

Abstract
Dihydropyrimidine dehydrogenase catalyzes the first step in pyrimidine degradation: the NADPH-dependent reduction of uracil and thymine to the corresponding 5,6-dihydropyrimidines. Its controlled inhibition has become an adjunct target for cancer therapy, since the enzyme is also responsible for the rapid breakdown of the chemotherapeutic drug 5-fluorouracil. The crystal structure of the homodimeric pig liver enzyme (2× 111 kDa) determined at 1.9 Å resolution reveals a highly modular subunit organization, consisting of five domains with different folds. Dihydropyrimidine dehydrogenase contains two FAD, two FMN and eight [4Fe–4S] clusters, arranged in two electron transfer chains that pass the dimer interface twice. Two of the Fe–S clusters show a hitherto unobserved coordination involving a glutamine residue. The ternary complex of an inactive mutant of the enzyme with bound NADPH and 5-fluorouracil reveals the architecture of the substrate-binding sites and residues responsible for recognition and binding of the drug.
Keywords: electron transfer/flavin/iron–sulfur clusters/protein crystallography/pyrimidine metabolism
Introduction
5-fluorouracil (5FU) has been used for the treatment of colorectal, breast and head/neck cancer for >40 years and is the third most commonly prescribed chemotherapeutic agent. This anti-neoplastic compound blocks DNA synthesis by inhibiting thymidylate synthase (Papamichael, 2000). However, >80% of the administered dose is rapidly degraded by the first and rate-limiting enzyme of the pyrimidine catabolic pathway: dihydropyrimidine dehydrogenase (DPD; EC 1.3.1.2.) (Heggie et al., 1987; Milano and Etienne, 1994). The degradation of 5FU by this pathway reduces its efficiency as a drug and requires the application of extremely high doses. In addition, the breakdown of 5FU leads to fluorinated products causing multiple side effects in the nervous system (Hull et al., 1988). Inhibition of the catabolic pyrimidine pathway could thus increase the clinical potential of 5FU and related compounds by increasing their efficacy (Ahmed et al., 1999; Brito et al., 1999), resulting in the application of lower doses and diminished drug side effects. DPD has therefore emerged as an important target for the improvement of cancer therapy and at least one DPD inhibitor (Porter et al., 1992) has reached phase II/III clinical trials (Mani et al., 2000).
The cytosolic enzyme, present in a variety of tissues with main activity found in liver (Ho et al., 1986), reduces the pyrimidines uracil and thymine in an NADPH-dependent reaction to the corresponding 5,6-dihydropyrimidines. After ring cleavage, ammonium ions and CO2 are formed by the action of the other two enzymes of the pathway, dihydropyrimidinase and β-alanine synthase, leaving as product β-alanine and β-aminoisobutyrate, respectively (Wasternack, 1980).
DPD has been purified from human, pig, rat and bovine sources (Shiotani and Weber, 1981; Podschun et al., 1989; Lu et al., 1992; Albin et al., 1996). The enzyme is a homodimer of 2× 111 kDa and contains a large number of different redox cofactors. Each subunit of 1025 amino acids carries one FAD, one FMN, 16 nonheme-iron and 16 acid-labile sulfur atoms (Rosenbaum et al., 1998). Analysis of the amino acid sequence led to the conclusion that the enzyme most likely contains four [4Fe–4S] clusters per subunit, two in an N-terminal Cys-rich region and two in a stretch of 50 amino acids at the C-terminus with sequence similarity to bacterial eight-iron ferredoxins (Yokota et al., 1994; Hagen et al., 2000).
The non-classical two-site ping-pong kinetic mechanism of the pig liver enzyme suggests two separate binding sites for the substrates NADPH/NADP+ and pyrimidine/5,6-dihydropyrimidine, respectively (Podschun et al., 1990). NADPH reduces FAD at site 1 and electrons are, most likely via Fe–S clusters, transferred to FMN in site 2 for reduction of uracil or thymine. It is, however, still unresolved as to whether or not all four Fe–S clusters participate in electron transfer (Hagen et al., 2000).
Here we report the crystal structures of recombinant pig liver DPD and a ternary complex of the enzyme with NADPH and 5FU. The structures reveal the modular organization of the DPD monomer, extended electron transfer pathways involving all prosthetic groups, as well as a hitherto unobserved coordination of a [4Fe–4S] cluster. The ternary complex shows the architecture of the substrate-binding site and residues responsible for binding of the drug. The three-dimensional (3D) structure also provides insights into the structural basis of DPD deficiencies caused by point mutations in the human DPD gene, which increase 5FU toxicity in certain cancer patients.
Results and discussion
Structure determination and overall structure
The structure of DPD was determined by multiple wavelength anomalous diffraction (MAD) in combination with single isomorphous replacement (SIR), and refined to 1.9 Å resolution. The final model of the native enzyme includes 4064 amino acids (residues 2–1017 for each chain), four FAD, four FMN, 16 [4Fe–4S] clusters and 3804 water molecules. Crystals of a ternary complex of DPD with its substrates were obtained by co-crystallization of the catalytically inactive mutant C671A with NADPH and 5FU. These crystals are isomorphous to the native crystals and the final model, refined to 2.0 Å resolution, contains the same number of prosthetic groups and 4076 amino acids (residues 2–1020 for each chain), as well as four molecules of the inhibitor 5FU, four NADPH and 4714 water molecules.
A view of the overall structure of the DPD monomer is shown in Figure 1A. The 1025 residues in the DPD monomer form five domains, different in size and folding pattern. The first four domains, comprising residues 27–847, build an elongated core with dimensions of ∼110 × 90 × 60 Å, whereas the fifth domain (residues 2–26 and 848–1020) is separated from this core by extended loop regions.

Fig. 1. Structure of pig liver DPD. (A) Schematic view of the subunit of DPD with the domains in different colors. The cofactors are shown as ball-and-stick models, iron ions in magenta and sulfur atoms in green. (B) The DPD dimer. The color codes for the domains of the first subunit are the same as in (A), the corresponding domains in the second subunit are shown in light green, brown, cyan, pink and light blue.
Domain structure and binding of prosthetic groups
Domain I contains an unusual Fe–S cluster. Domain I (residues 27–173) contains two of the Fe–S clusters and has exclusively α-helical secondary structure (Figures 2A and 3). A somewhat similar folding pattern has been observed for the second domain of the iron protein of Escherichia coli fumarate reductase, FR-IP, which also contains Fe–S clusters (Iverson et al., 1999). The sequence identity between the two enzymes is, however, negligible. Of the 52 matching residues [root mean square deviation (r.m.s.d.) 1.48 Å] only eight are identical, of which six are cysteine residues participating in the Fe–S cluster coordination (Figure 3). Four of the α-helices in the DPD domain have no equivalents in FR-IP, and Iα5 is positioned closer to the domain center than the corresponding FR-IP helix (Figure 2A). Furthermore, there are two [4Fe–4S] clusters bound instead of one [4Fe–4S] and one [3Fe–4S] as found in FR-IP. Cysteine residues 79, 82, 87 and 140 coordinate the first Fe–S cluster (nFeS1) in DPD. FR-IP contains cysteines at structurally equivalent positions, however with different spacing: (Cys-X2-Cys-X4-Cys + distal Cys) for DPD and (Cys-X2-Cys-X2-Cys + distal Cys) for FR-IP.

Fig. 2. Structure of the domains in DPD. (A) Domain I (green). Superimposed is the second domain of the E.coli fumarate reductase iron-protein (FR-IP, gray). The iron and sulfur atoms in DPD are shown in magenta and green, those of FR-IP in medium and dark gray. (B) Domains II (yellow) and III (orange). Superimposed is the structure of the Bos taurus adrenodoxin reductase (gray). For DPD, FAD and NADPH are shown in blue and cyan, those bound to adrenodoxin reductase in medium and dark gray, respectively. (C) Domain IV (red) superimposed on the structure of L.lactis dihydroorotate dehydrogenase (gray). Except for the barrel components, all secondary structure elements are labeled. For DPD, the cofactor FMN is shown in blue and the anti-cancer drug 5FU in cyan. FMN bound to DHOD(A) is colored dark-gray, the reaction product orotate is shown in medium gray. (D) Domain V (blue), superimposed on domain 5 of Desulfovibrio africanus pyruvate:ferredoxin oxidoreductase (gray). The iron and sulfur atoms in DPD are shown in magenta and green, those of pyruvate:ferredoxin oxidoreductase in medium and dark gray.

Fig. 3. Amino acid sequence of pig liver DPD. The secondary structure elements are indicated by arrows for β-strands and cylinders for α- and 310(γ)-helices, respectively. The colour code is as in Figure 1, i.e. green for domain I, yellow for domain II, orange for domain III, red for domain IV and blue for domain V. Underlined letters indicate structural similarities between domain I and the second domain of E.coli FR-IP, domains II and III and B.taurus adrenodoxin reductase, domain IV and L.lactis DHOD(A), and domain V and domain 5 of D.africanus pyruvate:ferredoxin oxidoreductase, respectively. Bold letters indicate sequence and structure conservation between DPD and these enzymes, lower case letters indicate differences between pig liver and human DPD.
The second cluster, nFeS2, shows a coordination that has not been observed in other Fe–S proteins so far (Figure 4). Three irons interact with cysteine residues 91, 130 and 136, whereas the Oε1 atom of Q156 coordinates the fourth iron. Due to the smaller atom radius of oxygen compared with sulfur, the Fe–Oε1 distance is significantly shorter (by 1.98 Å) than the average 2.30 Å found for the Fe–Sγ interactions in DPD. Whereas two of the ligands, C91 and C136, are conserved in FR-IP, C130 and Q156 are replaced by valine and isoleucine, respectively. Compared with the metal cluster in FR-IP, the DPD cluster is tilted around the bond between the iron ions interacting with C91 and C136 towards Q156, thus positioning this side chain within ligand distance to another iron ion. The cysteine residue binding to the corresponding iron in FR-IP has a counterpart in the sequence of DPD, the previously suggested fourth cluster ligand C126. The side chain of C126 is, however, 5.30 Å away from the closest iron ion in the cluster (binding to C130) and 6.50 Å away from that coordinated by Q156, and therefore does not act as a ligand in the Fe–S cluster.

Fig. 4. Unusual coordination of an Fe–S cluster in DPD. (A) Stereoview of the electron density map contoured around the Fe–S cluster with Q156 as iron ligand. Sulfur atoms are green, iron atoms magenta. (B) Sequence comparison of DPD domain I and the corresponding sequence of Amazona brasiliensis glutamate synthase β-subunit (numbering of the amino acids is according to the SwissProt database). Cluster coordinating residues are shown in magenta, the previously suggested cluster ligand C126 in green. Conserved residues are indicated in bold and asterisks denote the residues coordinating the iron ions of the first Fe–S cluster and # the residues coordinating the second Fe–S cluster.
Sequence comparisons revealed significant similarities between the DPD domains I, II and III, and the small (β) subunit of glutamate synthase (27% identity), suggesting a similar fold. The cysteine ligands and most of the residues that form a hydrophobic shell around the metal clusters in domain I of DPD are conserved or exchanged to residues with similar properties (Figure 4B). Glutamate synthase, however, has a glutamate residue at the position corresponding to Q156 in DPD.
Domains II and III bind FAD and NADPH. The FAD- and NADPH-binding domains II (residues 173–286, 442–524) and III (residues 287–441) together show highest structural similarity to adrenodoxin reductase (r.m.s.d. 1.65 Å, 247 equivalent Cα atoms out of 351) (Ziegler and Schulz, 2000). Therefore, the architecture of these DPD domains is related to the disulfide oxidoreductase family of flavoproteins. Both domains contain a central parallel β-sheet surrounded by α-helices, forming Rossman-type nucleotide-binding motifs that bind FAD and NADPH, respectively (Figure 2B).
Domain III is inserted between strand IIβ4 and helix IIα7 of domain II, and contains an additional three-stranded antiparallel β-sheet. With the exception of this additional sheet, the topologies of domains II and III resemble each other and might originate from gene duplication, as was proposed for the disulfide oxidoreductases (Schulz, 1980). The cofactor FAD is very well defined in the electron density map and its elongated configuration is virtually identical to that in adrenodoxin reductase. The adenosine moiety of FAD is deeply buried in domain II. Three residues interact directly with this part of the cofactor molecule (Figure 5A): the carboxyl group of E218 binds tightly to the ribose hydroxyl groups, and K219 and L261 interact via main chain atoms with the adenine atoms N6, N1 and N3. The diphosphate moiety binds to the main chain amides of L226, A198 and D481, as well as to four well defined water molecules, present in all four monomers in the asymmetric unit. D481 is also involved, through its side chain, in the coordination of one of the ribityl hydroxyl groups, whereas the other two are bridged by a water molecule. The isoalloxazine ring system of FAD points towards the NADPH-binding domain III, with the re-face exposed to solvent. Its O4 atom interacts with the side chain of R235 and two water molecules, from which one is also within hydrogen bonding distance of the N5 atom. The atoms O2 and N3 are stabilized by hydrogen bonds to a water molecule, the main chain amide of T489 and the main chain carboxyl oxygen of V129, respectively (Figure 5A).

Fig. 5. Ligand-binding sites. (A) Stereoview of the binding site for FAD and NADPH. The carbon atoms of the cofactor FAD are shown in gray, those of the cosubstrate NADPH in cyan. Side chains that are conserved between pig liver DPD and B.taurus adrenodoxin reductase are marked with an asterisk. (B) Stereoview of the substrate-binding site shows 5FU bound adjacent to the cofactor FMN in the C671A mutant. A 2|Fo| – |Fc| map is contoured at 1.2σ for 5FU. Side chains that are conserved between pig liver DPD and L.lactis DHOD(A) are marked with an asterisk.
DPD and adrenodoxin reductase lack the disulfide bonds characteristic for the other enzymes belonging to the disulfide oxidoreductase fold family, and the structures of DPD domains II and III were compared with those of glutathione reductase (Karplus and Schulz, 1989) and thioredoxin reductase (Lennon et al., 2000). Both enzymes use active-site dithiol–disulfide to transfer reducing equivalents from FAD to external substrates, but carry the active-site disulfide loop in different domains, i.e. in the NADPH-binding domain for thioredoxin reductase and in the FAD-binding domain for glutathione reductase, respectively. The corresponding DPD domains not only lack the cysteine residues at corresponding positions, they also show no structural similarities in their close neighborhood.
Domain IV binds FMN. Domain IV of DPD belongs to the glycolate oxidase family of flavoproteins, with the typical (α/β)8 barrel fold (Figures 2C and 3). This DPD domain closely resembles (r.m.s.d. 1.58 Å, 232 equivalent Cα atoms out of 322) the 3D structure of Lactococcus lactis dihydroorotate dehydrogenase class A, DHOD(A) (Rowland et al., 1998), an enzyme catalyzing a similar reaction, the oxidation of 5,6-dihydroorotate to orotate. The barrels forming β-strands and α-helices are termed IVβ1–IVβ8 and IVα1–IVα8, respectively. Two antiparallel β-strands (IVβa and IVβb) form the base of the barrel, preceded by the short 310-helix IVγa. Except for IVγf, another 310-helix at the C-terminus of domain IV, all other additional secondary structure elements are inserted at the top of the barrel, including a four-stranded antiparallel β-sheet (IVβc–βf). Two of these insertions contain cis-peptide bonds. The first is formed by S587 and P588 in loop IVγc–βc. Here domain IV interacts with domain V of the other subunit, in close proximity to cFeS2. The other cis-peptide bond between W751 and P752 is located in the center of the loop IVβe–βf. Both prolines involved in the formation of the cis-peptide bonds pack against each other. Domain IV contains a third non-proline cis-peptide bond between A734 and T735 at the end of the sixth barrel strand. With the exception of IVγa, IVγc and IVγf, all the secondary structure elements as well as two of the cis-peptide bonds (S587–P588 and A734–T735) are also found in DHOD(A).
FMN is bound at the C-terminal end of the barrel-forming strands, with the re-face of the isoalloxazine ring oriented towards the barrel. Most residues interacting with the cofactor via side chain atoms are conserved between DPD and DHOD(A) (Figure 5B), including two lysine residues binding to the isoalloxazine ring (K574 and K709). The positively charged lysine residue (K574) interacts with the redox active N5 atom of the FMN ring. However, the hydrogen-bonding pattern of K574 is slightly different from that of the corresponding K43 in DHOD(A). Both are involved in two bifurcated hydrogen bonds. The first to the isoalloxazine ring atoms N5 and O4 is found in both enzymes, but instead of two additional hydrogen bonds to water molecules, the Nε of K574 of DPD also interacts with the carboxyl group of E611 and the main chain oxygen of L612.
It is noteworthy that there are more active-site residues conserved between DPD and DHOD(A) from L.lactis than between all three families of DHODs. This observation suggests close evolutionary relationship and significant similarity in the catalytic mechanism of the two enzymes.
Domain V contains the second set of Fe–S clusters. Domain V comprises residues 1–26 and 848–1025 and contains two [4Fe–4S] clusters, coordinated by residues C953, 956, 959 and 996 for the first (cFeS1) and C963, 986, 989 and 992 for the second (cFeS2) metal cluster. The spacing in the sequence between the metal-binding cysteines as well as the fold of the domain core, with two α-helices on one side and a four-stranded antiparallel β-sheet on the other side of the clusters, resembles that of bacterial eight-iron ferredoxins (Bruschi and Guerlesquin, 1988). Pyruvate:ferredoxin oxidoreductase, i.e. its Fe–S cluster-binding domain 5, shows the highest structural similarity to the core of DPD domain V (r.m.s.d. 1.10 Å, 57 equivalent Cα atoms out of 116 in domain 5 of pyruvate:ferredoxin oxidoreductase) (Chabrière et al., 1999) (Figure 2D). All additional helices and β-strands in DPD have only few contacts to each other, to the domain core or to other domains in the subunit. The loop between helices Vα2 and Vα3 (residues 899–919) contains six prolines in a stretch of 14 amino acids and is located on the protein surface. This loop also contains the protease-labile site, which has been reported for DPD purified from pig liver (Podschun et al., 1989).
The two Fe–S clusters of domain V have no close contacts to the other redox cofactors in the subunit. The arrangement of the cofactors in the subunit of DPD thus results in a gap of ∼24 Å between the N-terminal Fe–S clusters and the FMN in the second binding site (Figure 1A).
Dimer assembly and electron transfer pathway
The two subunits in the DPD dimer are related by a non-crystallographic 2-fold axis (Figure 1B). With the exception of a few flexible surface loops, there are essentially no differences between the four molecules in the asymmetric unit, i.e. between or within the dimers.
The buried dimer interface is large, ∼10 800 Å2 per subunit, and covers 23.2% of the monomer surface area. Monomer–monomer interactions are mainly hydrophobic (62%), but also include >100 hydrogen bonds. Residues from all five domains participate in the formation of the interface. Domain I plays a central role in dimer formation, since it contacts all five domains of the opposite subunit, although interactions with domains III and IV are limited in number and exclusively hydrophobic.
In the dimer, the core of domain V packs more tightly to domains I and IV of the other subunit than to domains belonging to the same subunit. This has important consequences for the electron transfer pathway connecting the binding sites for NADPH and pyrimidine. The gap observed in the isolated subunit between the N-terminal Fe–S clusters and FMN is now closed by the two Fe–S clusters from domain V, but from the second subunit (Figures 1B and 6). As a result, the 12 redox cofactors of the DPD dimer are organized into two electron transfer chains passing the dimer interface twice. In these pathways, all redox partners are separated by ∼7.5–10.0 Å distances between their closest atoms, as commonly observed in multicenter electron transfer chains (Page et al., 1999). From this cofactor arrangement it can be concluded that DPD is active only as a dimer and that all four Fe–S clusters are participating in the transfer of electrons.

Fig. 6. Electron transfer pathways in DPD. Distances between closest atoms of the cofactors are indicated. The nicotinamide ring of NADPH is shown at its assumed position during electron transfer.
Substrate binding
Electrons are provided to the enzyme in the form of NADPH. The ternary complex of an inactive DPD mutant [with the catalytically crucial C671 (Rosenbaum et al., 1998) exchanged to alanine], NADPH and the anticancer drug 5FU provides a picture of the complete electron transfer chain. NADPH is bound to domain III, with the nicotinamide ring pointing towards the FAD isoalloxazine ring system (Figure 5A). Whereas the electron density for the adenine ribose-diphosphate moiety of NADPH is well defined, it is rather poor for the nicotinamide and the connected ribose, which prevents precise determination of the interaction of the nicotinamide ring with FAD and enzyme residues. There are some indications that in this inactive form, the nicotinamide ring is not placed parallel to the FAD isoalloxazine moiety, as required for electron transfer, but rather is flipped away from the active site onto the surface of the protein.
One of the hydroxyl groups of the connected ribose interacts via hydrogen bonds with the carboxyl group of D481 and a well defined water molecule. There are five additional water molecules participating in the binding of the co-substrate: three interact with the diphosphate, one forms a hydrogen bond to the N3 atom of the adenine, and the last water molecule coordinates the 2′-phosphate group. The positive charges of R364 and K365 neutralize the negative charge of this phosphate group, whereas the diphosphate moiety is stabilized by the interaction with R371 and the dipole field of helix IIIα3. One side of the adenine ring packs with the side chain of F438, the other side interacts with the guanidinium group of R364.
5FU is bound above the si-face of the FMN-isoalloxazine ring system (Figure 5B), almost parallel to the pyrimidine ring component. The O2 and O4 atoms of 5FU are positioned ∼3.8 Å away from the N10 and O2 atoms of FAD, respectively. The distance between the reactive centers of the C6 atom of 5FU and the N5 atom of FAD is also 3.8 Å. 5FU interacts via hydrogen bonds with three asparagine side chains (N609, N668, N736) and the side chains of T737 and S670. All three asparagines and the serine have counterparts in the active site of DHOD(A); the threonine is replaced by a serine. The fluor substituent of the drug is not involved in any direct contacts to the protein, but points into a small pocket of the substrate-binding site, surrounded by the side chains of M642, I613, T575 and N668 at >4 Å distance. The existence of this pocket explains why the enzyme can tolerate other, bulkier substituents at the 5-position of the pyrimidine ring (Naguib et al., 1987).
The active-site loop (IVβ4–IVγd) is in an open conformation, as seen in the unliganded enzyme, with the top of the loop (residues 675–679) being disordered. Therefore, the substrate-binding site is exposed to solvent (Figure 7) and the catalytically crucial residue, C671 (Rosenbaum et al., 1998), is positioned too far away to make contacts with the substrate (Figure 5B). This is different from the situation in DHOD(A), where binding of substrate induces a conformational change of this loop, which folds over and closes off the active site (Rowland et al., 1998) (Figure 2C). The reason for this difference is not yet clear, but the failure to trigger a putative conformational change as seen in DHOD(A) could be caused by the mutation of cysteine to alanine. Since the active-site loop contains a histidine residue, which most likely participates in substrate binding, the maintenance of the open loop conformation could also be due to the pH of crystallization (4.7), which is different from the pH optimum for catalytic activity (7.3).
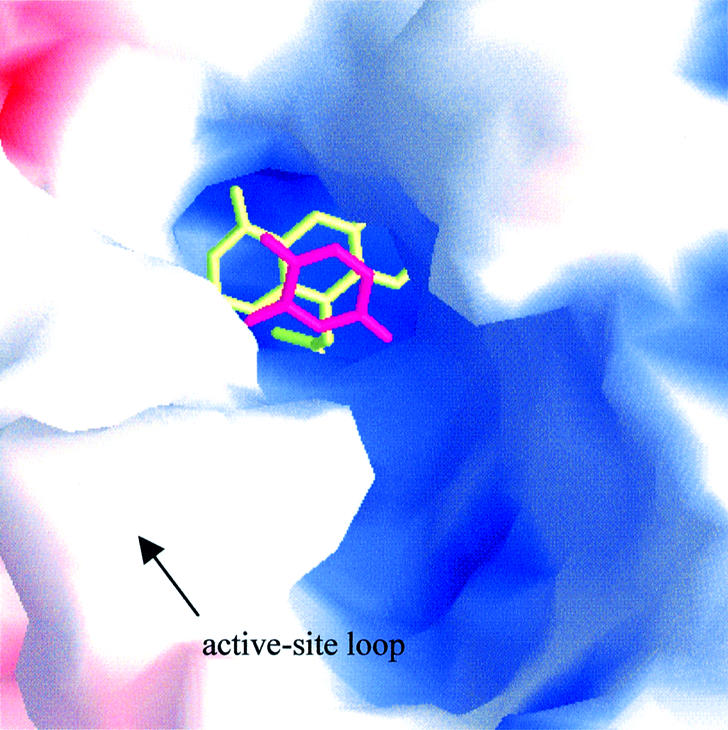
Fig. 7. Solvent accessibility of the pyrimidine-binding site. Molecular surface representation of the active site with bound FMN (yellow) and 5FU (magenta) as stick models. The surface is colored according to the electrostatic potential, as calculated with GRASP (Nicholls et al., 1991).
Naturally occurring point mutations and DPD deficiency
The amino acid sequences of pig and human DPD are 93% identical (Yokota et al., 1994). More than half of the total 74 amino acid exchanges are conservative and the majority of the non-conservative exchanges are located at solvent-exposed surface regions. All amino acids participating directly in cofactor or substrate binding are invariant for the two enzymes. Thus, the 3D structure of pig DPD will be a useful tool for structure-assisted design of new inhibitory compounds, which could be developed into drugs.
Several naturally occurring mutations in the gene coding for DPD in humans that result in decreased or abolished DPD activity and high risk for 5FU toxicity have been reported (Ridge et al., 1997; Kouwaki et al., 1998; Van Kuilenburg et al., 1999). Among these are point mutants R235W, V335L, D974V and V995F, which result in complete loss of enzyme activity. Mutations R235W and V995F interfere directly with cofactor and/or substrate binding. The guanidinium group of arginine 235 forms a hydrogen bond to the O4 atom of FAD, and the presence of a tryptophan side chain most likely weakens or prevents the binding of the isoalloxazine ring of the cofactor. The insertion of the larger phenylalanine side chain at position 995, located in direct neighborhood to C996, a ligand of one of the Fe–S clusters, has the same effect for the binding/assembly of the cluster. The V335L mutation, although conservative, disturbs packing interactions in the hydrophobic core formed by the β-sheet on one side and the helix IIIα3 on the other side of the Rossman-motif in domain III, thereby affecting NADPH binding. The effect of D974V is structural; the aspartic side chain is the N-cap residue at the entrance of a short α-helical turn.
Conclusions
The structure of the complex enzyme DPD, in the unliganded state as well as in complex with the anti-cancer drug 5FU and NADPH, has been determined by X-ray crystallography. It reveals a highly modular subunit organization. The single domains differ in size, folding pattern, nature of bound cofactor and therefore function, but show significant similarity to other proteins of known 3D structure. The assembly of the prosthetic groups to electron transfer chains connecting the two substrate-binding sites of a subunit requires the formation of the dimeric state, since each electron transfer chain involves Fe–S clusters from different subunits and passes the dimer interface twice. Furthermore, one of the metal clusters in each chain shows a hitherto unobserved coordination of one cluster-iron by a glutamine residue. In the ternary complex, electron density for NADPH has been observed close to the FAD in substrate-binding site 1. The second binding site contains one molecule of 5FU, and residues important for substrate binding could be identified. The 3D structure allows conclusions about the structural basis of DPD deficiencies seen in cancer patients, caused by point mutations in the human DPD gene. It also provides a framework for structure-assisted design of new anti-cancer drugs.
Materials and methods
Expression, crystallization and data collection
Pig liver DPD was produced as recombinant protein in E.coli (Rosenbaum et al., 1997). Details of purification and crystallization of the protein have been described previously (Dobritzsch et al., 2001). In brief, the protein was purified using an ion-exchange and a 2′5′-ADP Sepharose-affinity column. Crystals were grown aerobically by vapor diffusion in hanging drops over a reservoir solution of 19–22% (w/v) PEG 6000, 100 mM sodium citrate pH 4.7 at 20°C. A heavy atom derivative was prepared by soaking the native DPD crystals in mother liquor containing 5 mM K2IrCl6 for 36 h. A ternary complex of DPD was obtained by crystallization of the mutant C671A in the presence of 1 mM NADPH and 5 mM 5FU using identical conditions to those for the unliganded enzyme.
Before data collection, the crystals were transferred for 5 min in mother liquor containing 20% (v/v) glycerol and then flash-frozen in a nitrogen gas stream. X-ray diffraction data were collected at 100K at beamline 711 of MAX II laboratory (Lund, Sweden) with a MARResearch Imaging Plate. MAD data were collected at 100K at beamline BM14 of the European Synchrotron Radiation Facility (ESRF, Grenoble, France) with a MAR CCD detector. Data for MAD phasing were collected from a single crystal at three different wavelengths: λ1 = 1.7392 Å and λ2 = 1.7416 Å (to 2.9 Å resolution) near the Fe K-edge to maximize the anomalous f″ and the dispersive f′ signal, respectively, and λ3 = 0.9184 Å as a remote data set (1.9 Å resolution). All data sets were processed with DENZO and scaled with SCALEPACK (Otwinowski and Minor, 1997) or the CCP4 suite of programs (CCP4, 1994). Table I gives details of the data collection statistics. The crystals belong to space group P21 with cell dimensions of a = 82.0 Å, b = 159.3 Å, c = 163.6 Å, β = 96.1°, and contain two dimers per asymmetric unit.
Table I. Data collection and refinement statistics.
| Native MAD |
K2IrCl6 | Ternary complex | |||
|---|---|---|---|---|---|
| λ1 peak | λ2 inflection | λ3 remote | |||
| Wavelength (Å) | 1.7392 | 1.7416 | 0.9184 | 1.0180 | 0.9746 |
| X-ray source | BM14 ESRF | BM14 ESRF | BM14 ESRF | 711 Lund | 711 Lund |
| Resolution (Å) | 30.0–3.00 (3.05–3.00) | 30.0–3.00 (3.05–3.00) | 30.0–1.90 (1.97–1.90) | 35.0–3.0 (3.07–3.0) | 30.0–2.0 (2.1–2.0) |
| Mosaicity (°) | 0.16 | 0.15 | 0.17 | 1.21 | 0.41 |
| Rsym (%) | 4.6 (6.2) | 7.0 (7.5) | 6.6 (24.0) | 9.9 (20.9) | 6.3 (16.6) |
| I/σ(I) | 15.5 (11.9) | 9.3 (8.4) | 10.9 (4.0) | 8.1 (3.2) | 10.4 (4.4) |
| Completeness (%) | 96.0 (87.6) | 96.6 (90.8) | 97.4 (91.2) | 61.9 (32.6) | 99.0 (95.8) |
| Reflections (no.) | |||||
| overall | 387 678 | 336 664 | 2 358 836 | 805 143 | 1 471 519 |
| unique | 82 848 | 82 552 | 326 433 | 51 420 | 273 662 |
| Phasing power (iso/ano) | 4.06/1.30 | 4.13/1.08 | 0/1.00 | 1.04 | – |
| Rcullis (iso/ano) | 0.45/0.86 | 0.45/0.93 | 0/0.94 | 0.852 | – |
| Refinement statistics | ||
|---|---|---|
| Native | Ternary complex | |
| Reflections (no.) | ||
| working set | 317 244 | 268 205 |
| test set | 6435 | 5418 |
| Resolution (Å) | 25–1.90 | 25–2.0 |
| Number of atoms | 35 302 | 36 466 |
| R refinement (%) | 18.0 | 17.3 |
| Rfree (%) | 20.1 | 19.4 |
| B factor (Å2) protein atoms | 17.3 | 20.0 |
| B factor (Å2) cofactor/substrate atoms | 11.1 | 20.3 |
| B factor (Å2) water molecules | 27.7 | 32.1 |
| R.m.s.d. bond distance (Å) | 0.009 | 0.009 |
| R.m.s.d. bond angle (°) | 1.47 | 1.45 |
| Ramachandran plot, residues in | ||
| most favorable regions (%) | 89.4 | 89.0 |
| additional allowed regions (%) | 10.3 | 10.5 |
| disallowed regions (%) | 0.0 | 0.0 |
Structure determination and refinement
The structure was solved by MAD in combination with SIR. The parameters of the four iridium sites, identified using the program SOLVE (Terwilliger and Berendzen, 1999), were refined with the program SHARP (de la Fortelle and Bricogne, 1997). These initial phases were used to calculate a difference map to 5 Å resolution between the MAD data sets collected at the remote and the inflection point wavelength, which clearly showed the positions of 16 Fe–S clusters per asymmetric unit. Cluster positions and occupancies, as well as values for f′ and f″ for two wavelengths, were refined in SHARP. The refined positions were used for determination of the non-crystallographic symmetry (NCS) with FINDNCS (Lu, 1999). Density modification, NCS averaging and phase extension to 3 Å in the program DM (Cowtan and Main, 1996) resulted in a well defined electron density map used for initial tracing of the polypeptide chain. The protein model was built with the graphics program O (Jones et al., 1991) and refined to 1.9 Å resolution with CNS (Brünger et al., 1998), using the experimental phases to 3 Å and tight NCS restraints.
Positional and temperature factor refinement were repeated with manual adjustments in between refinement cycles. After the Rfree dropped below 32%, the NCS restraints were loosened for side chain atoms, and completely removed for all atoms of residues 48–54, 173–175, 320–328, 412–418, 673–681 and 899–910. All these stretches of amino acids belong to loops located on the protein surface, which show high mobility and/or different conformations between the molecules in the asymmetric unit. The values for Rcryst and Rfree after refinement are 18.0 and 20.1%, respectively, with the test set used for Rfree calculation containing 2% of the reflections (Table I). The final model shows good stereochemistry as defined in PROCHECK (Laskowski et al., 1993), with no residues in the disallowed regions of the Ramachandran plot. For the native enzyme, the model contains residues 2–1017 for each chain, four FAD, four FMN, 16 [4Fe–4S] clusters and 3804 water molecules.
Initial rigid body refinement in CNS for the X-ray data of the ternary complex, using the refined structure of the native enzyme but without waters as model, resulted in values of 26.6% for Rcryst and Rfree, respectively. A |Fo| – |Fc| map computed from this model showed clear electron density for 5FU and most of the NADPH molecules, located in binding sites 2 and 1, respectively. Substrate and water molecules were added to the model. Minor manual adjustments using the program O and following refinement with CNS resulted in Rcryst = 17.3% and Rfree = 19.4% (Table I). During the refinement, the occupancies for the nicotinamide ring atoms of the NADPH molecules were set to 0. The final model contains residues 2–1020 for each chain, four FAD, four FMN, 16 [4Fe–4S] clusters, four 5FU, four NADPH and 4714 water molecules.
Structural comparisons between DPD and fumarate reductase (Protein Data Bank code 1FUM), adrenodoxin reductase (1E1L), dihydroorotate dehydrogenase (2DOR), pyruvate:ferredoxin oxidoreductase (1B0P), glutathione reductase (1GRA) and thioredoxin reductase (1F6M), respectively, were accomplished using the program TOP (Lu, 1996) with default parameters.
Figure 7 was drawn using the program GRASP (Nicholls et al., 1991), all other figures were generated using BOBSCRIPT (Kraulis, 1991; Esnouf, 1997) and RASTER3D (Merritt and Bacon, 1997).
The crystallographic data have been deposited in the Protein Data Bank, with accession codes 1h7w for the native structure and 1h7X for the ternary complex.
Acknowledgments
Acknowledgements
We acknowledge access to synchrotron radiation at the ESRF, France and MAX laboratory, Sweden. We are grateful to Vivian Stojanoff and Tanja Sandalova for assistance with data collection. D.D. gratefully acknowledges a fellowship from the Wenner-Gren Foundations. This work was supported by grants from the Swedish Cancer Foundation.
REFERENCES
- Ahmed F.Y. et al. (1999) Eniluracil treatment completely inactivates dihydropyrimidine dehydrogenase in colorectal tumors. J. Clin. Oncol., 17, 2439–2445. [DOI] [PubMed] [Google Scholar]
- Albin N., Johnson,M.R. and Diasio,R.B. (1996) cDNA cloning of bovine liver dihydropyrimidine dehydrogenase. DNA Seq., 6, 243–250. [DOI] [PubMed] [Google Scholar]
- Brito R.A., Medgyesy,D., Zukowski,T.H., Royce,M.E., Ravandi-Kashani,F., Hoff,P.M. and Pazdur,R. (1999) Fluoropyrimidines: a critical evaluation. Oncology, 57 (suppl. 1), 2–8. [DOI] [PubMed] [Google Scholar]
- Brünger A.T. et al. (1998) Crystallography & NMR system: a new software suite for macromolecular structure determination. Acta Crystallogr. D, 54, 905–921. [DOI] [PubMed] [Google Scholar]
- Bruschi M. and Guerlesquin,F. (1988) Structure, function and evolution of bacterial ferredoxins. FEMS Microbiol. Rev., 54, 155–176. [DOI] [PubMed] [Google Scholar]
- Chabrière E., Charon,M.-H., Volbeda,A., Pieulle,L., Hatchikian,E.C. and Fontecilla-Camps,J.-C. (1999) Crystal structure of the key anaerobic enzyme pyruvate:ferredoxin oxidoreductase, free and in complex with pyruvate. Nature Struct. Biol., 6, 182–190. [DOI] [PubMed] [Google Scholar]
- Collaborative Computational Project No. 4 (1994) The CCP4 suite: programs for protein crystallography. Acta Crystallogr. D, 50, 760–763. [DOI] [PubMed] [Google Scholar]
- Cowtan K. and Main,P. (1996) Phase combination and cross validation in iterated density modification calculations. Acta Crystallogr. D, 52, 43–48. [DOI] [PubMed] [Google Scholar]
- de la Fortelle E. and Bricogne,G. (1997) Maximum-likelihood heavy-atom parameter refinement in the MIR and MAD methods. Methods Enzymol., 276A, 472–494. [DOI] [PubMed] [Google Scholar]
- Dobritzsch D., Persson,K., Schneider,G. and Lindqvist,Y. (2001) Crystallization and preliminary X-ray study of pig liver dihydropyrimidine dehydrogenase. Acta Crystallogr. D, 57, 153–155. [DOI] [PubMed] [Google Scholar]
- Esnouf R.M. (1997) An extensively modified version of MolScript that includes greatly enhanced coloring capabilities. J. Mol. Graph. Model., 15, 133–138. [DOI] [PubMed] [Google Scholar]
- Hagen W.R., Vanoni,M.A., Rosenbaum,K. and Schnackerz,K.D. (2000) On the iron–sulfur clusters in the complex redox enzyme dihydropyrimidine dehydrogenase. Eur. J. Biochem., 267, 3640–3646. [DOI] [PubMed] [Google Scholar]
- Heggie G.D., Sommadossi,J.-P., Cross,D.S., Huster,W.J. and Diasio,R.B. (1987) Clinical pharmacokinetics of 5-fluorouracil and its metabolites in plasma. Cancer Res., 47, 2203–2206. [PubMed] [Google Scholar]
- Ho D.H., Townsend,L., Luna,M.A. and Bodely,G.P. (1986) Distribution and inhibition of dihydrouracil dehydrogenase activities in human tissues using 5-fluorouracil as a substrate. Anticancer Res., 6, 781–784. [PubMed] [Google Scholar]
- Hull W.E., Port,R.E., Hermann,R., Britsch,B. and Kunz,W. (1988) Metabolites of 5-fluorouracil in plasma and urine, as monitored by 19F nuclear magnetic resonance spectroscopy, for patients receiving chemotherapy with and without methotrexate pretreatment. Cancer Res., 48, 1680–1688. [PubMed] [Google Scholar]
- Iverson T.M., Luna-Chavez,C., Cecchini,G. and Rees,D.C. (1999) Structure of the Escherichia coli fumarate reductase respiratory complex. Science, 284, 1961–1966. [DOI] [PubMed] [Google Scholar]
- Jones T.A., Zou,J.-Y., Cowan,S.W. and Kjeldgaard,M. (1991) Improved methods for building protein models in electron density maps and the location of errors in these models. Acta Crystallogr. A, 47, 110–119. [DOI] [PubMed] [Google Scholar]
- Karplus P.A. and Schulz,G.E. (1989) Substrate binding and catalysis by glutathione reductase as derived from refined enzyme: substrate crystal structures at 2 Å resolution. J. Mol. Biol., 210, 163–180. [DOI] [PubMed] [Google Scholar]
- Kouwaki M., Hamajima,N., Sumi,S., Nonaka,M., Sasaki,M., Dobashi,K., Kidouchi,K., Togari,H. and Wada,Y. (1998) Identification of novel point mutations in the dihydropyrimidine dehydrogenase gene in a Japanese patient with 5-fluorouracil toxicity. Clin. Cancer Res., 4, 2999–3004. [PubMed] [Google Scholar]
- Kraulis P.J. (1991) MOLSCRIPT: A program to produce both detailed and schematic plots of protein structures. J. Appl. Crystallogr., 24, 946–950. [Google Scholar]
- Laskowski R.A., MacArthur,M.W., Moss,D.S. and Thornton,J.M. (1993) PROCHECK: a program to check the stereochemical quality of protein structures. J. Appl. Crystallogr., 26, 283–291. [Google Scholar]
- Lennon B.W., Williams,C.H.,Jr and Ludwig,M.L. (2000) Twist in catalysis: alternating conformations of Escherichia coli thioredoxin reductase. Science, 289, 1190–1194. [DOI] [PubMed] [Google Scholar]
- Lu G. (1996) A WWW service system for automatic comparison of protein structures. Protein Data Bank Q. Newsl., 78, 10–11. [Google Scholar]
- Lu G. (1999) FINDNCS: A program to detect non-crystallographic symmetries in protein crystals from heavy-atom sites. J. Appl. Crystallogr., 32, 365–368. [Google Scholar]
- Lu Z.H., Zhang,R. and Diasio,R.B. (1992) Purification and characterization of dihydropyrimidine dehydrogenase from human liver. J. Biol. Chem., 267, 17102–17109. [PubMed] [Google Scholar]
- Mani S. et al. (2000) Multicenter phase II study to evaluate a 28-day regimen of oral fluorouracil plus eniluracil in the treatment of patients with previously untreated metastatic colorectal cancer. J. Clin. Oncol., 18, 2894–2901. [DOI] [PubMed] [Google Scholar]
- Merritt E.A. and Bacon,D.J. (1997) Raster3D: Photorealistic molecular graphics. Methods Enzymol., 277, 505–524. [DOI] [PubMed] [Google Scholar]
- Milano G. and Etienne,M.-C. (1994) Dihydropyrimidine dehydrogenase (DPD) and clinical pharmacology of 5-fluorouracil. Anticancer Res., 14, 2295–2297. [PubMed] [Google Scholar]
- Naguib F.N.M., El Kouni,M.H. and Cha,S. (1987) Structure–activity relationship of ligands of dihydrouracil dehydrogenase from mouse liver. Biochem. Pharmacol., 38, 1471–1480. [DOI] [PubMed] [Google Scholar]
- Nicholls A., Sharp,K.A. and Honig,B. (1991) Protein folding and association: insights from the interfacial and thermodynamic properties of hydrocarbons. Proteins, 11, 282–296. [DOI] [PubMed] [Google Scholar]
- Otwinowski Z. and Minor,W. (1997) Processing of X-ray diffraction data collected in oscillation mode. Methods Enzymol., 276A, 307–326. [DOI] [PubMed] [Google Scholar]
- Page C.C., Moser,C.C., Chen,X. and Dutton,P.L. (1999) Natural engineering principles of electron tunneling in biological oxidation–reduction. Nature, 402, 47–52. [DOI] [PubMed] [Google Scholar]
- Papamichael D. (2000) The use of thymidylate synthase inhibitors in the treatment of advanced colorectal cancer: current status. Stem Cells, 18, 166–175. [DOI] [PubMed] [Google Scholar]
- Podschun B., Wahler,G. and Schnackerz,K.D. (1989) Purification and characterization of dihydropyrimidine dehydrogenase from pig liver. Eur. J. Biochem., 185, 219–224. [DOI] [PubMed] [Google Scholar]
- Podschun B., Cook,P.F. and Schnackerz,K.D. (1990) Kinetic mechanism of dihydropyrimidine dehydrogenase from pig liver. J. Biol. Chem., 265, 12966–12972. [PubMed] [Google Scholar]
- Porter D.J.T., Chestnut,W.G., Merril,B.M. and Spector,T. (1992) Mechanism-based inactivation of dihydropyrimidine dehydrogenase by 5-ethynyluracil. J. Biol. Chem., 267, 5236–5242. [PubMed] [Google Scholar]
- Ridge S.A., Brown,O., McMurrough,J., Fernandez-Salguero,P., Gonzalez,F.J. and McLeod,H.L. (1997) Mutations at codon 974 of the DPYD gene are a rare event. Br. J. Cancer, 75, 178–179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rosenbaum K., Schaffrath,B., Hagen,W.R., Jahnke,K., Gonzalez,F.J., Cook,P.F. and Schnackerz,K.D. (1997) Purification, characterization, and kinetics of porcine recombinant dihydropyrimidine dehydrogenase. Protein Expr. Purif., 10, 185–191. [DOI] [PubMed] [Google Scholar]
- Rosenbaum K., Jahnke,K., Curti,B., Hagen,W.R., Schnackerz,K.D. and Vanoni,M.A. (1998) Porcine recombinant dihydropyrimidine dehydrogenase: comparison of the spectroscopic and catalytic properties of the wild-type and the C671A mutant enzymes. Biochemistry, 37, 17598–17609. [DOI] [PubMed] [Google Scholar]
- Rowland P., Björnberg,O., Nielsen,F.S., Jensen,K.F. and Larsen,S. (1998) The crystal structure of Lactococcus lactis dihydroorotate dehydrogenase A complexed with the enzyme reaction product throws light on its enzymatic function. Protein Sci., 7, 1269–1279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schulz G.E. (1980) Gene duplication in glutathione reductase. J. Mol. Biol., 138, 335–347. [DOI] [PubMed] [Google Scholar]
- Shiotani T. and Weber,G. (1981) Purification and properties of dihydrothymine dehydrogenase from rat liver. J. Biol. Chem., 256, 219–224. [PubMed] [Google Scholar]
- Terwilliger T.C. and Berendzen,J. (1999) Automated structure solution for MIR and MAD. Acta Crystallogr. D, 55, 849–861. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Van Kuilenburg A.B.B. et al. (1999) Genotype and phenotype in patients with dihydropyrimidine dehydrogenase defiency. Hum. Genet., 104, 1–9. [DOI] [PubMed] [Google Scholar]
- Wasternack C. (1980) Degradation of pyrimidines and pyrimidine analogs—pathways and mutual influences. Pharmacol. Ther., 8, 629–651. [DOI] [PubMed] [Google Scholar]
- Yokota H., Fernandez-Salguero,P., Furuya,H., Lin,K., McBride,O.W., Podschun,B., Schnackerz,K.D. and Gonzalez,F.J. (1994) cDNA cloning and chromosome mapping of human dihydropyrimidine dehydrogenase, an enzyme associated with 5-fluorouracil toxicity and congenital thymine uraciluria. J. Biol. Chem., 269, 23192–23196. [PubMed] [Google Scholar]
- Ziegler G.A. and Schulz,G.E. (2000) Crystal structures of adrenodoxin reductase in complex with NADP+ and NADPH suggesting a mechanism for the electron transfer of an enzyme family. Biochemistry, 39, 10986–10995. [DOI] [PubMed] [Google Scholar]