

Abstract
To understand the requirements for binding to G protein βγ subunits, phage-displayed random peptide libraries were screened using immobilized biotinylated βγ as the target. Selected peptides were grouped into four different families based on their sequence characteristics. One group (group I) had a clear conserved motif that has significant homology to peptides derived from phospholipase C β (PLC β) and to a short motif in phosducin that binds to G protein β subunits. The other groups had weaker sequence homologies or no homology to the group I sequences. A synthetic peptide from the strongest consensus group blocked activation of PLC by G protein βγ subunits. The peptide did not block βγ-mediated inhibition of voltage-gated calcium channels and had little effect on βγ-mediated inhibition of Gs-stimulated type I adenylate cyclase. Competition experiments indicated that peptides from all four families bound to a single site on βγ. These peptides may bind to a protein–protein interaction ‘hot spot’ on the surface of βγ subunits that is used by a subclass of effectors.
Keywords: G protein βγ subunits/G protein effectors/peptide/phage display/protein–protein interaction
Introduction
Heterotrimeric G proteins, consisting of multiple isoforms of distinct α, β and γ subunits, mediate the actions of a wide variety of cell surface receptors involved in processes ranging from phototransduction to neurotransmission (Hildebrandt, 1997; Sprang, 1997; Gutkind, 1998; Hamm, 1998). G protein-coupled receptors catalyze exchange of tightly bound GDP for GTP on the α subunit in a process that requires all three subunits. The binding of GTP results in activation of the G protein and dissociation of the α subunit from the βγ subunits. The α and βγ subunits both interact with effector molecules, such as phospholipases and ion channels, in a manner that leads to their activation or inhibition (Clapham and Neer, 1997). A variety of in vitro studies have shown that when βγ subunits are bound to α-GDP they are incapable of activating downstream effectors. Thus, activation and deactivation of βγ subunit-mediated signal transduction in cells are thought to rely on dissociation and reassociation of GTP- and GDP-bound α subunits, respectively.
G protein βγ subunit-mediated activation of effectors has diverse roles in the regulation of cell physiology. Some examples of the cellular processes regulated by βγ subunits are briefly described here. In excitable cells, including neurons and cardiac myocytes, βγ subunits that are released from Gi regulate inwardly rectifying K+ channels, so as to modulate membrane potential or heart rate (Clapham and Neer, 1997). Chemokine receptors, such as the interleukin-8 receptor and the co-receptors for entry of the AIDS virus into leukocytes, are coupled to the release of βγ subunits from Gi (Kuang et al., 1996; Littman, 1998). Recent evidence indicates that βγ-responsive phospholipase C (PLC) isoforms β2 and β3 play inhibitory roles in cell signaling. Mouse neutrophils, where βγ-responsive PLC β2 was eliminated by gene targeting, displayed increased chemotaxis in response to chemotactic peptides and the mice were more resistant to viral infection (Jiang et al., 1997). In transgenic mice lacking βγ-regulated PLC β3, morphine acting at Gi/o-linked opioid receptors produced pain-killing effects at much lower doses, correlating with greater inhibition of voltage-gated Ca2+ channels in dorsal root ganglion neurons (Xie et al., 1999). Activation of multiple Gi- and Gq-coupled receptors, including thrombin, lysophosphatidic acid (LPA) and acetylcholine receptors, results in a mitogenic response in several cell types. MAP kinases are critical components in the growth-promoting pathways regulated by these receptors. βγ subunits indirectly activate MAP kinase, suggesting that βγ subunits may mediate the growth-promoting effects of many G protein-coupled receptors (Gutkind, 1998).
The physiological processes regulated by βγ subunits are mediated by interactions between βγ subunits and a wide variety of diverse target molecules, ranging from inwardly rectifying K+ channels to soluble enzymes such as PLC (for review see Clapham and Neer, 1997). How βγ subunits regulate such a diverse range of effector molecules is not well understood, and a complete picture of how βγ subunits interact with various targets has not been forthcoming. Small synthetic peptide-based approaches have been used to dissect βγ–effector interactions. One such study used a peptide representing amino acids 956–984 of adenylate cyclase type 2 (QEHA peptide) (Chen et al., 1995). This peptide blocked activation of a number of effectors by βγ subunits, including adenylate cyclase 2, PLC β3, G protein-gated inwardly rectifying potassium channels (GIRK) and β-adrenergic receptor kinase (βARK) with an IC50 of 50–100 µM, and bound at a site overlapping the α subunit binding site on βγ (Weng et al., 1996). These data suggest that the peptide binds to a site shared by multiple effectors. Based on amino acid substitutions in the peptide and sequence comparison amongst adenylate cyclase, βARK and GIRK-1, the authors proposed a consensus βγ-binding sequence, QXXER. However, many proteins are regulated by βγ subunits but do not contain the QXXER motif [including phosphoinositide 3-kinase γ (PI3K γ) and PLC β2], so this may not be a universal motif.
As an alternative approach to defining requirements for binding to G protein βγ subunits, we have screened a panel of phage-displayed random peptide libraries to identify peptides that bind to βγ subunits. Using this approach we identified multiple peptides that bind specifically to βγ subunits. This has allowed us to further characterize the sequence requirements for binding to physiologically relevant sites on the surface of βγ subunits.
Results
Panning
To determine the types of amino acid sequence that can interact with βγ subunits, phage-displayed random peptide libraries were screened with β1γ2 that was bound to immobilized streptavidin via covalently attached biotin. We prepared biotinylated βγ subunits (b-βγ) using a modification of a previously published protocol (see Materials and methods) (Dingus et al., 1994). A key feature of this method is that βγ is chemically modified with an amine-specific reagent in the presence of α subunits to protect sites that are critical for protein– protein interactions. b-βγ is then purified on the basis of its ability to dissociate upon activation of α subunits with aluminum fluoride (Kozasa and Gilman, 1995). This procedure ensures that b-βγ is functional. The b-βγ prepared in this way activates PLC β2, PLC β3 and PI3K similarly to unmodified βγ (not shown). Previous work by others indicates that βγ subunits modified in this way can interact normally with α subunits (Kohnken and Hildebrandt, 1989; Dingus et al., 1994), adenylate cyclase (Kohnken and Hildebrandt, 1989) and phosducin (Satpaev and Slepak, 2000). This indicates that binding sites on βγ subunits, critical for interactions with α subunits and effectors, were available for interaction with phage-displayed peptides.
We screened 16 different phage-displayed libraries of various lengths, some of which had various constraints imposed by internal disulfide linkages (Bonnycastle et al., 1996) (Table I). The peptides were displayed as fusions with the pVIII coat protein of the f88-4 phage display vector; ∼10% of the coat proteins expressed have peptides fused (∼250 copies of peptide/phage particle). Each library has a diversity of 1 × 108–1 × 109 individual clones. All screening was performed in the presence of detergent, bovine serum albumin (BSA) and salt to minimize non-specific interactions.
Table I. Phage-displayed peptide libraries that were screened against G protein βγ subunits.
| Linear libraries | Disulfide-bridged-loop libraries with one-residue flanking regions: LXn | Disulfide-bridged-loop libraries with 4-5-residue flanking regions: Cys | Half-Cys libraries (having only one fixed Cys): | α-conotoxin library |
|---|---|---|---|---|
| 1) LX4 = XCX4CX | 1) Cys3 = X5CX3CX4 | |||
| 1) X6 | 2) LX6 = XCX6CX | 2) Cys4 = X4CX4CX4 | 1) X8CX8 | XCCX3CX5C4GIEG |
| 2) X15 | 3) LX8 = XCX8CX | 3) Cys5 = X4CX5CX4 | 2) X15CX | RG |
| 3) X30 | 4) LX10 = XCX10CX | 4) Cys6 = X4CX6CX4 | 3) X15CX | |
| 5) LX12 = XCX12CX |
In all cases, the libraries outlined above were fused to the N-terminus of the pVIII coat protein of the phage display vector f88-4. X stands for any of the 20 natural amino acids.
Multiple binding clones were obtained from several libraries and were characterized in various ways for binding specificity. For Figure 1A, two selected phage clones were compared with f88-4 in a phage enzyme-linked immunosorbent assay (ELISA) in which binding of phage was detected with a phage-specific antibody and absorbance at 405 nm was monitored after addition of a chromogenic substrate. Absorbance values for wild-type f88-4 phage in the absence of βγ represent non-specific binding of the phage (and/or the anti-phage antibody) to the plate and can be considered as background. In the presence of βγ subunits, f88-4 binding was not increased, indicating that the wild-type f88-4 phage did not bind to βγ. Binding of phage bearing the selected peptides in the absence of immobilized βγ was not significantly different from that for f88-4 (Figure 1A), indicating that the peptides did not interact non-specifically with the plate or the streptavidin. Only in the presence of immobilized βγ subunits was binding of the selected peptide-bearing phage clones significantly higher than that of wild-type phage (f88-4). These results clearly demonstrate that the peptides expressed as fusions with phage pVIII coat protein bind to βγ subunits.

Fig. 1. Phage bearing selected peptides bind specifically to immobilized βγ subunits and binding is inhibited by a synthetic peptide corresponding to one of the selected sequences. (A) 1 × 1010 f88-4 (black bars), P1 (white) or P21 (dark gray bars) phage were incubated in wells coated with streptavidin with or without prior addition of 50 nM b-βγ. (B) Binding of the same phage to 50 nM b-βγ was tested at the indicated concentrations of SIRK peptide.
Many of the binding clones were derived from disulfide-constrained libraries. To differentiate general non-specific hydrophobic interactions of these peptides with βγ subunits from interactions dependent on structure, we tested binding of phage to immobilized βγ after treatment of the phage with 10 mM dithiothreitol (DTT) to reduce intramolecular disulfide bonds. This treatment eliminated binding of all the tested phage derived from the disulfide-constrained libraries (Table II). This indicates that the disulfide constraint within the peptide is critical for binding to the βγ subunits. DTT treatment had no effect on binding of phage derived from the linear library, indicating that this treatment does not generally inhibit phage binding to βγ by some other mechanism. These data support the argument that binding of the peptides to βγ depends on the overall structure of the peptides, and not simply on general hydrophobic or charge characteristics.
Table II. Binding of phage bearing disulfide-constrained peptides to βγ in a phage ELISA is inhibited by reduction of disulfides.
|
A405 |
||||
|---|---|---|---|---|
| –βγ | +βγa | +βγ +10 mM DTTb | ||
| I | ACKRTKAQILLAAPCT | 0.129 | 0.595 | 0.187 |
| SIRKALNILGYPDYD | 0.118 | 0.245 | 0.299 | |
| SCEQTKTDRLLGNAC | 0.121 | 0.259 | 0.143 | |
| ACTLPGKPYSLLGIC | 0.121 | 0.492 | 0.178 | |
| II | TCQKLAWLTGKKEKCL | 0.129 | 0.744 | 0.330 |
| III | SCEKRYGIEFCT | 0.122 | 0.348 | 0.159 |
| SCEKRLGVRSCT | 0.128 | 0.717 | 0.150 | |
| IV | PTAVCNFFGQCPMEI | 0.116 | 0.560 | 0.147 |
| PSKVCAHFDICYTLS | 0.125 | 0.275 | 0.151 | |
aNinety nanograms of β1γ2 were immobilized in each well. Binding of phage was tested in a phage ELISA assay (see Materials and methods). Results are the mean A405 of duplicate determinations measured in a microplate reader. Similar experiments were performed three times.
bDTT (10 mM) was included in the binding solution during incubation with the indicated phage.
Sequence characteristics of the phage
Peptide sequences from specifically binding phage clones were grouped together into four classes based on shared sequence motifs (Figure 2A). The first group of phage had an apparent conserved sequence motif corresponding to KX3LLG. The second group had a similar motif that was not as strongly conserved, with substitutions at conserved positions possibly representing allowable variations of this motif. Groups III and IV had no obvious homology to these first two consensus groups. For the two clones in group III, there was significant internal sequence similarity, yielding a second consensus motif, EKRXGX3. The sole apparent unifying characteristic of group IV is the positioning of the cysteine residues, which were fixed before screening.
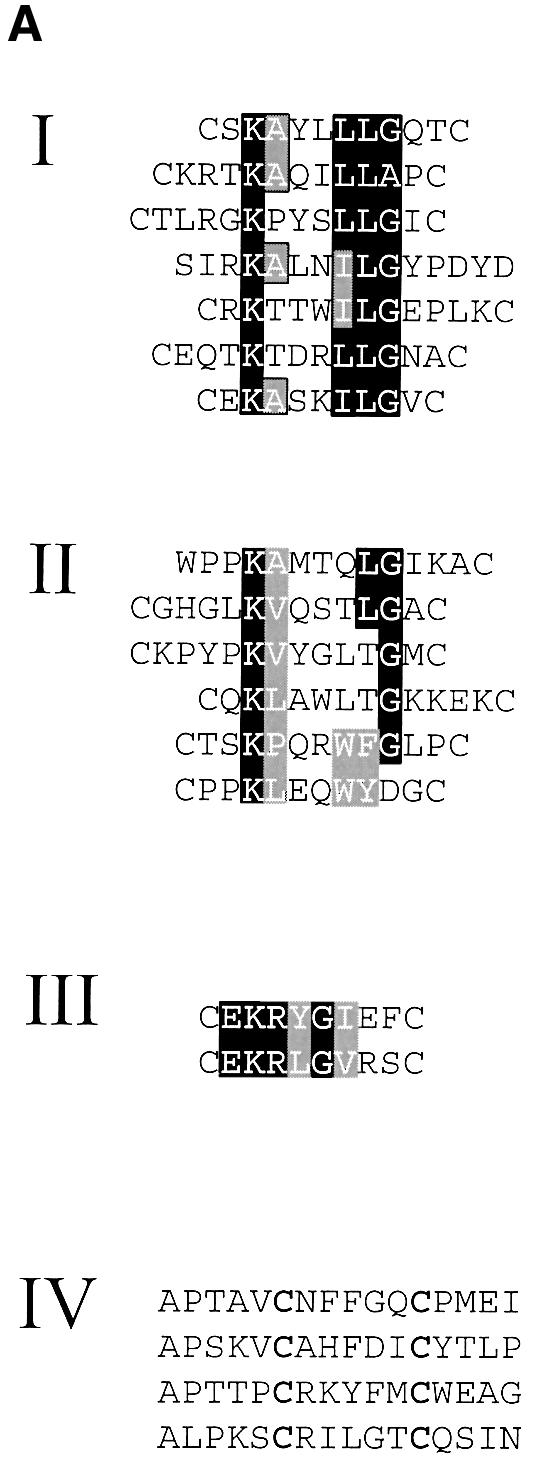

Fig. 2. Alignments of sequences obtained by random peptide phage display screening. (A) Sequences were placed into four groups based on sequence similarity. Black boxes indicate identities and gray boxes conservative substitutions. (B) Comparison of group A sequence with known βγ-binding sequences from PLC β2, rat and bovine phosducins and phosducin-like protein (PLP).
To determine whether the peptides displayed on the phage bound to βγ as isolated peptides and whether the displayed peptides had overlapping binding sites, we performed competition ELISA experiments with synthetic peptides corresponding to two of the phage sequences. In these experiments, binding of phage clones displaying various peptide sequences to immobilized βγ was monitored in the presence and absence of excess synthetic peptide. If the peptide bound to the βγ at the same site as the phage-borne peptide, it would be predicted to block binding of the phage. The first peptide that we tested in this analysis was the linear peptide that was derived from the screening: SIRKALNILGYPDYD (SIRK peptide). Results are shown in Figure 1B with two selected phage at various concentrations of peptide. The peptide had no effect on the background binding of f88-4, but inhibited binding of both of the selected phage in a concentration-dependent manner, indicating that the peptide binds to the βγ subunits at the same site as these two selected phage clones. The peptide was more potent at inhibiting binding of phage p21 (IC50 1–3 µM) than P1 (IC50 ∼10 µM) and may reflect differences in the affinity of these two phage clones for βγ. We predicted that SIRK peptide would block binding of all of the phage displaying the peptides in group I, since this peptide belongs to this consensus group, and that it would not block binding of phage in the other groups. We tested βγ binding of phage from each group at a fixed concentration of peptide (30 µM); the results are shown in Table III. The peptide blocked binding of phage in group I and, surprisingly, also inhibited binding of the phage in the other groups. This suggests that all of these disparate sequences bind to a single site or overlapping sites on βγ. To determine whether these effects are specific to peptides from group I, we tested another peptide from group III (SCERKLGVRSCT) (C5) and found that it inhibited binding of phage in group I and group II (Table IV); separate experiments showed that this peptide also inhibited binding of selected phage from group IV (not shown). When we initially performed the screening, we predicted that we would obtain sequences that bound to various sites on βγ subunits that would differ in sequence characteristics. Instead, it seems that there is a single site on βγ that dominated in the screening, and that it has the capacity to interact with a variety of sequences.
Table III. Synthetic peptide SIRK blocks binding to βγ in a phage ELISA.
|
A405 |
||||
|---|---|---|---|---|
| –βγ | +βγ | +βγ + 30 µM SIRKa | ||
| f88-4 | 0.237 ± 0.011 | 0.223 ± 0.002 | 0.206 ± 0.001 | |
| I | SGRLCSKAYLLLGQTCD | 0.216 ± 0.006 | 0.647 ± 0.006 | 0.180 ± 0.008 |
| ACKRTKAQILLAAPCT | 0.193 ± 0.009 | 0.427 ± 0.010 | 0.195 ± 0.002 | |
| ACTLPGKPYSLLGIC | 0.177 ± 0.002 | 0.427 ± 0.002 | 0.177 ± 0.003 | |
| SIRKALNILGYPDYD | 0.206 ± 0.009 | 0.663 ± 0.007 | 0.260 ± 0.003 | |
| II | DCKPYPKVYGLTGMC | 0.234 ± 0.020 | 0.726 ± 0.048 | 0.305 ± 0.022 |
| TCTSKPQRWFGLPC | 0.223 ± 0.005 | 0.463 ± 0.021 | 0.190 ± 0.006 | |
| GCPPKLEQWYDGCV | 0.215 ± 0.009 | 0.703 ± 0.043 | 0.270 ± 0.014 | |
| III | SCERKYGIEFCT | 0.192 ± 0.002 | 0.565 ± 0.065 | 0.220 ± 0.013 |
| SCEKRLGVRSCT | 0.199 ± 0.011 | 0.687 ± 0.002 | 0.289 ± 0.072 | |
| IV | PSKVCAHFDICYTLS | 0.192 ± 0.002 | 0.563 ± 0.017 | 0.210 ± 0.02 |
| PTTPCRKYFMCWEAG | 0.192 ± 0.004 | 0.672 ± 0.041 | 0.259 ± 0.036 | |
aPeptides were added to the wells just before addition of the phage. Results are the mean of duplicate determinations ± SE. Similar experiments were performed at least five times. The phage ELISA assay is the same as in Table I.
Table IV. Synthetic peptide C5 blocks binding of phage to βγ.
|
A405 |
|||
|---|---|---|---|
| –βγ | +βγ | +βγ + 30 µM C5a | |
| f88-4 | 0.165 ± 0.010 | 0.137 ± 0.002 | 0.137 ± 0.006 |
| DCKPYPKVYGLTGMC | 0.150 ± 0.009 | 0.735 ± 0.006 | 0.289 ± 0.021 |
| TCTSKPQRWFGLPC | 0.148 ± 0.003 | 0.350 ± 0.020 | 0.150 ± 0.004 |
| ACKRTKAQILLAAPCT | 0.161 ± 0.000 | 0.413 ± 0.010 | 0.149 ± 0.008 |
aSynthetic disulfide bridged peptides were dissolved in water at 1 mg/ml and allowed to oxidize overnight by exposure to air. The extent of cyclization was analyzed by reversed-phase HPLC.
Relevance of the selected binding site to βγ–effector interactions
The group I consensus sequence is similar to a sequence from PLC β2 that will also bind to βγ subunits as an isolated peptide (Sankaran et al., 1998). In fact, the first clone in group I (Table III) displays a high degree of similarity to the PLC β2 sequence in other positions too (Figure 2B). Based on this, we predicted that peptides from group I would bind to a site on βγ subunits recognized by PLC and might prevent activation of PLC by βγ subunits. We examined the ability of the SIRK peptide to inhibit activation of PLC β2 by βγ subunits. The peptide specifically inhibited PLC β2 activation by βγ subunits (Figure 3A) with an IC50 of ∼3 µM, without affecting basal PLC activity. Other peptides from this group, and from the other groups, also inhibited PLC β2 activation by βγ subunits with various potencies (Figure 3B and data not shown).

Fig. 3. Phage-displayed peptides inhibit activation of PLC β2 and PI3Kγ by βγ subunits. Synthetic peptides with the sequences (A) SIRKALNILGPDYD (linear) and (B) ACTLPGKPYSLLGICD (cyclic) were included in PLC assays at the indicated concentrations. Reactions included either no β1γ2 (filled triangles) or 100 nM β1γ2 (open triangles) and 1 ng of PLC β2, 100 nM free Ca2+; the reaction time was 5 min. Each data point is the mean of duplicate determinations and the data are representative of experiments repeated at least 10 times (A) and three times (B). (C) SIRK peptide inhibits activation of PI3kinase by βγ subunits. SIRK peptide was included at the indicated concentrations with 100 nM β1γ2 and 10 ng of a heterodimer of p110γ and EE-tagged p101 PI3K γ subunits purified from Sf9 cells (Stephens et al., 1997).
We also tested whether the SIRK peptide would inhibit interactions between βγ and other effectors. Activation of PI3K (Figure 3C) and PLC β3 (not shown) by βγ was blocked by peptide concentrations similar to that for PLC β2. On the other hand, SIRK peptide did not inhibit βγ-dependent inhibition of type I adenylate cyclase at concentrations up to 300 µM (Figure 4A). To confirm that PLC activation by βγ subunits is not inherently more sensitive to peptide inhibition than βγ-dependent inhibition of adenylate cyclase I in general, we tested a peptide previously shown to inhibit βγ interactions with type I adenylate cyclase, QEHA (Chen et al., 1995). This peptide inhibited PLC activation with the same potency as has been reported for its effects on βγ inhibition of adenylate cyclase (IC50 ∼100 µM) (not shown). This indicates that peptides, in general, are not inherently more potent at disrupting PLC interactions with βγ than adenylate cyclase–βγ interactions.

Fig. 4. SIRK peptide is a poor inhibitor of βγ-mediated inhibition of adenylate cyclase type I and voltage-gated Ca2+ channels. (A) SIRK peptide was included at the indicated concentrations with 5 µg of Sf9 membranes expressing type I adenylated cyclase and no βγ (open circles) or 20 nM βγ subunits (filled circles). Data points are the mean of duplicate determinations and the data are representative of experiments repeated three times. (B) SIRK peptide is a poor inhibitor of DAMGO-mediated inhibition of N-type Ca2+ channels. Black bars are control cells and gray bars are peptide-treated cells. Peptides at the indicated concentrations were included in the patch pipette and allowed to dialyze into the cell for 10 min before stimulation of the cells with 3 µM DAMGO. For 10 µM SIRK, there were 24 cells for control and 37 for peptide treated. For 100 µM SIRK, there were 13 cells for control and eight for peptide treated. For QEHA peptide, there were eight cells for control and 11 for peptide treated. Results are the mean ± SE. Treatments with SIRK (10 and 100 µM) were not statistically different from control treatments, while the QEHA treatment was significant with p <0.04 in a two-tailed paired t-test.
We also examined the effect of this peptide on opioid-mediated inhibition of voltage-gated Ca2+ currents in rat dorsal root ganglion neurons. The mechanism of channel inhibition, at least for N-type channels, involves in part direct binding of G protein βγ subunits to the α subunit of the channel (Herlitze et al., 1996, 1997; Qin et al., 1997; Samoriski and Gross, 2000). In these experiments, whole-cell Ca2+ currents were evoked by stepping to +10 mV from a holding potential of –80 mV in the presence and absence of the opioid agonist DAMGO, and in the absence or presence of selected peptides, dialyzed intracellularly via a patch pipette. Since the pipette contains an infinite reservoir of peptide, the intracellular solution should contain the same concentration of peptide as the patch pipette. Identical methods have been used to introduce Gαo antibodies into these cells to block inhibition of Ca2+ channels in this pathway (Wiley et al., 1997). The peptide apparently did not affect the DAMGO-dependent inhibition of the voltage-gated Ca2+ current (Figure 4B). There was no significant difference between mean current amplitudes in control and peptide-treated cells (not shown). To confirm that we could indeed inhibit βγ-mediated inhibition of Ca2+ currents with βγ-binding peptides, we performed the same experiments with the QEHA peptide derived from adenylyl cyclase (Chen et al., 1995). In these experiments, DAMGO-mediated Ca2+ channel inhibition was partially inhibited, indicating that βγ interactions with the Ca2+ channel can be inhibited by synthetic peptides. That only a portion of the receptor-mediated effects on Ca2+ currents were inhibited by QEHA may be because a submaximal dose of QEHA was used (IC50 ∼100 µM for inhibition of other processes) or because pathways other than the βγ-mediated one may be involved in regulating this Ca2+ channel. Based on more extensive experiments (Samoriski and Gross, 2000) we would predict only a partial block, as strong depolarization, which reverses βγ binding to the channels, only partially reverses DAMGO-induced current reductions.
Taking these data together, we conclude that this peptide-binding site is important for PLC β and PI3K interactions with βγ subunits, but perhaps not so important for interaction with adenylate cyclase I and voltage-gated Ca2+ channels.
Analysis of K+ channel interaction sites
To characterize the nature of the effector binding site on βγ further, we used the competition ELISA assay described earlier to determine whether peptides derived from another effector would prevent binding phage bearing the selected peptides. We tested four peptides derived from inwardly rectifying K+ channel subunits GIRK1 and GIRK4, which have previously been shown to inhibit interactions between βγ subunits and solubilized K+ channels, presumably through direct interactions with βγ subunits (Krapivinsky et al., 1998). GIRK1 (220–239) and the homologous GIRK4 (226–245) were reported to inhibit βγ–K+ interactions with relatively high IC50 values (70–100 µM) and we tested them in a competition ELISA at twice the IC50. GIRK1 (364–383) and GIRK4 (209–225) had lower reported IC50 values (8 and 0.6 µM, respectively), so we tested GIRK1 (364–383) at 10 times and GIRK4 (209–225) at 100 times their respective IC50 values. None of the peptides prevented binding of phage bearing the SIRK sequence (Figure 5). This suggests that these peptides and the regions that they represent on the K+ channel bind to other determinants on the βγ subunit.

Fig. 5. Peptides derived from inwardly rectifying K+ channels do not bind to the selected site on βγ subunits. Peptides derived from GIRK1 and GIRK4 that bind to βγ were tested for their ability to inhibit binding of phage displaying SCERKYGIEFCT at 1 × 1010 phage particles/well. Peptides were tested at the following concentrations: GIRK1 (220–239), 200 µM; GIRK1 (364–383), 100 µM; GIRK4 (209–225), 60 µM; scrambled GIRK4 (209–225), 60 µM; GIRK4 (226–245), 200 µM; scrambled GIRK4 (226–245), 200 µM; SIRK, 30 µM.
Analysis of the SIRK peptide
Based on the alignment of the sequences in group I, we predicted that the conserved amino acids would form critical binding determinants. To test this idea, we synthesized a set of ‘alanine scan’ analogs of the SIRK peptide. Sixteen peptides were synthesized, including wild-type peptide and peptides with successive alanine substitutions along the sequence. Where an alanine was already present in the sequence, it was substituted by glycine. Each synthetic peptide was purified to ≥80% homogeneity and analyzed by mass spectrometry to confirm its identity. Each peptide was tested for its ability to inhibit PLC activation by βγ subunits at various concentrations and IC50 values were estimated for each and are shown in Table V. The data strongly suggest that the predicted amino acids are important for binding. Residues at positions 8, 9 and 10, which correspond to {L/I}-LG in the predicted consensus, are clearly critical for peptide binding to βγ subunits and inhibition of PLC β2 activation. An amino acid that was, unexpectedly, important for binding of this peptide is the alanine at position 5. This amino acid was present in some of the peptide sequences, but not others. While mutation of the lysine at position 4 reduced the binding by ∼10-fold, this amino acid is apparently not as critical for binding to βγ as the LLG sequence. Interestingly, substitution of alanine for asparagine at position 7 increased the apparent binding of the peptide to βγ. Given all the positions that are required for binding of this peptide, why do peptides from group II bind where there are variations at all of the positions except position 4? Perhaps there are allowable substitutions for these amino acids in the peptide (just not alanine) or changes at other positions that are compensating for the loss of binding energy.
Table V. Alanine scan analogs of the SIRK peptide sequence and their ability to inhibit activation of PLC by βγ.
| Inhibition of βγ-PLC activation: IC50 (µM)a | Position of alanine or glycine substitution | |
|---|---|---|
| SIRKALNILGYPDYD | 5 | – |
| AIRKALNILGYPDYD | 5 | 1 |
| SARKALNILGYPDYD | 20 | 2 |
| SIAKALNILGYPDYD | 10 | 3 |
| SIRAALNILGYPDYD | 60 | 4 |
| SIRKGLNILGYPDYD | 100 | 5 |
| SIRKAANILGYPDYD | 65 | 6 |
| SIRKALAILGYPDYD | 0.5 | 7 |
| SIRKALNALGYPDYD | 200 | 8 |
| SIRKALNIAGYPDYD | 300 | 9 |
| SIRKALNILAYPDYD | 70 | 10 |
| SIRKALNILGAPDYD | 5 | 11 |
| SIRKALNILGYADYD | 5 | 12 |
| SIRKALNILGYPAYD | 5 | 13 |
| SIRKALNILGYPDAD | 5 | 14 |
| SIRKALNILGYPDYA | 5 | 15 |
aResults are representative data from titrations performed two or three times for each peptide at at least five different concentrations of peptide. Wild-type peptide was included in all titrations to control for experimental variation. Titrations were performed using methods identical to those used for Figure 3 and are described in Materials and methods.
Discussion
We have used random peptide phage display screening to identify amino acid motifs involved in binding to G protein βγ subunits. We predicted that we would obtain sequence groups that bound to different surfaces of the βγ subunit and that we would be able to identify groups of consensus sequence that would be used by different effectors. Instead, the screen yielded four distinct groups of binding sequences, most of which were derived from disulfide-constrained libraries that appeared to bind to the same site on βγ subunits. Group I sequences had a clear consensus that was definable on the basis of seven distinct sequences, K-X3-{I,L}-L-G. Based on competition analysis in the phage ELISA assay, all of the peptides share a single binding site. This single binding site appears to be shared by several effectors that bind βγ subunits including PLC β and PI3K γ. Critical binding determinants predicted from the apparent homology of the selected peptides was confirmed by scanning alanine mutagenesis. Most of the critical binding amino acids were non-polar, suggesting that hydrophobic interactions could be a major energetic driving force for binding of βγ subunits to effectors.
Protein–protein interactions are often driven by burial of exposed hydrophobic surfaces. A series of studies has been performed using phage display library screening to study protein–protein interactions. Repeatedly, such screens yield peptides that bind to relevant protein–protein interaction sites on the surfaces of proteins, despite the fact that the libraries screened are entirely random (Wrighton et al., 1996; Fairbrother et al., 1998; Lowman et al., 1998). It has been suggested that phage display methods tend to identify ‘hot spots’ on proteins that form the energetically critical contacts in a protein–protein interaction. A number of studies have analyzed such ‘hot spots’ in detail. For example, for human growth hormone (hGH) >30 contact sites have been identified in the three-dimensional co-crystal (Wells and de Vos, 1996). Alanine scanning mutagenesis of these contacts indicates that only five of these residues contribute to >85% of the binding energy and that these critical interactions are primarily non-polar. This has implications for the specificity of the interaction. Non-polar interactions do not have the strict stereochemical requirements found in electrostatic or hydrogen-bond-driven interactions and thus may accommodate a number of different sequences. This is different from what has been suggested for other peptides that bind to βγ. In particular, the QEHA peptide derived from adenylate cyclase and a peptide from the C terminus of βARK (Koch et al., 1993; Touhara et al., 1995) are both charged and have been proposed to bind through electrostatic interactions to surfaces on β subunits (Weng et al., 1996). Thus, effector interactions with βγ subunits may occur through hydrophobic interactions. At least one peptide that we have found, SIRK, has a 10- to 100-fold higher affinity for βγ than the βARK and QEHA peptides.
A number of βγ-regulated effectors (PLC β, phosducin and phosducin-like protein) have sequences with similarity to the sequence we derived (Figure 2B). The co-crystal structure of phosducin and βγ has been determined (Gaudet et al., 1996). In this structure, amino acids 193–198 of phosducin (the region that has similarity to the group I consensus) form a loop between two β strands with a lysine and two adjacent leucine residues analogous to the KX3LLG amino acids in direct contact with β subunit amino acids (Figure 6A and B). It is tempting to speculate that our peptides bind to this same region on βγ. Since many of the peptides we found are disulfide constrained with short inter-cysteine sequences, they are predicted to form a loop. The region that this sequence binds to on the β subunit undergoes major conformational rearrangement when phosducin binds between blades 6 and 7. It is possible that these peptides also cause this conformational change to occur (Loew et al., 1998). We emphasize that the idea that the phage-displayed peptides bind to the site on βγ shown for phosducin binding in Figure 6 is highly speculative, and further experiments are needed to demonstrate more rigorously the structural basis for interactions between these peptides and βγ subunits.

Fig. 6. Model of the interaction between phosducin 193–198 and β1γ1. (A) Top view of the β subunit bound to a loop from 189 to 201 between β strands S4 and S5 of phosducin [Sigler nomenclature (Gaudet et al., 1996)]. The β subunit propeller ribbons are in yellow, the β N-terminal helix is in red and the γ subunit is dark green. The β subunit amino acids that contact the K-X3-LL sequence in phosducin are shown in spacefill representation in CPK colors. The amino acids from phosducin, K193, L197 and L198 are turquoise, while other amino acids in the region from 189 to 201 are displayed only as a ribbon and colored light green. (B) Close-up view of amino acids involved in direct interaction between the K-X3-LL sequence in phosducin and the β subunit. Amino acids from phosducin are labeled with a ‘p’ prefix. The models were constructed from coordinates deposited in the SwissProt data bank (Gaudet et al., 1996) using RasTop version 1.3 developed by P.Valadon, A.Meuller and R.Sayle (http://www.bernstein-plus-sons.com/software/RasMol_2.7.1/).
Our results suggest that the peptides we identified bind to a unique site on βγ subunits that is not targeted by the QEHA and βARK peptides. In support of this, the binding site for the peptides we have found seems only to be shared by some effectors. The peptides block activation of PLC and PI3K, but are relatively ineffective at blocking inhibition of adenylate cyclase and Ca2+ channels. All the peptides previously found to bind to βγ subunits and block effector activation seem to do so in a general way, in that they block activation of all the βγ-regulated effectors that have been tested. The SIRK peptide is the first peptide that binds to βγ subunits and inhibits effectors with this kind of selectivity. This has important implications for potential pharmacological manipulation of βγ-regulated pathways. Various studies with β subunit mutants (Li et al., 1998) or the β5-subunit isoform (Yoshikawa et al., 2000) have suggested that effectors can bind to βγ subunits in different ways.
In the analysis presented here, we have begun to define consensus amino acids that could be used by effectors for interactions with βγ subunits. While a consensus did arise from our studies, there was a striking variation in the types of sequence that interacted with the same site on βγ. This suggests that one possible reason for the ability of βγ subunits to interact with a wide diversity of effectors is that there is a binding site on βγ that can accommodate a diverse range of sequences. This is clearly not the whole story, because the peptides we defined only block some effectors. Thus, there must be other binding sites on βγ subunits that are used by other effectors. It is interesting that our screen did not identify peptides that bind to these other binding sites nor did it identify sequences with homology to other known βγ-binding peptides such as the QEHA or the βARK peptides. We suggest that the binding site on βγ for the peptides we selected is a dominant binding determinant on βγ subunits and that the peptides that bound there were specifically enriched in the selection process at the expense of weaker binding peptides that bind at other sites. Further screening in the presence of peptides that block this dominant binding site may reveal peptides that bind at other sites.
Materials and methods
βγ biotinylation
Biotinylated β1γ2 subunits were prepared using a modification of the method developed by Dingus et al. (1994). Sf9 insect cells were infected with His6-αi1, -β1 and -γ2 as described previously (Kozasa and Gilman, 1995). Proteins were extracted from the membrane fraction and bound to a nickel NTA–agarose column. Heterotrimeric α1β1γ2 was eluted from the column in 4 ml of 20 mM HEPES pH 8.0, 100 mM NaCl, 0.1% polyoxyethylene 10 lauryl ether (C12E10), 10 µM GDP, 150 mM imidazole. The eluted protein was diluted to 1 mg protein/ml with 20 mM HEPES pH 8.0, 1 mM EDTA, 1 mM DTT, 100 mM NaCl, 10 µM GDP; the final detergent concentration was adjusted to 0.05% C12E10. NHS-LC-biotin (Pierce) was added from a 20 mM stock in dimethylsulfoxide to give a final concentration of 1 mM. The reaction was allowed to proceed for 30 min at room temperature, followed by addition of 10 mM ethanolamine pH 8.0 from a 200 mM stock and incubation on ice for 10 min. The sample was diluted to 90 ml with dilution buffer (20 mM HEPES pH 8.0, 100 mM NaCl, 10 µM GDP, 0.5% C12E10). Washed nickel NTA–agarose (2 ml) was added and incubated at 4°C, with mixing, overnight. The mixture was poured through a column and washed with 20 ml of dilution buffer. The column was warmed to room temperature for 15 min and washed with 5 ml of wash buffer (20 mM HEPES pH 8.0, 100 mM NaCl, 10 µM GDP, 1% Na-cholate). βγ subunits were eluted in wash buffer plus 10 mM MgCl2, 10 mM NaF, 30 µM AlCl3. Biotinylation was confirmed by electrophoresis and blotting with streptavidin–horseradish peroxidase and by showing that all of the b-βγ could be bound to streptavidin–agarose. b-βγ was capable of activating PLC β2 and β3 with a potency and efficacy similar to those of unmodified βγ (not shown).
Phage screening
The experiment was designed with several libraries (Bonnycastle et al., 1996); the Cys libraries were kindly donated by G.P.Smith (University of Missouri-Columbia) and combined in single wells of the microtiter plate such that 16 different libraries were screened in seven wells. The libraries that were screened are shown in Table I. The panning process we describe is the standard method for selecting binding phage (Smith and Scott, 1993; Bonnycastle et al., 1996). Briefly, 1012 virions were incubated with b-βγ subunits (100 ng) that had been bound to immobilized streptavidin on a microtiter plate. The wells were washed thoroughly to remove unbound phage. Non-specific binding of phage, either to the plate or to the βγ subunits, was minimized by performing the incubations and washes with buffers containing 150 mM NaCl, 0.5% Tween-20 and 2 mg/ml BSA. Bound phage were eluted at pH 2.2, and the eluates were neutralized and used to infect Escherichia coli cells that amplified the phage. Panning of each library mix was repeated three times with each to enrich for βγ-binding phage.
After panning, the pools of phage were tested for binding to the target using a phage ELISA protocol (Smith and Scott, 1993), in which b-βγ was bound to immobilized streptavidin and reacted with the amplified phage pools. The purpose of this step was to ensure that the pools contained phage that bound specifically to βγ subunits. Phage binding was detected with anti-phage antibody conjugated to horseradish peroxidase. From each of the selected pools of phage, individual clones were isolated, purified and tested for binding, and the DNA encoding the peptide was sequenced.
Phage preparation
Isolated colonies of E.coli infected with phage were picked from NZY-tet (Gibco-BRL) plates and grown overnight in 1.5 ml at 37°C. Phage were purified from the supernatant of the overnight cultures by precipitation with 20% PEG 8000 and heat treatment at 70°C. Phage concentrations were estimated by agarose gel electrophoresis of the purified phage by comparison with a standard curve generated with f88-4 phage that had been grown on a large scale, purified twice by PEG precipitation and quantified spectrophotometrically (Smith and Scott, 1993).
DNA sequencing
Single-stranded DNA was isolated from an aliquot of the purified phage preparation by standard methods (Maniatis et al., 1991). Sequencing reactions were performed according to the protocol supplied with Big Dye Sequencing Kit (Perkin-Elmer) using the primer CTGAGTTCATTAAGACG; reactions were analyzed by the automated DNA sequencing core facility at the University of Rochester.
Phage ELISA
b-βγ (100 ng) was immobilized in streptavidin-coated wells of a 96-well plate. Phage clones (1 × 1010) were incubated with immobilized βγ in TBS with 0.1% Tween-20 for 1–4 h. For competition ELISAs, peptides were added just before incubation with the phage. After washing to remove unbound phage, anti-M13 antibody linked to horseradish peroxidase was added for 1 h followed by a chromogenic dye. The extent of color reaction was monitored in a microplate reader. The absorbance values (A405) shown are a crude measure of the strength of phage binding to βγ.
PLC and PI3K assays
PLC assays were performed as described (Romoser et al., 1996) with specific details given in the figure legends. Briefly, purified PLC β2 was mixed with sonicated phospholipid vesicles containing 50 µM brain phosphatidylinositol 4,5-bisphosphate (PIP2), 200 µM liver phosphatidylethanolamine (PE) (Avanti Polar Lipids, Inc.) and [3H]inositol-PIP2 (New England Nuclear) (6000–8000 c.p.m./assay), with or without 100 nM purified β1γ2 and peptides at the indicated concentrations. Reactions were allowed to proceed for 10–30 min at 30°C in the presence of ∼100 nM free Ca2+. Intact lipids and proteins were precipitated with BSA and 10% trichloroacetic acid, and removed by centrifugation. Supernatant containing soluble [3H]inositol 1,4,5-trisphosphate was removed and analyzed by liquid scintillation counting.
PI3K assays were performed as described (Parish et al., 1995). Briefly, purified p101–p110γ heterodimer was mixed with sonicated phospholipid vesicles containing 300 µM liver phosphatidylinositol (Avanti Polar Lipids, Inc.) and 600 µM PE, with or without 100 nM purified β1γ2 and peptides at the indicated concentrations. Reactions were initiated by the addition of 10 µM ATP containing [γ-32P]ATP at 100 000 c.p.m./assay. Reactions were allowed to proceed for 30 min at 30°C. Lipids were extracted and analyzed by liquid scintillation counting.
Adenylate cyclase assays
Adenylyl cyclase activity was measured as described (Smigel, 1986). All assays were performed for 10 min at 30°C in a final volume of 100 µl containing 50 mM sodium HEPES pH 8.0, 500 µM ATP, 0.6 mM EDTA, 3 mM K2-phosphoenolpyruvate, 10 mM MgCl2, 500 µM 3-isobutyl-1-methylxanthine (IBMX), 0.1 µg/µl BSA, 1 µg/µl pyruvate kinase, 5 µg of membrane protein. Activated G protein α subunits were diluted into 50 mM sodium HEPES pH 8.0, 5 mM MgSO4, 1 mM EDTA, 1 mM DTT and mixed with membranes before the start of the assay.
Peptide synthesis
The alanine scan mutants of SIRK peptide were synthesized manually using Chiron multipin methodology (Geysen et al., 1984). The 16 peptides shown in Table V were synthesized simultaneously on Fmoc-Rink amide handle-crowns (the substitution level of each crown was 6.9 µmol). Each peptide was synthesized on an individual crown and in a separate well. Each impure peptide was purified by HPLC using a semi-preparative Zorbax C-8 column (9.4 × 250 mm, 5 µm) using a linear gradient from 5 to 70% acetonitrile in 0.05% trifluoroacetic acid over 50 min. The fraction corresponding to the required peptide was checked by matrix-assisted laser desorption/ionization-time of flight (MALDI-TOF) mass spectroscopy, concentrated and lyophilized.
Measurement of DAMGO inhibition of N-type Ca2+ channels
Electrophysiological measurements of dorsal root ganglion neurons were made as in Samoriski and Gross (2000).
Acknowledgments
Acknowledgements
The authors would like to thank Grigory Krapivinsky for the generous gift of K+ channel peptides and Karen Bresciano for technical assistance. This work was supported by grants from the National Institutes of Health #GM60286 to A.V.S., #GM53645 to R.T., and a Natural Sciences and Engineering Research Council of Canada grant to J.K.S.
REFERENCES
- Bonnycastle L.L.C., Mehroke,J.S., Rashed,M., Gong,X. and Scott,J.K. (1996) Probing the basis of antibody reactivity with a panel of constrained peptide libraries displayed by filamentous phage. J. Mol. Biol., 258, 747–762. [DOI] [PubMed] [Google Scholar]
- Chen J. et al. (1995) A region of adenylyl cyclase 2 critical for regulation by G protein βγ subunits. Science, 268, 1166–1169. [DOI] [PubMed] [Google Scholar]
- Clapham D.E. and Neer,E.J. (1997) G protein βγ subunits. Annu. Rev. Pharmacol. Toxicol., 37, 167–203. [DOI] [PubMed] [Google Scholar]
- Dingus J., Wilcox,M.D., Kohnken,R. and Hildebrandt,J.D. (1994) Synthesis and use of biotinylated βγ complexes prepared from bovine brain G proteins. Methods Enzymol., 237, 457–471. [DOI] [PubMed] [Google Scholar]
- Fairbrother W.J. et al. (1998) Novel peptides selected to bind vascular endothelial growth factor target the receptor-binding site. Biochemistry, 37, 17754–17764. [DOI] [PubMed] [Google Scholar]
- Gaudet R., Bohm,A. and Sigler,P.B. (1996) Crystal structure at 2.4 Å resolution of the complex of transducin βγ and its regulator, phosducin. Cell, 87, 577–588. [DOI] [PubMed] [Google Scholar]
- Geysen H.M., Meloen,R.H. and Barteling,S.J. (1984) Use of peptide synthesis to probe viral antigens for epitopes to a resolution of a single amino acid. Proc. Natl Acad. Sci. USA, 81, 3998–4002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gutkind J.S. (1998) The pathways connecting G protein coupled receptors to the nucleus through divergent mitogen-activated protein kinase cascades. J. Biol. Chem., 273, 1839–1842. [DOI] [PubMed] [Google Scholar]
- Hamm H.E. (1998) The many faces of G protein signaling. J. Biol. Chem., 273, 669–672. [DOI] [PubMed] [Google Scholar]
- Herlitze S., Garcia,D.E., Mackie,K., Hille,B., Scheuer,T. and Catterall,W.A. (1996) Modulation of Ca2+ channels by G-protein βγ subunits. Nature, 380, 258–262. [DOI] [PubMed] [Google Scholar]
- Herlitze S., Hockerman,G.H., Scheuer,T. and Catterall,W.A. (1997) Molecular determinants of inactivation and G protein modulation in the intracellular loop connecting domains I and II of the calcium channel α1A subunit. Proc. Natl Acad. Sci. USA, 94, 1512–1516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hildebrandt J.D. (1997) Role of subunit diversity in signaling by heterotrimeric G proteins. Biochem. Pharmacol., 54, 325–339. [DOI] [PubMed] [Google Scholar]
- Jiang H., Kuang,Y., Wu,Y., Xie,W., Simon,M.I. and Wu,D. (1997) Roles of phospholipase C β2 in chemoattractant-elicited responses. Proc. Natl Acad. Sci. USA, 94, 7971–7975. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koch W.J., Inglese,J., Stone,W.C. and Lefkowitz,R.J. (1993) The binding site for the βγ subunits of heterotrimeric G proteins on the β-adrenergic receptor kinase. J. Biol. Chem., 268, 8256–8260. [PubMed] [Google Scholar]
- Kohnken R.E. and Hildebrandt,J.D. (1989) G protein subunit interactions: studies with biotinylated G protein subunits. J. Biol. Chem., 264, 20688–20696. [PubMed] [Google Scholar]
- Kozasa T. and Gilman,A.G. (1995) Purification of recombinant G proteins from Sf9 cells by hexahistidine tagging of associated subunits. Characterization of α12 and inhibition of adenylyl cyclase by αz. J. Biol. Chem., 270, 1734–1741. [DOI] [PubMed] [Google Scholar]
- Krapivinsky G., Kennedy,M.E., Nemec,J., Medina,I., Krapivinsky,L. and Clapham,D.E. (1998) Gβ binding to GIRK4 subunit is critical for G protein-gated K+ channel activation. J. Biol. Chem., 273, 16946–16952. [DOI] [PubMed] [Google Scholar]
- Kuang Y., Wu,Y., Jiang,H. and Wu,D. (1996) Selective G protein coupling by C-C chemokine receptors. J. Biol. Chem., 271, 3975–3978. [DOI] [PubMed] [Google Scholar]
- Li Y., Sternweis,P.M., Charnecki,S., Smith,T.F., Gilman,A.G., Neer,E.J. and Kozasa,T. (1998) Sites for G-α binding on the G protein β subunit overlap with sites for regulation of phospholipase C β and adenylyl cyclase. J. Biol. Chem., 273, 16265–16272. [DOI] [PubMed] [Google Scholar]
- Littman D.R. (1998) Chemokine receptors: keys to AIDS pathogenesis? Cell, 93, 677–680. [DOI] [PubMed] [Google Scholar]
- Loew A., Ho,Y.K., Blundell,T. and Bax,B. (1998) Phosducin induces a structural change in transducin βγ. Structure, 6, 1007–1019. [DOI] [PubMed] [Google Scholar]
- Lowman H.B., Chen,Y.M., Skelton,N.J., Mortensen,D.L., Tomlinson,E.E., Sadick,M.D., Robinson,I.C. and Clark,R.G. (1998) Molecular mimics of insulin-like growth factor 1 (IGF-1) for inhibiting IGF-1: IGF-binding protein interactions. Biochemistry, 37, 8870–8878. [DOI] [PubMed] [Google Scholar]
- Maniatis T., Fritsch,E. and Sambrook,J. (1991) Molecular Cloning: A Laboratory Manual. Cold Spring Harbor Laboratory Press, Cold Spring Harbor, NY.
- Parish C.A., Smrcka,A.V. and Rando,R.R. (1995) Functional significance of βγ subunit carboxymethylation for the activation of phospholipase C and phosphoinositide 3-kinase. Biochemistry, 34, 7722–7727. [DOI] [PubMed] [Google Scholar]
- Qin N., Platano,D., Olcese,R., Stefani,E. and Birnbaumer,L. (1997) Direct interaction of G βγ with a C-terminal G βγ-binding domain of the Ca2+ channel α1 subunit is responsible for channel inhibition by G protein-coupled receptors. Proc. Natl Acad. Sci. USA, 94, 8866–8871. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Romoser V., Ball,R. and Smrcka,A.V. (1996) Phospholipase C β2 association with phospholipid interfaces assessed by fluorescence resonance energy transfer. G protein βγ subunit-mediated translocation is not required for enzyme activation. J. Biol. Chem., 271, 25071–25078. [DOI] [PubMed] [Google Scholar]
- Samoriski G.M. and Gross,R.A. (2000) Functional compartmentalization of opioid desensitization in primary sensory neurons. J. Pharmacol. Exp. Ther., 294, 500–509. [PubMed] [Google Scholar]
- Sankaran B., Osterhout,J., Wu,D. and Smrcka,A.V. (1998) Identification of a structural element in phospholipase C β2 that interacts with G protein βγ subunits. J. Biol. Chem., 273, 7148–7154. [DOI] [PubMed] [Google Scholar]
- Satpaev D.K. and Slepak,V.Z. (2000) Analysis of protein–protein interactions in phototransduction cascade using surface plasmon resonance. Methods Enzymol., 316, 20–40. [DOI] [PubMed] [Google Scholar]
- Smigel M.D. (1986) Purification of the catalyst of adenylate cyclase. J. Biol. Chem., 261, 1976–1982. [PubMed] [Google Scholar]
- Smith G.P. and Scott,J.K. (1993) Libraries of peptides and proteins displayed on filamentous phage. Methods Enzymol., 217, 228–257. [DOI] [PubMed] [Google Scholar]
- Sprang S.R. (1997) G protein mechanisms: insights from structural analysis. Annu. Rev. Biochem., 66, 639–678. [DOI] [PubMed] [Google Scholar]
- Stephens L.R. et al. (1997) The Gβγ sensitivity of a PI3K is dependent upon a tightly associated adaptor, p101. Cell, 89, 105–114. [DOI] [PubMed] [Google Scholar]
- Touhara K., Koch,W.J., Hawes,B.E. and Lefkowitz,R.J. (1995) Mutational analysis of the pleckstrin homology domain of the β-adrenergic receptor kinase: differential effects on G βγ and phosphatidylinositol 4,5-bisphosphate binding. J. Biol. Chem., 270, 17000–17005. [DOI] [PubMed] [Google Scholar]
- Wells J.A. and de Vos,A.M. (1996) Hematopoietic receptor complexes. Annu. Rev. Biochem., 65, 609–634. [DOI] [PubMed] [Google Scholar]
- Weng G.Z., Li,J.R., Dingus,J., Hildebrandt,J.D., Weinstein,H. and Iyengar,R. (1996) G-β subunit interacts with a peptide encoding region 956–982 of adenylyl cyclase 2: cross-linking of the peptide to free G βγ but not the heterotrimer. J. Biol. Chem., 271, 26445–26448. [DOI] [PubMed] [Google Scholar]
- Wiley J.W., Moises,H.C., Gross,R.A. and Macdonald,R.L. (1997) Dynorphin A-mediated reduction in multiple calcium currents involves a G(o) α-subtype G protein in rat primary afferent neurons. J. Neurophysiol., 77, 1338–1348. [DOI] [PubMed] [Google Scholar]
- Wrighton N.C. et al. (1996) Small peptides as potent mimetics of the protein hormone erythropoietin. Science, 273, 458–464. [DOI] [PubMed] [Google Scholar]
- Xie W. et al. (1999) Genetic alteration of phospholipase Cβ3 expression modulates behavioral and cellular responses to µ opioids. Proc. Natl Acad. Sci. USA, 96, 10385–10390. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yoshikawa D.M., Hatwar,M. and Smrcka,A.V. (2000) G protein β5 subunit interactions with α subunits and effectors. Biochemistry, 39, 11340–11347. [DOI] [PubMed] [Google Scholar]