

Abstract
The heat shock proteins ClpC and ClpP are subunits of an ATP-dependent protease of Bacillus subtilis. Under non-stressed conditions, transcription of the clpC and clpP genes is negatively regulated by CtsR, the global repressor of clp gene expression. Here, CtsR was proven to be a specific substrate of the ClpCP protease under stress conditions. Two proteins of former unknown function, McsA and McsB, which are also encoded by the clpC operon, act as modulators of CtsR repression. McsA containing zinc finger motifs stabilizes CtsR under non-stressed conditions. McsB, a putative kinase, can inactivate CtsR by modification to remove the repressor from the DNA and to target CtsR for degradation by the ClpCP protease during stress. Thus, clp gene expression in Gram-positive bacteria is autoregulated by a novel mechanism of controlled proteolysis, a circuit of down-regulation by stabilization and protection of a transcription repressor, and induction by presenting the repressor to the protease. Thereby, the ClpC ATPase, a member of the Hsp100 family, was identified as a positive regulator of the heat shock response.
Keywords: Bacillus subtilis/ClpCP protease/heat shock/Hsp100/proteolysis
Introduction
Protein degradation plays a key role in cell physiology of all organisms by regulating the availability of certain short-lived regulatory proteins or preventing the accumulation of abnormal proteins. Energy-dependent proteases of Escherichia coli, namely the Clp proteases ClpAP (Ti), ClpXP and ClpYQ (HslUV), and also Lon (La) and FtsH (HflB), have been studied extensively and serve as models of how proteases participate in regulatory cascades (reviewed in Gottesman, 1996).
Clp protease complexes of E.coli consist in general of a regulatory ATPase subunit (ClpA, ClpX or ClpY) and a proteolytic component (ClpP or ClpQ) that assemble into structures similar to the eukaryotic proteasome (reviewed in Gottesman, 1996, 1997a,b; Wickner et al., 1999; Bochtler et al., 2000). Besides degradation of misfolded proteins, several specific substrates were found for Clp proteases. Targets degraded by ClpXP are exemplified by the stationary phase transcription factor σS, several phage proteins and the heterodimeric form of the UmuD protein involved in SOS mutagenesis. Certain β-galactosidase fusion proteins following the N-end rule, and the host addiction protein MazE have been described as substrates for ClpAP (reviewed in Gottesman 1996; Varshavsky, 1996).
Tagging systems for unstable proteins are crucial for selectivity of proteases. In eukaryotes, the ubiquitin system mediates the targeting of short-lived or misfolded proteins and directs them for degradation by the 26S proteasome (reviewed in Ciechanover, 1998; Laney and Hochstrasser, 1999). A system for tagging of unstable proteins in prokaryotes is the SsrA-mediated co-translational addition of a peptide signal to the C-terminus of incompletely synthesized polypeptides (Keiler et al., 1996). SsrA-tagged proteins are targets of both the ClpAP and ClpXP proteases (Gottesmann et al., 1998). Substrate specificity is assigned to the ATPase components. They appear to direct substrate recognition, binding, unfolding and translocation into the protease cavity that requires ATP hydrolysis (Levchenko et al., 1997; Hoskins et al., 1998; Pak et al., 1999; Weber-Ban et al., 1999). It has been accepted that Clp-ATPases belonging to the Hsp100 family conserved in all organisms can function either as molecular chaperones or as proteolysis regulators (reviewed in Squires and Squires, 1992; Schirmer et al., 1996; Gottesman et al., 1997a,b; Wickner et al., 1999).
Although Clp proteins are highly conserved and ubiquitous in bacteria and higher organisms, only a few data are available about the importance of Clp-mediated proteolysis in organisms other than E.coli. For example, the ClpXP protease is involved in the cell cycle control of Caulobacter crescentus (Jenal and Fuchs, 1998). In the Gram-positive soil bacterium Bacillus subtilis, the presence of ClpC, ClpP or ClpX in the cell is essential for tolerance to many forms of stress. Furthermore, B.subtilis Clp proteins were found to be required for several cellular processes such as cell division, motility and degradative enzyme synthesis, and for developmental processes such as sporulation and genetic competence (Kong and Dubnau, 1994; Krüger et al., 1994; Msadek et al., 1994; Turgay et al., 1997, 1998; Gerth et al., 1998; Msadek et al., 1998; Nanamiya et al., 1998).
Expression mechanisms for heat-inducible ATP-dependent protease genes differ significantly in E.coli and B.subtilis. Induction of heat shock genes including the clp genes in E.coli depends mainly on the level of the heat shock transcription factor σ32 regulating the gene expression positively. A higher intracellular concentration of σ32 after a temperature upshift is caused by increased synthesis, stability and activity. The DnaK chaperone machine plays a central role in the control of σ32 activity as a negative modulator and in targeting of σ32 for ATP-dependent degradation by FtsH (reviewed in Bukau, 1997). In contrast, expression of the hexacistronic clpC operon and the clpP gene in B.subtilis is directed by two stress induction pathways relying either on positive control by the alternative σ factor, σB, or on a dominant stress induction mechanism acting at a vegetative σA-like promoter (Krüger et al., 1996; Gerth et al., 1998; Msadek et al., 1998). Recently, this mechanism was determined as the negative regulation by the CtsR repressor, which is encoded by the first gene of the clpC operon (Krüger and Hecker, 1998; Derré et al., 1999a). Additionally, ClpE, a novel type of Hsp100 ATPase, is part of the CtsR heat shock regulon (Derré et al., 1999b). Products of the second and third genes of the clpC operon, Orf2 and Orf3, were suggested to have regulatory function in clp gene expression (Krüger and Hecker, 1998; Figure 1A).
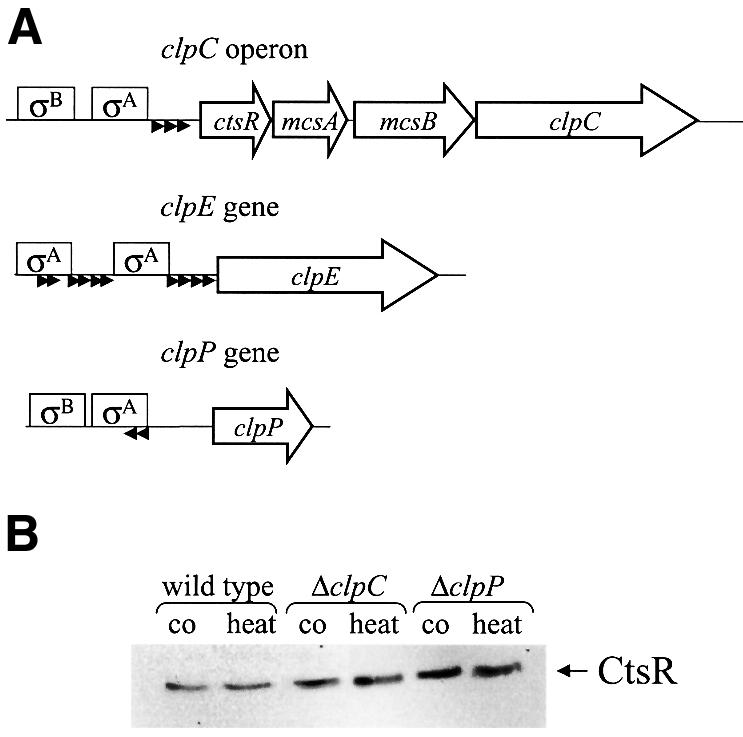
Fig. 1. (A) Schematic representation of the clpC operon, and the clpE and clpP genes. The genes (coding sequence) are indicated by open arrows, the σA- and σB-dependent promoters by boxes and the CtsR-binding sites by small black arrowheads. (B) The amount of CtsR in wild type, and clpC and clpP mutant strains during exponential growth and after heat shock. Samples were taken before (co) and 15 min after 50°C heat shock (heat), and analyzed by western blotting using antibodies against CtsR.
Induction patterns of CtsR-dependent genes led to the postulation that the CtsR repressor has to be inactivated and removed during several stresses such as heat shock, puromycin or ethanol stress (Krüger et al., 1996; Gerth et al., 1998). However, the exact mechanism of this step for induction of the essential ClpCP-mediated proteolysis under such conditions remained unknown. In this study, investigation of this phenomenon reveals that the CtsR repressor is labile under stress conditions and becomes a substrate of the ClpCP protease in vivo and in vitro. Studies of mutants lacking the Orf2 and Orf3 proteins provide evidence that they play essential roles in modulating CtsR stability. In an opposite mode of action, Orf2 and Orf3, now referred to as McsA and McsB (modulator of CtsR repression), ensure a system of clp gene repression and induction by protecting CtsR or modifying the repressor to target it for proteolysis. Thus, expression of a key enzyme involved in many cellular processes is autoregulated by controlled proteolysis.
Results
CtsR protein levels do not correlate with transcription of the clpC operon
Certain N-terminal amino acid sequences confer instability to proteins when examined as β-galactosidase fusions, a phenomenon called N-end rule (reviewed in Varshavsky, 1996). Originally, a translational fusion of the first five codons of the ctsR gene to lacZ driven by the transcriptional and translational control sequences of the clpC operon was constructed to study its regulation (Krüger et al., 1996). Unexpectedly, the level of CtsR–LacZ did not increase after glucose starvation. This result was not in agreement with our mRNA data showing that the clpC operon is induced under these conditions (Krüger et al., 1994). However, by assaying LacZ of a transcriptional fusion containing the same DNA fragment of the clpC operon regulatory region, a normal stationary phase induction was verified, suggesting a post-transcriptional control of CtsR (Table I). To determine whether the B.subtilis Clp proteins play a role in the stability of CtsR–LacZ, we introduced deletions of either clpC or clpP into the strain carrying the translational fusion. Both LacZ assays and mRNA measurements (data not shown) of the ctsR–lacZ transcription of glucose-starved cells were done in parallel with these strains. Transcription analysis revealed an ∼4-fold lacZ induction after entry into stationary phase, which is consistent with our previous data (Krüger et al., 1994). None of the clp deletions notably affected the ctsR–lacZ mRNA induction ratio under starvation conditions. However, β-galactosidase activity of the translational ctsR–lacZ fusion was significantly increased in clpC or clpP deletion mutants (Table I). In strains lacking either ClpC or ClpP, the CtsR–LacZ fusion protein became more active than in the wild type, probably due to its higher stability. These data strongly suggested that the CtsR–LacZ fusion protein is unstable and may be degraded by the ClpCP protease.
Table I. β-galactosidase activity of transcriptional and translational ctsR–lacZ fusions.
| Strain | β-galactosidase activity (Miller units) | |
|---|---|---|
| Exponential phase | Stationary phase | |
| amyE::cat (ctsR–lacZ) transcriptional fusion | 9 | 41 |
| amyE::aphA3 (ctsR–′lacZ) translational fusion | 3 | 7 |
| ΔclpC::spcR amyE::aphA3 (ctsR–′lacZ) translational fusion | 9 | 28 |
| ΔclpP::spcR amyE::aphA3 (ctsR–′lacZ) translational fusion | 5 | 25 |
Studies of the clpC operon revealed a strong induction of the polycistronic mRNA (Figure 1A) specifically after certain stress conditions that are known to produce non-native proteins in the cell (Krüger et al., 1996; Mogk et al., 1998). For ClpC, a strict correlation between mRNA induction ratio and increased protein synthesis has been shown for various stress conditions (Krüger et al., 1994; Bernhardt et al., 1999). To know more about the intracellular concentrations of the CtsR repressor after heat shock, western blot experiments were performed with protein extracts of control and heat-shocked cells of the wild type, or of clpC or clpP mutants. Surprisingly, there was no obvious difference in the level of the CtsR protein content in non-stressed and heat-shocked cells (Figure 1B), although our studies revealed a strong induction of ctsR gene transcription by heat shock using either mRNA analysis (data not shown) or β-galactosidase assays of a ctsR–bgaB reporter gene fusion (see below; Table II). In contrast, a clear heat induction for ClpC encoded by the same operon was observed previously and shown in western blot experiments (Krüger et al., 1994, 2000; see below; Figure 5C). However, the CtsR content in clpC or clpP mutants was slightly increased, indicating that ClpC and ClpP influence the CtsR level in the cell (Figure 1B).
Table II. Influence of deletions for different regulatory protein genes on the expression of the clpC operon, and the clpE and clpP genes.
| Strains | β-galactosidase activity (Miller units) |
|||
|---|---|---|---|---|
| Exponential growth (control) | 30 min heat shock at 50°C | Induction | Derepression of the basal level | |
| BHL1 (clpE–bgaB, wild type) | 2 | 507 | 254-fold | no |
| BHL5 (clpE–bgaB, ΔctsR) | 430 | 411 | 1-fold | 215-fold |
| BHL3 (clpE–bgaB, ΔmcsA) | 436 | 501 | 1.1-fold | 218-fold |
| BHL4 (clpE–bgaB, ΔmcsB) | 11 | 37 | 3.4-fold | 5.5-fold |
| BHL6 (clpE–bgaB, ΔclpC) | 11 | 159 | 14-fold | 5.5-fold |
| BHL7 (clpE–bgaB, ΔclpP) | 13 | 76 | 5.8-fold | 6.5-fold |
| BDZ2 (clpE–bgaB, ΔmcsA ΔclpP) | 204 | 225 | no | 102-fold |
| BDZ3 (clpE–bgaB, ΔmcsB ΔclpP) | 5 | 19 | 3.8-fold | 2.5-fold |
| BDZ4 (clpE–bgaB, ΔmcsA ΔmcsB) | 20 | 79 | 4-fold | 10-fold |
| BEK49 (ctsR–bgaB, wild type) | 5 | 178 | 35.6-fold | no |
| BEK86 (ctsR–bgaB, ΔctsR) | 59 | 188 | 3.2-fold | 11.8-fold |
| BEK91 (ctsR–bgaB, ΔmcsA) | 64 | 164 | 2.6-fold | 12.8-fold |
| BEK92 (ctsR–bgaB, ΔmcsB) | 9 | 28 | 3.1-fold | 1.8-fold |
| BEK93 (ctsR–bgaB, ΔclpC) | 15 | 91 | 6-fold | 3-fold |
| BEK94 (ctsR–bgaB, ΔclpP) | 8 | 65 | 8-fold | 1.6-fold |
| BUG3 (clpP–bgaB, wild type) | 14 | 214 | 15.3-fold | no |
| BEK95 (clpP–bgaB, ΔctsR) | 69 | 189 | 2.7-fold | 5-fold |
| BEK96 (clpP–bgaB, ΔmcsA) | 75 | 201 | 2.7-fold | 5.4-fold |
| BEK97 (clpP–bgaB, ΔmcsB) | 12 | 86 | 7.2-fold | no |
| BEK98 (clpP–bgaB, ΔclpC) | 30 | 122 | 4-fold | 2-fold |
| BEK99 (clpP–bgaB, ΔclpP) | 11 | 65 | 5.9-fold | no |
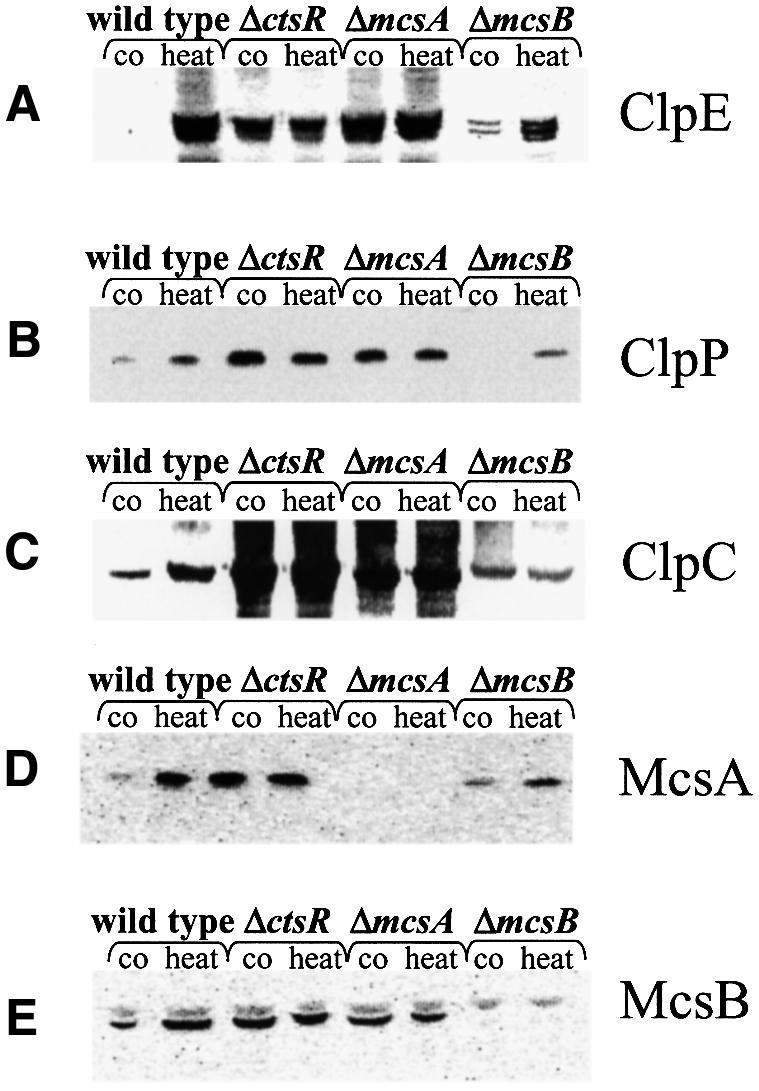
Fig. 5. The amount of CtsR-dependent heat shock proteins in strains lacking regulatory proteins. The amount of ClpE (A), ClpP (B), ClpC (C), McsA (D) and McsB (E) in strains lacking either CtsR, McsA or McsB during exponential growth and after heat shock. Samples were taken before (co) and 15 min after 50°C heat shock (heat) and analyzed by western blotting using antibodies against ClpE (A), ClpP (B) ClpC (C), McsA (D) and McsB (E).
In vivo stability of CtsR
From the data obtained by β-galactosidase assays and immunoblots with CtsR antibodies (see above), we concluded that the CtsR repressor itself may be a labile protein, whose degradation is probably mediated by the ClpCP protease. To analyze the in vivo stability of the CtsR repressor, either of the antibiotics chloramphenicol or puromycin were added to exponentially growing cells of a B.subtilis wild-type strain to cut off translation. Cellular extracts of chloramphenicol- and puromycin-treated cells were studied in parallel by western blotting with CtsR antibodies. In cells treated with chloramphenicol, which does not induce the clpC operon, intracellular levels of CtsR did not change significantly during a time course of 30 min at 37°C (Figure 2A). After puromycin stress that induced the clpC operon transcription >40-fold (Krüger et al., 1996), the content of CtsR decreased significantly in wild-type cells (Figure 2B). Similar results could be achieved by chloramphenicol treatment under heat shock conditions (data not shown). During exponential growth, CtsR was rather stable, whereas the in vivo half-life of this protein under stress conditions was <2 min (Figure 2A and B).

Fig. 2. Stability of the CtsR repressor in vivo. The antibiotics chloramphenicol (100 µg/ml) (A) and puromycin (60 µg/ml) (B–E) were added to exponentially growing cultures of different B.subtilis strains to stop the bacterial translation (chloramphenicol) and additionally to induce the heat shock response (puromycin). Samples were taken before (control), and 10, 20 and 30 min after the addition of chloramphenicol (A) or 2, 4 and 8 min after the addition of puromycin (B–E). Protein extracts of the wild type (A and B) and the clpX (C), clpC (D) and clpP (E) mutant strains were analyzed for CtsR amount by western blotting.
To resolve whether the Clp proteins play a role in the stability of the repressor protein CtsR, these immunoblot experiments were repeated with cellular extracts of clpC, clpP and clpX mutant strains. Analysis of the protein extracts of clpX mutant cells also revealed a strong decrease in CtsR levels after puromycin stress similar to the wild type (Figure 2C). In contrast, CtsR was much more stable in the clpC or clpP mutants (Figure 2D and E). These results show that ClpC and ClpP, but not ClpX, are involved in the degradation of CtsR under stress. As a consequence, CtsR did not accumulate after heat shock because of its rapid degradation.
In vitro stability of CtsR
The substrate specificity of Clp proteases is thought to be provided by the regulatory ATPase subunit. Both the substrate unfolding and translocation into the protease cavity as well as the regulation of the accessibility to proteolytic sites require ATP hydrolysis during the regulation of ATP-dependent protein degradation (reviewed in Gottesman et al., 1997a,b). Since it turned out that CtsR is a substrate of the ClpCP protease, we further examined whether the ATPase activity of ClpC could be stimulated in the presence of CtsR or CtsR plus ClpP in vitro. For these experiments, all three proteins were purified as His6-tag derivatives and used in colorimetric ATPase assays. ClpC alone exhibited a relatively low ATPase activity as also found previously (Turgay et al., 1997; Figure 3A). No ATPase activity was observed using either ClpP or CtsR alone (data not shown). Therefore, ATP hydrolysis seemed to be clearly related to ClpC. Addition of ClpP in an equimolar range to the reaction mixture did not result in a considerable increase in ATPase activity (Figure 3A). In the presence of CtsR, the ClpC ATPase activity was stimulated >5-fold, most probably due to a chaperone-like activity of ClpC. A further increase in CtsR-stimulated ClpC ATPase activity was achieved by adding ClpP to the reaction (Figure 3A), which may reflect the coupling of ATP hydrolysis to degradation. These experiments not only support the idea of an interaction of the ClpC ATPase with CtsR, but also indicate a degradation of the CtsR repressor by the ClpCP protease (see below).

Fig. 3. ClpC ATPase activity and stability of the CtsR repressor in vitro. (A) ClpC ATPase activity is dependent on the presence of CtsR and ClpP. The specific ATPase activity of ClpC (ATPase units are µmol released of phosphate per µg of enzyme) was determined colorimetrically as described in Materials and methods. Symbols: ClpC alone (C; closed squares), ClpC in the presence of CtsR (CR; triangles), ClpC in the presence of ClpP (CP; circles), ClpC in the presence of CtsR and ClpP (CPR; open squares). (B) The stability of the CtsR repressor is dependent on the presence of ClpC and ClpP. Purified CtsR was incubated with or without ClpC, ClpP and ATP and with the ATP-generating system as described in Materials and methods. After the indicated times, the samples were analyzed by western blotting using anti-CtsR antibodies.
To test this, the purified proteins were used to demonstrate an ATP-dependent degradation of CtsR by ClpP in association with ClpC in vitro. ATPase assays as well as gel mobility shift experiments with His6-CtsR and its DNA target (Krüger and Hecker, 1998) showed that the His6-tagged proteins were active. Purified ClpC, ClpP and CtsR proteins were incubated in the presence of ATP and an ATP-generating system, and analyzed by western blotting with CtsR antibodies. Indeed, the band intensities corresponding to CtsR decreased significantly over a period of 45 min, most probably due to degradation of CtsR by the ClpCP protease (Figure 3B). When ATP, ClpC or ClpP was omitted, no degradation occurred, showing that the in vitro stability of CtsR is dependent on ATP and the presence of both components of the ClpCP protease (Figure 3B). Quantitative evaluation of these data revealed an in vitro half-life of CtsR of ∼30 min (data not shown). Remarkably, the in vivo half-life is only 2 min, which may indicate that the presence of ATP and the association of ClpC and ClpP are essential but not sufficient for the degradation of the CtsR repressor under stress conditions.
Influence of deletions in the mcsA and mcsB genes on clp gene expression
Regulatory function has been suggested for gene products of the second and third genes of the clpC operon (Krüger and Hecker, 1998; see Figure 1A). In extensive database analysis, similarities to a large family of eukaryotic regulatory proteins were noted for the product of the second gene (Krüger et al., 1997). The amino acid sequence of McsA predicts two potential Cys2–Cys2 zinc finger motifs (reviewed in Berg, 1990; Mackay and Crossley, 1998). mcsB encodes a putative protein with a domain that is highly conserved among ATP:guanidino phosphotransferases such as arginine kinases or creatine kinases (Krüger et al., 1997). Here we addressed the question of whether homologs of McsA and McsB might also be present in other Gram-positive bacteria, as already shown for CtsR and its DNA target site (Derré et al., 1999a). Database comparisons allowed us to identify both genes in a number of other low G+C Gram-positive bacteria, e.g. Listeria monocytogenes (Rouquette et al., 1996), Bacillus anthracis, Clostridium acetobutylicum, Clostridium difficile and Staphylococcus aureus (Figure 4A and B), as well as partial sequences of both genes for Streptococcus pyogenes, Streptococcus pneumoniae, Enterococcus faecalis and Lactobacillus sake (databases of unfinished microbial genomes). Remarkably, the zinc finger motifs of McsA and the kinase domain of McsB, as well as other domains of as yet unknown function have been conserved in all species examined (Figure 4A and B).


Fig. 4. Conservation of McsA and McsB among the low G+C Gram-positive bacteria. Alignment of the amino acid sequences of McsA (A) and McsB (B) of B.subtilis (B.su) with those of B.anthracis (B.an), C.difficile (C.di), C.acetobutylicum (C.ac), L.monocytogenes (L.mo; DDBJ/EMBL/GenBank accession No. U40604) and S.aureus (S.au). Sequences from unfinished bacterial genomes were obtained from the public websites of TIGR (http://www.tigr.org). Multiple alignments were performed using the Clustal_W program with the default parameter values provided. Identical residues are shaded in black and similar residues in gray. The conserved CXXC Zn finger motifs and the kinase domains typical for ATP:guanidino phosphotransferases (residues 119–252) are indicated by brackets.
For a detailed characterization of putative regulatory roles of McsA and McsB, non-polar in-frame deletions in both genes were constructed. Deletions in the respective genes were combined with transcriptional fusions of the regulatory regions of either the clpC operon (ctsR–bgaB), or the clpP or clpE gene to the bgaB β-galactosidase reporter gene integrated at the amyE site of the chromosome. Such combination allowed measurement of the influence of a deficiency in CtsR, McsA, McsB, ClpC or ClpP on transcription of CtsR-dependent heat shock genes before (control) and 30 min after 50°C heat shock (summarized in Table II). The expression of clpE–bgaB, which is solely dependent on CtsR, can be considered as the most representative fusion. Under non-stressed conditions in wild-type cells, the clpE–bgaB, ctsR–bgaB or clpP–bgaB fusions displayed low β-galactosidase activities, but a strong increase in BgaB activity was observed after heat shock. Deletion of mcsA resulted in high bgaB expression not only after heat shock, but also at 37°C under non-stressed conditions for all gene fusions tested. Interestingly, this phenotype has already been shown for a ctsR deletion (Krüger and Hecker, 1998; Derré et al., 1999a,b). A slight induction by heat shock remained for the clpP gene and the clpC operon, probably due to the additional regulation by σB. CtsR-dependent repression was not restored in a clpP/mcsA double mutant. Conversely, the control levels resembled that of the wild-type strains in mcsB mutants, whereas lower heat shock levels were measured. Consistent with a stabilization of CtsR in strains lacking ClpC or ClpP (see above), a deletion of clpC or clpP led to prolonged repression of the reporter gene system even after heat shock, similar to that obtained for mcsB mutants. The mcsB phenotype seems to be epistatic in both a clpP/mcsB and a mcsA/mcsB double mutant, indicating that McsB may act upstream of CtsR degradation. Furthermore, these experiments demonstrate that CtsR is hyperactivated in a mcsB mutant under all circumstances.
To confirm these effects at the protein level, western blot experiments with protein extracts of the wild type, and the ctsR, mcsA and mcsB mutants before and 15 min after heat shock were processed with antibodies raised against the CtsR-dependent proteins ClpE, ClpP, ClpC, McsA or McsB. As shown for the reporter gene fusions, in cells of the ctsR and mcsA mutants the contents of CtsR-dependent heat shock proteins under non-stressed conditions were increased compared with the wild-type control. The respective levels after heat shock correlated with the heat shock levels of the wild type (Figure 5A–E). In contrast, the content of CtsR-dependent heat shock proteins in the mcsB deletion mutant was considerably decreased (Figure 5A–E). This is underlined by the stress-sensitive phenotype of mutants lacking McsB, most probably due to the depletion of ClpC and ClpP (data not shown). Consequently, these data suggested that the product of mcsA acts as a positive regulator of CtsR repression whereas the putative kinase McsB influences the CtsR-mediated repression negatively. Since both proteins were shown to modulate the repression by CtsR, we renamed the genes mcsA and mcsB (modulators of CtsR repression).
Modulation of CtsR repression by McsA and McsB
Two possibilities were considered to explain how the McsA and McsB proteins may modulate CtsR-dependent stress gene expression. They can either function directly as regulators at the DNA level, competing with CtsR for binding at the regulatory region, or these two proteins may modulate the activity of CtsR. The possible DNA binding activity of McsA and McsB was investigated by gel mobility shift experiments. A DNA fragment containing the regulatory parts of the clpC operon was incubated with either purified recombinant CtsR, McsA or McsB (Figure 6A). Low amounts of CtsR led to efficient retardation of the DNA fragment, with two retardation signals because of the two CtsR-binding sites, as described (Krüger and Hecker, 1998; lanes 2 and 3). Neither for McsA nor for McsB could retardation be observed, excluding the possibility that these proteins compete with CtsR for DNA binding (lanes 4 and 5). Therefore, we further tested whether these proteins may modulate CtsR directly by promoting or inhibiting its DNA binding capacity. CtsR was incubated with the DNA in combination with either McsA or McsB. Addition of McsA does not influence the CtsR DNA binding activity (lane 6). Excess (12-fold) McsA could promote CtsR-dependent DNA binding (lane 7). The combination with McsB resulted in a concentration-dependent decrease in the amount of DNA retarded by CtsR, suggesting that McsB is able to abolish the DNA binding capacity of CtsR (lanes 8–10). McsB action predominates over McsA action, since addition of McsA cannot reverse the effects of McsB (lane 11; see also Table II).
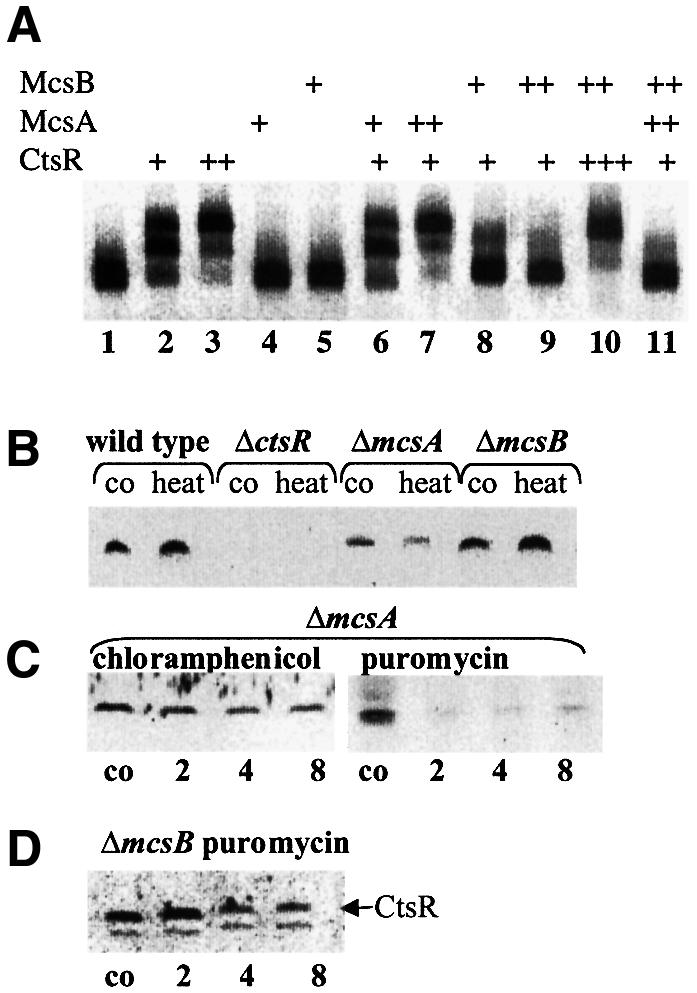
Fig. 6. Modulation of the CtsR activity by McsA or McsB. (A) Gel mobility shift experiments with CtsR alone or in combination with McsA or McsB. Target DNA (0.5 µg of a 171 bp fragment containing the entire regulatory part of the clpC operon) was incubated with 0.025–0.3 nM CtsR in combination with 0.025–0.1 nM McsB or 0.1–0.3 nM McsA. Lanes were loaded as follows (DNA incubated with): 1, free DNA; 2, 0.025 nM CtsR; 3, 0.1 nM CtsR; 4, 0.1 nM McsA; 5, 0.1 nM McsB; 6, 0.025 nM CtsR and 0.1 nM McsA; 7, 0.025 nM CtsR and 0.3 nM McsA; 8, 0.025 nM CtsR and 0.025 nM McsB; 9, 0.025 nM CtsR and 0.1 nM McsB; 10, 0.3 nM CtsR and 0.1 nM McsB; and 11, 0.025 nM CtsR, 0.3 nM McsA and 0.1 nM McsB. The figure shows an inverse image of the ethidium bromide stain. (B) Content of CtsR in the wild type, and mcsA and mcsB mutants before and after heat shock. (C) Stability of CtsR in mcsA mutants after chloramphenicol and puromycin treatment. (D) Stability of CtsR in mcsB mutants after puromycin treatment. Samples were taken before (co), 15 min after 50°C heat shock (heat) or 2, 4 and 8 min after the addition of chloramphenical (100 µg/ml) or puromycin (60 µg/ml) and analyzed by western blotting using antibodies against CtsR.
To determine whether McsA and McsB are also related to CtsR stability, we investigated the intracellular level of CtsR in mcsA and mcsB mutants by western blot experiments with antibodies against CtsR. In non-stressed mcsA mutant cells, the CtsR level was lower than it was in the wild type. After exposure to heat shock, the CtsR content in the mcsA mutant even decreased in comparison with the wild type as well as with the respective CtsR control level in the mcsA mutant (Figure 6B). This result suggested that CtsR is less stable in cells lacking McsA. To prove this, the in vivo stability of CtsR in the mcsA mutant strain was determined using chloramphenicol and puromycin treatment. As expected, the half-life of CtsR was lower in cells lacking McsA than in the corresponding wild-type strain, indicating that the in vivo function of McsA is to stabilize CtsR under non-stressed conditions (Figure 6C). The CtsR level in cells lacking McsB resembled that of the wild-type strain, although CtsR synthesis should be repressed by its autoregulatory function (Figure 6B). To investigate whether this is due to a better stability of the CtsR protein in mcsB mutant cells, we compared the in vivo stability of CtsR in the mcsB mutant strain with that in wild-type cells. The half-life of the CtsR repressor was increased in mcsB mutant cells, indicating that the function of McsB is to destabilize CtsR probably by abolishing its binding to DNA (Figure 6D).
Since McsB shows similarities to kinases (Krüger et al., 1997), it was tempting to speculate that McsB modifies CtsR to remove the repressor from its DNA-binding site before it is degraded by the ClpCP protease. In order to prove this assumption, protein extracts of wild-type and mcsB mutant cells prepared before and after 15 min of heat shock at 50°C were separated by two-dimensional SDS–PAGE and immunodetected with antibodies against CtsR. Two protein spots corresponding to CtsR with different isoelectric points (pI) were observed in these experiments under normal growth and heat shock conditions for wild-type and mcsB mutant cells. However, the relationship between the spots differed in both strains depending on the growth conditions (Figure 7). In wild-type cells under non-stressed conditions, the more basic spot predominated (Figure 7A), whereas the acidic form of CtsR was more intense after heat shock (Figure 7B). These data showed that under heat shock conditions, CtsR is most likely to be modified by phosphorylation, conferring an acidic charge to proteins. This modification did not occur in mcsB mutants, since the spot with the more basic pI predominated in extracts of non-stressed and heat-shocked cells (Figure 7C and D). In summary, these results suggested that CtsR is removed from the DNA via a modification by the putative kinase McsB and thereby targeted for its degradation by the ClpCP protease under stress conditions. Under non-stressed conditions, the degradation of CtsR may be protected by the McsA factor.
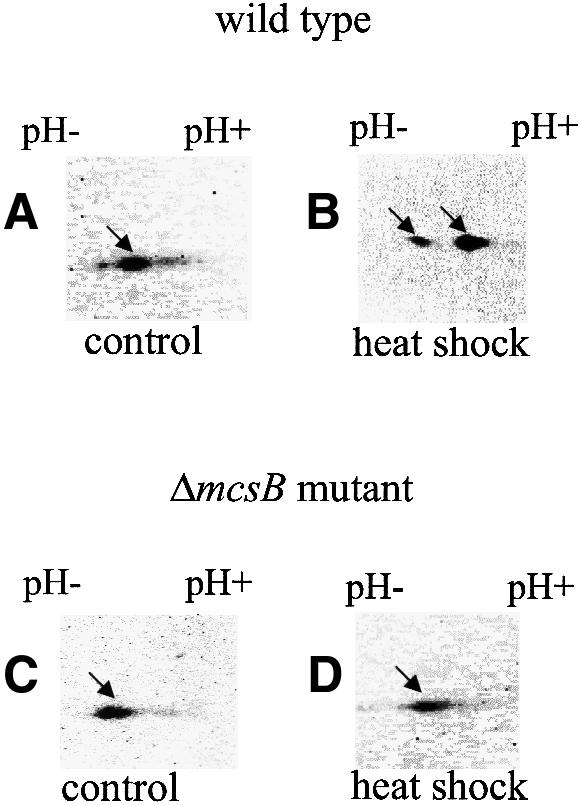
Fig. 7. Modification of CtsR by McsB. Protein extracts of wild-type (A and B) and mcsB mutant cells (C and D) prepared before (A and C) and after 15 min heat shock at 50°C (B and D) were separated on two-dimensional gels and processed in immunoblots with antibodies against CtsR. The spots corresponding to both CtsR forms are indicated by arrows.
Discussion
Current knowledge about the regulation, targets and molecular steps of energy-dependent proteolysis in bacteria is based mainly on studies of the E.coli proteases ClpAP, ClpXP, ClpYQ, Lon and FtsH (reviewed in Gottesman, 1996, 1997a,b). Recently, we found a direct participation in overall proteolysis of misfolded proteins for the ClpCP protease in B.subtilis (Krüger et al., 2000). Besides this, involvement of ClpC and ClpP of B.subtilis in a variety of cellular processes implies that there exist different regulatory proteins as potential specific substrates of the Clp protease (Kong and Dubnau, 1994; Krüger et al., 1994; Msadek et al., 1994, 1998; Gerth et al., 1998; Nanamiya et al., 1998). Nevertheless, to date, only for control of genetic competence has a defined function been characterized in detail for the ClpCP protease that degrades the competence transcription factor ComK (Turgay et al., 1998).
A temperature upshift in B.subtilis stimulates the transient induction of heat shock gene expression by at least three different mechanisms. Class I heat shock genes, such as the classical chaperone genes dnaK and groEL, were shown to be under negative control of the HrcA repressor interacting with its operator site, CIRCE. HrcA activity is modulated by the GroE chaperonin machine (reviewed in Hecker et al., 1996; Mogk et al., 1997). In addition to heat shock, the large group of class II genes are induced in response to various other stresses such as energy starvation, and require the alternative sigma factor σB for induction (reviewed in Hecker et al., 1996; Hecker and Völker, 1998). Class III genes are negatively regulated by the CtsR repressor and remain inducible in the absence of σB and CIRCE. They encode predominantly ATP-dependent protease components exemplified by ClpC, ClpP and ClpE; the function of the latter is unknown (Krüger and Hecker, 1998; Derré et al., 1999a,b). A directly repeated heptad sequence (A/GGTCAAANANA/GGTCAAA) has been determined as the target sequence of CtsR repression (Derré et al., 1999a).
A decisive step in the induction of Clp proteins of B.subtilis to ensure stress tolerance and degradation of heat-damaged proteins is the inactivation and removal of the CtsR repressor. This study clearly shows CtsR to be a labile protein under stress conditions whose stability is strongly dependent on ClpC and ClpP in vivo and in vitro. Similarly to the E.coli ClpAP protease, which degrades certain β-galactosidase fusion proteins as N-end rule targets (Tobias et al., 1991), ClpC of B.subtilis seems to recognize the N-terminus of a CtsR–LacZ fusion protein (see Table I). The selectivity of the E.coli ClpAP (Ti) and Lon proteases has been shown to be accompanied by significant stimulation of ATPase activity by specific substrates (Waxmann and Goldberg, 1986; Hwang et al., 1987). The stimulation of ClpC ATPase activity by CtsR can be interpreted by ClpC action in substrate unfolding and translocation into the protease cavity requiring ATP hydrolysis, as found for ClpA (Hoskins et al., 1998; Pak et al., 1999; Weber-Ban et al., 1999; Wickner et al., 1999). A further increase of released orthophosphate in the presence of ClpP (Figure 3) is probably due to ATP hydrolysis needed for the accessibility to proteolytic sites (reviewed in Gottesman et al., 1997a). The previously proposed non-functional state of the CtsR repressor is probably caused by its inactivation and rapid degradation during stress. Thus, energy-dependent degradation required for an intracellular balance in protein homeostasis is autoregulated by proteolysis. Furthermore, our data propose that two other class III heat shock proteins, sharing similarities to zinc finger proteins (McsA) or arginine kinases (McsB), play an important role in the control of CtsR function.
Zinc finger proteins not only function as DNA-binding proteins, but also participate in the mediation of protein– protein interaction. Several eukaryotic zinc finger proteins were shown to be implicated in mediating homodimerization (reviewed in Berg, 1990; Mackay and Crossley, 1998). Very recently, the studies of Derré et al. (2000) provided strong evidence that CtsR acts as a dimer. It is therefore tempting to speculate that McsA may, perhaps in combination with other factors, assist CtsR in adopting and stabilizing its active conformation (Figure 6A, lane 7). This active form of CtsR seems not to be accessible for proteolytic degradation by ClpCP. To ensure basal level transcription of CtsR-dependent genes, the repressor level is thought to be controlled by ClpXP proteolysis (Derré et al., 2000). The putative kinase McsB is able to abolish CtsR DNA binding activity by modification (i.e. phosphorylation), which thereby targets the repressor for ClpCP-mediated proteolysis (Figure 8). An increased amount of McsB might be the trigger, since McsB action predominates over that of McsA (Table II; Figure 6A). Our mRNA induction kinetics for the clpC operon suggest that the system returns to equilibrium after 15–30 min (depending on the stimulus), although the stress stimulus is still present (Krüger et al., 1994, 1996). If CtsR is inactivated and the operon transcription is at a high level, CtsR is also synthesized at higher levels. The ClpCP protease controlling the accumulation of CtsR is titrated by the increased pool of non-native proteins in the cell and CtsR becomes able to repress once again. Our model suggests a novel heat induction mechanism of controlled proteolysis, a circuit of down-regulation by stabilization and protection of a transcription repressor, and induction by presenting it to the protease. The exact determination of biochemical interactions and the function of the structural domains of proteins involved in this mode of regulation will be the subject of future studies.

Fig. 8. Model of the regulation cycle for CtsR repression. In non-stressed cells, the active CtsR dimer is positively influenced and stabilized by McsA, thereby repressing transcription of class III heat shock genes. After stress, McsB modifies CtsR, negatively affecting its active conformation, and targets the repressor for degradation by ClpCP. Transcription of class III heat shock genes is induced. The arrow symbolizes positive regulation, the cross symbolizes blocked degradation, an asterisk symbolizes modification, and a dashed line instability.
CtsR-dependent control of heat shock gene expression is highly conserved among low G+C Gram-positive bacteria. This heat shock regulation mechanism cannot be assigned only to clp genes but also to chaperone genes encoding the DnaK chaperone machine or chaperone genes of the hsp18 class encoding small heat shock proteins (Derré et al., 1999a; Nair et al., 2000). Along with the conservation of CtsR and its DNA target site, our database analysis revealed that the modulators of CtsR repression, the zinc finger protein McsA and the putative kinase McsB, have also been conserved in this group of bacteria (Figure 4). Low G+C Gram-positive bacteria comprise many pathogens, among them the human pathogen L.monocytogenes, whose Clp proteins were found to be closely related to virulence (Rouquette et al., 1996, 1998; Nair et al., 1999).
A new heat stress regulation mechanism in Gram-positive bacteria of the low G+C branch has been described here comprising negative control by CtsR and McsA. However, heat-inducible clp gene expression is positively autoregulated by promoting the degradation of the CtsR repressor. The association of energy-dependent proteolysis with the regulation of the heat shock response has been discovered recently for eukaryotic cells. Inhibition of the ubiquitin–proteasome pathway leads to increased amounts and activation of the mammalian heat shock factor 2 (HSF2) (Mathew et al., 1998). Activation of eukaryotic HSFs also involves molecular chaperones and negative regulators (reviewed in Morimoto, 1998). Consequently, there are remarkable parallels in the control of heat shock gene induction in prokaryotes and eukaryotes. Although the regulatory proteins differ, the heat shock regulation mechanism by molecular chaperones, negative regulators and proteolysis has been conserved. Altogether this is, to our knowledge, the first report to identify a member of the conserved Hsp100 ATPase family as a positive regulator of the heat shock response. Thus, elucidation of this novel mode of regulation can add to the understanding of the nature of the heat shock response in general as well as virulence in Gram-positive bacteria.
Materials and methods
Bacterial strains and culture conditions
The bacterial strains are listed in Table III. Escherichia coli and B.subtilis cells were cultivated routinely under agitation at 37°C in Luria–Bertani (LB) medium. The different stress conditions were provoked as described earlier (Völker et al., 1994). The culture was divided during exponential growth, and one half of the culture was grown at 37°C (control), whereas the other half was exposed to stress. Glucose starvation was accomplished by cultivating the bacteria in synthetic medium with limiting amounts of glucose [0.05% (w/v)] and addition of 0.05% (w/v) yeast extract to avoid growth defects of clp mutant cells (Stülke et al., 1993; Kunst et al., 1994). Media were supplemented with the following antibiotics if necessary: ampicillin (100 µg/ml), chloramphenicol (5 µg/ml for B.subtilis or 25 µg/ml for E.coli), kanamycin (10 µg/ml), erythromycin (2 µg/ml), lincomycin (25 µg/ml) or spectinomycin (100 µg/ml). For determination of the in vivo stability of CtsR, 100 µg/ml chloramphenicol or 60 µg/ml the aminoacyl-tRNA analog puromycin was added to exponentially growing cultures to stop translation, and samples were taken at the times indicated.
Table III. Bacterial strains used in this study.
| Strain | Genotype | Source or reference |
|---|---|---|
| E.coli | ||
| RR1 | F– mcrB mrr hsdS20 (rB–, mB–) ara14 proA2 lacY1 leu B2 galK2 rpsL20 (Smr) xyl5 mtl1 supE44 | Bolivar et al. (1977) |
| DH5α | F– Φ 80dlacZΔ M15Δ(lacZYA-argF)U169 deoR recA1 endA1 hsdR17 (rK–,mK+) supE44 thi-1 gyrA96 | Sambrook et al. (1989) |
| BL21(DE3)PLysS | F– lon hsdSB (rB–, mB–) with DE3, a λ prophage carrying the T7 RNA polymerase gene and pLysS plasmid containing the T7 phage lysozyme gene | Studier et al. (1990) |
| B.subtilis | ||
| IS58 | trpC2 lys-3 | Smith et al. (1980) |
| BD2790 | his leu met amyE::cat (comG-lacZ) clpC::clpC-his6 aphA3 | Turgay et al. (1997) |
| BUG1 | trpC2 lys-3 ΔclpP::spec | Gerth et al. (1998) |
| BUG2 | trpC2 lys-3 clpX::pMUTIN4 | Gerth et al. (1998) |
| BUG3 | trpC2 lys-3 amyE::cat (clpP–′bgaB) | Gerth et al. (1998) |
| BEO3 | trpC2 lys-3 amyE::cat (ctsR–lacZ) | pDB3→IS58 |
| BEK4 | trpC2 lys-3 ΔclpC::spec | Krüger and Hecker (1998) |
| BEK26 | trpC2 lys-3 amyE::aphA3 (ctsR–′lacZ) ΔclpC::spec | BEK4→BEK56 |
| BEK49 | trpC2 lys-3 amyE::cat (ctsR–′bgaB) | Krüger and Hecker (1998) |
| BEK56 | trpC2 lys-3 amyE::aphA3 (ctsR–′lacZ) | Krüger et al. (1996) |
| BEK86 | trpC2 lys-3 amyE::cat (ctsR–′bgaB) Ω aphA3 ctsR::Δ | Krüger and Hecker (1998) |
| BEK88 | trpC2 lys-3 ΔmcsA:: aphA3 | pDORF2→IS58 |
| BEK89 | trpC2 lys-3 ΔmcsB:: aphA3 | pDORF3→IS58 |
| BEK90 | trpC2 lys-3 amyE::aphA3 (ctsR–′lacZ) ΔclpP::spec | BUG1→BEK56 |
| BEK91 | trpC2 lys-3 amyE::cat (ctsR–′bgaB) ΔmcsA:: aphA3 | BEK88→BEK49 |
| BEK92 | trpC2 lys-3 amyE::cat (ctsR–′bgaB) ΔmcsB:: aphA3 | BEK89→BEK49 |
| BEK93 | trpC2 lys-3 amyE::cat (ctsR–′bgaB) ΔclpC::spec | BEK4→BEK49 |
| BEK94 | trpC2 lys-3 amyE::cat (ctsR–′bgaB) ΔclpP::spec | BUG1→BEK49 |
| BEK95 | trpC2 lys-3 amyE::cat (clpP–′bgaB) Ω aphA3 ctsR::Δ | BEK86→BUG3 |
| BEK96 | trpC2 lys-3 amyE::cat (clpP–′bgaB) ΔmcsA:: aphA3 | BEK88→BUG3 |
| BEK97 | trpC2 lys-3 amyE::cat (clpP–′bgaB) ΔmcsB:: aphA3 | BEK89→BUG3 |
| BEK98 | trpC2 lys-3 amyE::cat (clpP–′bgaB) ΔclpC::spec | BEK4→BUG3 |
| BEK99 | trpC2 lys-3 amyE::cat (clpP–′bgaB) ΔclpP::spec | BUG1→BUG3 |
| BHL1 | trpC2 lys-3 amyE::cat (clpE–′bgaB) | PDLCLPE→IS58 |
| BHL3 | trpC2 lys-3 amyE::cat (clpE–′bgaB) ΔmcsA:: aphA3 | BEK88→BHL1 |
| BHL4 | trpC2 lys-3 amyE::cat (clpE–′bgaB) ΔmcsB:: aphA3 | BEK89→BHL1 |
| BHL5 | trpC2 lys-3 amyE::cat (clpE–′bgaB) Ω aphA3 ctsR::Δ | BEK86→BHL1 |
| BHL6 | trpC2 lys-3 amyE::cat (clpE–′bgaB) ΔclpC::spec | BEK4→BHL1 |
| BHL7 | trpC2 lys-3 amyE::cat (clpE–′bgaB) ΔclpP::spec | BUG1→BHL1 |
| BDZ2 | trpC2 lys-3 amyE::cat (clpE–′bgaB) ΔmcsA:: aphA3ΔclpP::spec | BUG1→BHL3 |
| BDZ3 | trpC2 lys-3 amyE::cat (clpE–′bgaB) ΔmcsB:: aphA3ΔclpP::spec | BUG1→BHL4 |
| BDZ4 | trpC2 lys-3 amyE::cat (clpE–′bgaB) ΔmcsA:: aphA3ΔmcsB::aphA3 | PDORF2/3→BHL1 |
Construction of mutant strains and reporter gene fusions
Construction of plasmid pDORF2 was performed by cloning a 642 bp HindIII–EcoRI PCR fragment with primers TM-120 (AAGAAGCTTAAAGGAGGGGGTTGAGTGGGAC) and PORF2DUPR (GAAGAATTCATAGAGAAACCCTGATTTGC), covering the upstream and N-terminal region of mcsA, into pBluescriptSK. Behind this fragment, a 495 bp PstI–BamHI PCR fragment with primers PORF2DELDF (CTGCTGCAGAATCCTTAATCCATCAAG) and PORF2DELDR (GGAGGATCCTAATATGGTCCTCTTCATTTAG) was inserted, covering the C-terminal region of mcsA and the downstream area. For selection in B.subtilis, the coding sequence without the regulatory sequences of the kanamycin resistance determinant aphAIII (Trieu-Cout and Courvalin, 1983) was amplified by PCR as an EcoRI–PstI fragment using primers PAPHAF (GAAGAATTCGTAGA AAAGAGGAAGGAAAAT) and PAPHAR (CTGCTGCAGGGTACTAAAACAATTCATCC). This fragment was cloned in-frame into the EcoRI–PstI sites between the two up- and downstream fragments of mcsA, thereby generating an in-frame fusion of aphAIII with the remaining parts of mcsA. Plasmid pDORF3 was constructed in a similar way by cloning a 1055 bp HindIII–EcoRI PCR fragment (primers PORF1iF AAGAAGCTATTTCAAATGCGTTCCTTCCC and PORFXUPR GAAGAATTCGAACCGAATATGCTCAAAG), covering the upstream and the N-terminal part of mcsB, and a 986 bp PstI–BamHI fragment (primers PORFXDF CTGCTGCAGATCGCTCAACGGATAAAAGC and PORFXDR GGAGGATCCGTATTTTGTGCCGGCAACAAC), covering the C-terminal region of mcsB and the downstream area, into pBluescriptSK. Thereafter, an in-frame fusion of the aphAIII cassette with the remaining parts of mcsB was achieved by cloning the 745 bp PstI PCR fragment of the aphAIII coding sequence between the mcsB (see above). For the mcsA/mcsB double mutant, plasmid pDORF2/3 was constructed by cloning the 642 bp HindIII–EcoRI PCR fragment of plasmid pDORF2 into plasmid pDORF3. Non-polar in-frame deletions of the mcsA and mcsB genes were generated by transforming B.subtilis IS58 cells with plasmids pDORF2 and pDORF3, generating strains BEK88 (mcsA deletion) and BEK89 (mcsB deletion), respectively. Positive candidates were selected for kanamycin resistance and proven by PCR. Combinations of the mcsA and mcsB deletions with other strains were achieved by transforming the respective strains with chromosomal DNA of strains BEK88 and BEK89
A transcriptional fusion of ctsR with lacZ was constructed using plasmid pDH32M (Kraus et al., 1994). The 271 bp EcoRI–BamHI fragment of pMEC56 (Krüger et al., 1996) was cloned into pDH32M, giving plasmid pDP3, and integrated into the amyE site of the chromosome, giving strain BEO3. Transformants carrying chromosomal lacZ fusions in amyE were screened for expression of β-galactosidase activity and deficiency of α-amylase activity. Transformants were picked on LB-agar plates containing 1% (w/v) starch, and starch degradation was detected by sublimating iodine onto the plates. LacZ activity was visualized on plates by 5-bromo-4-indolyl-β-d-galactoside (X-gal, 100 µg/ml) hydrolysis. Deletions in clpC and clpP were introduced into strain BEK56 by transformation with chromosomal DNA of BEK4 or BUG1, generating strains BEK26 and BEK90. A transcriptional fusion of clpE with bgaB was constructed using plasmid pDL and an EcoRI– BamHI PCR fragment (primers EBFOR1E GAAGAATTCCACAAAAGCAAGCTGACTTGC and PCLPEBR GGAGGATCCTCGTTTTGATGACAATGTGG) covering the clpE regulatory region.
General methods
DNA manipulations and transformation of E.coli were done according to standard protocols (Sambrook et al., 1989). Transformation of B.subtilis with plasmid or chromosomal DNA was carried out using a two-step protocol (Hoch, 1991). Analysis of transcription by mRNA slot-blotting was described previously (Krüger et al., 1994). For biochemical analysis, cells were resuspended in disruption buffer (10 mM Tris pH 8.0, 1 mM EDTA) containing protease inhibitor (Complete, Roche Diagnostics) and disrupted by a French press. The protein concentrations of crude extracts were determined by the Bio-Rad protein assay (Bradford, 1976). Two-dimensional PAGE was carried out as described earlier by separation on Immobiline dry strips (IPG pH 3–11, Amersham Pharmacia Biotech) in the first dimension (Bernhardt et al., 1999). Gel shift analysis was done as described in Krüger and Hecker (1998). Protein extracts were separated by standard SDS–PAGE (Laemmli, 1975). Western blotting and immunodetection was performed by the protocol described in Krüger et al. (2000). The Lumi-Imager system (Roche Diagnostics) was used for documentation and quantitation.
Purification of proteins and antibody production
For overproduction and purification of McsA and McsB in E.coli, the entire genes were amplified by PCR using primers EWORF2AF2 (GGAGGATCCTTGATTTGTCAAGAGTGCCA) and EWORF2ABR (GAAGAATTCTTACTCCTGTTCCTCCTCAC) as well as EWORF3AF (CGCGGATCCATGTCGCTAAAGCATTTTAT) and EWORF3AR (CGGGGTACCCATATCGATTCATCCTCCTG), and cloned as a BamHI–EcoRI or BamHI–KpnI fragment into pRSETA (InVitrogen). Recombinant N-terminal His-tagged proteins (His6-CtsR, His6-ClpP, His6-McsA and His6-McsB) were purified by standard procedures according to the manufacturer (Qiagen) or as described (Krüger and Hecker, 1998; Krüger et al., 2000). His6-McsA, His6-McsB and the His6-CtsR proteins were used for custom antibody production in rabbits (Eurogentec, Belgium). The ClpC-His6 protein was purified using heat-shocked cells of B.subtilis strain BD2790 as described (Turgay et al., 1997).
Enzyme assays
For assaying both LacZ and BgaB activities, B.subtilis cells were grown in LB or synthetic medium as indicated in Results. Samples (1 ml) were taken during exponential growth, after entry into stationary phase or 30 min after heat shock at 50°C. β-galactosidase activities were determined after permeabilizing the cells with chloroform and SDS according to the method of Kenney and Moran (1987) and calculated as Miller units per optical density. For measurement of BgaB activity, the assay buffer was modified as described in Krüger and Hecker (2000).
ATPase activity was measured by an assay based on the colorimetric determination of released orthophosphate as described (Turgay et al., 1997). The assay was carried out with the addition of 0.3 µM ClpC, ClpP and CtsR in a final buffer composition of 100 mM KCl, 25 mM Tris–HCl pH 8.0, 5 mM MgCl2, 0.1 mM EDTA, 0.5 mg/ml bovine serum albumin (BSA), 2 mM ATP and 0.5 mM dithiothreitol (DTT).
An in vitro degradation assay was used to determine the in vitro stability of CtsR. ClpC (0.6 µM), Clp (0.6 µM) and CtsR (0.4 µM) were incubated for 45 min at 37°C in a final volume of 50 µl of the same buffer as shown above containing 4 mM ATP, 2 mM phosphoenol pyruvate and 1.17 µM pyruvate kinase (Roche Diagnostics) as an energy-regenerating system. Samples of 10 µl were taken at the times indicated in Results and added to the same amount of 2× sample buffer to stop the reaction. The mixture was boiled for 5 min, and 10 µl were applied to a 12% SDS–polyacrylamide gel and analyzed by western blotting with anti-CtsR antibodies.
Acknowledgments
Acknowledgements
We thank Kürsad Turgay and David Dubnau for frequent exchange of data and material as well as valuable discussions. We are also grateful to Tarek Msadek for providing strain QB4847 (orf3::pHT181) allowing first analysis of an mcsB mutant. Annette Tschirner and Britta Girbardt are acknowledged for excellent technical assistance, and Jörg Mostertz for support in protein purification. This work was supported by grants from the Deutsche Forschungsgemeinschaft and the Fonds der Chemischen Industrie to M.H.
REFERENCES
- Berg J.M. (1990) Zinc fingers and other metal-binding domains. Elements for interaction between macromolecules. J. Biol. Chem., 265, 6513–6516. [PubMed] [Google Scholar]
- Bernhardt J., Büttner,K., Scharf,C. and Hecker,M. (1999) Dual channel imaging of two-dimensional electropherograms in Bacillus subtilis.Electrophoresis, 20, 2225–2240. [DOI] [PubMed] [Google Scholar]
- Bochtler M., Hartmann,C., Song,H.K., Bourenkov,G.P., Bartunik,H.D. and Huber,R. (2000) The structures of HslU and the ATP-dependent protease HslU-HslV. Nature, 403, 800–805. [DOI] [PubMed] [Google Scholar]
- Bolivar F., Rodrigues,R.L., Greener,P.J., Betlach,M.C., Heyneker,H.L., Boyer,H.W., Crosa,J.H. and Falkow,S. (1977) Construction and characterization of new cloning vehicles II. A multipurpose cloning system. Gene, 2, 95–133. [PubMed] [Google Scholar]
- Bradford M.M. (1976) A rapid and sensitive method for quantitation of microgram quantities of protein utilizing the principle of protein–dye binding. Anal. Biochem., 72, 148–154. [DOI] [PubMed] [Google Scholar]
- Bukau B. (1997) The heat shock response in Escherichia coli. In Gething,M.J. (ed.), Guidebook to Molecular Chaperones and Protein-folding Catalysts. Oxford University Press, Oxford, UK, pp. 525–528.
- Ciechanover A. (1998) The ubiquitin–proteasome pathway: on protein death and cell life. EMBO J., 17, 7151–7160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Derré I., Rappoport,G. and Msadek,T. (1999a) CtsR, a novel regulator of stress and heat shock response, controls clp and molecular chaperone gene expression in Gram-positive bacteria. Mol. Microbiol., 31, 117–131. [DOI] [PubMed] [Google Scholar]
- Derré I., Rappoport,G., Devine,K., Rose,M. and Msadek,T. (1999b) ClpE, a novel type of the HSP100 ATPase, is part of the CtsR heat shock regulon of Bacillus subtilis.Mol. Microbiol., 32, 581–593. [DOI] [PubMed] [Google Scholar]
- Derré I., Rappoport,G. and Msadek,T. (2000) The CtsR regulator of stress response is active as a dimer and specifically degraded in vivo at 37°C. Mol. Microbiol., 38, 335–347. [DOI] [PubMed] [Google Scholar]
- Gerth U., Krüger,E., Derré,I., Msadek,T. and Hecker,M. (1998) Stress induction of the Bacillus subtilis clpP gene encoding the proteolytic component of the Clp protease and involvement of ClpP and ClpX in stress tolerance. Mol. Microbiol., 28, 787–802. [DOI] [PubMed] [Google Scholar]
- Gottesman S. (1996) Proteases and their targets in Escherichia coli.Annu. Rev. Genet., 30, 465–506. [DOI] [PubMed] [Google Scholar]
- Gottesman S., Maurizi,M.R. and Wickner,S. (1997a) Regulatory subunits of energy-dependent proteases. Cell, 91, 435–438. [DOI] [PubMed] [Google Scholar]
- Gottesman S., Wickner,S. and Maurizi,M.R. (1997b) Protein quality control: triage by chaperones and proteases. Genes Dev., 11, 815–823. [DOI] [PubMed] [Google Scholar]
- Gottesman S., Roche,E., Zhou,Y.N. and Sauer,R.T. (1998) The ClpXP and ClpAP proteases degrade proteins with carboxy-terminal peptide tails added by the SsrA tagging-system. Genes Dev., 12, 1338–1347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hecker M. and Völker,U. (1998) Non-specific, general and multiple stress resistance of growth restricted Bacillus subtilis cells by the expression of the σB-regulon. Mol. Microbiol., 29, 1129–1136. [DOI] [PubMed] [Google Scholar]
- Hecker M., Schumann,W. and Völker,U. (1996) Heat shock and general stress response in Bacillus subtilis.Mol. Microbiol., 19, 417–428. [DOI] [PubMed] [Google Scholar]
- Hoch J.A. (1991) Genetic analysis in Bacillus subtilis. Methods Enzymol., 204, 305–320. [DOI] [PubMed] [Google Scholar]
- Hoskins J.R., Pak,M., Maurizi,M.R. and Wickner,S. (1998) The role of the ClpA chaperone in proteolysis by ClpAP. Proc. Natl Acad. Sci. USA, 95, 12135–12140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hwang B.J., Park,W.J., Chung,C.H. and Goldberg,A.L. (1987) Escherichia coli contains a soluble ATP-dependent protease (Ti) distinct from protease La. Proc. Natl Acad. Sci. USA, 84, 5550–5554. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jenal U. and Fuchs,T. (1998) An essential protease involved in bacterial cell cycle control. EMBO J., 17, 5658–5669. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Keiler K.C., Waller,P.R.H. and Sauer,R.T. (1996) Role of a peptide tagging system in degradation of proteins synthesized from damaged messenger RNA. Science, 271, 990–993. [DOI] [PubMed] [Google Scholar]
- Kenney T.J. and Moran,C.P.,Jr (1987) Organization and regulation of an operon that encodes a sporulation-essential σ factor in Bacillus subtilis.J. Bacteriol., 169, 3329–3339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kong L. and Dubnau,D. (1994) Regulation of competence-specific gene expression by Mec-mediated protein–protein interaction in Bacillus subtilis. Proc. Natl Acad. Sci. USA, 91, 5793–5797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kraus A., Hueck,C., Gärtner,D. and Hillen,W. (1994) Catabolite repression of the Bacillus subitilis xyl operon involves a cis element functional in the context of an unrelated sequence and glucose exerts additional xylR-dependent repression. J. Bacteriol., 176, 1738–1745. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krüger E. and Hecker,M. (1998) The first gene of the clpC-operon in Bacillus subtilis, ctsR, encodes a negative regulator of its own operon and other class III heat shock genes. J. Bacteriol., 180, 6681–6688. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krüger E., Völker,U. and Hecker,M. (1994) Stress induction of clpC in Bacillus subtilis and its involvement in stress tolerance. J. Bacteriol., 176, 3360–3367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krüger E., Msadek,T. and Hecker,M. (1996) Alternate promoters direct stress induced transcription of the Bacillus subtilis clpC operon. Mol. Microbiol., 20, 713–723. [DOI] [PubMed] [Google Scholar]
- Krüger E., Msadek,T., Ohlmeier,S. and Hecker,M. (1997) The Bacillus subtilis clpC operon encodes DNA repair and competence proteins. Microbiology, 143, 1309–1316. [DOI] [PubMed] [Google Scholar]
- Krüger E., Witt,E., Ohlmeier,S., Hanschke,R. and Hecker,M. (2000) The Clp proteases of Bacillus subtilis are directly involved in degradation of misfolded proteins. J. Bacteriol., 182, 3259–3265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kunst F., Msadek,T. and Rapoport,G. (1994) Signal transduction network controlling degradative enzyme synthesis and competence in Bacillus subtilis. In Piggot,P.G., Moran,C.P.,Jr and Youngman,P. (eds), Regulation of Bacterial Differentiation. American Society for Microbiology, Washington, DC, pp. 1–20.
- Laemmli U.K. (1975) Cleavage of structural proteins during the assembly of the head of bacteriophage T4. Nature, 227, 680–685. [DOI] [PubMed] [Google Scholar]
- Laney J.D. and Hochstrasser,M. (1999) Substrate targeting in the ubiquitin system. Cell, 97, 427–430. [DOI] [PubMed] [Google Scholar]
- Levchenko I., Smith,C.K., Walsh,N.P., Sauer,R.T. and Baker,T. (1997) PDZ-like domains mediate binding specificity in the Clp/Hsp100 family of chaperones and protease regulatory subunits. Cell, 91, 939–947. [DOI] [PubMed] [Google Scholar]
- Mackay J.P. and Crossley,M. (1998) Zinc fingers are sticking together. Trends Biochem. Sci., 23, 1–4. [DOI] [PubMed] [Google Scholar]
- Mathew A., Mathur,S.K. and Morimoto,R.I. (1998) Heat shock response and protein degradation: regulation of HSF2 by the ubiquitin–proteasome pathway. Mol. Cell. Biol., 18, 5091–5098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mogk A., Homuth,G., Scholz,C., Kim,L., Schmid,F.X. and Schumann,W. (1997) The GroE chaperone machine is a major modulator of the CIRCE heat shock regulon of Bacillus subtilis. EMBO J., 16, 4579–4590. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mogk A., Völker,A., Engelmann,S., Hecker,M., Schumann,W. and Völker,U. (1998) Nonnative proteins induce expression of the Bacillus subtilis CIRCE regulon. J. Bacteriol., 180, 2895–2900. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morimoto R.I. (1998) Regulation of the heat shock transcription response: cross talk between a family of heat shock factors, molecular chaperones and negative regulators. Genes Dev., 12, 3788–3796. [DOI] [PubMed] [Google Scholar]
- Msadek T., Kunst,F. and Rapoport,G. (1994) MecB of Bacillus subtilis, a member of the ClpC ATPase family, is a pleiotropic regulator controlling competence gene expression and survival at high temperature. Proc. Natl Acad. Sci. USA, 91, 5788–5792. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Msadek T., Dartois,V., Kunst,F., Herbaud,M.L., Denizot,F. and Rapoport,G. (1998) ClpP of Bacillus subtilis is required for competence development, degradative enzyme synthesis, motility, growth at high temperature and sporulation. Mol. Microbiol., 27, 899–914. [DOI] [PubMed] [Google Scholar]
- Nair S., Frehel,C., Nguyen,L., Escuyver,V. and Berche,P. (1999) ClpE, a novel member of the Hsp100 family, is involved in cell division and virulence of Listeria monocytogens. Mol. Microbiol., 31, 185–196. [DOI] [PubMed] [Google Scholar]
- Nair S., Derré,I., Msadek,T., Gaillot,O. and Berche,P. (2000) CtsR controls class III heat shock gene expression in the human pathogen Listeria monocytogenes. Mol. Microbiol., 35, 800–811. [DOI] [PubMed] [Google Scholar]
- Nanamiya H., Ohashi,Y., Asai,K., Moriya,S., Ogasawara,N., Fujita,M., Sadiae,Y. and Kawamura,F. (1998) ClpC regulates the fate of a sporulation initiation σ factor, σH protein, in Bacillus subtilis at elevated temperatures. Mol. Microbiol., 29, 505–513. [DOI] [PubMed] [Google Scholar]
- Pak M., Hoskins,J.R., Singh,S.K., Maurizi,M.R. and Wickner,S. (1999) Concurrent chaperone and protease activities of ClpAP and requirement for the N-terminal ClpA ATP binding site for chaperone activity. J. Biol. Chem., 265, 12536–12545. [DOI] [PubMed] [Google Scholar]
- Rouquette C., Ripio,M.T., Pellegrini,E., Bolla,J.M., Tascon,R.I., Vázquez-Boland,J.A. and Berche,P. (1996) Identification of a ClpC ATPase required for stress tolerance and in vivo survival of Listeria monocytogenes. Mol. Microbiol., 21, 977–987. [DOI] [PubMed] [Google Scholar]
- Rouquette C., de Chastellier,C., Nair,S. and Berche,P. (1998) The ClpC ATPase of Listeria monocytogenes is a general stress protein required for virulence and promoting of early bacterial escape from the phagosome of macrophages. Mol. Microbiol., 27, 1235–1245. [DOI] [PubMed] [Google Scholar]
- Sambrook J., Fritsch,E.F. and Maniatis,T. (1989) Molecular Cloning: A Laboratory Manual, 2nd edn. Cold Spring Harbor Laboratory Press, Cold Spring Harbor, NY.
- Schirmer E.C., Glover,J.R., Singer,M.A. and Lindquist,S. (1996) HSP 100/Clp proteins: a common mechanism explains diverse functions. Trends Biochem. Sci., 21, 289–296. [PubMed] [Google Scholar]
- Smith I., Paress,P., Cabane,K. and Dubnau,E. (1980) Genetics and physiology of the rel system of Bacillus subtilis. Mol. Gen. Genet., 178, 271–279. [DOI] [PubMed] [Google Scholar]
- Squires C. and Squires,C.L. (1992) The Clp proteins—proteolysis regulators or molecular chaperones. J. Bacteriol., 174, 1081–1085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Studier F.W., Rosenberg,A.H., Dunn,J.J. and Dubendorff,J.W. (1990) Use of T7 RNA polymerase to direct expression of cloned genes. Methods Enzymol., 185, 60–89. [DOI] [PubMed] [Google Scholar]
- Stülke J., Hanschke,R. and Hecker,M. (1993) Temporal activation of β-glucanase synthesis in Bacillus subtilis is mediated by the GTP pool. J. Gen. Microbiol., 139, 2041–2045. [DOI] [PubMed] [Google Scholar]
- Tobias J.W., Shrader,T.E., Rocap,G. and Varshavsky,A. (1991) The N-end rule in bacteria. Science, 254, 1374–1377. [DOI] [PubMed] [Google Scholar]
- Trieu-Cout P. and Courvalin,P. (1983) Nucleotide sequence of the Streptococcus faecalis plasmid gene encoding the 3′5′′aminoglycoside phosphotransferase type III. Gene, 23, 331–341. [DOI] [PubMed] [Google Scholar]
- Turgay K., Hamoen,L., Venema,G. and Dubnau,D. (1997) Biochemical characterization of a molecular switch involving the heat shock protein ClpC, which controls the activity of ComK, the competence transcription factor of Bacillus subtilis. Genes Dev., 11, 119–128. [DOI] [PubMed] [Google Scholar]
- Turgay K., Hahn,J., Burghorn,J. and Dubnau,D. (1998) Competence in Bacillus subtilis is controlled by regulated proteolysis of a transcription factor. EMBO J., 17, 6730–6738. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Varshavsky A. (1996) The N-end rule: functions, mysteries, uses. Proc. Natl Acad. Sci. USA, 93, 12142–12149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Völker U., Engelmann,S., Maul,B., Riethdorf,S., Völker,A., Schmid,R., Mach,H. and Hecker,M. (1994) Analysis of the induction of general stress proteins of Bacillus subtilis. Microbiology, 140, 741–752. [DOI] [PubMed] [Google Scholar]
- Waxmann L. and Goldberg,A.L. (1986) Selectivity of intracellular proteolysis: protein substrates activate the ATP-dependent protease (La). Science, 232, 500–503. [DOI] [PubMed] [Google Scholar]
- Weber-Ban E.U., Reid,B.G., Miranker,A.D. and Horwich,A.L. (1999) Global unfolding of a substrate protein by the Hsp100 chaperone ClpA. Nature, 401, 90–93. [DOI] [PubMed] [Google Scholar]
- Wickner S., Maurizi,M.R. and Gottesman, S (1999) Posttranslational quality control: folding, refolding and degrading proteins. Science, 286, 1888–1893. [DOI] [PubMed] [Google Scholar]