, uracil-containing DNA was first treated with UDG (Gibco-BRL) to generate an AP site on each DNA fragment. Reaction mixtures (20 µl) containing APE (8 ng), DNA pol β (8 ng) and 50 U of T4 DNA ligase (New England Biolabs) and p53, as indicated, were incubated with 50 ng of AP-DNA in 0.1 M Tris–HCl pH 7.5, 5 mM MgCl2, 1 mM dithiothreitol (DTT), 0.1 mM EDTA, 2 mM ATP, 5 mM creatine phosphate, 0.1 mg/ml creatine phosphokinase and 1 µCi of [α-32P]dCTP at 37°C for 10 min. Reactions were then terminated by the addition of SDS to 0.6% and EDTA to 25 mM. For BER in cell extracts, each reaction mixture containing 3 µg of cell extract, 3 µg of bovine serum albumin (BSA) and p53 as indicated was incubated and processed as above. BER reaction products were analyzed by autoradiography after electrophoretic separation in a 12% polyacrylamide gel.
Immunodepletion
Purified PAb421 was mixed with Dynabeads M-280 (Dynal) at 4°C overnight. After washing four times with phosphate-buffered saline (PBS)/BSA, the Dynabeads were collected with a magnetic stand (Dynal MPC) and excess liquid was removed. The PAb421-coated beads were then mixed with whole-cell extracts at 4°C for 2 h. The supernatants were collected after removal of the beads and used for BER reactions and western blotting.
Far-western blotting, co-immunoprecipitation and protein cross-linking
Far-western blotting was performed as described (Jayaraman et al., 1998). One microgram of full-length HDM2, a truncated HDM2 (dl166) that does not bind to p53, DNA pol β and molecular weight marker polypeptides were resolved by SDS–PAGE and transferred to a nitrocellulose filter. The filter was then incubated with human p53 protein (1 µg in 5 ml) followed by detection with PAb421. Co-immunoprecipitation was performed as follows: purified baculovirus-expressed p53, DNA pol β and PAb421 cross-linked protein A–Sepharose (Pierce) were incubated in BER reaction buffer at 4°C for 2 h. After washing three times, Sepharose beads were resuspended in protein loading buffer (50 mM Tris–HCl pH 6.8, 100 mM DTT, 2% SDS, 10% glycerol, 0.1% bromophenol blue). Proteins bound to the beads were examined by SDS–PAGE and western blotting using PAb421 and antiserum generated against DNA pol β (a gift from B.Demple). Co-immunoprecipitation with magnetic beads coated with 18S monoclonal antibody against DNA pol β was performed as described by the manufacturer (Dynal). Protein cross-linking was performed as described (Craig, 1988). Six hundred nanograms of purified DNA pol β and 600 ng of purified baculovirus-expressed p53 were incubated with 8 mM glutaraldehyde at 15°C for 1 h. Cross-linking products were separated in a step-gradient SDS–PAGE gel (4% stacking gel, 6 and 10% separation gel) and the gel was silver stained.
Electrophoretic mobility shift assay
EMSA was performed as described (Bennett et al., 1997). For a typical reaction (20 µl), 0.04 pmol of end-labeled AP-DNA were mixed (where indicated) with APE, DNA pol β and p53 in 50 mM HEPES–KOH pH 7.5, 100 mM KCl, 10% glycerol, and either 10 mM MgCl2 (Mg2+ buffer) or 4 mM EDTA (EDTA buffer). The mixture was incubated on ice for 20 min and resolved on an 8% non-denaturing polyacrylamide gel followed by autoradiography. For the competition/dissociation assay, EMSA reaction mixtures (in Mg2+ buffer), containing 20 ng of APE, 20 ng of DNA pol β and end-labeled AP-DNA, contained or lacked 80 ng of p53. Thirty-fold excess unlabeled competitor AP-DNA was added to each EMSA reaction mixture at time zero. After the indicated time, mixtures were loaded onto a running 8% polyacrylamide gel.
Acknowledgments
Acknowledgements
We are grateful to Ella Freulich for excellent technical assistance. We thank B.Demple for rabbit antiserum against DNA pol β and helpful suggestions. G.Wahl is thanked for suggesting the use of fibroblasts from p53-null and mutant p53 (L25Q/W26S) mice as well as for providing cells to carry out these experiments. This work was supported by NIH grant CA58316 to C.P.
REFERENCES
- Bakalkin G. et al. (1995) p53 binds single-stranded DNA ends through the C-terminal domain and internal DNA segments via the middle domain. Nucleic Acids Res., 23, 362–369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Banin S. et al. (1998) Enhanced phosphorylation of p53 by ATM in response to DNA damage. Science, 281, 1674–1677. [DOI] [PubMed] [Google Scholar]
- Barzilay G. and Hickson,I.D. (1995) Structure and function of apurinic/apyrimidinic endonucleases. BioEssays, 17, 713–719. [DOI] [PubMed] [Google Scholar]
- Bennett R.A., Wilson,D.M.,III, Wong,D. and Demple,B. (1997) Inter action of human apurinic endonuclease and DNA polymerase β in the base excision repair pathway. Proc. Natl Acad. Sci. USA, 94, 7166–7169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bogenhagen D.F. and Pinz,K.G. (1998) The action of DNA ligase at abasic sites in DNA. J. Biol. Chem., 273, 7888–7893. [DOI] [PubMed] [Google Scholar]
- Caldecott K.W., McKeown,C.K., Tucker,J.D., Ljungquist,S. and Thompson,L.H. (1994) An interaction between the mammalian DNA repair protein XRCC1 and DNA ligase III. Mol. Cell. Biol., 14, 68–76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Caldecott K.W., Tucker,J.D., Stanker,L.H. and Thompson,L.H. (1995) Characterization of the XRCC1–DNA ligase III complex in vitro and its absence from mutant hamster cells. Nucleic Acids Res., 23, 4836–4843. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Canman C.E., Lim,D.S., Cimprich,K.A., Taya,Y., Tamai,K., Sakaguchi,K., Appella,E., Kastan,M.B. and Siliciano,J.D. (1998) Activation of the ATM kinase by ionizing radiation and phosphorylation of p53. Science, 281, 1677–1679. [DOI] [PubMed] [Google Scholar]
- Chehab N.H., Malikzay,A., Appel,M. and Halazonetis,T.D. (2000) Chk2/hCds1 functions as a DNA damage checkpoint in G1 by stabilizing p53. Genes Dev., 14, 278–288. [PMC free article] [PubMed] [Google Scholar]
- Chen D.S., Herman,T. and Demple,B. (1991) Two distinct human DNA diesterases that hydrolyze 3′-blocking deoxyribose fragments from oxidized DNA. Nucleic Acids Res., 19, 5907–5914. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen X., Ko,L.J., Jayaraman,L. and Prives,C. (1996) p53 levels, functional domains and DNA damage determine the extent of the apoptotic response of tumor cells. Genes Dev., 10, 2438–2451. [DOI] [PubMed] [Google Scholar]
- Courtemanche C. and Anderson,A. (1999) The p53 tumor suppressor protein reduces point mutation frequency of a shuttle vector modified by the chemical mutagens (+/–)7,8-dihydroxy-9,10-epoxy-7,8,9,10-tetrahydrobenzo[a]pyrene, aflatoxin B1 and meta-chloroperoxy benzoic acid. Oncogene, 18, 4672–4680. [DOI] [PubMed] [Google Scholar]
- Craig W.S. (1988) Determination of quaternary structure of an active enzyme using chemical cross-linking with glutaraldehyde. Methods Enzymol., 156, 333–345. [DOI] [PubMed] [Google Scholar]
- Date T., Yamaguchi,M., Hirose,F., Nishimoto,Y., Tanihara,K. and Matsukage,A. (1988) Expression of active rat DNA polymerase β in Escherichia coli. Biochemistry, 27, 2983–2990. [DOI] [PubMed] [Google Scholar]
- Demple B. and Harrison,L. (1994) Repair of oxidative damage to DNA: enzymology and biology. Annu. Rev. Biochem., 63, 915–948. [DOI] [PubMed] [Google Scholar]
- Demple B., Herman,T. and Chen,D.S. (1991) Cloning and expression of APE, the cDNA encoding the major human apurinic endonuclease: definition of a family of DNA repair enzymes. Proc. Natl Acad. Sci. USA, 88, 11450–11454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dianov G. and Lindahl,T. (1994) Reconstitution of the DNA base excision-repair pathway. Curr. Biol., 4, 1069–1076. [DOI] [PubMed] [Google Scholar]
- Ford J.M., Baron,E.L. and Hanawalt,P.C. (1998) Human fibroblasts expressing the human papillomavirus E6 gene are deficient in global genomic nucleotide excision repair and sensitive to ultraviolet irradiation. Cancer Res., 58, 599–603. [PubMed] [Google Scholar]
- Fornace A.J. Jr, Zmudzka,B., Hollander,M.C. and Wilson,S.H. (1989) Induction of β-polymerase mRNA by DNA-damaging agents in Chinese hamster ovary cells. Mol. Cell. Biol., 9, 851–853. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Friedberg E.C., Walker,G.C. and Siede,W. (1995) DNA Repair and Mutagenesis. American Society for Microbiology, Washington, DC.
- Frosina G., Fortini,P., Rossi,O., Carrozzino,F., Raspaglio,G., Cox,L.S., Lane,D.P., Abbondandolo,A. and Dogliotti,E. (1996) Two pathways for base excision repair in mammalian cells. J. Biol. Chem., 271, 9573–9578. [DOI] [PubMed] [Google Scholar]
- Gaiddon C., Moorthy,N.C. and Prives,C. (1999) Ref-1 regulates the transactivation and pro-apoptotic functions of p53 in vivo. EMBO J., 18, 5609–5621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Giaccia A.J. and Kastan,M.B. (1998) The complexity of p53 modulation: emerging patterns from divergent signals. Genes Dev., 12, 2973–2983. [DOI] [PubMed] [Google Scholar]
- Gu W., Shi,X.L. and Roeder,R.G. (1997) Synergistic activation of transcription by CBP and p53. Nature, 387, 819–823. [DOI] [PubMed] [Google Scholar]
- Harvey M. et al. (1993) In vitro growth characteristics of embryo fibroblasts isolated from p53-deficient mice. Oncogene, 8, 2457–2467. [PubMed] [Google Scholar]
- Hollstein M., Sidransky,D., Vogelstein,B. and Harris,C.C. (1991) p53 mutations in human cancers. Science, 253, 49–53. [DOI] [PubMed] [Google Scholar]
- Hollstein M. et al. (1994) Database of p53 gene somatic mutations in human tumors and cell lines. Nucleic Acids Res., 22, 3551–3555. [PMC free article] [PubMed] [Google Scholar]
- Huang P. (1998) Excision of mismatched nucleotides from DNA: a potential mechanism for enhancing DNA replication fidelity by the wild-type p53 protein. Oncogene, 17, 261–270. [DOI] [PubMed] [Google Scholar]
- Hwang B.J., Ford,J.M., Hanawalt,P.C. and Chu,G. (1999) Expression of the p48 xeroderma pigmentosum gene is p53-dependent and is involved in global genomic repair. Proc. Natl Acad. Sci. USA, 96, 424–428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jayaraman J. and Prives,C. (1995) Activation of p53 sequence-specific DNA binding by short single strands of DNA requires the p53 C-terminus. Cell, 81, 1021–1029. [DOI] [PubMed] [Google Scholar]
- Jayaraman L. and Prives,C. (1999) Covalent and noncovalent modifiers of the p53 protein. Cell. Mol. Life Sci., 55, 76–87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jayaraman L., Murthy,K.G., Zhu,C., Curran,T., Xanthoudakis,S. and Prives,C. (1997) Identification of redox/repair protein Ref-1 as a potent activator of p53. Genes Dev., 11, 558–570. [DOI] [PubMed] [Google Scholar]
- Jayaraman L., Moorthy,N.C., Murthy,K.G., Manley,J.L., Bustin,M. and Prives,C. (1998) High mobility group protein-1 (HMG-1) is a unique activator of p53. Genes Dev., 12, 462–472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jimenez G.S., Nister,M., Stommel,J.M., Beeche,M., Barcarse,E.A., Zhang,X.Q., O’Gorman,S. and Wahl,G.M. (2000) A transactivation-deficient mouse model provides insights into trp53 regulation and function. Nature Genet., 26, 37–43. [DOI] [PubMed] [Google Scholar]
- Khanna K.K. et al. (1998) ATM associates with and phosphorylates p53: mapping the region of interaction. Nature Genet., 20, 398–400. [DOI] [PubMed] [Google Scholar]
- Klungland A. and Lindahl,T. (1997) Second pathway for completion of human DNA base excision-repair: reconstitution with purified proteins and requirement for DNase IV (FEN1). EMBO J., 16, 3341–3348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ko L.J. and Prives,C. (1996) p53: puzzle and paradigm. Genes Dev., 10, 1054–1072. [DOI] [PubMed] [Google Scholar]
- Ko L.J., Shieh,S.Y., Chen,X., Jayaraman,L., Tamai,K., Taya,Y., Prives,C. and Pan,Z.Q. (1997) p53 is phosphorylated by CDK7–cyclin H in a p36MAT1-dependent manner. Mol. Cell. Biol., 17, 7220–7229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kubota Y., Nash,R.A., Klungland,A., Schar,P., Barnes,D.E. and Lindahl,T. (1996) Reconstitution of DNA base excision-repair with purified human proteins: interaction between DNA polymerase β and the XRCC1 protein. EMBO J., 15, 6662–6670. [PMC free article] [PubMed] [Google Scholar]
- Kunkel T.A. (1985) The mutational specificity of DNA polymerase-β during in vitro DNA synthesis. Production of frameshift, base substitution and deletion mutations. J. Biol. Chem., 260, 5787–5796. [PubMed] [Google Scholar]
- Lane D.P. (1992) Cancer. p53, guardian of the genome. Nature, 358, 15–16. [DOI] [PubMed] [Google Scholar]
- Lee J.M., Abrahamson,J.L., Kandel,R., Donehower,L.A. and Bernstein,A. (1994) Susceptibility to radiation-carcinogenesis and accumulation of chromosomal breakage in p53 deficient mice. Oncogene, 9, 3731–3736. [PubMed] [Google Scholar]
- Lee S., Elenbaas,B., Levine,A. and Griffith,J. (1995) p53 and its 14 kDa C-terminal domain recognize primary DNA damage in the form of insertion/deletion mismatches. Cell, 81, 1013–1020. [DOI] [PubMed] [Google Scholar]
- Liebermann D.A., Hoffman,B. and Steinman,R.A. (1995) Molecular controls of growth arrest and apoptosis: p53-dependent and independent pathways. Oncogene, 11, 199–210. [PubMed] [Google Scholar]
- Lin J., Chen,J., Elenbaas,B. and Levine,A.J. (1994) Several hydrophobic amino acids in the p53 amino-terminal domain are required for transcriptional activation, binding to mdm-2 and the adenovirus 5 E1B 55-kD protein. Genes Dev., 8, 1235–1246. [DOI] [PubMed] [Google Scholar]
- Lin J., Teresky,A.K. and Levine,A.J. (1995) Two critical hydrophobic amino acids in the N-terminal domain of the p53 protein are required for the gain of function phenotypes of human p53 mutants. Oncogene, 10, 2387–2390. [PubMed] [Google Scholar]
- Lindahl T. (1993) Instability and decay of the primary structure of DNA. Nature, 362, 709–715. [DOI] [PubMed] [Google Scholar]
- Lindahl T., Satoh,M.S. and Dianov,G. (1995) Enzymes acting at strand interruptions in DNA. Philos. Trans. R. Soc. Lond. B Biol. Sci., 347, 57–62. [DOI] [PubMed] [Google Scholar]
- Lloyd D.R. and Hanawalt,P.C. (2000) p53-dependent global genomic repair of benzo[a]pyrene-7,8-diol-9,10-epoxide adducts in human cells. Cancer Res., 60, 517–521. [PubMed] [Google Scholar]
- Lu H. and Levine,A.J. (1995) Human TAFII31 protein is a transcriptional coactivator of the p53 protein. Proc. Natl Acad. Sci. USA, 92, 5154–5158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matsumoto Y. and Bogenhagen,D.F. (1991) Repair of a synthetic abasic site involves concerted reactions of DNA synthesis followed by excision and ligation. Mol. Cell. Biol., 11, 4441–4447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matsumoto Y. and Kim,K. (1995) Excision of deoxyribose phosphate residues by DNA polymerase β during DNA repair. Science, 269, 699–702. [DOI] [PubMed] [Google Scholar]
- Mummenbrauer T., Janus,F., Muller,B., Wiesmuller,L., Deppert,W. and Grosse,F. (1996) p53 protein exhibits 3′-to-5′ exonuclease activity. Cell, 85, 1089–1099. [DOI] [PubMed] [Google Scholar]
- Nicholl I.D., Nealon,K. and Kenny,M.K. (1997) Reconstitution of human base excision repair with purified proteins. Biochemistry, 36, 7557–7566. [DOI] [PubMed] [Google Scholar]
- Nigro J.M. et al. (1989) Mutations in the p53 gene occur in diverse human tumour types. Nature, 342, 705–708. [DOI] [PubMed] [Google Scholar]
- Offer H., Wolkowicz,R., Matas,D., Blumenstein,S., Livneh,Z. and Rotter,V. (1999) Direct involvement of p53 in the base excision repair pathway of the DNA repair machinery. FEBS Lett., 450, 197–204. [DOI] [PubMed] [Google Scholar]
- Osheroff W.P., Jung,H.K., Beard,W.A., Wilson,S.H. and Kunkel,T.A. (1999) The fidelity of DNA polymerase β during distributive and processive DNA synthesis. J. Biol. Chem., 274, 3642–3650. [DOI] [PubMed] [Google Scholar]
- Piersen C.E., Prasad,R., Wilson,S.H. and Lloyd,R.S. (1996) Evidence for an imino intermediate in the DNA polymerase β deoxyribose phosphate excision reaction. J. Biol. Chem., 271, 17811–17815. [DOI] [PubMed] [Google Scholar]
- Plug A.W., Clairmont,C.A., Sapi,E., Ashley,T. and Sweasy,J.B. (1997) Evidence for a role for DNA polymerase β in mammalian meiosis. Proc. Natl Acad. Sci. USA, 94, 1327–1331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prives C. (1998) Signaling to p53: breaking the MDM2–p53 circuit. Cell, 95, 5–8. [DOI] [PubMed] [Google Scholar]
- Prives C. and Hall,P.A. (1999) The p53 pathway. J. Pathol., 187, 112–126. [DOI] [PubMed] [Google Scholar]
- Reed M., Woelker,B., Wang,P., Wang,Y., Anderson,M.E. and Tegtmeyer,P. (1995) The C-terminal domain of p53 recognizes DNA damaged by ionizing radiation. Proc. Natl Acad. Sci. USA, 92, 9455–9459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seeberg E., Eide,L. and Bjoras,M. (1995) The base excision repair pathway. Trends Biochem. Sci., 20, 391–397. [DOI] [PubMed] [Google Scholar]
- Shieh S.Y., Ikeda,M., Taya,Y. and Prives,C. (1997) DNA damage-induced phosphorylation of p53 alleviates inhibition by MDM2. Cell, 91, 325–334. [DOI] [PubMed] [Google Scholar]
- Shieh S.Y., Ahn,J., Tamai,K., Taya,Y. and Prives,C. (2000) The human homologs of checkpoint kinases Chk1 and Cds1 (Chk2) phosphorylate p53 at multiple DNA damage-inducible sites. Genes Dev., 14, 289–300. [PMC free article] [PubMed] [Google Scholar]
- Singhal R.K., Prasad,R. and Wilson,S.H. (1995) DNA polymerase β conducts the gap-filling step in uracil-initiated base excision repair in a bovine testis nuclear extract. J. Biol. Chem., 270, 949–957. [DOI] [PubMed] [Google Scholar]
- Sobol R.W., Horton,J.K., Kuhn,R., Gu,H., Singhal,R.K., Prasad,R., Rajewsky,K. and Wilson,S.H. (1996) Requirement of mammalian DNA polymerase-β in base-excision repair. Nature, 379, 183–186. [DOI] [PubMed] [Google Scholar]
- Srivastava D.K., Berg,B.J., Prasad,R., Molina,J.T., Beard,W.A., Tomkinson,A.E. and Wilson,S.H. (1998) Mammalian abasic site base excision repair. Identification of the reaction sequence and rate-determining steps. J. Biol. Chem., 273, 21203–21209. [DOI] [PubMed] [Google Scholar]
- Tang J.Y., Hwang,B.J., Ford,J.M., Hanawalt,P.C. and Chu,G. (2000) Xeroderma pigmentosum p48 gene enhances global genomic repair and suppresses UV-induced mutagenesis. Mol. Cell, 5, 737–744. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walker L.J., Robson,C.N., Black,E., Gillespie,D. and Hickson,I.D. (1993) Identification of residues in the human DNA repair enzyme HAP1 (Ref-1) that are essential for redox regulation of Jun DNA binding. Mol. Cell. Biol., 13, 5370–5376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Y., Reed,M., Wang,P., Stenger,J.E., Mayr,G., Anderson,M.E., Schwedes,J.F. and Tegtmeyer,P. (1993) p53 domains: identification and characterization of two autonomous DNA-binding regions. Genes Dev., 7, 2575–2586. [DOI] [PubMed] [Google Scholar]
- Wilson D.M. III, Takeshita,M. and Demple,B. (1997) Abasic site binding by the human apurinic endonuclease, Ape and determination of the DNA contact sites. Nucleic Acids Res., 25, 933–939. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wood R.D., Robins,P. and Lindahl,T. (1988) Complementation of the xeroderma pigmentosum DNA repair defect in cell-free extracts. Cell, 53, 97–106. [DOI] [PubMed] [Google Scholar]
- Xanthoudakis S. and Curran,T. (1992) Identification and characterization of Ref-1, a nuclear protein that facilitates AP-1 DNA-binding activity. EMBO J., 11, 653–665. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xanthoudakis S., Miao,G., Wang,F., Pan,Y.C. and Curran,T. (1992) Redox activation of Fos–Jun DNA binding activity is mediated by a DNA repair enzyme. EMBO J., 11, 3323–3335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xanthoudakis S., Miao,G.G. and Curran,T. (1994) The redox and DNA-repair activities of Ref-1 are encoded by nonoverlapping domains. Proc. Natl Acad. Sci. USA, 91, 23–27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yoo M.A., Lee,W.H., Ha,H.Y., Ryu,J.R., Yamaguchi,M., Fujikawa,K., Matsukage,A., Kondo,S. and Nishida,Y. (1994) Effects of DNA polymerase β gene over-expressed in transgenic Drosophila on DNA repair and recombination. Jpn. J. Genet., 69, 21–33. [DOI] [PubMed] [Google Scholar]
- Yu Y., Li,C.Y. and Little,J.B. (1997) Abrogation of p53 function by HPV16 E6 gene delays apoptosis and enhances mutagenesis but does not alter radiosensitivity in TK6 human lymphoblast cells. Oncogene, 14, 1661–1667. [DOI] [PubMed] [Google Scholar]