

Abstract
Three classes of mammalian phosphoinositide-specific phospholipase C (PLC) have been characterized, PLCβ, PLCγ and PLCδ, that are differentially regulated by heterotrimeric G-proteins, tyrosine kinases and calcium. Here we describe a fourth class, PLCε, that in addition to conserved PLC domains, contains a GTP exchange factor (GRF CDC25) domain and two C-terminal Ras-binding (RA) domains, RA1 and RA2. The RA2 domain binds H-Ras in a GTP-dependent manner, comparable with the Ras-binding domain of Raf-1; however, the RA1 domain binds H-Ras with a low affinity in a GTP-independent manner. While Gαq, Gβγ or, surprisingly, H-Ras do not activate recombinant purified protein in vitro, constitutively active Q61L H-Ras stimulates PLCε co-expressed in COS-7 cells in parallel with Ras binding. Deletion of either the RA1 or RA2 domain inhibits this activation. Site-directed mutagenesis of the RA2 domain or Ras demonstrates a conserved Ras–effector interaction and a unique profile of activation by Ras effector domain mutants. These studies identify a novel fourth class of mammalian PLC that is directly regulated by Ras and links two critical signaling pathways.
Keywords: GTP-binding protein Ras/GTP exchange factor/phospholipase C/Raf-1/Ras-binding domain
Introduction
Phosphoinositide-specific phospholipase C (PLC) is a critical signaling enzyme that hydrolyzes membrane phospholipids to generate inositol trisphosphate (IP3), which binds to IP3 receptors and stimulates increases in intracellular Ca2+, and diacylglycerol (DAG), which activates specific isoforms of protein kinase C (PKC). Much elegant work has characterized this effector in many tissues (Rhee and Choi, 1992; Noh et al., 1995; Exton, 1997). In all, 10 PLC isoforms have been cloned in mammals. Each isoform has conserved catalytic X and Y, and C2 domains. Based on their functional and structural characteristics, they have been grouped into three classes: PLCβ1–4, PLCδ1–4 and PLCγ1–2. PLCβ isoforms are activated by heterotrimeric G-protein α-subunits of the Gq family and by Gβγ subunits. PLCγ isoforms contain Src homology (SH2 and SH3) domains and are activated by receptor or cytosolic tyrosine kinases. The mechanism of regulation of PLCδ is less clear, but changes in intracellular Ca2+ may be involved.
Ras is a monomeric GTP-binding protein that is involved in many cellular processes including proliferation, differentiation, migration, the cell cycle and apoptosis (Herrmann and Nassar, 1996; Wittinghofer and Nassar, 1996; Bos, 1998). Three highly conserved family members have been identified, H-, K- and N-Ras (Harvey, Kirsten and neuroblastoma, respectively). Ras is activated by binding GTP which induces a conformational change in two conserved regions, switch I (residues 30–37) and switch II (residues 60–76) (Milburn et al., 1990). A region determined by mutational studies encompassing switch I, the effector domain (residues 32–40), is responsible for the GTP-dependent interaction of Ras with its effectors (Sigal et al., 1986; Stone et al., 1988). The molecular basis of this interaction has been examined by NMR spectroscopy and X-ray crystallography (Emerson et al., 1995; Herrmann and Nassar, 1996; Wittinghofer and Nassar, 1996; Vetter et al., 1999). Ras also undergoes post-translational modification, including farnesylation and carboxymethylation at the C-terminal Caax domain (cysteine, two aliphatic amino acids and any amino acid). This modification is necessary for Ras activation of its effectors, possibly through membrane localization (Leevers et al., 1994; Stokoe et al., 1994). There is also evidence, however, that Ras participates directly in the activation of its effectors (Hu et al., 1995; Drugan et al., 1996; Morrison and Cutler, 1997; Inouye et al., 2000; Rizzo et al., 2000)
Multiple Ras effectors have been identified including Raf, a serine-threonine kinase and an integral member of the MAP kinase pathway; RalGDS, a Ral GTP exchange factor; and phosphatidylinositol 3-kinase (PI 3-kinase), an enzyme that generates phospholipids that activate protein kinase B (Herrmann and Nassar, 1996; Wittinghofer and Nassar, 1996; Bos, 1998). Each of these effectors plays a unique role in Ras-induced cellular transformation as determined by elegant studies using specific Ras effector domain mutants (Joneson et al., 1996; White et al., 1995, 1996; Rodriguez-Viciana et al., 1997). These studies demonstrated that T35S oncogenic Ras activates Raf-1 but not RalGDS or PI 3-kinase, whereas E37G and Y40C specifically activate RalGDS and PI 3-kinase, respectively. While each mutant Ras alone cannot induce full oncogenic transformation, a combination that activates each effector simultaneously induces transformation similar to that induced by oncogenic Ras.
Previous studies have suggested that Ras may regulate mammalian PLC activity (Wakelam et al., 1986; Hancock et al., 1988). To date, however, a PLC isoform that is regulated by Ras has not been identified. Recently, a novel PLC was cloned from the nematode Caenorhabditis elegans (Shibatohge et al., 1998). This PLC, named PLC210, contains an N-terminal GRF CDC25-like domain that has structural homology to guanine nucleotide exchange factors (GEFs) for Ras and two C-terminal Ras-binding (RA; RalGDS/AF6; Ras-associating) domains, suggesting that it may be regulated by Ras. In the present study, we have cloned a novel PLC, named PLCε, which is the rat homolog of PLC210 and is distinct from all known mammalian isoforms. We demonstrate that PLCε is directly regulated by Ras and represents a novel Ras effector.
Results
Cloning and expression of rat PLCε
We have cloned a novel mammalian PLC, PLCε (Figure 1). The cloned sequence is 8358 bp and was submitted to the DDBJ/EMBL/GenBank databases under accession No. AF323615. This sequence length is consistent with a predominant 8.5 kb message found by northern blot analysis to be ubiquitously expressed but expressed mostly in the liver, lung, heart, skeletal muscle and small intestine (Figure 2). The largest open reading frame determined by an upstream, in-frame stop codon is 6846 bp, which encodes a predicted protein of 2281 amino acid residues (Figure 1B). The calculated mol. wt of this protein is 255 kDa, which is consistent with the observed molecular weight from western blot analysis of FLAG-tagged PLCε overexpressed in COS-7 cells (Figure 1C) and the Coomassie blue-stained gel of purified protein (Figure 4A).


Fig. 1. PLCε represents a novel class of PLC. (A) Four classes of mammalian PLC isoforms, rat PLCβ1, human PLCγ1, rat PLCδ1 and PLCε, and C.elegans PLC210 are illustrated. Domains identified include PLC catalytic X and Y domains; C2, Ca2+-dependent lipid-binding domain; SH2 and SH3, Src homology 2 and 3 domains; GRF CDC25, GTP/GDP exchange factor domain; and two Ras-binding domains, RA1 and RA2. The percentage identity with PLCε is shown above each domain. (B) Predicted amino acid sequence of the rat PLCε. Domain regions are boxed. (C) Autoradiograph of a western blot stained with anti-FLAG of a COS-7 cell lysate overexpressing PLCε (pCMV-rPLCε-FLAG). Precision molecular weight markers (Bio-Rad Laboratories, Hercules, CA) were used to determine the relative molecular weight.

Fig. 2. Tissue distribution of PLCε. A 376 bp probe encompassing the C2 domain was used to probe a rat multitissue northern blot (top panel). Hybridization to a β-actin probe is shown in the bottom panel.

Fig. 4. In vitro regulation of purified PLCε. (A) Rat His6-tagged PLCε was purified from SF9 cells. A Coomassie blue-stained gel showing purified PLCε and precision molecular weight markers. (B) Effect of free Ca2+ on PLC enzymatic activity. Purified PLCε and PLCβ3 were incubated with increasing concentrations of free Ca2+, and PLC enzymatic activity was determined. (C) Effect of Gαq-AlF4– on PLCε and PLCβ3. Increasing concentrations of Gαq were incubated with 10 ng of PLCε and 5 ng of PLCβ3. The effect on PI hydrolysis is shown. (D) Effect of Gβγ on PLCε and PLCβ2. Increasing amounts of PLCε or 1 ng of PLCβ2 were incubated with 100 nM Gβγ. The effect on enzyme activity is illustrated. Values are the mean ± SD of duplicate determinations and representative of 3–5 similar experiments.
Analysis of the protein sequence, using the ExPASy Molecular Biology Server and searching Prosite and Pfam with Profilescan, as well as comparisons with other GenBank sequences, demonstrates the presence of centrally located X and Y domains that form the catalytic core of PLC, and a C2 domain which is a Ca2+-dependent phospholipid-binding domain that is also essential for PLC catalytic activity. In addition, an N-terminal GRF CDC25 domain and two C-terminal RA domains, RA1 and RA2, are identified (Figure 1A and B). The arrangement of these domains is the same as in C.elegans PLC210, suggesting that PLCε is the rat homolog of this gene. Using MegAlign (DNASTAR, Madison, WI), a comparison of the domains of PLCε with PLC210 (T42440), rat PLCβ1 (P10687), rat PLCδ1 (8393981) and human PLCγ1 (4505869) was made (Figure 1A). The percentage identity, a quantitation of the percentage of identical amino acids shared between two protein sequences in a pairwise alignment, with PLCε is shown above each domain in Figure 1A. There is a high degree of conservation within these domains across mammalian isoforms and between PLCε and PLC210.
The PLCε gene is also highly conserved across mammalian species. The available amino acid sequence of the mouse (X domain to the C-terminus, aaf40208) and the human sequence, obtained from the Human Genome Project database on chromosome 10, are 94 and 84% identical to the rat sequence, respectively. There is greater divergence of the rat and human sequences between the N-terminus and the CDC25 domain, with an identity of 66% (remainder 90%). This region is also different from PLC210 that is truncated to 66 amino acids compared with 528 and 532 in rat and human PLCε, respectively. Within this N-terminal region in rat, but not human, two WNT domains were identified by searching Pfam (residues 123–136, n = 9.5 and considered significant; and 248–264, n = 6.9), but not Prosite. WNT proteins are secreted proteins that are involved in embryogenesis and cell proliferation (Peifer and Polakis, 2000). Secreted phospholipase A2 has a region that is conserved with WNT and involved in phospholipid binding (Reichsman et al., 1999), and this region has similarity to the second motif identified on PLCε. Their significance remains to be determined.
Binding of H-Ras to the RA domains of PLCε
To examine whether the RA domains of PLCε bind Ras, three GST fusion proteins were generated and Ras binding was determined in a pull-down assay. The sizes and locations of these fusion proteins are diagrammed in Figure 3A. GST–CT PLCε (1942–2281) contains both RA domains, GST–RA1 PLCε (1942–2111) encompasses the entire RA1 domain and GST–RA2 PLCε (2111–2281) encompasses the entire RA2 domain. Figure 3B shows binding of purified GTPγS- and GDP-bound H-Ras, and lysates of constitutively active Q61L H-Ras and dominant-negative S17N H-Ras to these fusion proteins. Both GST–CT PLCε and GST–RA2 PLCε bound active GTP-bound more than GDP-bound H-Ras, indicating that Ras binds to the RA2 domain in a GTP-dependent manner. Binding of activated Ras to the RA2 domain was similar to binding to the well-characterized Ras-binding domain of Raf-1, GST–RBD Raf-1 (51–131) (de Rooij and Bos, 1997). Similarly to H-Ras, Rap1A also bound to the RA2 domain in a GTP-dependent manner (data not shown). Under these conditions, however, Ras binding to the RA1 domain was not observed (Figure 3B).

Fig. 3. H-Ras binding to RA domains of PLCε and the Ras-binding domain (RBD) of Raf-1. (A) Schematic diagram of regions of PLCε used in the GST fusion proteins. (B) H-Ras binding to RA domains of PLCε and the RBD of Raf-1. GST pull-down assays were used to determine Ras binding. GST and GST fusion proteins, as indicated, were incubated with GTPγS- or GDP-loaded H-Ras (upper two panels) or lysates from COS-7 cells overexpressing mutant active (Q61L H-Ras) and inactive (S17N H-Ras) H-Ras, all shown with arrows. An image of an autoradiograph of a typical western blot stained with anti-Ras is shown. Input, 1:4-fold dilution, is the amount of Ras added per lane. Approximately 100 pmol of each fusion protein was loaded on each lane. Representative of three similar experiments. (C) GTP-independent binding of Ras to the RA1 domain of PLCε. Binding of GTPγS- and GDP-loaded H-Ras to increasing concentrations of GST or GST–RA1 PLCε fusion protein is shown. The input was diluted 20-fold compared with the input in (B). Representative of two similar experiments.
In order to determine if the RA1 domain could bind H-Ras under different conditions, a pull-down assay was performed with increasing amounts of fusion protein. Both GTPγS-loaded and GDP-loaded forms of H-Ras bound equally well to high concentrations of GST–RA1 PLCε (Figure 3C). These studies demonstrate that the RA1 domain has a markedly lower affinity than the RA2 domain for Ras and that the binding is GTP independent.
In addition to the above fusion proteins, two smaller fusions of RA2 were assayed for Ras binding to define better the Ras-binding domain. A fusion protein (2111–2268), that still encompassed the entire RA2 domain but was 69 amino acid residues shorter on the C-terminal end, bound H-Ras similarly to the longer RA2 fusion. However, a fusion protein (2111–2212) that removed four amino acid residues at the C-terminal end of the putative RA2 domain was unable to bind H-Ras (data not shown).
Regulation of purified PLCε activity in vitro
To examine the catalytic properties of PLCε and to establish direct regulatory mechanisms, the enzyme was purified to near homogeneity from the cytoplasm of Sf9 cells overexpressing full-length PLCε with a C-terminal His6 tag (Figure 4A). The molecular weight of the purified protein, ∼255 kDa, is the same as the predicted molecular weight, confirming that the full-length protein was purified. Purified PLCε hydrolyzed phosphatidylinositol 4,5-bisphosphate in a Ca2+-dependent manner (Figure 4B). Maximum activity was observed at a free calcium concentration of 800 nM with a specific activity of 2.4 µmol/mg/min. This activity is significantly greater than that seen for PLCβ3 in this same assay (Figure 4B) but is lower than for PLCβ2 (data not shown). Thus the purified full-length enzyme has catalytic activity that is comparable with what is seen for other PLC isoforms.
We next determined if PLCε is regulated by the heterotrimeric G-protein subunits Gαq and Gβγ that regulate PLCβ isoforms since this is the class of enzymes most closely related to PLCε. In contrast to the PLCβ2 and PLCβ3 isoforms, PLCε was not stimulated by either Gαq or Gβγ subunits (Figure 4C and D).
Since PLCε possesses two RA domains and binds Ras, activation of purified PLCε by purified GTPγS-bound H-Ras was examined in this same assay. Multiple conditions were tested in the presence of GTPγS-H-Ras, including varied Ca2+ concentrations, co-stimulation with Gαq and Gβγ subunits, addition of cellular lysate or addition of Sf9 membranes expressing H-Ras, but no stimulation was observed (data not shown). Functional activation of the purified H-Ras by GTPγS was confirmed in RA domain binding studies shown in Figure 3B, where the same H-Ras used for the activation studies was shown to bind to the RA2 domain of PLCε in a GTPγS-dependent manner. Thus GTPγS-H-Ras can bind to PLCε, but it does not regulate the enzyme activity in this assay where allosteric regulation of other PLC isoforms can be demonstrated.
Activation of PLCε by H-Ras co-expressed in COS-7 cells
Only a few studies have been able to demonstrate activation of Raf-1 by Ras in vitro, and those studies have demonstrated that additional proteins are required (Morrison and Cutler, 1997; Inouye et al., 2000). We therefore undertook studies to determine if Ras could activate PLCε when co-expressed within the cell. FLAG-tagged PLCε and vector or constitutively active Q61L H-Ras were co-expressed in COS-7 cells and enzyme activity was measured. Figure 5A demonstrates that active Q61L H-Ras significantly (P <0.04) stimulated PLCε activity >5.5-fold above PLCε basal activity. In contrast, Q61L H-Ras did not significantly stimulate PLCγ1. Western blot analysis revealed no significant difference in levels of expression for either PLC enzyme co-expressed with Q61L H-Ras or an effect of either PLC on Ras expression (Figure 5B). Thus, while Ras activation of PLCε in vitro was not detected, constitutively active Ras markedly stimulates the enzyme when co-expressed within the cell, suggesting that, similarly to Raf-1, other cellular factors may be required for stimulation.
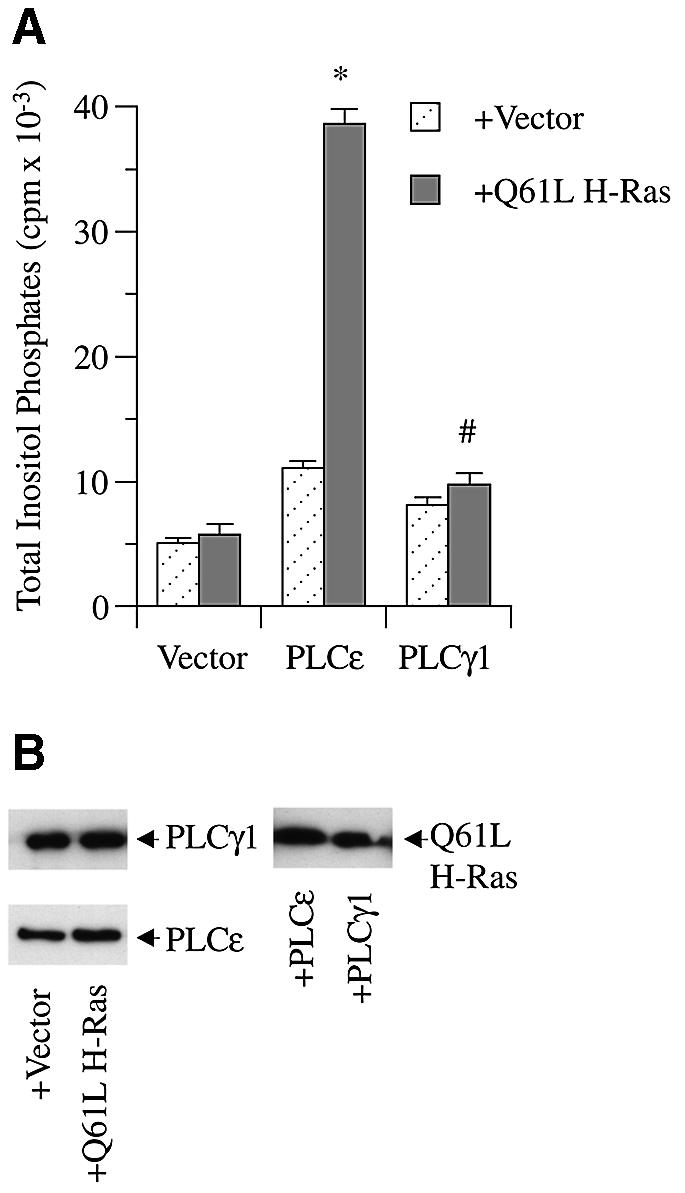
Fig. 5. Activation of PLCε by constitutively active Q61L H-Ras. (A) Effect of constitutively active H-Ras on rat PLCε and PLCγ1. COS-7 cells were transiently transfected with 0.5 µg of vector (pCMV-LacZ), rat PLCε (pCMV-rPLCε-FLAG), or rat PLCγ1 (pNeoSRα2-rPLCγ1) and vector (pRSV-LacZ) or Q61L H-Ras (pRSV-Leu61), and PLC activity was determined. Values are the mean ± SEM of two experiments performed in duplicate. *P <0.04 compared with PLCε + vector. #P = NS compared with PLCγ1 + vector. (B) Expression of PLCε, PLCγ1 and Ras. Image of an autoradiograph of a western blot stained with anti-PLCγ, and anti-FLAG left panels, and anti-Ras right panel. Arrows indicate overexpressed construct and antibody used for detection. Blots are representative of three similar experiments.
RA domain deletions prevent H-Ras activation of PLCε
To demonstrate that the PLC stimulation observed in transfected cells was mediated by a direct interaction of Ras with the RA domains of PLCε, portions of each RA domain were deleted and Ras activation of PLC activity was determined. Three deletion constructs of the FLAG-tagged PLCε expression plasmid were generated as diagrammed in Figure 6A: RA1-deletion (19 amino acids), RA2-deletion (76 amino acids) and RA1/2-deletion. Figure 6B shows that the basal activity and Q61L H-Ras stimulation of each deletion mutant was decreased significantly as compared with the respective studies using PLCε (P at least <0.03). Western blot analysis of the deletion mutants showed similar expression of wild-type, and RA1 and RA1/2 deletion mutants but higher levels of expression of the RA2 deletion mutant, for reasons that are unclear but unimportant because PLC activity remained low (Figure 6C). These studies indicate that both the RA1 and RA2 domains are required for Ras activation of PLCε.

Fig. 6. Effect of RA domain deletion mutations on activation of PLCε. (A) Schematic diagram of regions in the RA domains deleted by restriction digestion. (B) Effect of constitutively active H-Ras on RA domain deletion mutants. COS-7 cells were transiently transfected with 0.5 µg of wild-type PLCε (pCMVscript-PLCε FLAG), RA1-deletion (2064–2082), RA2-deletion (2136–2211) or RA1/2-deletion (2065–2211) with either vector (pRSVneo-LacZ) or Q61L H-Ras (pRSV-Leu 61), and PLC activity was determined. Values are the mean ± SEM of 2–6 experiments performed in duplicate. *P at least <0.01 compared with each deletion mutant + Q61L H-Ras. #P = NS compared with each deletion mutant + Q61L H-Ras. (C) Expression levels of transfected PLCε, RA1-deletion, RA2-deletion, RA1/2-deletion and Q61L H-Ras constructs. A western blot was stained concurrently with FLAG (PLCε) and Ras (Q61L H-Ras) antibodies as indicated by arrows. The blot is representative of two similar experiments.
PLCε RA2 mutants affect binding and activation of H-Ras
Because deletion mutations can have non-specific effects due to conformational changes of the protein or loss of regulatory sites, specific point mutations were introduced into the RA2 domain of PLCε. Figure 7D shows an alignment of the RA2 domain of PLCε with RA domains of other Ras effectors. This alignment reveals conserved regions that are important for Ras binding at the molecular level (Emerson et al., 1995; Herrmann and Nassar, 1996; Wittinghofer and Nassar, 1996; Vetter et al., 1999). A conserved lysine residue (K2150) within the RA2 domain of PLCε was identified as potentially important for Ras binding (Figure 7D). Mutations in the equivalent lysine in both RalGDS (rat K816; equivalent to human K249) and RGL (mouse K685) impair Ras binding (Shirouzu et al., 1999). To demonstrate further that Ras directly interacts with PLCε to regulate PLC activity and in a manner common to other Ras effectors, we mutated K2150 to an alanine (K2150A). Because Shirouzu et al. (1999) demonstrated that mutating this positively charged lysine to a negatively charged glutamic acid almost completely inhibits Ras binding to RGL, we also introduced a similar mutation in PLCε (K2150E). To determine the role of the adjacent lysine, K2152, we also mutated this amino acid alone (K2152A and K2152E) or in addition to the K2150 mutations (K2150A/K2152A and K2150E/K2152E). In all, six mutants were generated.

Fig. 7. Effect of specific RA2 mutations on Ras binding and activation of PLCε. (A) Binding of activated H-Ras to wild-type and mutant PLCε RA2 domains. Mutations in the RA2 domain of GST–RA2 PLCε as shown in (D) were introduced by site-directed mutagenesis. Binding was determined by GST pull-down assays using lysate from COS-7 cells overexpressing constitutively active Q61L H-Ras (input) as detailed in Figure 3 and Materials and methods. An image of an autoradiograph of a western blot stained with anti-Ras (arrow) is shown. Upper panel: densitometry of multiple exposures and dilutions was performed to quantitate autoradiographs. (B) Effect of RA2 mutations on PLCε activation by activated Ras. COS-7 cells were transiently transfected with 0.5 µg of vector (pCMV-LacZ), wild-type PLCε (pCMV-PLCε-FLAG), or PLCε with the indicated RA2 mutation and vector (pRSV-LacZ) or Q61L H-Ras (pRSV-Leu61). Values are the mean ± SEM of four experiments performed in duplicate. *P at least <0.016 compared with all mutants + Q61L H-Ras. (C) Expression of the indicated construct in the presence of Q61L H-Ras. A western blot was stained concurrently with FLAG (PLCε) and Ras (Q61L H-Ras) antibodies as indicated by arrows. The blot is representative of three similar experiments. (D) Alignment of the Ras-binding domains of rat PLCε (rPLCε af323615), mouse PLCε (mPLCε aaf40208), human PLCε (hPLCε ab040949), C.elegans PLC210 (cPLC210 aac38963), human AF6 (hAF6 430933), human RalGDS (hRalGDS u14417) and human c-Raf-1 (hc-Raf-1 x03484). Clustal alignment was performed with MegAlign (DNASTAR, Madison, WI) and adjusted manually. Black shading represents amino acid identity with rat PLCε. The locations of amino acid mutations are indicated by arrows. K2150 and K2152 (lysines at position 2150 and 2152) were mutated to A (alanine) or E (glutamic acid).
The effect of these RA2 domain point mutations on Ras binding and activation of PLC activity is shown in Figure 7. All six mutants exhibited reduced binding of Q61L H-Ras in a GST pull-down assay (Figure 7A). Mutating the conserved lysine at position 2150 to an alanine (K2150A) inhibited binding by 59%, whereas mutating the same amino acid to a positively charged glutamate (K2150E) almost completely abolished binding (>99%). Mutating the adjacent lysine (K2152A or K2152E) also inhibited binding, but to a lesser extent (19 and 67%, respectively). This lysine, K2152, may have a similar role to that of the histidine adjacent to the lysine in RalGDS and RGL, that when mutated also inhibits Ras binding (Shirouzu et al., 1999). The combined mutations K2150A/K2152A and K2150E/K2152E almost completely inhibited binding (>97%).
Concurrently, we examined the effect of these mutations on Ras activation of PLC activity. While these mutations had no effect on expression (Figure 7C), all six mutants significantly (P at least <0.016) inhibited the ability of Q61LH-Ras to stimulate PLCε activity in a manner that paralleled the ability of the RA2 domain mutants to bind Ras (Figure 7B). The K2150A and K2150E mutations inhibited Ras stimulation of PLCε by 51 and 94%, respectively, whereas the K2152A and K2152E mutations inhibited by 30 and 47%, respectively. The dual mutants K2150A/K2152A and K2150E/K2152E also effectively blocked the Ras stimulation by 86 and 94%, respectively. These findings demonstrate that the Ras-binding domain of PLCε is conserved with other Ras effectors and that Ras binding to the RA2 domain is required for activation.
H-Ras mutants differentially affect RA2 binding and PLCε activity
Ras mutants have also been used effectively to elucidate the Ras–effector interface (Sigal et al., 1986; Stone et al., 1988) and to determine mechanisms of effector activation (Leevers et al., 1994; Stokoe et al., 1994; Hu et al., 1995). Therefore, we next examined the effect of specific mutations on the ability of Ras to bind and activate PLCε. All mutations were introduced into the same Q61L H-Ras plasmid to decrease the variability of expression. E37G, T35S, Y40C and D38N are effector domain (amino acid residues 32–40) mutations and N26G is an activator domain (amino acid residues 26–28 and 40–49) mutation. An additional mutant, C186S, was generated to examine the role of post-translational modification (i.e. farnesylation) on Ras activation of PLCε.
The effect of these Ras mutants on binding to the RA2 domain and the RBD of Raf-1 and activation of PLCε is shown in Figure 8. The RA2 domain of PLCε bound to the effector domain mutants E37G and D38N (73 and 40% of control, respectively) but minimally to T35S and Y40C (7 and 1% of control, respectively). The RBD of Raf-1, however, bound minimally to T35S and E37G (9 and 3% of control, respectively) and not to Y40C or D38N, similar to results of other studies except that greater binding (∼40% of control) of the T35S mutant has been observed (Shirouzu et al., 1999). Activation of PLC activity paralleled binding (Figure 8B). Thus, the E37G mutant bound and activated PLCε similarly to control, and the D38N mutant also effectively bound and stimulated PLC activity. The T35S and the Y40C mutations, on the other hand, significantly inhibited Ras activation by 77% (P <0.004) and 85% (P <0.0001), respectively.

Fig. 8. Effect of specific Ras mutations on binding to the RA2 domain of PLCε and activation of PLC activity. (A) Ras mutant binding to the RA2 domain of PLCε and RBD of Raf-1. Site-directed mutagenesis was used to introduce the indicated mutations into constitutively active Q61L H-Ras (pRSV-Leu61). COS-7 cell lysates overexpressing each mutant (input; bottom arrow) were incubated with GST fusion proteins to the RA2 domain of PLCε (GST–RA2 PLCε; top arrow) and the RBD of Raf-1 (GST–RBD Raf-1; middle arrow). Ras binding and GST pull-down assay were performed as detailed in Figure 3 and Materials and methods. An image of an autoradiograph of a western blot stained with anti-Ras is shown. Autoradiographs were quantitated by densitometry and are shown in the top panel. (B) Regulation of PLCε activity by H-Ras mutants. COS-7 cells were transiently transfected with 0.5 µg of vector (pRSV-LacZ), Q61L H-Ras (pRSV-Leu61) or the indicated mutant and vector (pCMV-LacZ) or rat PLCε (pCMV-PLCε-FLAG). Values are the mean ± SEM of five experiments performed in duplicate. *P at least <0.05 compared with stimulation of PLCε with each Ras mutant except E37G. (C) Expression of PLCε and H-Ras mutants. A western blot was stained concurrently with anti-FLAG (PLCε; arrow) and anti-Ras (Q61L H-Ras; arrow). The blot is representative of three similar experiments.
The N26G activator domain mutant bound equally well to RA2 of PLCε and the RBD of Raf-1 and only slightly less than control. PLC stimulation by the activator domain mutant N26G was also partially inhibited (39%; P <0.03). Interestingly, binding of the effector domain mutant D38N was inhibited more than the N26G mutant despite statistically similar inhibition of PLC activity. This suggests that the inhibitory effect of the N26G mutation may not be mediated by decreased binding to the high affinity Ras-binding domain, similarly to the findings with Raf-1 (Hu et al., 1995).
Despite binding to the RA2 domain, the C186S mutant was unable to stimulate PLC activity, indicating that post-translational modification is required for Ras activation of PLCε.
Discussion
We have cloned a novel PLC that is structurally different from all know mammalian PLC isoforms. In addition to prototypical PLC catalytic X and Y domains and a Ca2+-dependent lipid-binding C2 domain (Rhee and Choi, 1992), this PLC has an N-terminal GRF CDC25 domain (Boguski and McCormick, 1993) and two C-terminal RA domains, RA1 and RA2, conserved among other Ras effectors (Ponting and Benjamin, 1996). This PLC is the mammalian homolog of C.elegans PLC210 (Shibatohge et al., 1998) and, because of its unique structure, represents a novel fourth class of mammalian PLC isoforms, named PLCε.
Our studies demonstrate on multiple levels that Ras directly regulates the function of PLCε by a molecular mechanism that is conserved among other Ras–effector interactions (Emerson et al., 1995; Herrmann and Nassar, 1996; Wittinghofer and Nassar, 1996; Shirouzu et al., 1999; Vetter et al., 1999). First, Ras binds to the RA2 domain of PLCε in a GTP-dependent manner. Secondly, the RA2 domain is a typical Ras-binding domain that binds Ras through amino acid residues predicted to be important by structural and functional studies of other Ras effectors (Emerson et al., 1995; Shirouzu et al., 1999; Vetter et al., 1999). Thus, a single lysine that is conserved in RA domains of other Ras effectors (Ponting and Benjamin, 1996) and shown to be critical for binding (Shirouzu et al., 1999), when mutated in the RA2 domain of PLCε, inhibits Ras binding. Thirdly, like its binding to other Ras effectors (Sigal et al., 1986; Stone et al., 1988), Ras binds to the RA2 domain of PLCε through its effector domain since mutations in this region, T35S and Y40C, inhibit binding. Importantly, at each level, mutational effects on binding directly correlate with changes in Ras stimulation of PLC enzymatic activity. These studies provide strong evidence that PLCε is a novel Ras effector. Thus, PLCε joins a select group, including Raf-1, RalGDS and PI-3 kinase, where direct regulation of enzymatic activity by Ras has been shown (Herrmann and Nassar, 1996; Bos, 1998).
While the Ras–PLCε interaction is conserved, our studies also demonstrate a unique profile for Ras mutant activation of PLCε. Thus, the E37G mutant activated PLCε similarly to controls, and the D38N mutant, considered to be a universal inhibitor, significantly stimulated PLC activity (50–60%) while the T35S and Y40C mutants had little effect on binding or activation. The E37G mutant has been used previously as a specific activator of RalGDS (White et al., 1996). In addition to the T35S and Y40C mutants which specifically activate Raf-1 and PI-3 kinase, respectively, these mutants have been used to determine the role of each effector in Ras transformation (White et al., 1995, 1996; Joneson et al., 1996; Rodriguez-Viciana et al., 1997). Our studies would indicate that the E37G mutant is not specific for RalGDS, and co-activation of PLCε will need to be addressed in future studies. On the other hand, the D38N effector mutant specifically activates PLCε and may provide a useful tool to examine the cellular role of this effector.
The precise mechanism by which Ras activates its effectors is unclear. Initial evidence suggested that post-translationally modified Ras recruited Raf-1 to the plasma membrane near its activators (Willumsen et al., 1984; Leevers et al., 1994; Stokoe et al., 1994). Thus, the C186S Ras mutant, which has a serine replacing the cysteine in the Caax box to prevent farnesylation, does not activate or localize Raf-1 to the membrane. Our studies with this mutant also demonstrated that post-translational modification is required for Ras-mediated activation of PLCε. Thus, Ras may activate PLCε by associating it with the plasma membrane near its substrate or activators. This would be consistent with our observation that additional cellular factors, that may be membrane associated, are required for Ras to stimulate PLCε.
However, recent evidence has accumulated that demonstrates that Ras participates directly in the activation of its effectors and does not simply mediate membrane association. Rizzo et al. (2000) showed that phosphatidic acid, and not Ras, recruits Raf-1 to membranes, but that Ras is required for activation at the membrane. The role of the farnesyl group in dimerization of Ras and activation of Raf-1 has also been demonstrated (Inouye et al., 2000). Furthermore, evidence has accumulated that suggests that Ras is directly involved in the activation of Raf-1 by binding to two Ras-binding domains (Hu et al., 1995; Drugan et al., 1996; Morrison and Cutler, 1997; Inouye et al., 2000). In addition to the high affinity RBD (residues 51–131), Raf-1 also has an adjacent cysteine-rich region or domain (CRD) (residues 139–184) that binds Ras in a GTP-independent manner and requires post-translational modification for binding (Hu et al., 1995). It has been proposed that Ras interaction with this domain is critical for Ras activation of Raf-1 (Hu et al., 1995; Drugan et al., 1996; Morrison and Cutler, 1997). Similarly, Kataoka and co-workers (Shima et al., 2000) have also provided evidence for two Ras-binding sites that are required for Ras activation of yeast adenylyl cyclase and have suggested that dual binding may be important in effector regulation.
While the role of the RA1 domain is unclear, this domain may function as a second Ras-binding domain. Similarly to the CRD, the RA1 domain binds Ras with a low affinity and in a GTP-independent manner. Moreover, deleting 19 amino acids of this domain prevented Q61L H-Ras stimulation of PLC activity, suggesting that it is important for Ras activation. While other explanations are possible, it is interesting to note that the structural motif of tandem RA domains is present in other Ras effectors including DAGK (Prosite) and AF6 (Yamamoto et al., 1999). Furthermore, like PLCε, Ras binds with a high affinity to only one of the two RA domains of AF6 (Yamamoto et al., 1999). It is conceivable that tandem RA domains function in a manner similar to the dual Ras-binding domains of Raf-1 and yeast adenylyl cyclase.
While the cellular role of PLCε is not known, its regulation by Ras would potentially link it to one of the many pathways that activate Ras signaling in the cell. The prototypical PLC that is activated by growth factors is PLCγ (Rhee and Choi, 1992; Noh et al., 1995). Given our findings, it is conceivable that activation of Ras through growth factor receptors would also activate PLCε. To this end, we have observed that epidermal growth factor (EGF) stimulates PLCε overexpressed in COS-7 cells (G.G.Kelley and S.E.Reks, unpublished data). Thus, when considering EGF and possibly other growth factor stimulation of PLC, activation of PLCε and PLCγ may need to be addressed.
Another important question is the function of the GRF CDC25 domain of PLCε. A functional GEF domain would make PLCε a bifunctional enzyme. While the GTPase that is activated by this domain is not known, it is possible that it functions as a Ras GEF. It is conceivable, therefore, that it activates Ras and autoregulates its own PLC activity. On the other hand, Ras may regulate the GEF activity of this domain in a manner similar to its regulation of the Ral GEF domain of RalGDS. Interestingly, deletion mutants of this domain inhibit Q61L H-Ras activation of PLC activity (G.G.Kelley and S.E.Reks, unpublished data), suggesting that it has a regulatory role.
In summary, our studies demonstrate that PLCε is a novel Ras effector. Ras binds to the RA2 domain of PLCε in a GTP-dependent manner through its effector domain with a unique Ras mutant profile, and this binding directly correlates with stimulation of PLC activity. The mechanism of this activation may involve translocation and/or activation via the tandem RA domains. In addition, other cellular components are involved in regulating its function since Ras does not activate PLCε in vitro. Because Ras is a critical molecular switch regulating cellular proliferation, differentiation, migration, the cell cycle and apoptosis (Herrmann and Nassar, 1996; Wittinghofer and Nassar, 1996; Morrison and Cutler, 1997; Bos, 1998) and PLC is one of the major cellular signaling effectors (Rhee and Choi, 1992; Noh et al., 1995; Exton, 1997), it is likely that PLCε will play an important role in cellular function.
Materials and methods
Cloning of PLCε
Human expressed sequence tags (ESTs), W24408, R13311 and AA351454, with homology to PLC210 were identified. Using this sequence, a rat probe was generated and used to screen an oligo(dT)-primed rat brain phage library (Stratagene, La Jolla, CA). A clone of 2.3 kb (clone 5-2) was isolated and sequenced. To obtain the 5′ sequence, four iterations of 5′ RACE using rat heart total RNA (Ambion, Austin, TX) were performed following the protocol described by Zhang and Frohman (1997).
Plasmids
A full-length clone of PLCε was generated by RT–PCR using Advantage DNA polymerase mix (Clontech, Palo Alto, CA), and cloned into pCMVscript (Stratagene, La Jolla, CA), generating pCMVscript-PLCε. pCMVscript-PLCε was tagged with FLAG (pCMVscript-PLCεFLAG) or His6 (pCMVscript-PLCεHis) epitopes at the C-terminus by PCR techniques. A baculovirus construct was generated by cloning PLCεHis into pFASTBAC1 (Life Technologies, Rockville, MD). RA domain deletion constructs were created by restriction digestion of pCMVscript-PLCεFLAG to generate: pCMVscript-PLCεFLAG-RA1 (RA1-deletion; 2064–2082); pCMVscript-PLCεFLAG-RA1/2 (RA1/2-deletion; 2065– 2211); and pCMVscript-PLCεFLAG-RA2 (RA2-deletion; 2136–2211). Point mutations (K2150A, K2152A, K2150A/K2152A, K2150E, K2152E and K2150E/K2152E; labeled as XnY; X representing the wild-type amino acid, n the residue position and Y the amino acid substitution) were introduced into the RA2 domain of pGEX-RA2 by site-directed mutagenesis (Quick Mutagenesis Kit, Stratagene, La Jolla, CA) and then subcloned into pCMVscript-PLCεFLAG. GST fusion constructs were generated by subcloning restriction fragments of PLCε into pGEX-6P (Amersham Pharmacia Biotech, Piscataway, NJ): the C-terminus of PLCε, pGEX-EcoRVCT (1942–2281); the RA1 domain, pGEX-RA1 (1942–2111); and RA2, pGEX-RA2 ( 2111–2281). A Raf-1–GST fusion construct encoding residues 51–131 of the human Raf-1 RBD was generously provided by Johannes L.Bos. Ras mutations D38N, E37G, N26G, T35S, C186S and Y40C were introduced into pRSV-Leu61 (Q61L H-Ras expression vector generously provided by Johannes L.Bos) by site-directed mutagenesis.
Northern blot analysis
A 376 bp 32P-random primed RT–PCR C2 domain probe of PLCε was hybridized to a rat multiple tissue northern blot containing 2 µg of poly(A)+ RNA per lane (Origene Technologies, Inc., Rockville, MD). The hybridization was carried out overnight at 42°C in Rapid-Hyb buffer (Ambion, Inc., Austin, TX) with 1 × 106 c.p.m./ml of the radiolabeled probe, washed and exposed to X-ray film. A random primed β-actin probe was used as a control.
Ras-binding assay
Escherichia coli DH5α transformed with pGEX plasmids were induced with 100 µM isopropyl-β-d-thiogalactopyranoside (IPTG) and cultures were incubated at room temperature for 4 h except GST–RBD Raf-1 and pGEX vector which were induced at 37°C for 2 h. After harvesting, bacterial pellets were stored at –70°C for <1 month. Sarkosyl-solubilized GST fusion proteins were isolated using glutathione–Sepharose 4B (Amersham Pharmacia Biotech, Piscataway, NJ) according to the manufacturer’s instructions and as previously described (Youssoufian, 1998).
Freshly purified GST fusion proteins were then resuspended to a 50% slurry using RIPA buffer [10% glycerol, 1% NP-40, 50 mM Tris–HCl, pH 7.5, 200 mM NaCl, 2 mM MgCl2, 1 mM phenylmethylsulfonyl fluoride (PMSF), 5 µg/ml aprotinin, 5 µg/ml leupeptin, 10 µg/ml soybean trypsin inhibitor (SBTI)]. A 20 µl portion of this slurry (typically 500 pmol of fusion protein) was then incubated for 2 h or overnight with either purified His6-tagged H-Ras (15 pmol) loaded with guanosine nucleotides (Camp and Hofmann, 1993) or the equivalent H-Ras mutant lysate, 200–400 µg of a COS-7 cell lysate overexpressing mutant Ras, at 4°C. Following the incubation, the reaction was centrifuged at 500 g for 7 min and then washed three times with 600 µl of RIPA buffer. A 40 µl aliquot of sample buffer was added and 10 µl was loaded on a 12% polyacrylamide gel and transferred to Immobilon-P (Millipore Co., Bedford, MA). Ras binding was determined by ECL detection using a Ras antibody [Transduction Labs (R02120) Lexington, KY].
PLCε purification from Sf9 cells
Viruses for expression of PLCε-His6 were prepared from pFASTBAC1-PLCεHis using the FASTBAC system (Life Technologies, Rockville, MD) according to the manufacturer’s instructions. Sf9 cells were grown at 27°C in Sf900 medium (Life Technologies). Sf9 cells (800 ml) were infected and expression was allowed to proceed for 48 h.
The infected cells were harvested and then lysed by freezing and thawing the cell pellet four times in 15 ml of lysis buffer [50 mM Na-HEPES, pH 7.4, 0.1 mM EGTA, 0.1 mM EDTA, 0.1 mM dithiothreitol (DTT), 100 mM NaCl and protease inhibitors: 133 µM PMSF, 21 µg/ml tosyllysyl chloromethyl ketone, 21 µg/ml tosylphenylalanyl chloromethyl ketone, 0.5 µg/ml aprotonin, 0.2 µg/ml leupeptin, 1 µg/ml pepstatin A, 42 µg/ml tosylarginine methyl ester and 10 µg/ml SBTI]. The lysate was adjusted to 100 ml with lysis buffer, centrifuged at 100 000 g for 45 min and the supernatant applied to a 4 ml column of Ni-NTA agarose (Qiagen). The column was washed with 80 ml of wash buffer [10 mM Na-HEPES pH 8.0, 0.1 mM EGTA, 0.1 mM EDTA, 800 mM NaCl, 0.5% polyoxethylene 10 lauryl ether (C12E10), 15 mM imidazole, plus protease inhibitors) followed by 10 ml of wash buffer lacking C12E10 and SBTI and containing 100 mM NaCl. Protein was eluted with six 4 ml applications of 10 mM Na-HEPES, pH 8.0, 0.1 mM EDTA, 0.1 mM EGTA, 50 mM NaCl, 125 mM imidazole, and protease inhibitors without SBTI. Fractions were analyzed by SDS–PAGE followed by staining with Coomassie blue.
After Ni-NTA chromatography, the PLC was bound to and eluted from a 1 ml HiTrap heparin Sepharose (Amersham Pharmacia Biotech) column with a gradient from 0 to 800 mM NaCl in 20 mM Na-HEPES pH 8.0, 0.1 mM EDTA, 0.1 mM EGTA, 1 mM DTT and protease inhibitors without SBTI. Fractions were analyzed by SDS–PAGE and those containing pure protein were pooled, aliquoted and frozen at –70°C. Protein concentrations were determined by an amido black protein assay (Schaffner and Weissmann, 1973).
PLC activity and other proteins from Sf9 cells
In vitro PLC assays were performed as described (Romoser et al., 1996). Expression and purification of β1γ2 from Sf9 cells has been described previously (Kozasa and Gilman, 1995; Romoser et al., 1996). N-terminally His6-tagged Ras was purified and activated with GTPγS as previously described (Camp and Hofmann, 1993).
Transfection of COS-7 cells and PLC activity assay
COS-7 cells were maintained in Dulbecco’s modified Eagle’s medium (DMEM) supplemented with 10% fetal bovine serum. For transfection, cells were seeded into 12-well tissue culture plates at a density of 1.6 × 105 and grown overnight. Cells were then transfected in conditioned medium with 0.5 µg of each type of cDNA construct using 2 µl of Lipofectamine 2000 reagent (Gibco-BRL, Grand Island, NY) per well following the manufacturer’s protocol. After exposure to the DNA–liposome complex overnight, the cells to be assayed for PLC activity were labeled overnight with 4 µCi of [3H]inositol in inositol-free, serum-free medium. Expression was determined in parallel wells without radioactivity by western blot analysis using anti-PLCγ (UBI, Lake Placid, NY), anti-FLAG (Sigma, St Louis, MO) and anti-Ras.
To determine PLC activity, transfected cells were incubated in 600 µl of fresh serum-free, inositol-free medium with 20 mM lithium chloride for 60 min. The reaction was stopped with the addition of 1.2 ml of ice-cold 4.5% perchloric acid and maintained on ice for 15 min. Samples were then removed and centrifuged at 13 000 g for 12 min. The supernatants were neutralized with 0.5 M KOH/9 mM borax and the precipitated potassium perchlorate was pelleted by centrifugation. The supernatant was removed and 10 ml of ice-cold water were added to the sample. Total inositol phosphates were separated by column chromatography using AG 1-X8 200–400 mesh, formate form. Inositol and glycerophosphoinositol were eluted by three 5 ml washes with 60 mM ammonium formate/5 mM sodium tetraborate. Total inositol phosphates (IP1–3) were then eluted with 5 ml of 1.2 M ammonium formate/0.1 M formic acid and quantitated by liquid scintillation counting.
Statistics
Paired or unpaired Student t-tests were performed where appropriate. A P-value <0.05 was considered significant.
Acknowledgments
Acknowledgements
Special thanks to Johannes L.Bos for generously providing GST–RBD Raf-1 and Q61L H-Ras plasmids, Sue Goo Rhee for PLCγ1, and Sandra Hofmann for His-tagged Ras baculovirus. Technical assistance by Henry Putz is greatly appreciated. We also thank Dr Richard Wojcikievicz for critical reading of the manuscript. These studies were supported by the National Institute of Health, DK56294 (G.G.K.) and GM53536 (A.V.S.), and the American Diabetes Association (G.G.K.).
Note added in proof
While this paper was in preparation, two papers appeared as Journal of Biological Chemistry papers in press (e-pubs): Song et al. ‘Regulation of novel human phospholipase C, PLC-ε, through membrane targeting by Ras’ and Lopez et al. ‘A novel bifunctional phospholipase C that is regulated by Gα12 and stimulates the Ras/Map kinase pathway’. Both papers describe the cloning and regulation of the human isoform of this novel PLC. Song et al. demonstrate that constitutively active G12V H-Ras or Rap1A induce the translocation of green fluorescent protein (GFP)-tagged PLCε from the cytosol to the plasma membrane or perinuclear region, respectively, and show that epidermal growth factor (EGF) may act as a physiological stimulator targeting this PLC to these regions. Lopez et al., on the other hand, provide evidence that PLCε and the N-terminal GRF CDC25-like domain stimulate MAP kinase and Ras GDP/GTP exchange and show that Gα12, but not G12V H-Ras, regulates the PLC activity of this enzyme. These studies complement our studies and emphasize the complexity and uniqueness of this novel Ras effector.
REFERENCES
- Boguski M.S. and McCormick,F. (1993) Proteins regulating Ras and its relatives. Nature, 366, 643–654. [DOI] [PubMed] [Google Scholar]
- Bos J.L. (1998) All in the family? New insights and questions regarding interconnectivity of Ras, Rap1 and Ral. EMBO J., 17, 6776–6782. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Camp L.A. and Hofmann,S.L. (1993) Purification and properties of a palmitoyl-protein thioesterase that cleaves palmitate from H-Ras. J. Biol. Chem., 268, 22566–22574. [PubMed] [Google Scholar]
- de Rooij J. and Bos,J.L. (1997) Minimal Ras-binding domain of Raf1 can be used as an activation- specific probe for Ras. Oncogene, 14, 623–625. [DOI] [PubMed] [Google Scholar]
- Drugan J.K., Khosravi-Far,R., White,M.A., Der,C.J., Sung,Y.J., Hwang,Y.W. and Campbell,S.L. (1996) Ras interaction with two distinct binding domains in Raf-1 may be required for Ras transformation. J. Biol. Chem., 271, 233–237. [DOI] [PubMed] [Google Scholar]
- Emerson S.D., Madison,V.S., Palermo,R.E., Waugh,D.S., Scheffler,J.E., Tsao,K.L., Kiefer,S.E., Liu,S.P. and Fry,D.C. (1995) Solution structure of the Ras-binding domain of c-Raf-1 and identification of its Ras interaction surface. Biochemistry, 34, 6911–6918. [DOI] [PubMed] [Google Scholar]
- Exton J.H. (1997) Cell signalling through guanine-nucleotide-binding regulatory proteins (G proteins) and phospholipases. Eur. J. Biochem., 243, 10–20. [DOI] [PubMed] [Google Scholar]
- Hancock J.F., Marshall,C.J., McKay,I.A., Gardner,S., Houslay,M.D., Hall,A. and Wakelam,M.J. (1988) Mutant but not normal p21 ras elevates inositol phospholipid breakdown in two different cell systems. Oncogene, 3, 187–193. [PubMed] [Google Scholar]
- Herrmann C. and Nassar,N. (1996) Ras and its effectors. Prog. Biophys. Mol. Biol., 66, 1–41. [DOI] [PubMed] [Google Scholar]
- Hu C.D., Kariya,K., Tamada,M., Akasaka,K., Shirouzu,M., Yokoyama,S. and Kataoka,T. (1995) Cysteine-rich region of Raf-1 interacts with activator domain of post-translationally modified Ha-Ras. J. Biol. Chem., 270, 30274–30277. [DOI] [PubMed] [Google Scholar]
- Inouye K., Mizutani,S., Koide,H. and Kaziro,Y. (2000) Formation of the Ras dimer is essential for Raf-1 activation. J. Biol. Chem., 275, 3737–3740. [DOI] [PubMed] [Google Scholar]
- Joneson T., White,M.A., Wigler,M.H. and Bar-Sagi,D. (1996) Stimulation of membrane ruffling and MAP kinase activation by distinct effectors of RAS. Science, 271, 810–812. [DOI] [PubMed] [Google Scholar]
- Kozasa T. and Gilman,A.G. (1995) Purification of recombinant G proteins from Sf9 cells by hexahistidine tagging of associated subunits. Characterization of α12 and inhibition of adenylyl cyclase by αz. J. Biol. Chem., 270, 1734–1741. [DOI] [PubMed] [Google Scholar]
- Leevers S.J., Paterson,H.F. and Marshall,C.J. (1994) Requirement for Ras in Raf activation is overcome by targeting Raf to the plasma membrane. Nature, 369, 411–414. [DOI] [PubMed] [Google Scholar]
- Milburn M.V., Tong,L., deVos,A.M., Brunger,A., Yamaizumi,Z., Nishimura,S. and Kim,S.H. (1990) Molecular switch for signal transduction: structural differences between active and inactive forms of protooncogenic ras proteins. Science, 247, 939–945. [DOI] [PubMed] [Google Scholar]
- Morrison D.K. and Cutler,R.E. (1997) The complexity of Raf-1 regulation. Curr. Opin. Cell Biol., 9, 174–179. [DOI] [PubMed] [Google Scholar]
- Noh D.Y., Shin,S.H. and Rhee,S.G. (1995) Phosphoinositide-specific phospholipase C and mitogenic signaling. Biochim. Biophys. Acta, 1242, 99–113. [DOI] [PubMed] [Google Scholar]
- Peifer M. and Polakis,P. (2000) Wnt signaling in oncogenesis and embryogenesis—a look outside the nucleus. Science, 287, 1606–1609. [DOI] [PubMed] [Google Scholar]
- Ponting C.P. and Benjamin,D.R. (1996) A novel family of Ras-binding domains. Trends Biochem. Sci., 21, 422–425. [DOI] [PubMed] [Google Scholar]
- Reichsman F., Moore,H.M. and Cumberledge,S. (1999) Sequence homology between Wingless/Wnt-1 and a lipid-binding domain in secreted phospholipase A2. Curr. Biol., 9, R353–R355. [DOI] [PubMed] [Google Scholar]
- Rhee S.G. and Choi,K.D. (1992) Regulation of inositol phospholipid-specific phospholipase C isozymes. J. Biol. Chem., 267, 12393–12396. [PubMed] [Google Scholar]
- Rizzo M.A., Shome,K., Watkins,S.C. and Romero,G. (2000) The recruitment of Raf-1 to membranes is mediated by direct interaction with phosphatidic acid and is independent of association with Ras. J. Biol. Chem., 275, 23911–23918. [DOI] [PubMed] [Google Scholar]
- Rodriguez-Viciana P., Warne,P.H., Khwaja,A., Marte,B.M., Pappin,D., Das,P., Waterfield,M.D., Ridley,A. and Downward,J. (1997) Role of phosphoinositide 3-OH kinase in cell transformation and control of the actin cytoskeleton by Ras. Cell, 89, 457–467. [DOI] [PubMed] [Google Scholar]
- Romoser V., Ball,R. and Smrcka,A.V. (1996) Phospholipase C β2 association with phospholipid interfaces assessed by fluorescence resonance energy transfer. G protein βγ subunit-mediated translocation is not required for enzyme activation. J. Biol. Chem., 271, 25071–25078. [DOI] [PubMed] [Google Scholar]
- Schaffner W. and Weissmann,C. (1973) A rapid, sensitive and specific method for the determination of protein in dilute solution. Anal. Biochem., 56, 502–514. [DOI] [PubMed] [Google Scholar]
- Shibatohge M., Kariya,K., Liao,Y., Hu,C.D., Watari,Y., Goshima,M., Shima,F. and Kataoka,T. (1998) Identification of PLC210, a Caenorhabditis elegans phospholipase C, as a putative effector of Ras. J. Biol. Chem., 273, 6218–6222. [DOI] [PubMed] [Google Scholar]
- Shima F. et al. (2000) Association of yeast adenylyl cyclase with cyclase-associated protein CAP forms a second Ras-binding site which mediates its Ras-dependent activation. Mol. Cell. Biol., 20, 26–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shirouzu M., Hashimoto,K., Kikuchi,A. and Yokoyama,S. (1999) Double-mutant analysis of the interaction of Ras with the Ras-binding domain of RGL. Biochemistry, 38, 5103–5110. [DOI] [PubMed] [Google Scholar]
- Sigal I.S., Gibbs,J.B., D’Alonzo,J.S. and Scolnick,E.M. (1986) Identification of effector residues and a neutralizing epitope of Ha-ras-encoded p21. Proc. Natl Acad. Sci. USA, 83, 4725–4729. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stokoe D., Macdonald,S.G., Cadwallader,K., Symons,M. and Hancock,J.F. (1994) Activation of Raf as a result of recruitment to the plasma membrane. Science, 264, 1463–1467. [DOI] [PubMed] [Google Scholar]
- Stone J.C., Vass,W.C., Willumsen,B.M. and Lowy,D.R. (1988) p21-ras effector domain mutants constructed by ‘cassette’ mutagenesis. Mol. Cell. Biol., 8, 3565–3569. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vetter I.R., Linnemann,T., Wohlgemuth,S., Geyer,M., Kalbitzer,H.R., Herrmann,C. and Wittinghofer,A. (1999) Structural and biochemical analysis of Ras–effector signaling via RalGDS. FEBS Lett., 451, 175–180. [DOI] [PubMed] [Google Scholar]
- Wakelam M.J., Davies,S.A., Houslay,M.D., McKay,I., Marshall,C.J. and Hall,A. (1986) Normal p21N-ras couples bombesin and other growth factor receptors to inositol phosphate production. Nature, 323, 173–176. [DOI] [PubMed] [Google Scholar]
- White M.A., Nicolette,C., Minden,A., Polverino,A., Van Aelst,L., Karin,M. and Wigler,M.H. (1995) Multiple Ras functions can contribute to mammalian cell transformation. Cell, 80, 533–541. [DOI] [PubMed] [Google Scholar]
- White M.A., Vale,T., Camonis,J.H., Schaefer,E. and Wigler,M.H. (1996) A role for the Ral guanine nucleotide dissociation stimulator in mediating Ras-induced transformation. J. Biol. Chem., 271, 16439–16442. [DOI] [PubMed] [Google Scholar]
- Willumsen B.M., Christensen,A., Hubbert,N.L., Papageorge,A.G. and Lowy,D.R. (1984) The p21 ras C-terminus is required for transformation and membrane association. Nature, 310, 583–586. [DOI] [PubMed] [Google Scholar]
- Wittinghofer A. and Nassar,N. (1996) How Ras-related proteins talk to their effectors. Trends Biochem. Sci., 21, 488–491. [DOI] [PubMed] [Google Scholar]
- Yamamoto T., Harada,N., Kawano,Y., Taya,S. and Kaibuchi,K. (1999) In vivo interaction of AF-6 with activated Ras and ZO-1. Biochem. Biophys. Res. Commun., 259, 103–107. [DOI] [PubMed] [Google Scholar]
- Youssoufian H. (1998) Immunoaffinity purification of antibodies against GST fusion proteins. Biotechniques, 24, 198–200, 202. [DOI] [PubMed] [Google Scholar]
- Zhang Y. and Frohman,M.A. (1997) Using rapid amplification of cDNA ends (RACE) to obtain full-length cDNAs. In Cowell,I.G. (ed.), cDNA Library Protocols; Methods in Molecular Biology. Human Press Inc., Totowa, NJ, Vol. 69, pp. 61--87. [DOI] [PubMed]