

Abstract
Multiple apoptotic pathways release cytochrome c from the mitochondrial intermembrane space, resulting in the activation of downstream caspases. In vivo activation of Fas (CD95) resulted in increased permeability of the mitochondrial outer membrane and depletion of cytochrome c stores. Serial measurements of oxygen consumption, NADH redox state and membrane potential revealed a loss of respiratory state transitions. This tBID-induced respiratory failure did not require any caspase activity. At early time points, re-addition of exogenous cytochrome c markedly restored respiratory functions. Over time, however, mitochondria showed increasing irreversible respiratory dysfunction as well as diminished calcium buffering. Electron microscopy and tomographic reconstruction revealed asymmetric mitochondria with blebs of herniated matrix, distended inner membrane and partial loss of cristae structure. Thus, apoptogenic redistribution of cytochrome c is responsible for a distinct program of mitochondrial respiratory dysfunction, in addition to the activation of downstream caspases.
Keywords: apoptosis/cytochrome c/Fas/mitochondria
Introduction
The mitochondrion is a critical organelle during apoptosis, being required for cell death following certain death stimuli (Green and Reed, 1998). Mitochondrial dysfunction is a prominent component of apoptosis. A common manifestation is the release of cytochrome c from the intermembrane space where it normally shuttles electrons between cytochrome reductase (complex III) and cytochrome oxidase (complex IV). Following its release, cytochrome c triggers the central death pathway by promoting oligomerization of a cytochrome c/Apaf-1/caspase-9 complex resulting in the activation of caspase-9, which cleaves and activates downstream effector caspases-3 and -7 (Liu et al., 1996; Li et al., 1997). Once the release of cytochrome c is initiated, it is released rapidly and completely (Goldstein et al., 2000; Martinou et al., 2000).
Activation of the cell surface death receptor, Fas (CD95), initiates one of the best defined death pathways. Trimerization of Fas recruits and activates caspase-8, which then cleaves downstream substrates including BID, a member of the ‘BH3 domain only’ subset of pro-apoptotic BCL-2 family members. Inactive, cytosolic BID (22 kDa) is cleaved, generating an active truncated molecule (tBID; 15 kDa), which rapidly relocates to the mitochondrial outer membrane (Li et al., 1998; Luo et al., 1998; Gross et al., 1999). Immunodepletion of BID and Bid-deficient mice indicate that BID is required for the release of cytochrome c (Li et al., 1998; Luo et al., 1998; Gross et al., 1999; Yin et al., 1999). The precise mechanism whereby tBID or other signals result in the release of cytochrome c remains under active investigation. One model holds that an exposed BH3 domain of tBID binds to and induces an allosteric, conformational activation of BAK, a resident mitochondrial BCL-2 member, which oligomerizes, forming a pore which transports cytochrome c (Saito et al., 2000; Wei et al., 2000). Other studies of these pro-apoptotic molecules suggest that they result in more global permeability of the outer mitochondrial membrane, releasing multiple intermembrane space proteins (Jurgensmeier et al., 1998; Basanez et al., 1999; Kluck et al., 1999). Alternative theories promote BCL-2 members interacting with resident mitochondrial proteins such as the voltage-dependent anion channel (VDAC) or adenine nucleotide translocase (ANT), either releasing cytochrome c or leading to a change in permeability, which causes mitochondria to swell, resulting in rupture of the outer membrane (Marzo et al., 1998; Shimizu et al., 1999). Yet another model holds that growth factor withdrawal leads to defects in ADP exchange, promoting hyperpolarization of the membrane, expanded matrix volume and non-specific rupture of the outer membrane (Vander Heiden et al., 1997, 1999). Whatever the precise mechanism(s) of cytochrome c release, Fas-dependent hepatocyte death uses tBID as a death ligand and distinct initiating point for mitochondrial dysfunction (Wei et al., 2000; Yin et al., 1999). Studies of cell lines and Bid-deficient mice indicate that certain cell types do not require mitochondria to be killed by Fas activation, whereas others, including hepatocytes, require a BID-mediated mitochondrial amplification loop for cell death (Scaffidi et al., 1999; Yin et al., 1999).
While caspases are of well-established importance in the apoptotic cascade, multiple lines of evidence now indicate that, following certain stimuli, cell death can proceed in a caspase-independent fashion (for review see Green and Kroemer, 1998). Enforced dimerization or inducible expression of BAX or BAK results in mitochondrial dysfunction with altered transmembrane potential, release of cytochrome c and activation of downstream caspases. However, cell death still occurs in the presence of broad caspase inhibitors (Xiang et al., 1996; Hirsch et al., 1997; Lesage et al., 1997; McCarthy et al., 1997; Ohta et al., 1997; Amarante-Mendes et al., 1998; Gross et al., 1999; Woo et al., 1999). Caspase-independent death occurs over a longer time interval and demonstrates an altered morphology in which the nucleus is principally spared, while mitochondrial alterations and plasma membrane permeability proceed. Both BAX and BAK can also induce mitochondrial dysfunction and kill yeast that lack endogenous caspases (Greenhalf et al., 1996; Zha et al., 1996; Ink et al., 1997; Gross et al., 2000; Harris et al., 2000). Although cell lines lacking cytochrome c display attenuated apoptotic responses to multiple stress stimuli, they are still killed by tumor necrosis factor α (TNF-α) (Li et al., 2000). Thus, there are several examples where mitochondrial dysfunction ensures cell death, independent of cytochrome c-mediated activation of downstream caspases.
Several studies have investigated the mechanism and kinetics of cytochrome c release and its downstream effects on caspase activation (Green and Reed, 1998; Goldstein et al., 2000; Martinou et al., 2000). Less is known about the caspase-independent portion of mitochondrial dysfunction. In particular, it is uncertain whether the program of mitochondrial dysfunction results exclusively from a respiratory blockade due to loss of cytochrome c or whether other irreversible events occur. To characterize mitochondrial function systematically during apoptosis, we returned to in vivo Fas-activated hepatocytes. Measurement of several mitochondrial parameters indicated that the loss of cytochrome c caused respiratory inhibition, reflecting a blockade between respiratory complexes III and IV. Respiratory function was reversed dramatically by adding exogenous cytochrome c at early time points. At later time points, however, exogenous cytochrome c only partially rescued mitochondrial respiratory function, indicating that a prolonged lack of cytochrome c leads to irreversible mitochondrial damage. Thus, during apoptosis mitochondria experience a progressive dysfunction.
Results
Fas activation changes mitochondrial outer membrane permeability and leads to cytochrome c release
We used an in vivo system to study mitochondrial physiology following Fas-induced apoptosis (Ogasawara et al., 1993). Mice were injected with either saline (control) or anti-Fas monoclonal antibody and euthanized 30, 60 or 90 min after injection. Hepatocyte mitochondria were isolated for biochemical and physiological experiments. Previous studies indicate that by 2 h after Fas activation, liver cells demonstrate redistribution of cytochrome c, as assessed by in situ immunohistochemistry (Yin et al., 1999). To ascertain whether the hepatocyte mitochondria used here had depleted cytochrome c, we incubated isolated mitochondria in a physiological respiratory buffer and assessed the amount of cytochrome c in the mitochondrial pellet (P) and supernatant (S) fractions. Nearly all of the cytochrome c was released from hepatocyte mitochondria isolated from mice 90 min after Fas activation, but not from the mitochondria of mice injected with saline (Figure 1A). Mitochondria isolated from Bid–/– mice 90 min after injection of anti-Fas antibody showed minimal release of cytochrome c, consistent with previous studies (Yin et al., 1999). Cytochrome c appears to be rapidly and completely released in a cell line following death stimuli (Goldstein et al., 2000), consistent with the current in vivo results.

Fig. 1. Outer membrane permeability and loss of cytochrome c in mitochondria from Fas-activated hepatocytes. (A) Western blot analysis of the mitochondrial pellet (P) and supernatant (S) fractions from liver mitochondria of wild-type and Bid–/– mice 90 min after injection of saline or anti-Fas antibody. Hypotonically lysed control mitochondria are included for comparison of complete release. (B) Mitochondrial outer membrane permeability assay as developed by Colombini and colleagues (Lee et al., 1994). Exogenous cytochrome c will stimulate respiration if the cytochrome c can traverse the outer membrane and access cytochrome c oxidase. Hypotonically lysed mitochondria (mitoplasts) have a ruptured outer membrane and display a burst in oxygen consumption in response to added cytochrome c, demonstrating the utility of the assay. (C) Oxygen consumption following the addition of exogenous cytochrome c to mitochondria isolated from liver of wild-type mice at 30, 60 or 90 min after injection of anti-Fas antibody, as well as mitochondria from Bid–/– mice 90 min after Fas treatment.
Previous studies have shown that activation of the pro-apoptotic BCL-2 members can permeabilize the mitochondrial outer membrane (Kluck et al., 1999; Eskes et al., 2000; Wei et al., 2000). We performed an outer membrane permeability assay to measure the accessibility of complex IV to exogenous cytochrome c (Lee et al., 1994). In this assay, mitochondria are placed in a buffer containing excess inorganic phosphate, ascorbate and ADP, and oxygen consumption is monitored. Addition of exogenous cytochrome c, which is reduced by ascorbate (confirmed by spectrophotometry; data not shown), will stimulate oxygen consumption if it is able to traverse the outer membrane and access cytochrome oxidase, complex IV. Correspondingly, cytochrome c-stimulated oxygen consumption is significantly greater in hypotonically lysed mitochondria (mitoplasts) than in control mitochondria, owing to the rupture of the outer membrane and immediate accessibility of cytochrome oxidase to the added cytochrome c (Figure 1B).
A time-dependent increase in outer membrane permeability was seen after Fas activation (Figure 1C). No significant permeability was evident 30 min after injection of anti-Fas antibody, but by 90 min the permeability approached that of hypotonically lysed mitochondria. Bid–/– liver mitochondria were not permeant even 90 min after treatment with the agonistic antibody (Figure 1C). These findings of permeabilization of the mitochondrial outer membrane from in vivo activated cells are consistent with previous studies in vitro (Kluck et al., 1999).
Fas signaling blunts mitochondrial respiratory state transitions
Isolated mitochondria incubated with excess carbon substrate and inorganic phosphate will undergo transitions from state 4 to state 3 respiration upon the addition of ADP (Figure 2) (Chance and Williams, 1955). State 4 respiration results when reducing equivalents and inorganic phosphate are in excess, but no ADP is available to stimulate respiration. During state 4 respiration, membrane potential is high, NADH is chemically reduced and oxygen consumption is minimal. Addition of ADP drives the F1F0-ATPase and results in state 3 respiration, where the membrane potential decreases, NADH becomes oxidized and oxygen consumption increases. Once the added ADP is converted to ATP, state 4 respiration returns. For mitochondria to undergo respiratory state transitions, the electron transport chain, the F1F0ATPase, the ANT and the inner mitochondrial membrane must be intact. Respiratory state transitions can be monitored experimentally by following pyridine nucleotide (NADH) fluorescence, oxygen consumption and mitochondrial membrane potential (Δψm).

Fig. 2. Overview of mitochondrial oxidative phosphorylation and respiratory states. Abbreviations: UQ, ubiquinone; c, cytochrome c; IM, mitochondrial inner membrane; OM, mitochondrial outer membrane; Δψm, mitochondrial membrane potential; VDAC, voltage-dependent anion channel; KCN, potassium cyanide; VO2, oxygen consumption.
Addition of carbon substrate (glutamate + malate, a complex I reducing agent) to control mitochondria in an isotonic respiratory buffer containing inorganic phosphate led to rapid chemical reduction of pyridine nucleotides until a steady state (state 4) NADH level was attained (Figure 3A). Addition of ADP initiates state 3 respiration and, because the electron transport chain is functionally intact, the pyridine nucleotides are transiently oxidized until the added ADP is converted to ATP, at which time state 4 respiration returns. Subsequent addition of cyanide (KCN), which inhibits cytochrome c oxidase, results in the further reduction of pyridine nucleotides. Bid–/– mitochondria, isolated 90 min after anti-Fas antibody injection, displayed an NADH response (Figure 3B) indistinguishable from that of control wild-type mitochondria (Figure 3A). Mitochondria isolated from wild-type livers following Fas activation underwent a blunted NADH reduction in response to glutamate + malate (Figure 3C). However, they failed entirely to respond to ADP, indicative of a respiratory blockade. This pattern of inhibition was mimicked by the addition of cyanide to control mitochondria, where addition of carbon substrate led to pyridine nucleotide reduction, but ADP no longer stimulated oxygen consumption or NADH oxidation due to the respiratory blockade (Figure 3D). Chemical reduction of pyridine nucleotides by carbon substrates does not require electron transport between complexes III and IV, whereas ADP-stimulated transitions from state 4 to state 3, where glutamate + malate is the carbon source, requires a fully intact electron transport chain.

Fig. 3. Loss of NADH redox transitions in mitochondria damaged by the Fas pathway resembling cyanide-treated mitochondria. (A) Mitochondria from wild-type mice injected with saline were monitored for NADH fluorescence after the addition of carbon substrate (5 mM glutamate + 5 mM malate), a pulse of ADP (100 nmol) and finally cyanide (KCN, 1 mM). (B) Mitochondria from Bid–/– liver isolated 90 min after injection of anti-Fas antibody respond to carbon substrate (glutamate + malate) and ADP, similar to wild-type controls in (A). (C) Fas-activated mitochondria from wild-type mice were provided carbon substrate (glutamate + malate) and two subsequent additions of ADP (100 nmol), and NADH fluorescence was plotted. (D) Control wild-type mitochondria were incubated with cyanide (1 mM) at time 0. Carbon substrate (glutamate + malate) and two subsequent ADP (100 nmol) additions were made, and NADH fluorescence was monitored.
We next assessed whether re-addition of cytochrome c would be sufficient to restore respiratory state transitions in mitochondria damaged by the Fas pathway. Mitochondria were incubated in respiratory buffer with excess carbon substrate (glutamate + malate) and monitored for NADH redox transitions in response to ADP. The transitions were robust in untreated, control mitochondria from saline-injected mice (Figure 4A), but were blunted in mitochondria obtained 60 min after Fas activation in vivo (Figure 4B) and essentially absent in mitochondria 90 min after Fas activation (Figure 4C). The same mitochondrial preparations were monitored after the addition of 10 µM exogenous cytochrome c (Figure 4D–F). Control mitochondria continued to undergo classic NADH transitions in the presence of added cytochrome c (Figure 4D), indicating that exogenous cytochrome c does not interfere with respiratory transitions in intact mitochondria. Added cytochrome c enhanced redox transitions in mitochondria obtained 60 min after Fas activation (Figure 4E). Remarkably, the lack of respiratory transitions in mitochondria 90 min after Fas activation was restored by exogenous cytochrome c (Figure 4F). In this set of experiments, mitochondrial respiration is supported by the complex I carbon substrate, glutamate + malate, as no ascorbate was added. Spectrophotometric analysis of the added cytochrome c revealed that it was fully oxidized in the experimental buffer. Thus, the exogenous cytochrome c is reduced by an electron transport chain that is intact from complex I, forward. The reduced cytochrome c must then transfer its electrons to cytochrome c oxidase to restore responsiveness to ADP.

Fig. 4. NADH respiratory transitions rescued by exogenous cytochrome c. Freshly isolated mitochondria were incubated initially with complex I carbon substrate (5 mM glutamate + 5 mM malate). Liver mitochondria were obtained from mice following saline injection (A and D), or 60 min (B and E) and 90 min (C and F) after injection of anti-Fas antibody. Mitochondria are shown either in the absence (A–C) or presence (D–F) of 10 µM cytochrome c. (A) Mitochondria from saline-treated mice undergo classic NADH respiratory transitions in response to added ADP (100 nmol) pulses. (B) Sixty minutes after injection, the NADH redox transitions are blunted and take longer. (C) Ninety minutes after treatment, there is no response to added ADP and no recovery in NADH levels is observed. (D) An experiment identical to that in (A), except that it was performed in the presence of 10 µM cytochrome c, demonstrates that saline-treated control mitochondria continue to undergo NADH transitions even in the presence of cytochrome c. Cytochrome c quenches NADH fluorescence, so the y-axis has been rescaled in experiments performed in the presence of cytochrome c (D–F). (E) Addition of cytochrome c deepens the state 3 NADH redox transient and improves the recovery when compared with mitochondria seen in (B). (F) Mitochondria isolated 90 min after treatment previously (E) did not undergo NADH transitions but, with exogenous cytochrome c, now undergo robust redox transitions, indicating that the respiratory chain can still be rescued.
To define further the mitochondrial dysfunction and its correction, we continuously monitored oxygen consumption and the distribution of tetraphenylphosphonium (TPP+) (Figure 5). TPP+ is a lipophilic cation which distributes across the inner mitochondrial membrane in a Nernstian fashion and provides qualitative information about membrane potential. Wild-type mitochondria from saline-treated mice (Figure 5A) and Fas-activated Bid-deficient mitochondria (Figure 5B) generated a transmembrane potential in response to the addition of the complex I carbon substrate glutamate + malate. They also underwent rapid membrane depolarization and increased oxygen consumption [respiratory control ratio (RCR) = 6.0] in response to added ADP. In contrast, mitochondria isolated 60 min after Fas activation in wild-type mice failed to enhance membrane potential (Figure 5C). They underwent a diminished transition from state 4 to state 3 respiration with the addition of ADP, and failed to return to a higher state 4 membrane potential. These mitochondria failed to respond to a second pulse of ADP and there was almost no increase in oxygen consumption following ADP addition (RCR = 1.1). When the same experiment was repeated in the presence of exogenous cytochrome c (Figure 5D), the mitochondria were able to generate state 4 membrane potential in response to added carbon substrate (glutamate + malate) and they underwent transitions from state 4 to state 3 respiration with added ADP. The oxygen consumption trace demonstrates a modest recovery of respiratory control (RCR = 2.8). Mitochondria isolated 90 min after Fas activation in wild-type mice (Figure 5E) failed to enhance membrane potential with the addition of carbon substrate and exhibited poor respiratory control (RCR = 1.3). This lack of response is reminiscent of mitochondria following treatment with cyanide (data not shown). Addition of exogenous cytochrome c to the Fas-activated mitochondria restored their ability to generate a membrane potential upon addition of carbon substrate (glutamate + malate) (Figure 5F). Moreover, the membrane depolarized slightly in response to a pulse of ADP; this was accompanied by a concomitant increase in oxygen consumption and respiratory control (RCR = 2.5).

Fig. 5. Fas-induced defects in mitochondrial respiration and membrane potential partially rescued by exogenous cytochrome c. Freshly isolated mitochondria (after injection of saline or anti-Fas antibody) were placed in a respiratory chamber in which oxygen consumption (gray) and TPP+ concentration (black) were measured simultaneously. (A) Control mitochondria develop a membrane potential in response to an added carbon substrate (5 mM glutamate + 5 mM malate). Upon addition of ADP (100 nmol), mitochondria transiently depolarize and undergo a burst of oxygen consumption (state 3 respiration) until added ADP is converted to ATP. These mitochondria continue to respond to added ADP, demonstrating intact respiratory control. (B) Bid–/– liver mitochondria isolated 90 min after injection of anti-Fas antibody also exhibit high respiratory control and membrane potential responses to carbon substrate and ADP, indistinguishable from normal wild-type mitochondria in (A). (C) Mitochondria obtained 60 min after injection of anti-Fas antibody exhibit minimal responsiveness to added carbon substrate (glutamate + malate), but a slight membrane depolarization is seen with ADP addition. (D) Fas-pathway-damaged mitochondria as in (C) were incubated with 10 µM exogenous cytochrome c at time 0. Membrane potential now responds to carbon substrate as well as two pulses of ADP. Oxygen consumption also responds to ADP pulses, indicating partial recovery of respiratory control. (E) Mitochondria obtained 90 min after Fas activation display a membrane potential which does not increase with addition of glutamate + malate. Oxygen consumption and membrane potential fail to respond to ADP additions, indicating a loss of respiratory control. (F) Fas-pathway-damaged mitochondria as in (E) were incubated with 10 µM exogenous cytochrome c at time 0. Membrane potential now increases in response to glutamate + malate, while membrane potential drops with a concomitant increase in oxygen consumption after addition of ADP, indicating some recovery of respiratory control by adding cytochrome c. Later in the experiment, after oxygen consumption has slowed and membrane potential has recovered modestly, the mitochondria are still able to depolarize in response to a second pulse of ADP.
In contrast, addition of bovine serum albumin did not rescue respiratory transitions, suggesting that the rescue did not reflect a nonspecific protein effect (data not shown). Addition of ascorbate alone could not rescue these transitions either, indicating that the recovery was not due to modulating the redox state of any endogenous cytochrome c (data not shown). To explore whether caspase activity was required for the mitochondrial respiratory dysfunction, we performed in vitro experiments in which purified liver mitochondria were exposed to tBID, as previously described (Wei et al., 2000), in the presence or absence of zVAD-fmk, a caspase inhibitor, at a concentration (50 µM) that does not inhibit respiratory complexes. Caspase inhibition did not substantially alter the respiratory defect due to cytochrome c release. Re-addition of exogenous cytochrome c and repletion of NADH produced a comparable recovery of oxygen consumption with or without the caspase inhibitor (Figure 6). The RCR after tBID treatment (2.2) was not affected by the presence of the caspase inhibitor.
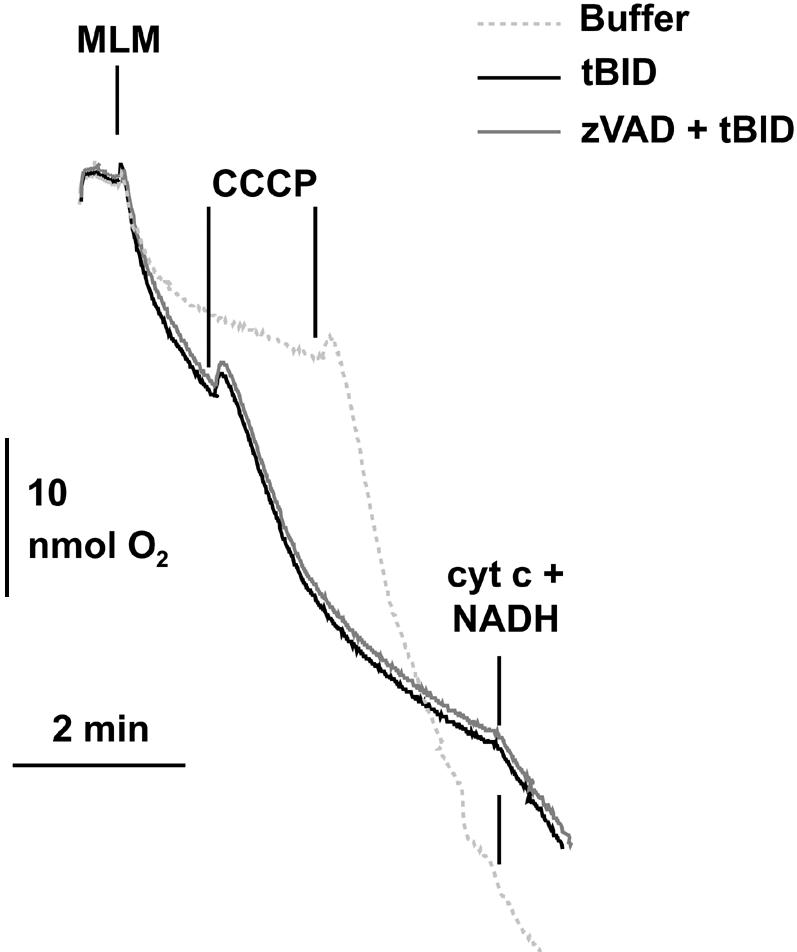
Fig. 6. Caspase inhibition does not prevent tBID-induced respiratory defect. Mitochondria isolated from untreated wild-type mouse livers (MLM) were incubated with 320 pmol tBID/mg mitochondrial protein (black line) or tBID plus 50 µM zVAD-fmk (dark gray line), or were left untreated (light gray line) in respiratory buffer. Oxygen consumption was measured with a Clarke-type electrode. Uncoupled respiration was started by adding 200 pmol carbonyl cyanide m-chlorophenylhydrazone (CCCP)/mg mitochondrial protein (lines). Where indicated, 20 µmol NADH/mg mitochondrial protein and 10 µmol cytochrome c/mg mitochondrial protein were added.
This indicates that tBID does not require caspase activity to induce a respiratory defect in mitochondria. Collectively, these experiments indicate that by 60 min after activation by the Fas pathway, addition of cytochrome c produces a substantial but still incomplete recovery of mitochondrial respiratory function.
Fas activation results in mitochondrial swelling and altered morphology
To determine whether there was gross alteration of mitochondrial structure after Fas activation, we obtained transmission electron microscopic images of liver sections from mice injected with an anti-Fas antibody (Figure 7A). We also employed high-voltage electron microscopic (HVEM) tomography (Figure 7B–D), which provides valuable information about the three-dimensional organization of mitochondrial compartments and the relationship between the inner and outer mitochondrial membranes (Mannella et al., 1994; Frey and Mannella, 2000). Hepatocytes 90 min after Fas activation showed a high frequency of nuclear condensation typical of apoptosis. Fas-treated cells had pale, swollen mitochondria with partial loss of the cristae structure (Figure 7A). A number of mitochondria displayed blebs, with an asymmetrical herniation of the matrix, distension of the inner membrane and loss of tubular cristae (Figure 7A–D). Mitochondrial morphology was not completely destroyed, as tomographic reconstruction indicated that the inner membrane was intact, consistent with previous studies of Fas-mediated apoptosis (Krippner et al., 1996; Feldmann et al., 2000). In a number of mitochondria from Fas-activated cells, while the inner membrane was completely intact, electron microscopy and tomography revealed partial rupture of the outer membrane, exposing the inner membrane (Figure 7C and D). Typically, there were few if any cristae in the swollen (bleb) inner membrane region, suggesting that the blebs derive from localized matrix swelling and unfolding of the inner membrane.

Fig. 7. Electron microscopy and tomographic reconstruction of Fas-injured mitochondria. (A) Representative field in a liver section obtained 90 min after anti-Fas antibody treatment, demonstrating mitochondria with large blebs (arrows). (B) Cross-sectional slice (6 nm thickness) from the electron tomographic reconstruction of a semi-thick (0.22 µM) section of a mitochondrion with a bleb. (C and D) Two surface-rendered views of the reconstructed mitochondrion, showing ruptured outer membrane (red), inner surface membrane (IM) with large bleb (yellow) and selected cristae (green), with narrow tubular junctions to the IM space (arrows).
Fas activation alters calcium buffering capacity
To pursue these observations further, 90° side scatter measurements of isolated mitochondria were performed over time (Figure 8) (Nicholls and Ferguson, 1992). Control mitochondria (Figure 8A) or Fas-activated, Bid-deficient mitochondria (Figure 8B) both swelled in response to a super-physiological pulse of calcium. However, wild-type mitochondria isolated 60 min after Fas activation exhibited a blunted swelling response to calcium (Figure 8C). By 90 min following Fas activation, isolated mitochondria were more swollen and failed to swell at all in response to added calcium (Figure 8D).

Fig. 8. Mitochondrial swelling and loss of calcium buffering capacity. (A) Ninety degree side scatter provides a measure of mitochondrial size for control mitochondria purified from livers of saline-injected mice. Control mitochondria swell in response to a pulse of calcium (20 nmol CaCl2). (B) The ability to swell in response to Ca2+ is preserved in mitochondria from Bid–/– mice isolated 90 min after injection of anti-Fas antibody. (C) Sixty minutes after injection of Fas antibody, wild-type liver mitochondria exhibit a blunted swelling response to calcium addition. (D) By 90 min, mitochondria are initially more swollen and exhibit no response to added calcium. (E) Extramitochondrial calcium was measured fluorimetrically with the calcium indicator Calcium Green 5N. Control mitochondria can buffer multiple pulses (10 nmol CaCl2) of added calcium (arrowheads). (F) Mitochondria isolated 90 min after Fas activation cannot buffer even a single pulse of calcium.
We next examined calcium homeostasis as an additional parameter of mitochondrial function. Mitochondria can sequester calcium via the electrophoretic uniporter, which is critical for cellular calcium buffering (Gunter and Gunter, 1994). Previous studies in hematopoietic cells have shown that withdrawal of interleukin 3 results in alterations in calcium partitioning (Baffy et al., 1993), and investigations in neuronal cells have shown that BCL-2 results in increased mitochondrial calcium buffering capacity (Murphy et al., 1996). The ability of mitochondria to take up calcium can be assessed by measuring the disappearance of extramitochondrial free Ca2+ from the medium, following the addition of CaCl2 pulses. Normal mitochondria buffered multiple additions of CaCl2 (Figure 8E), whereas Fas-activated mitochondria showed additional evidence of defects, displaying a substantially diminished buffering capacity (Figure 8F).
Discussion
The role of cytochrome c in mitochondrial oxidative phosphorylation has long been established (Salemme, 1977), while its participation in apoptosis has been appreciated more recently (Liu et al., 1996). A number of death stimuli result in the release of cytochrome c, which activates the effector caspases of the core apoptotic pathway (Green and Reed, 1998). Evidence also exists for defects in mitochondrial function during apoptosis that appear to be distinct from cytochrome c-induced caspase activity. Previous reports have demonstrated that the outer mitochondrial membrane becomes permeable in cells undergoing apoptosis. The addition of ascorbate, which directly reduces exogenous cytochrome c, has revealed that mitochondria with permeable outer membranes have an intact complex IV during apoptosis (Krippner et al., 1996; Kluck et al., 1999) (Figure 1). With this background, we investigated the serial time course of defects in mitochondrial respiratory function that follow the in vivo induction of apoptosis in hepatocytes activated by Fas. We found that 30 min after injection of anti-Fas antibody, the mitochondrial outer membrane and respiratory function were intact. However, by 60 min after Fas activation the outer membrane was permeable and there were defects in mitochondrial respiration. Several parameters of mitochondrial physiology, including NADH redox state and oxygen consumption, defined a defect in respiratory state transitions. We assessed the entire electron transport chain by using glutamate + malate as a complex I carbon source without ascorbate present. Specifically, at 60 min the state 4/state 3 respiratory transitions were partially blunted as assessed by NADH measurements. The marked correction of respiratory transitions by the re-addition of cytochrome c indicates that complexes I, III and IV, as well as the F1F0-ATPase and the ANT, are intact and competent for electron transfer at this time point. At these early time points, mitochondrial respiratory function was nearly completely reversible with added cytochrome c. The earliest definable defect in mitochondrial respiration appears to be due to the loss of cytochrome c in that it is reversible, being corrected by exogenous cytochrome c. By 90 min after Fas activation, when there was loss of transmembrane potential and no respiratory response to ADP, there was only modest recovery from added cytochrome c. There is a significant irreversible component of mitochondrial respiratory dysfunction, and mitochondria exhibited defective calcium buffering, swelling and partial outer membrane rupture by this time point.
Several possibilities may account for the irreversible damage. The loss of cytochrome c inhibits respiration with complete loss of oxidative phosphorylation. ATP produced by glycolysis can initially support mitochondrial homeostasis. The F1F0-ATPase is known to be reversible, and ATP can be hydrolyzed to drive the extrusion of protons across the inner mitochondrial membrane to support Δψm. However, continued ATP-consuming reactions can threaten the integrity of the mitochondrion. Mitochondrial calcium uptake occurs via the electrophoretic uniporter, but calcium is extruded via ATP-dependent H+/Ca2+ and Na+/Ca2+ exchangers on the inner membrane (Bernardi, 1999). Energetic collapse from ATP-consuming reactions could, therefore, result in the mitochondrial calcium accumulation and osmotic swelling observed here.
Alternatively, these events might reflect activation of the permeability transition (PT) pore (as reviewed in Bernardi, 1999), as suggested in previous in vivo studies in which Fas-mediated mitochondrial matrix expansion, outer membrane rupture and membrane potential collapse could be partially prevented by pre-treatment with cyclosporin A (Feldmann et al., 2000). However, BID does not appear to require permeability transition for the initial release of cytochrome c in in vitro studies (von Ahsen et al., 2000; Wei et al., 2000), raising the possibility that PT activation is a subsequent event. Respiratory blockade between complexes III and IV would be expected to increase production of reactive oxygen species with subsequent lipid peroxidation, which has been noted in several cell death systems (Hockenbery et al., 1993; Kane et al., 1993; Cai and Jones, 1998).
Caspase-induced mitochondrial dysfunction is yet another possible cause for the irreversible damage. However, the mitochondrial defects induced by Fas activation were prevented in experiments using Bid–/– mice (Figures 1, 3, 4 and 6). In these mice an initiator caspase, caspase-8, which is upstream of BID, is activated and, in a subset of Bid–/– hepatocytes, a diminished amount of effector caspase-3 appears to be directly activated by caspase-8 (Srinivasula et al., 1996; Yin et al., 1999). In addition, we performed in vitro experiments in which the function of isolated mitochondria in the presence or absence of caspase inhibitors was monitored following the addition of recombinant tBID. Pre-treatment with the inhibitor zVAD-fmk did not appreciably alter the tBID-induced defect in mitochondrial respiratory control. Thus, tBID does not appear to require caspase activity to induce mitochondrial dysfunction.
Considerable attention has focused on whether caspases or mitochondrial damage represents the point of no return during apoptotic cell death (Green and Kroemer, 1998). Prior studies in cell lines indicate that caspase inhibitors, while preventing DNA fragmentation, do not prevent mitochondrial dysfunction and cell death following certain stimuli (Xiang et al., 1996; McCarthy et al., 1997). In contrast, studies of neurons indicate that caspase inhibitors can prevent factor deprivation deaths even after the release of cytochrome c, as long as mitochondrial membrane potential is maintained (Martinou et al., 1999; Deshmukh et al., 2000). Perhaps the noted dependence of post-mitotic neurons upon glycolysis provides an explanation for their apparent difference from hepatocytes and cell lines.
Overall, these studies indicate that the release of cytochrome c results in two distinct downstream pathways of cellular damage, caspase activation and respiratory inhibition. The reversibility of the initial mitochondrial respiratory defect argues that the loss of cytochrome c is principally responsible. Over time, progressive damage to mitochondria becomes irreversible, thereby ensuring cell death. In this context, strategies that seek to block cell death may need to consider maintaining mitochondrial function as well as inhibiting caspases.
Materials and methods
Animal handling and treatment
All mice were handled according to Dana Farber Cancer Institute and Harvard Medical School institutional guidelines on animal care. C57BL/6 females aged 6–8 weeks were injected intravenously via tail vein with anti-Fas antibody (clone Jo2; Pharmingen) at a dose of 0.5 µg/g body weight or with 200 µl normal saline for control experiments.
Isolation of mitochondria
Mitochondria were isolated as previously described (Gross et al., 1999) and stored on ice at a concentration of 10 mg/ml in isolation buffer (200 mM mannitol, 70 mM sucrose, 10 mM HEPES pH 7.5, 1 mM EGTA) until used for experiments. Mitochondrial protein content was quantified using the Bio-Rad protein assay.
Western blot analysis for cytochrome c release
Isolated mitochondria (0.4 mg/ml) were placed in respiratory buffer (137 mM KCl, 10 mM HEPES pH 7.2, 2.5 mM MgCl2) supplemented with 5 mM glutamate, 5 mM malate, 3 mM K2HPO4, and stirred at room temperature (24°C) for 10 min. Mitochondrial pellet (P) and supernatant (S) fractions were obtained by spinning the mixture at 8000 g for 5 min. Hypotonically lysed mitochondria were used as positive controls; they were prepared as previously described (Lee et al., 1994) by placing 0.2 mg mitochondria in 375 µl of water for 10 min at 4°C and then combining this volume with 125 µl of 4× respiratory buffer. Cytochrome c immunoblotting of the two fractions was performed with a commercially available anti-cytochrome c monoclonal antibody (Pharmingen).
Outer membrane permeability assay
Intactness of the outer membrane was measured as previously described (Lee et al., 1994) by measuring cytochrome c-dependent oxygen consumption. Exogenously added ascorbate-reduced cytochrome c must pass through the outer membrane to be oxidized by cytochrome c oxidase on the outer surface of the inner mitochondrial membrane. Mitochondria (0.4 mg/ml) were placed in the respiratory chamber (see below) containing 0.5 ml permeability buffer (200 mM mannitol, 4 mM NaH2HPO4 pH 7.2, 5 mM MgCl2, 10 mM KCl) supplemented with 1 mM ascorbate and 900 µM ADP. Cytochrome c-dependent oxygen consumption was initiated by adding 0.6 mg cytochrome c (Sigma) at room temperature (24°C). Control experiments verified that all of the cytochrome c-induced respiration was sensitive to 1 mM KCN, and absorbance spectroscopy revealed that stock cytochrome c was immediately and fully reduced when incubated with 1 mM ascorbate. The solubility of oxygen in water at 24°C was assumed to be 245 nmol oxygen/ml H2O.
Absorbance spectroscopy
Cytochrome c redox state was monitored at A540 using an absorbance spectrophotometer. Cytochrome c (0.6 mg) was added to a quartz cuvette containing 500 µl of respiratory buffer. The addition of 1 mM ascorbate immediately resulted in an absorbance change equivalent to that achieved with sodium hydrosulfite, demonstrating that 1 mM ascorbate immediately and completely reduces this quantity of cytochrome c.
Measurement of oxygen consumption and TPP+ concentration
Oxygen consumption and TPP+ concentration were measured simultaneously as previously described (Mootha et al., 1996, 1997) in a 500 µl stirred respiratory chamber fitted with oxygen and TPP+ electrodes (Microelectrodes, Inc., Londonderry, NH). The TPP+ electrode monitors the extramitochondrial concentration and, because the cation distributes in a Nernstian fashion, higher TPP+ concentrations in the bath are reflective of a lower membrane potential. However, since mitochondrial matrix volume appears to vary following Fas activation (Figures 7 and 8), we did not formally calculate mV units.
Experiments were performed in 500 µl respiratory buffer containing 2 mM K2HPO4. Mitochondria (0.8 mg/ml) were added to the buffer, 5 mM glutamate + 5 mM malate was used as the carbon substrate, and state 3 respiration was initiated by adding ADP. TPP+ measurement was initiated with only the experimental buffer in the apparatus. Three pulses of TPP+ were added (to a final concentration of 30 µM) and with each pulse a steady state recording was achieved, enabling calibration of the electrode recording to TPP+ concentration.
For tBID and caspase inhibitor experiments, 1 mg of wild-type mouse liver mitochondria was incubated in a respiratory buffer containing 5 mM glutamate and 2.5 mM malate, in the presence or absence of zVAD-fmk and in the presence or absence of tBID (Wei et al., 2000) for 30 min at 25°C. Mitochondria were then centrifuged at 7000 g for 10 min at 4°C, resuspended in 10 µl isolation buffer and immediately used for respiration studies in the respiratory chamber. Corrections were not made for non-specific TPP+ binding or for electrode drift.
Pyridine nucleotide fluorescence measurement
Pyridine nucleotide (NADH) fluorescence measurements were made in a Perkin Elmer LS50B spectrophotometer using an excitation of 366 ± 10 nm and an emission of 455 ± 10 nm as previously described (Mootha et al., 1997). This fluorescence is a measure of the reduced pyridine nucleotide pool. Experiments were performed in a well-stirred quartz cuvette at 24°C. Mitochondria (0.4 mg/ml) were placed in 500 µl respiratory buffer with 5 mM glutamate + 5 mM malate as the carbon substrate and 2 mM K2HPO4. State 3 respiration was initiated with ADP addition.
Mitochondria side scatter
Ninety degree side scatter serves as a useful measure of isolated mitochondria volume (Nicholls and Ferguson, 1992). Mitochondria (40 µg/ml) were placed in a stirred cuvette containing 500 µl respiratory buffer supplemented with 5 mM glutamate, 5 mM malate and 2 mM K2HPO4; 90° side scatter was monitored continuously at 520 ± 2.5 nm using a Perkin Elmer LS50B spectrophotometer.
Calcium buffering capacity of mitochondria
Mitochondrial uptake of calcium was determined fluorimetrically as previously described (Murphy et al., 1996) using the calcium indicator Calcium Green 5N (Molecular Probes, Inc.). Briefly, mitochondria (0.4 mg/ml) were added to a well-stirred quartz cuvette at 24°C containing 1 ml of respiratory buffer supplemented with 5 mM glutamate, 5 mM malate, 2 mM K2HPO4 and 0.1 µM Calcium Green 5N. The cuvette was placed in a spectrophotometer and extramitochondrial calcium was monitored by following the fluorescence with an excitation of 506 ± 2.5 nm and emission of 531 ± 5.0 nm. CaCl2 stocks were prepared using deionized distilled water.
Electron microscopy and tomographic reconstruction
Liver tissue was fixed with aldehyde and osmium tetroxide, embedded in plastic, sectioned, and stained with uranyl acetate and lead citrate. Thin sections were imaged on a Zeiss 910 TEM at 80 kV. For tomography, colloidal gold particles were applied to one side of 250-nm-thick sections as alignment markers. A tilt series of 122 images was recorded on the Albany AEI EM7 MkII HVEM, operated at an acceleration voltage of 1000 kV. The images were recorded around two orthogonal tilt axes, each over an angular range of 120° with a 2° tilt interval. The double-tilt images were aligned as previously described (Penczek et al., 1995) and a tomographic reconstruction was made using the weighted back-projection method (Radermacher, 1992). Image processing was done using the SPIDER system (Frank et al., 1996). The reconstructed volume had dimensions of 512 × 512 × 60 pixels, with a pixel size of 3.1 nm. A surface-rendered model was made using Sterecon (Marko and Leith, 1996) to segment the volume and Iris Explorer (NAG, Downers Grove, IL) for rendering.
Acknowledgments
Acknowledgements
We are indebted to M.Colombini for helpful discussions regarding the outer membrane permeability assay; to G.P.Wong for technical assistance in constructing the electrode rig; to S.Zinkel and X.Yin for providing Bid–/– mice; to B.Gewurz, S.Tavazoie and S.F.Tavazoie for careful review of the manuscript; and to E.Smith for assistance with manuscript preparation. Electron tomography work at the Resource for the Visualization of Biological Complexity is supported by the National Center for Research Resources, NIH grant #RR01219. M.C.W. is supported in part by NIH Training Grant #5T32AT09361. L.S. is a recipient of Human Frontier Science Program Organization Fellowship #LT0302/2000-M. This work is supported in part by NIH grant #CA50239-13.
REFERENCES
- Amarante-Mendes G.P., Finucane,D.M., Martin,S.J., Cotter,T.G., Salvesen,G.S. and Green,D.R. (1998) Anti-apoptotic oncogenes prevent caspase-dependent and independent commitment for cell death. Cell Death Differ., 5, 298–306. [DOI] [PubMed] [Google Scholar]
- Baffy G., Miyashita,T., Williamson,J.R. and Reed,J.C. (1993) Apoptosis induced by withdrawal of interleukin-3 (IL-3) from an IL-3-dependent hematopoietic cell line is associated with repartitioning of intracellular calcium and is blocked by enforced Bcl-2 oncoprotein production. J. Biol. Chem., 268, 6511–6519. [PubMed] [Google Scholar]
- Basanez M.G., Nechushtan,A., Drozhinin,O., Chanturiya,A., Choe,E., Tutt,S., Wood,K.A., Hsu,Y., Zimmerberg,J. and Youle,R.J. (1999) Bax, but not Bcl-xL, decreases the lifetime of planar phospholipid bilayer membranes at subnanomolar concentrations. Proc. Natl Acad. Sci. USA, 96, 5492–5497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bernardi P. (1999) Mitochondrial transport of cations: channels, exchangers and permeability transition. Physiol. Rev., 79, 1127–1155. [DOI] [PubMed] [Google Scholar]
- Cai J. and Jones,D.P. (1998) Superoxide in apoptosis. Mitochondrial generation triggered by cytochrome c loss. J. Biol. Chem., 273, 11401–11404. [DOI] [PubMed] [Google Scholar]
- Chance B. and Williams,G.R. (1955) Respiratory enzymes in oxidative phosphorylation. I. Kinetics of oxygen utilization. J. Biol. Chem., 217, 383–393. [PubMed] [Google Scholar]
- Deshmukh M., Kuida,K. and Johnson,E.M.,Jr (2000) Caspase inhibition extends the commitment to neuronal death beyond cytochrome c release to the point of mitochondrial depolarization. J. Cell Biol., 150, 131–143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eskes R., Desagher,S., Antonsson,B. and Martinou,J.C. (2000) Bid induces the oligomerization and insertion of Bax into the outer mitochondrial membrane. Mol. Cell. Biol., 20, 929–935. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feldmann G., Haouzi,D., Moreau,A., Durand-Schneider,A.M., Bringuier,A., Berson,A., Mansouri,A., Fau,D. and Pessayre,D. (2000) Opening of the mitochondrial permeability transition pore causes matrix expansion and outer membrane rupture in Fas-mediated hepatic apoptosis in mice. Hepatology, 31, 674–683. [DOI] [PubMed] [Google Scholar]
- Frank J., Radermacher,M., Penczek,P., Zhu,P., Li,Y., Ladjadj,M. and Leith,L. (1996) SPIDER and WEB: processing and visualization of images in 3D electron microscopy and related fields. J. Struct. Biol., 116, 190–199. [DOI] [PubMed] [Google Scholar]
- Frey T.G. and Mannella,C.A. (2000) The internal structure of mitochondria. Trends Biochem. Sci., 25, 319–324. [DOI] [PubMed] [Google Scholar]
- Goldstein J.C., Waterhouse,N.J., Juin,P., Evan,G.I. and Green,D.R. (2000) The coordinate release of cytochrome c during apoptosis is rapid, complete and kinetically invariant. Nature Cell Biol., 2, 156–162. [DOI] [PubMed] [Google Scholar]
- Green D. and Kroemer,G. (1998) The central executioners of apoptosis: caspases or mitochondria? Trends Cell Biol., 8, 267–271. [DOI] [PubMed] [Google Scholar]
- Green D.R. and Reed,J.C. (1998) Mitochondria and apoptosis. Science, 281, 1309–1312. [DOI] [PubMed] [Google Scholar]
- Greenhalf W., Stephan,C. and Chaudhuri,B. (1996) Role of mitochondria and C-terminal membrane anchor of Bcl-2 in Bax induced growth arrest and mortality in Saccharomyces cerevisiae. FEBS Lett., 380, 169–175. [DOI] [PubMed] [Google Scholar]
- Gross A., Yin,X.M., Wang,K., Wei,M.C., Jockel,J., Milliman,C., Erdjument-Bromage,H., Tempst,P. and Korsmeyer,S.J. (1999) Caspase cleaved BID targets mitochondria and is required for cytochrome c release, while BCL-XL prevents this release but not tumor necrosis factor-R1/Fas death. J. Biol. Chem., 274, 1156–1163. [DOI] [PubMed] [Google Scholar]
- Gross A., Pilcher,K., Blachly-Dyson,E., Basso,E., Jockel,J., Bassik,M.C., Korsmeyer,S.J. and Forte,M. (2000) Biochemical and genetic analysis of the mitochondrial response of yeast to BAX and BCL-XL. Mol. Cell. Biol., 20, 3125–3136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gunter K.K. and Gunter,T.E. (1994) Transport of calcium by mitochondria. J. Bioenerg. Biomembr., 26, 471–485. [DOI] [PubMed] [Google Scholar]
- Harris M.H., Vander Heiden,M.G., Kron,S.J. and Thompson,C.B. (2000) Role of oxidative phosphorylation in Bax toxicity. Mol. Cell. Biol., 20, 3590–3596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hirsch T., Marchetti,P., Susin,S.A., Dallaporta,B., Zamzami,N., Marzo,I., Geuskens,M. and Kroemer,G. (1997) The apoptosis–necrosis paradox. Apoptogenic proteases activated after mitochondrial permeability transition determine the mode of cell death. Oncogene, 15, 1573–1581. [DOI] [PubMed] [Google Scholar]
- Hockenbery D.M., Oltvai,Z.N., Yin,X.M., Milliman,C.L. and Korsmeyer,S.J. (1993) Bcl-2 functions in an antioxidant pathway to prevent apoptosis. Cell, 75, 241–251. [DOI] [PubMed] [Google Scholar]
- Ink B., Zornig,M., Baum,B., Hajibagheri,N., James,C., Chittenden,T. and Evan,G. (1997) Human Bak induces cell death in Schizosaccharomyces pombe with morphological changes similar to those with apoptosis in mammalian cells. Mol. Cell. Biol., 17, 2468–2474. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jurgensmeier J.M., Xie,Z., Deveraux,Q., Ellerby,L., Bredesen,D. and Reed,J.C. (1998) Bax directly induces release of cytochrome c from isolated mitochondria. Proc. Natl Acad. Sci. USA, 95, 4997–5002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kane D.J., Sarafian,T.A., Anton,R., Hahn,H., Gralla,E.B., Valentine,J.S., Ord,T. and Bredesen,D.E. (1993) Bcl-2 inhibition of neural death: decreased generation of reactive oxygen species. Science, 262, 1274–1277. [DOI] [PubMed] [Google Scholar]
- Kluck R.M. et al. (1999) The pro-apoptotic proteins, Bid and Bax, cause a limited permeabilization of the mitochondrial outer membrane that is enhanced by cytosol. J. Cell Biol., 147, 809–822. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krippner A., Matsuno-Yagi,A., Gottlieb,R.A. and Babior,B.M. (1996) Loss of function of cytochrome c in Jurkat cells undergoing Fas-mediated apoptosis. J. Biol. Chem., 271, 21629–21636. [DOI] [PubMed] [Google Scholar]
- Lee A.C., Zizi,M. and Colombini,M. (1994) β-NADH decreases the permeability of the mitochondrial outer membrane to ADP by a factor of 6. J. Biol. Chem., 269, 30974–30980. [PubMed] [Google Scholar]
- Lesage S., Steff,A.M., Philippoussis,F., Page,M., Trop,S., Mateo,V. and Hugo,P. (1997) CD4+ CD8+ thymocytes are preferentially induced to die following CD45 cross-linking, through a novel apoptotic pathway. J. Immunol., 159, 4762–4771. [PubMed] [Google Scholar]
- Li H., Zhu,H., Xu,C.J. and Yuan,J. (1998) Cleavage of BID by caspase 8 mediates the mitochondrial damage in the Fas pathway of apoptosis. Cell, 94, 491–501. [DOI] [PubMed] [Google Scholar]
- Li K., Li,Y., Shelton,J.M., Richardson,J.A., Spencer,E., Chen,Z.J., Wang,X. and Williams,R.S. (2000) Cytochrome c deficiency causes embryonic lethality and attenuates stress-induced apoptosis. Cell, 101, 389–399. [DOI] [PubMed] [Google Scholar]
- Li P., Nijhawan,D., Budihardjo,I., Srinivasula,S.M., Ahmad,M., Alnemri,E.S. and Wang,X. (1997) Cytochrome c and dATP-dependent formation of Apaf-1/caspase-9 complex initiates an apoptotic protease cascade. Cell, 91, 479–489. [DOI] [PubMed] [Google Scholar]
- Liu X., Kim,C.N., Yang,J., Jemmerson,R. and Wang,X. (1996) Induction of apoptotic program in cell-free extracts: requirement for dATP and cytochrome c. Cell, 86, 147–157. [DOI] [PubMed] [Google Scholar]
- Luo X., Budihardjo,I., Zou,H., Slaughter,C. and Wang,X. (1998) Bid, a Bcl2 interacting protein, mediates cytochrome c release from mitochondria in response to activation of cell surface death receptors. Cell, 94, 481–490. [DOI] [PubMed] [Google Scholar]
- Mannella C.A., Marko,M., Penczek,P., Barnard,D. and Frank,J. (1994) The internal compartmentation of rat-liver mitochondria: a tomographic study using the high-voltage transmission electron microscope. Microsc. Res. Tech., 27, 278–283. [DOI] [PubMed] [Google Scholar]
- Marko M. and Leith,A. (1996) Sterecon—three-dimensional reconstruction from stereoscopic contouring. J. Struct. Biol., 117, 24–35. [DOI] [PubMed] [Google Scholar]
- Martinou I., Desagher,S., Eskes,R., Antonsson,B., Andre,E., Fakan,S. and Martinou,J.C. (1999) The release of cytochrome c from mitochondria during apoptosis of NGF-deprived sympathetic neurons is a reversible event. J. Cell Biol., 144, 883–889. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martinou J.C., Desagher,S. and Antonsson,B. (2000) Cytochrome c release from mitochondria: all or nothing. Nature Cell Biol., 2, E41–E43. [DOI] [PubMed] [Google Scholar]
- Marzo I. et al. (1998) Bax and adenine nucleotide translocator cooperate in the mitochondrial control of apoptosis. Science, 281, 2027–2031. [DOI] [PubMed] [Google Scholar]
- McCarthy N.J., Whyte,M.K., Gilbert,C.S. and Evan,G.I. (1997) Inhibition of Ced-3/ICE-related proteases does not prevent cell death induced by oncogenes, DNA damage, or the Bcl-2 homologue Bak. J. Cell Biol., 136, 215–227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mootha V.K., French,S. and Balaban,R.S. (1996) Neutral carrier-based ‘Ca2+-selective’ microelectrodes for the measurement of tetraphenylphosphonium. Anal. Biochem., 236, 327–330. [DOI] [PubMed] [Google Scholar]
- Mootha V.K., Arai,A.E. and Balaban,R.S. (1997) Maximum oxidative phosphorylation capacity of the mammalian heart. Am. J. Physiol., 272, H769–H775. [DOI] [PubMed] [Google Scholar]
- Murphy A.N., Bredesen,D.E., Cortopassi,G., Wang,E. and Fiskum,G. (1996) Bcl-2 potentiates the maximal calcium uptake capacity of neural cell mitochondria. Proc. Natl Acad. Sci. USA, 93, 9893–9898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nicholls D.G. and Ferguson,S.J. (1992) Bioenergetics II. Academic Press, Boston, MA, pp. 34–37.
- Ogasawara J., Watanabe-Fukunaga,R., Adachi,M., Matsuzawa,A., Kasugai,T., Kitamura,Y., Itoh,N., Suda,T. and Nagata,S. (1993) Lethal effect of the anti-Fas antibody in mice. Nature, 364, 806–809. [DOI] [PubMed] [Google Scholar]
- Ohta T., Kinoshita,T., Naito,M., Nozaki,T., Masutani,M., Tsuruo,T. and Miyajima,A. (1997) Requirement of the caspase-3/CPP32 protease cascade for apoptotic death following cytokine deprivation in hematopoietic cells. J. Biol. Chem., 272, 23111–23116. [DOI] [PubMed] [Google Scholar]
- Penczek P., Marko,M., Buttle,K. and Frank,J. (1995) Double-tilt electron microscopy. Ultramicroscopy, 60, 393–410. [DOI] [PubMed] [Google Scholar]
- Radermacher M. (1992) Weighted back-projection methods. In Frank,J. (ed.), Electron Tomography. Plenum, New York, NY, pp. 91–115.
- Saito M., Korsmeyer,S.J. and Schlesinger,P.H. (2000) BAX-dependent transport of cytochrome c reconstituted in pure liposomes. Nature Cell Biol., 2, 553–555. [DOI] [PubMed] [Google Scholar]
- Salemme F.R. (1977) Structure and function of cytochromes c. Annu. Rev. Biochem., 46, 299–329. [DOI] [PubMed] [Google Scholar]
- Scaffidi C., Schmitz,I., Zha,J., Korsmeyer,S.J., Krammer,P.H. and Peter,M.E. (1999) Differential modulation of apoptosis sensitivity in CD95 type I and type II cells. J. Biol. Chem., 274, 22532–22538. [DOI] [PubMed] [Google Scholar]
- Shimizu S., Narita,M. and Tsujimoto,Y. (1999) Bcl-2 family proteins regulate the release of apoptogenic cytochrome c by the mitochondrial channel VDAC. Nature, 399, 483–487. [DOI] [PubMed] [Google Scholar]
- Srinivasula S.M., Manzoor,A., Fernandes-Alnemri,T., Litwack,G. and Alnemri,E.S. (1996) Molecular ordering of the Fas-apoptotic pathway: the Fas/APO-1 protease Mch5 is a CrmA-inhibitable protease that activates multiple Ced-3/ICE-like cysteine proteases. Proc. Natl Acad. Sci. USA, 93, 14486–14491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vander Heiden M.G., Chandel,N.S., Williamson,E.K., Schumacker,P.T. and Thompson,C.B. (1997) Bcl-xL regulates the membrane potential and volume homeostasis of mitochondria. Cell, 91, 627–637. [DOI] [PubMed] [Google Scholar]
- Vander Heiden M.G., Chandel,N.S., Schumacker,P.T. and Thompson,C.B. (1999) Bcl-xL prevents cell death following growth factor withdrawal by facilitating mitochondrial ATP/ADP exchange. Mol. Cell, 3, 159–167. [DOI] [PubMed] [Google Scholar]
- von Ahsen O., Renken,C., Perkins,G., Kluck,R.M., Bossy-Wetzel,E. and Newmeyer,D.D. (2000) Preservation of mitochondrial structure and function after Bid- or Bax-mediated cytochrome c release. J. Cell Biol., 150, 1027–1036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wei M.C., Lindsten,T., Mootha,V.K., Weiler,S., Gross,A., Ashiya,M., Thompson,C.B. and Korsmeyer,S.J. (2000) tBID, a membrane-targeted death ligand, oligomerizes BAK to release cytochrome c. Genes Dev., 14, 2060–2071. [PMC free article] [PubMed] [Google Scholar]
- Woo M., Hakem,A., Elia,A.J., Hakem,R., Duncan,G.S., Patterson,B.J. and Mak,T.W. (1999) In vivo evidence that caspase-3 is required for Fas-mediated apoptosis of hepatocytes. J. Immunol., 163, 4909–4916. [PubMed] [Google Scholar]
- Xiang J., Chao,D.T. and Korsmeyer,S.J. (1996) BAX-induced cell death may not require interleukin 1β-converting enzyme-like proteases. Proc. Natl Acad. Sci. USA, 93, 14559–14563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yin X.M., Wang,K., Gross,A., Zhao,Y., Zinkel,S., Klocke,B., Roth,K.A. and Korsmeyer,S.J. (1999) Bid-deficient mice are resistant to Fas-induced hepatocellular apoptosis. Nature, 400, 886–891. [DOI] [PubMed] [Google Scholar]
- Zha H., Fisk,H.A., Yaffe,M.P., Mahajan,N., Herman,B. and Reed,J.C. (1996) Structure–function comparisons of the proapoptotic protein Bax in yeast and mammalian cells. Mol. Cell. Biol., 16, 6494–6508. [DOI] [PMC free article] [PubMed] [Google Scholar]