

Abstract
During early postnatal development, midbrain dopamine (DA) neurons display anomalous firing patterns and amphetamine response. Spontaneous miniature hyperpolarizations (SMHs) are observed in DA neurons during the same period but not in adults. These hyperpolarizations have been shown to be dependent on the release of Ca2+ from internal stores and the subsequent activation of Ca2+-sensitive K+ channels. However, the triggering mechanism and the functional significance of SMHs remain poorly understood. To address these issues, using brain slices, we recorded spontaneous miniature outward currents (SMOCs) in DA neurons of neonatal rats. Two types of SMOCs were identified based on the peak amplitude. Both types were suppressed by intracellular dialysis of ruthenium red, a ryanodine receptor (RyR) antagonist, yet none of the known Ca2+-releasing messengers were involved. T-type Ca2+ channel blockers (Ni2+ and mibefradil) inhibited large-amplitude SMOCs without affecting the small-amplitude ones. The voltage dependence of SMOCs displayed a peak of approximately -50 mV, consistent with the involvement of low-threshold T-type Ca2+ channels. Blockade of SMOCs with cyclopiazonic acid or ryanodine converted the irregular firing of DA neurons in neonatal rats into an adult-like pacemaker pattern. This effect was reversed by the injection of artificial currents mimicking SMOCs. Finally, amphetamine inhibited SMOCs and transformed the irregular firing pattern into a more regular one. These data demonstrate that Ca2+ influx through T-type Ca2+ channels, followed by Ca2+-induced Ca2+ release via RyRs, contributes to the generation of SMOCs. We propose that SMOCs-SMHs may underlie the anomalous firing and amphetamine response of DA neurons during the postnatal developmental period.
Keywords: intracellular Ca2+ signaling, T-type Ca2+ channels, SK channels, ryanodine receptors, dopamine neurons, firing pattern
Introduction
Dopaminergic projections from the ventral midbrain to the frontal cortex and the striatum, termed the mesocorticolimbic dopamine (DA) system, play a major role in controlling voluntary movements, motivated behaviors, and reward processing. Pathological changes in this pathway are associated with the etiology of Parkinson's disease, schizophrenia, and drug addiction. Neurodevelopmental deficits in the mesocorticolimbic DA system have also been implicated in the pathogenesis of attention deficit/hyperactivity disorder (Kinsbourne, 1973; Castellanos and Tannock, 2002). The synaptic connectivity of DA terminals in projection areas matures into an adult-like pattern over the early postnatal period (∼4 weeks) (Loizou, 1972; Voorn et al., 1988). During this postnatal development of the DA circuitry, the basic electrophysiological properties of DA neurons also undergo significant changes. In vivo recording studies have shown that DA neurons fire much more slowly and irregularly in neonatal rats (1-2 weeks of age) than in adults (Pitts et al., 1990; Tepper et al., 1990; Wang and Pitts, 1994). Furthermore, psychostimulant amphetamine, which inhibits DA neuronal activity in adults, produces a paradoxical increase in the firing rate of DA neurons in neonates (Trent et al., 1991). The firing pattern of neurons is determined by the interplay between their intrinsic properties and the afferent inputs. Although the reason for the differential firing patterns of DA neurons in neonates and adults is not known, it is possible that some intrinsic mechanisms unique to DA neurons in the early postnatal period contribute to this difference.
Intracellular Ca2+ controls a multitude of neuronal processes, ranging from membrane excitability to plasticity and formation of synapses (Berridge, 1998). The highly specialized spatial organization between Ca2+ sources and targets is thought to dictate the specificity of these “Ca2+-gated” processes (Augustine et al., 2003). In DA neurons, the influx of Ca2+ through voltage-gated Ca2+ channels triggered by action potentials leads to the activation of small-conductance Ca2+-sensitive K+ (SK) channels, resulting in the generation of large afterhyperpolarizations (AHPs) that dominate the interspike interval (ISI) of pacemaker firing (Wolfart and Roeper, 2002). Furthermore, synaptic activation of metabotropic glutamate receptors (mGluRs) elicits mobilization of Ca2+ from intracellular stores and the subsequent activation of SK channels, producing a slow IPSP and a prolonged pause of DA neuronal firing (Fiorillo and Williams, 1998; Morikawa et al., 2003). Recently, spontaneous miniature hyperpolarizations (SMHs) have been reported in DA neurons of neonatal rats, but not adult rats, using intracellular recordings in brain slices (Seutin et al., 1998). SMHs are also dependent on internal Ca2+ mobilization and SK channel activation (Seutin et al., 2000). However, the triggering mechanism and the functional significance of these SMHs are not well understood.
In this study, we performed whole-cell voltage-clamp recordings of spontaneous miniature outward currents (SMOCs) in DA neurons of neonatal rats to delineate the cellular mechanism underlying SMHs. We found that the generation of SMOCs involves the selective coupling of T-type Ca2+ channels, ryanodine receptors (RyRs), and SK channels. Our results also show that SMOCs-SMHs may contribute to the anomalous firing pattern and amphetamine response of DA neurons observed during the early postnatal period.
Materials and Methods
Slices and solutions. Horizontal slices (200-220 μm) of the ventral midbrain were prepared from neonatal Wistar rats [postnatal day 6 (P6)-P12]. Some of the experiments were done in slices from neonatal Sprague Dawley rats (P6-P12), which gave equivalent results, and the data were pooled from both strains of rats. Slices were cut using a vibratome (VT1000S; Leica, Nussloch, Germany) in ice-cold (4°C) physiological saline containing the following (in mm): 126 NaCl, 2.5 KCl, 1.2 NaH2PO4, 1.2 MgCl2, 2.4 CaCl2, 11 glucose, and 21.4 NaHCO3, pH 7.4 (saturated with 95% O2 and 5% CO2, 300 mOsm/kg). Slices were then stored in the same solution, warmed to 35°C for ≥30 min. For recordings, a single slice was placed in a recording chamber and superfused with warmed (35°C) physiological saline at 2.5-3 ml/min. Unless noted otherwise, pipette solutions used for whole-cell and cell-attached recordings contained the following (in mm): 115 K-methylsulfate, 20 KCl, 1.5 MgCl2, 10 HEPES, 0.025 EGTA, 2 Mg-ATP, 0.2 Na2-GTP, and 10 Na2 phosphocreatine, pH 7.3 (280 mOsm/kg).
Whole-cell recordings. All of the recordings were performed in DA neurons identified by their large cell bodies (∼20 μm), spontaneous firing at 1-5 Hz, and the presence of large hyperpolarization-activated cation currents (>200 pA), evoked by a 1.5 sec hyperpolarizing step from -55 to -105 mV. Cells were visualized using a 60× water-immersion objective on an upright microscope (BX51WI; Olympus Optical, Tokyo, Japan) with infrared-differential interference contrast optics. Whole-cell pipettes had resistances of 1.8-2.4 MΩ. Voltage-clamp recordings were made at a holding potential of -55 mV unless stated otherwise. A MultiClamp 700A amplifier (Axon Instruments, Union City, CA) was used to record the data, which were filtered at 1 kHz, digitized at 2 kHz, and collected on a personal computer using AxoGraph 4.9 (Axon Instruments).
Cell-attached recordings. The firing was monitored using the cell-attached configuration because the spontaneous firing of dopamine neurons was significantly distorted with standard whole-cell recordings (Morikawa et al., 2003). The use of low-resistance pipettes (1.5-1.8 MΩ), together with the formation of a large Ω-shaped membrane invagination, created some access to the cell interior to monitor the membrane potential in current clamp without intentional break-in. Although the recording of membrane potential with this configuration was crude and inaccurate in terms of the measured value and the kinetics of changes in membrane potential, it allowed us to determine the spike timing and to inject some currents into the recorded cell.
In experiments in which we injected artificial currents that simulated SMOCs, we used a double-exponential current waveform with rise and decay time constants of 10 and 25 msec to mimic the average kinetics of SMOCs. The peak amplitude of the injected waveform was 20-40 pA, although the actual amounts of current injected into the cell are not known because of the limited and unknown access associated with these recording conditions. These waveforms were injected at 0.8-1 Hz with random timing, using the simulation function in AxoGraph 4.9.
Aspartate iontophoresis. mGluR-mediated outward currents were evoked by iontophoresis of aspartate (Morikawa et al., 2003). Iontophoresis was performed with a MultiClamp 700A amplifier (50-200 nA ejection current of 50-200 msec duration and 5 nA backing current), using small-tipped pipettes (40-100 MΩ) containing l-aspartate (1 m) and HEPES (10 mm), pH 7.4. Iontophoretic pipettes were placed within 5 μm of the soma. Experiments were done in slices pretreated with (5S,10R)-(+)-5-methyl-10,11-dihydro-5H-dibenzo[a,d]cyclohepten-5,10-imine (MK-801) (50 μm) to block NMDA-mediated currents.
Drugs. Drugs were applied by either bath perfusion or intracellular dialysis through the whole-cell pipette. When drugs were applied by intracellular dialysis, at least one recording using a control internal solution without drugs was made in a slice from the same rat. Data from these recordings were used as control (see Fig. 2C).
Figure 2.

SMOCs are dependent on Ca2+ release from internal stores via RyRs. A, Summary time graph showing the effect of CPA (10 μm) on SMOCs (n = 4). B, Summary time graph illustrating the effect of ryanodine (Ryn) (20 μm) on SMOCs (n = 6). Both CPA and ryanodine eliminated SMOCs in ∼5 min. C1, Representative traces of SMOCs with a control internal solution or with a solution containing ruthenium red (100 μm). Traces are shown at 1 and 5 min after entering the whole-cell configuration. C2, The SMOC frequency is plotted against time after going into the whole-cell configuration. Recordings were made with a pipette containing a control internal solution (n = 22), ruthenium red (RR) (100 μm; n = 11), both heparin (Hep) (1 mg/ml) and 8-NH2-cADPR (8AcADPR) (50 μm) (n = 8), and NAADP (1 mm; n = 9). Only ruthenium red induced significant inhibition of SMOCs. *p < 0.05; **p < 0.01 versus control.
2,3-Dioxo-6-nitro-1,2,3,4-tetrahydrobenzo[f]quinoxaline-7-sulfonamide (NBQX), MK-801, and α-methyl-4-carboxyphenylglycine (MCPG) were obtained from Tocris Cookson (Ellisville, MO). Heparin, ruthenium red, and ryanodine were purchased from Calbiochem (La Jolla, CA). Tetrodotoxin (TTX) and SNX-482 were obtained from Alomone Labs (Jerusalem, Israel). 8-NH2-cyclic ADP ribose (cADPR) was from Molecular Probes (Eugene, OR). All of the other chemicals were obtained from Sigma (St. Louis, MO). Peptide Ca2+ channel blockers were applied in the presence of cytochrome c (0.1 mg/ml) to block nonspecific binding sites.
Data analysis. Occasionally, there were rats in which none of the recorded DA neurons displayed SMOCs. Data from these rats were not used for the analysis. A 30 sec sweep of SMOCs was recorded every minute. SMOCs were detected with a sliding template algorithm using AxoGraph 4.9 (Clements and Bekkers, 1997), and their amplitude and frequency were obtained. When drugs were applied by bath perfusion, the SMOC frequency was normalized to the average value preceding the drug application over a duration that matched the duration of drug application (5-10 min). Amplitude histograms were routinely constructed from five sweeps, except for the experiments testing the effect of Cd2+ (three sweeps) (see Fig. 3A2) and the voltage dependence (four sweeps) (see Fig. 4 A2).
Figure 3.

Large-amplitude SMOCs are triggered by Ca2+ influx through T-type Ca2+ channels. A1, Representative traces of SMOCs, mGluR-induced outward currents, and AHP currents before and during a brief perfusion (5 min) of Cd2+ (200 μm). Aspartate iontophoresis was made at the time indicated by the arrow to evoke mGluR-mediated currents. Slices were pretreated with MK-801 (50 μm) to block NMDA-mediated currents. A2, Superimposed amplitude histograms of SMOCs before and during Cd2+ application from the same experiment as in A1. A3, Summary bar graph depicting the effects of Cd2+ on the SMOC frequency (n = 7), the mGluR-induced current amplitude (n = 5), and the AHP current amplitude (n = 6). Cd2+ significantly inhibited SMOCs and AHP currents but had a marginal effect on mGluR-induced currents. B1, C1, Representative traces of SMOCs showing the effects of Ni2+ (100 μm) and mibefradil (100 μm) on SMOCs. B2, C2, Superimposed amplitude histograms of SMOCs from the same experiments as in B1 and C1, respectively. D, Summary bar graph showing the effects of Ni2+ (100 μm; n = 9), mibefradil (Mib) (100 μm; n = 8), nifedipine (Nif) (10 μm; n = 5), ω-conotoxin GVIA (Cono) (1 μm; n = 3), ω-agatoxin IVA (Aga) (200 nm; n = 3), and SNX-482 (SNX) (200 nm; n = 3) on SMOCs. *p < 0.05; **p < 0.01; ***p < 0.001.
Figure 4.

Voltage dependence of SMOCs. A1, Representative traces of SMOCs at three different holding potentials. All of the traces are from the same cell. A2, Superimposed amplitude histograms of SMOCs from the same recording as in A1. B, Summary graph showing the voltage dependence of SMOC frequency (n = 26). The frequency was normalized to the value at -55 mV in each cell. Each data point, except for the one at -55 mV, represents data from 7-18 cells. The frequency-voltage relationship displayed a bell-shaped curve that peaked at -55 to -45 mV.
ISI histograms and autocorrelograms of the firing pattern were constructed from 90 sec sweeps obtained before and during the bath application of drugs. Autocorrelograms were constructed using the event autocorrelation program in AxoGraph 4.9. The number of peaks in the autocorrelogram, which occur at integral multiples of the mean ISI, represents an index of the regularity of firing (Paladini et al., 2001). Data are expressed as means ± SEM. Statistical significance was determined by paired or unpaired Student's t test or one-way ANOVA followed by post hoc Dunnett's test. The difference was considered significant at p < 0.05.
Results
Two types of SMOCs in DA neurons of neonatal rats
Whole-cell voltage-clamp recordings (holding potential at -55 mV) were made from DA neurons in midbrain slices obtained from neonatal rats (P6-P12). SMOCs were detected in 125 of 160 cells (78%) recorded from 89 rats (Fig. 1A1,B1). They are mostly absent in DA neurons from adult rats (>3 weeks of age) under the same recording conditions (Morikawa et al., 2003). These events appeared to occur in a completely random, or stochastic, manner. The frequency of SMOCs was 0.2-2.3 Hz (0.83 ± 0.04 Hz; n = 125). In a total of 9366 events detected over a 90 sec period in each of these 125 cells, the amplitude ranged from 4.7 to 75.2 pA (21.6 ± 0.1 pA). The rise time constant and the half-width were 11.4 ± 0.2 and 38.3 ± 0.2 msec, respectively. Of these 125 cells, 74 cells displayed two identifiable peaks in their amplitude histograms, with the first and second peaks located at 12 ± 0.4 pA (6-22 pA) and 28 ± 0.9 pA (14-48 pA), respectively (Fig. 1A2), whereas 51 cells showed single-peak amplitude histograms, with the peak located at 19 ± 0.9 pA (10-34 pA) (Fig. 1B2). No significant correlation was found between the amplitude and the rise time constant of SMOCs in any of the recorded cells (r ≤ 0.3) (Fig. 1A3). However, the average rise time constant of large-amplitude SMOCs (L-SMOCs) was significantly smaller than small-amplitude SMOCs (S-SMOCs) in 13 of 18 cells whose amplitude histograms had two distinct populations with minimal overlap between the two (Fig. 1A4). Although the rise time constant of L-SMOCs was invariably smaller than that of S-SMOCs, the difference was not significant in the remaining five cells.
Figure 1.
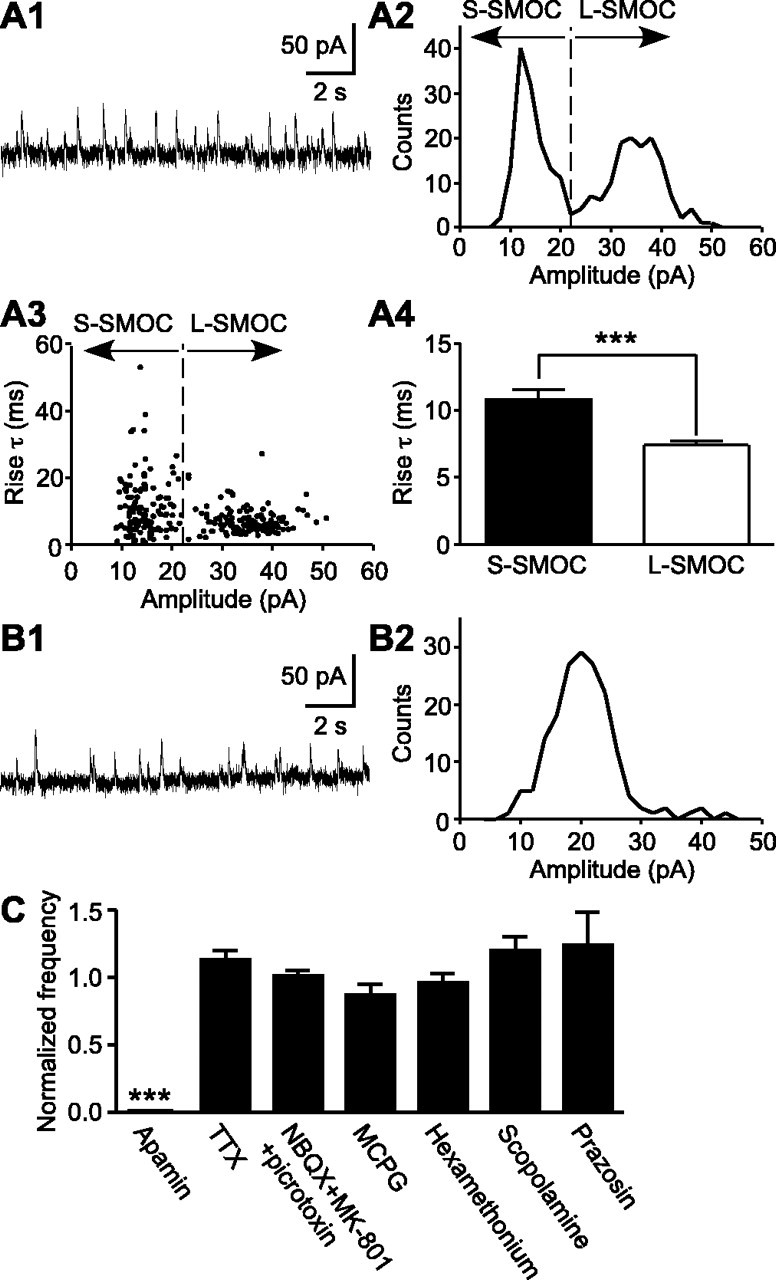
Basal properties of SMOCs. A1, A2, A representative trace of SMOCs (A1) and their amplitude histogram (A2) from a DA neuron showing two peaks in the SMOC amplitude distribution. A3, Relationship between the amplitude and the rise time constant of SMOCs from the same recording as in A1 and A2. There was no correlation between the amplitude and the rise time constant (r = 0.25). A4, Bar graph depicting the average rise time constant of S-SMOCs (<22 pA) and L-SMOCs (≥22 pA) from the same recording. The line dividing S-SMOCs and L-SMOCs is shown in A2 and A3. B1, B2, Representative trace of SMOCs (B1) and their amplitude histogram (B2) from a DA neuron showing only one peak in the amplitude distribution. C, Summary bar graph depicting the effects of apamin, TTX, and various neurotransmitter antagonists on SMOCs. The SMOC frequency in the presence of drugs was normalized to the control frequency before drug application. The drugs tested included apamin (100 nm; n = 5), TTX (300 nm; n = 4), a mixture of NBQX (AMPA antagonist; 5 μm), MK-801 (NMDA antagonist; 50 μm), and picrotoxin (GABAA antagonist; 100 μm) (n = 4), MCPG (mGluR antagonist; 1 mm; n = 4), hexamethonium (nicotinic acetylcholine receptor antagonist; 200 μm; n = 4), scopolamine (muscarinic acetylcholine receptor antagonist; 1 μm; n = 7), and prazosin (α1 adrenergic receptor antagonist; 100 nm; n = 3). Only apamin suppressed SMOCs. ***p < 0.001.
SMOCs were not affected by bath application of TTX (300 nm) or antagonists of major postsynaptic neurotransmitter receptors present in DA neurons (Fig. 1C), showing that they are intrinsically generated. Consistent with the previous report on SMHs in DA neurons (Seutin et al., 1998), SMOCs were completely eliminated by apamin (100 nm), a selective blocker of SK channels (n = 5) (Fig. 1C). Furthermore, perfusion of cyclopiazonic acid (CPA) (10 μm), which depletes endoplasmic reticulum Ca2+ stores (Seidler et al., 1989), abolished SMOCs in ∼5 min (n = 4) and confirmed the involvement of Ca2+ mobilization from internal stores (Fig. 2A).
SMOCs are dependent on RyRs
Subsequently, we tested the effect of ryanodine, which locks RyR channels in a subconductance open state and thus can deplete Ca2+ stores expressing RyRs (Smith et al., 1988; Zucchi and Ronca-Testoni, 1997; Morikawa et al., 2000). Bath perfusion of ryanodine (20 μm) nearly eliminated SMOCs in ∼5 min (18 ± 3% of control; n = 6) (Fig. 2B). To examine further whether the effect of ryanodine was attributable to the direct blockade of RyRs or to depletion of RyR-expressing stores, we tested ruthenium red, which inhibits the opening of RyR channels (Smith et al., 1988). Intracellular dialysis of ruthenium red (100 μm) significantly attenuated SMOCs within 1 min after the onset of whole-cell recordings and nearly abolished SMOCs in 3-5 min (n = 11) (Fig. 2C). Therefore, Ca2+ release via RyRs is essential for the generation of SMOCs.
Known Ca2+-mobilizing messengers are not involved
Ca2+ mobilization from internal stores is generally induced by intracellular messengers (Galione and Churchill, 2002). Thus, it is possible that these messengers trigger Ca2+ mobilization underlying the generation of SMOCs. Inositol 1,4,5-triphosphate (IP3) and cADPR are two well established Ca2+-mobilizing messengers (Galione, 1994; Berridge, 1995). In particular, cADPR can directly activate RyRs (Meszaros et al., 1993). To test the involvement of these two messengers, a combination of heparin, a competitive IP3 receptor antagonist (Ghosh et al., 1988), and 8-NH2-cADPR, an inhibitor of cADPR signaling (Walseth and Lee, 1993), was used to simultaneously block both messenger pathways. This combination can eliminate the mGluR-induced Ca2+ mobilization and outward currents in DA neurons (Morikawa et al., 2003). Inclusion of both heparin (1 mg/ml) and 8-NH2-cADPR (50 μm) in the pipette failed to inhibit SMOCs for ≤20 min of whole-cell recordings (n = 8) (Fig. 2C2 shows the first 5 min). There was a significant increase in the SMOC frequency after 4 min with the combination of heparin and 8-NH2-cADPR. This may be attributable to the stimulatory effect of heparin on RyRs (Ehrlich et al., 1994).
Nicotinic acid ADP (NAADP) is another newly emerging Ca2+-mobilizing messenger (Lee and Aarhus, 1995; Cancela et al., 1999; Patel et al., 2001). Several lines of evidence suggest that NAADP plays an important role in neuronal Ca2+ signaling (Bak et al., 1999; Petersen and Cancela, 1999; Patel et al., 2000; Brailoiu et al., 2003). Thus, we examined the potential involvement of NAADP. It has been shown that high concentrations of NAADP can self-desensitize the NAADP-induced Ca2+ release mechanism (Cancela et al., 1999; Berg et al., 2000). Accordingly, we used a very high concentration of NAADP (1 mm) to fully inactivate the NAADP-mediated Ca2+ signaling. Intracellular dialysis of NAADP had no significant effect on SMOCs after ≤20 min of whole-cell recordings (n = 9) (Fig. 2C2 shows the first 5 min). Together, these results suggest that intracellular Ca2+-mobilizing messengers do not play a role in the release of Ca2+, which triggers SMOCs in DA neurons.
Ca2+ influx through T-type Ca2+ channels triggers large-amplitude SMOCs
Ca2+ influx through voltage-gated Ca2+ channels can activate RyRs and trigger Ca2+-induced Ca2+ release (CICR) from internal stores (Berridge, 1998). To examine whether this mechanism underlies the generation of SMOCs, we used a panel of Ca2+ channel blockers. All types of Ca2+ channels (L-, N-, P/Q-, R-, and T-type) are known to be expressed in DA neurons (Cardozo and Bean, 1995; Wolfart and Roeper, 2002). First, we tested Cd2+, a nonselective Ca2+ channel blocker. To rule out the possibility that the effect of Cd2+ was a result of the depletion of internal Ca2+ stores, we also measured the peak amplitude of mGluR-mediated outward currents evoked by the iontophoretic application of aspartate. These mGluR currents are totally dependent on internal Ca2+ stores, with no contribution of Ca2+ influx from the extracellular space (Morikawa et al., 2003). Brief perfusion (3-5 min) of Cd2+ (200 μm) reduced dramatically the frequency of SMOCs to 39 ± 6% of control (n = 7; p < 0.05) but had no significant effect on mGluR-mediated currents (82 ± 14% of control; n = 5; p = 0.23) (Fig. 3A1,A3). Cd2+ also significantly inhibited AHP currents evoked by a 100 msec depolarization to 0 mV (27 ± 2% of control; n = 6; p < 0.001), consistent with the blockade of Ca2+ influx by Cd2+. Subsequently, we tested Ni2+, which preferentially blocks T- and R-type Ca2+ channels (Zamponi et al., 1996; Lee et al., 1999; Perez-Reyes, 2003). Ni2+ (100 μm) produced a significant reduction in SMOC frequency (38 ± 6% of control; n = 9; p < 0.001) (Fig. 3B1,D). A similar result was obtained with mibefradil (100 μm), a T-type Ca2+ channel blocker (Martin et al., 2000) (48 ± 10% of control; n = 8; p < 0.01) (Fig. 3C1,D). A common feature shared by Cd2+, Ni2+, and mibefradil was the selective inhibition of large-amplitude SMOCs in cells with both two-population amplitude histograms (Fig. 3A2,B2) and one-population histograms (Fig. 3C2). We also tested several other specific Ca2+ channel blockers. These included nifedipine (10 μm; n = 5), ω-conotoxin GVIA (1 μm; n = 3), ω-agatoxin IVA (200 nm; n = 3), and SNX-482 (200 nm; n = 3), which selectively block L-, N-, P/Q-, and R-type channels, respectively (Bean, 1989; Boland et al., 1994; Randall and Tsien, 1995; Newcomb et al., 1998). None of these drugs caused suppression of SMOCs (Fig. 3D). These results suggest that Ca2+ influx through T-type Ca2+ channels triggers the Ca2+ mobilization that induces L-SMOCs but not S-SMOCs.
Subsequently, we examined the voltage dependence of SMOCs. Depolarizing the holding potential above -40 mV to inactivate T-type channels completely eliminated L-SMOCs (Fig. 4A1,A2). Likewise, when the cell was hyperpolarized to -60 or -65 mV, which is near the threshold for T-type channel activation, the frequency of L-SMOCs was dramatically reduced, together with a leftward shift in the peak of L-SMOC amplitude distribution, resulting from a reduction in the driving force for K+ ions. The S-SMOC frequency was also reduced by both depolarization and hyperpolarization. This was most likely attributable to an increase in the background noise level from the activation of voltage-dependent K+ conductances in the case of depolarization and to a reduction in the K+ ion driving force in the case of hyperpolarization, thus compromising the detection of S-SMOCs in both cases. Overall, when the holding potential was varied from -75 to -35 mV, the voltage dependence of SMOC frequency exhibited a bell-shaped curve (n = 26) (Fig. 4B). The optimal potential was -55 to -45 mV, a voltage range in which low-threshold T-type channels but not other high-threshold Ca2+ channels expressed in DA neurons, can be activated (Cardozo and Bean, 1995; Randall and Tsien, 1997).
SMOCs modulate the firing of DA neurons
To examine whether SMOC-induced transient hyperpolarizations (i.e., SMHs) can modulate the firing of DA neurons, we tested the effect of CPA and ryanodine, which can effectively block the occurrence of SMOCs (Fig. 2A,B). The spontaneous firing of DA neurons was monitored with a cell-attached configuration, which produced small access to the cell interior without intentional break-in (see Materials and Methods). The firing pattern was irregular [the coefficient of variation (CV) of the ISI ≥ 20%] in 25 of 42 cells recorded from 16 rats. Perfusion of either CPA (10 μm) or ryanodine (20 μm) transformed the irregular firing pattern into a regular one (Fig. 5A). Accordingly, the CV of the ISI was reduced from 34 ± 2 to 13 ± 1% (n = 4; p < 0.01) (Fig. 5B). The firing rate was increased from 1.1 ± 0.1 to 1.6 ± 0.3 Hz; however, this increase was not statistically significant (p = 0.14). The change in the firing pattern was also reflected in the autocorrelogram, which displayed an increased number of identifiable peaks after treatment with CPA or ryanodine (Fig. 5C). Strikingly, injection of artificial currents simulating SMOCs at 0.8-1 Hz, with random timing, restored the original irregular firing pattern in CPA-treated cells or made the firing irregular in cells that had a regular firing pattern in control, producing a significant increase in the CV of the ISI from 11 ± 2 to 22 ± 3% (n = 5; p < 0.05). These data demonstrate the effectiveness of SMOC-induced hyperpolarizations in modulating the firing of DA neurons.
Figure 5.

SMOCs contribute to the irregular firing pattern of DA neurons in neonatal rats. A, Representative traces of spontaneous firing before and during perfusion of CPA (10 μm). The bottom trace was obtained when simulated SMOCs were injected during CPA treatment. B, ISI histograms from the corresponding recordings in A. Note then arrow peak with a Gaussian distribution in CPA. C, Autocorrelograms from the corresponding recordings in A. Note multiple identifiable peaks in CPA.
Amphetamine inhibits SMOCs via α1 adrenergic receptors
It has been shown that amphetamine inhibits mGluR-mediated outward currents via release of DA, which activates postsynaptic α1 adrenergic receptors (α1ARs) (Paladini et al., 2001). Partial depletion of intracellular Ca2+ stores was suggested to mediate this α1AR-induced heterologous desensitization of the mGluR response. We thus examined whether Ca2+ store depletion induced by amphetamine can also affect SMOCs. Bath application of amphetamine (10 μm) reduced the SMOC frequency to 44 ± 6% of control (n = 9) (Fig. 6A,B). This inhibition was reversed by coapplication of prazosin (100 nm), an α1AR antagonist (n = 4), but not by eticlopride (200 nm), a D2 DA receptor antagonist (n = 5) (data not shown). Similar inhibition was observed with the perfusion of DA (100 μm; 57 ± 5% of control; n = 4) and phenylephrine (30 μm), a selective α1AR agonist (61 ± 6% of control; n = 5). The amplitude histogram revealed that amphetamine reduced the frequency of both L-SMOCs and S-SMOCs, eliminating larger events in each group (Fig. 6A2). The reduced Ca2+ release, attributable to partial store depletion, most likely curtailed the CICR process underlying SMOC generation. Altogether, these results are consistent with the idea that α1AR activation mediates the depletion of internal Ca2+ stores and suggest that amphetamine-induced Ca2+ store depletion may serve as a common mechanism by which amphetamine affects the activity of DA neurons.
Figure 6.

Amphetamine suppresses SMOCs via activation of α1 adrenergic receptors. A1, Representative traces of SMOCs in a control extracellular solution, amphetamine (Amph) (10 μm), and amphetamine plus prazosin (100 nm). A2, Superimposed amplitude histograms of SMOCs from the same experiment as in A1. B, Summary time graph illustrating the effect of amphetamine on SMOCs and its reversal by prazosin (n = 4). C, Representative traces of firing before and during perfusion of amphetamine (10 μm).
In addition, we tested the effect of amphetamine on the firing. The experiments were done in the presence of eticlopride (200 nm) or sulpiride (50 nm) to block the D2 receptor-mediated inhibition. Bath application of amphetamine (10 μm) reduced the CV of the ISI from 35 ± 8 to 19 ± 3% (n = 5; p < 0.05), thus converting the irregular firing pattern into a more regular one (Fig. 6C). Amphetamine also increased the firing rate from 1.2 ± 0.3 to 2.0 ± 0.6 Hz (p < 0.05). Therefore, it is suggested that amphetamine affects the firing of DA neurons in neonatal rats by suppressing the occurrence of SMOCs.
Discussion
In the present study, two types of SMOCs (L-SMOCs and S-SMOCs) were identified based on the difference in their amplitude. Although both types are dependent on the release of Ca2+ via RyRs and the subsequent activation of SK channels, an initial trigger by spontaneous opening of T-type Ca2+ channels is necessary to produce the larger amount of Ca2+ release underlying L-SMOCs (Fig. 7). In this scenario, Ca2+ influx through individual or a small packet of T-type Ca2+ channels provides the initial Ca2+ signal, which by itself is not sufficient to activate SK channels but can stimulate Ca2+ release from internal stores via Ca2+-activated RyRs (CICR). After the amplification by CICR, the local Ca2+ signal was then large enough to activate nearby SK channels. Furthermore, we found that SMOCs can modulate the firing pattern of DA neurons, suggesting a functional role that SMOCs may play during early postnatal development.
Figure 7.

Proposed model illustrating two types of SMOCs. L-SMOCs are initially triggered by Ca2+ influx through T-type Ca2+ channels (1), which by itself is not sufficient to activate SK channels but which can activate RyRs in close apposition to the plasma membrane and induce CICR. The Ca2+ signal amplified by CICR (2) is now sufficient to activate nearby SK channels. S-SMOCs are also produced by Ca2+ release via RyRs. However, the opening of RyRs is not triggered by T-type channel-mediated Ca2+ influx in S-SMOCs. A larger amount of Ca2+ is mobilized in L-SMOCs than in S-SMOCs.
Two types of SMOCs
Two distinct populations of SMOCs with different amplitudes could be identified in more than one-half of the recorded cells, whereas only one population could be resolved in the SMOC amplitude histogram from the rest of the cells. However, T-type Ca2+ channel blockers, which selectively suppressed L-SMOCs in cells with two-population amplitude histograms, also selectively eliminated large-amplitude events in cells with one-population histograms. Therefore, it is likely that L-SMOCs and S-SMOCs had a significant overlap in amplitude distribution in cells with one-population histograms. Moreover, the application of amphetamine, which suppressed SMOCs by partial depletion of internal Ca2+ stores, frequently uncovered the presence of two populations in these cells (data not shown), supporting further the idea that SMOCs comprise two populations in all cells.
L-SMOCs had a smaller rise time constant than that of S-SMOCs in individual cells, suggesting that larger Ca2+ mobilization, which would produce faster activation of target SK channels, underlies the larger outward currents for L-SMOCs. However, the amplitude was not correlated with the rise time constant within each population (Fig. 1A3). Thus, activation of different numbers of SK channels is likely responsible for the amplitude distribution of L-SMOCs and S-SMOCs in each cell. The number of open SK channels at the peak of each SMOC can be estimated from the equation I = γN(Vm - Veq), where I is the peak current amplitude; γ is the single-channel conductance; N is the total number of open channels; Vm is the membrane potential (-55 mV in the present study); and Veq is the K+ equilibrium potential (approximately -105 mV, based on the Nernst equation). The peak amplitude of L-SMOCs and S-SMOCs generally ranged from 20 to 50 and 5 to 20 pA in individual cells. Assuming a single-channel conductance of ∼10 pS (Bond et al., 1999), there should be ∼40-100 and 10-40 SK channels open at the peak of each L-SMOC and S-SMOC.
Triggering mechanism
SMOCs or SMHs are observed in various types of neurons (Brown et al., 1983; Mathers and Barker, 1984; Satin and Adams, 1987; Berridge, 1998; Seutin et al., 1998; Arima et al., 2001), as well as in smooth muscle cells, in which they are usually called spontaneous transient outward currents (STOCs) (Bolton and Imaizumi, 1996). SMOCs-STOCs are mediated by the activation of Ca2+-sensitive K+ channels. Focal release of Ca2+ from internal stores is generally thought to provide the source of Ca2+ (Satin and Adams, 1987; Berridge, 1998; Merriam et al., 1999). However, the triggering mechanism for spontaneous Ca2+ mobilization has not been clear and might vary in different types of cells. Our results ruled out the involvement of known endogenous Ca2+-mobilizing molecules (IP3, cADPR, and NAADP) in DA neurons. In contrast, Cd2+, a general Ca2+ channel blocker, suppressed L-SMOCs without depleting internal Ca2+ stores, demonstrating that the spontaneous opening of Ca2+ channels most likely triggers the generation of L-SMOCs. In line with this view, TTX-insensitive spontaneous Ca2+ spikes mediated by voltage-gated Ca2+ channels have been observed in cerebellar Purkinje neurons (Womack and Khodakhah, 2004).
The process of CICR ensuing Ca2+ influx through plasma membrane Ca2+ channels is well documented in neurons (Verkhratsky and Shmigol, 1996). Indeed, this mechanism has been used to explain the generation of SMOCs in mudpuppy parasympathetic cardiac neurons (Merriam et al., 1999) and rat Meynert neurons (Arima et al., 2001). One unique property of SMOCs in DA neurons lies in the voltage dependence. The frequency of SMOCs was highest at approximately -50 mV in DA neurons, whereas it peaked at -20 mV or more depolarized potentials in other types of cells (Satin and Adams, 1987; Fletcher and Chiappinelli, 1992; Merriam et al., 1999). This difference can be accounted for by the selective coupling of low-threshold T-type Ca2+ channels with SMOCs in DA neurons but not in other cells (Merriam et al., 1999). The activation threshold for T-type channels is -70 to -60 mV, whereas they become fully activated at -40 to -30 mV (Randall and Tsien, 1997; Perez-Reyes, 2003). Therefore, there is a voltage window between -70 and -30 mV in which T-type channels can be tonically active. The firing of DA neurons is characterized by a large AHP, followed by a slow ramp-like voltage trajectory from approximately -60 mV until it reaches the action potential threshold (approximately -40 mV) (Wilson and Callaway, 2000). This will give an opportunity for spontaneous T-type channel opening to take place during the tonic firing of DA neurons.
The coupling of T-type Ca2+ channels and SK channels required the RyR-mediated amplification of Ca2+ signals, suggesting that the spatial organization of these three channels may be critically important (Fig. 7). SK channels can be activated by submicromolar concentrations of Ca2+ (Xia et al., 1998), whereas 1-100 μm Ca2+ is required to fully activate RyRs (Bezprozvanny et al., 1991). This implies that RyRs are in closer proximity to T-type channels than SK channels are. In fact, the endoplasmic reticulum and plasma membrane can be as close as 20 nm or even in direct contact with each other in neurons (Berridge, 1998).
We do not know what triggers the opening of RyR channels for S-SMOCs. Partial depletion of internal Ca2+ stores by amphetamine reduced the frequency of S-SMOCs by eliminating the relatively larger events. This suggests that the generation of S-SMOCs also involves the CICR process, which can be abrogated if the amount of RyR-mediated Ca2+ release is reduced.
Functional significance
SMOCs and STOCs have been shown to play a role in the relaxation of smooth muscles (Porter et al., 1998) and to affect the action potential threshold in mudpuppy cardiac neurons (Parsons et al., 2002). In this study, we showed that SMOCs can modulate the firing pattern of DA neurons and may contribute to the slow and irregular firing observed during the early postnatal period (Pitts et al., 1990; Tepper et al., 1990; Wang and Pitts, 1994). The irregular firing pattern in central neurons is thought to reflect the response to correlated and uncorrelated synaptic inputs (Hausser and Clark, 1997; Carter and Regehr, 2002; Kononenko and Dudek, 2004). In DA neurons, the synaptic release of glutamate transforms the regular pacemaker-like firing, observed routinely in a slice preparation, into a burst-pause pattern (Morikawa et al., 2003). The findings in this study provide evidence that, in addition to modulation by synaptic inputs, DA neurons in neonates also have an intrinsic mechanism to generate an irregular firing pattern. SMOCs occurring at the slow ramp-like depolarization during the interspike interval would produce a small delay in the action potential timing. A similar delay in spike timing can be elicited by single GABAergic inputs occurring during the interspike interval in cerebellar Purkinje neurons, thus generating an irregular firing pattern (Hausser and Clark, 1997). DA release in the projection area is highly dependent on the rate and pattern of DA neuronal firing (Gonon, 1988; Montague et al., 2004). It should also be noted that DA neurons corelease glutamate at the terminal to induce fast excitatory neurotransmission (Chuhma et al., 2004). Therefore, even a small change in the spike timing may have a significant functional consequence in terms of signal output.
SMOCs, which act to disrupt the pacemaker firing in an input-independent manner, may actually represent the functional immaturity of DA neurons. Behavioral responses to psychostimulant amphetamine develops into an adult-like pattern after the third postnatal week (Kolta et al., 1990). In the present study, amphetamine inhibited SMOCs via activation of α1ARs and “corrected” the irregular firing pattern to a more regular and faster one. In line with this observation, an in vivo recording study showed that amphetamine can produce a paradoxical increase in DA neuronal firing in neonatal rats (Trent et al., 1991). Another possibility is that SMOCs, which are observed only during the early postnatal period, may be involved in the development of the DA system. DA neurons themselves and their connection with target neurons undergo significant changes over the first couple of postnatal weeks (Voorn et al., 1988). Spontaneous Ca2+ transients are known to regulate the development of CNS circuitry (Gomez and Spitzer, 1999; Tang et al., 2003). Therefore, transient Ca2+ elevations underlying SMOCs may play an active role in the wiring of the DA network.
Footnotes
This work was supported by National Institutes of Health Grant DA015687. We thank Drs. R. Adron Harris and Nace Golding for comments on this manuscript and Dr. Carlos Paladini for assistance with the autocorrelation analysis.
Correspondence should be addressed to Hitoshi Morikawa, Waggoner Center for Alcohol and Addiction Research, University of Texas, 2500 Speedway, Molecular Biology Building 1.150A, Austin, TX 78712. E-mail: morikawa@mail.utexas.edu.
Copyright © 2004 Society for Neuroscience 0270-6474/04/2411079-09$15.00/0
References
- Arima J, Matsumoto N, Kishimoto K, Akaike N (2001) Spontaneous miniature outward currents in mechanically dissociated rat Meynert neurons. J Physiol (Lond) 534: 99-107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Augustine GJ, Santamaria F, Tanaka K (2003) Local calcium signaling in neurons. Neuron 40: 331-346. [DOI] [PubMed] [Google Scholar]
- Bak J, White P, Timar G, Missiaen L, Genazzani AA, Galione A (1999) Nicotinic acid adenine dinucleotide phosphate triggers Ca2+ release from brain microsomes. Curr Biol 9: 751-754. [DOI] [PubMed] [Google Scholar]
- Bean BP (1989) Classes of calcium channels in vertebrate cells. Annu Rev Physiol 51: 367-384. [DOI] [PubMed] [Google Scholar]
- Berg I, Potter BV, Mayr GW, Guse AH (2000) Nicotinic acid adenine dinucleotide phosphate (NAADP(+)) is an essential regulator of T-lymphocyte Ca(2+)-signaling. J Cell Biol 150: 581-588. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berridge MJ (1995) Inositol trisphosphate and calcium signaling. Ann NY Acad Sci 766: 31-43. [DOI] [PubMed] [Google Scholar]
- Berridge MJ (1998) Neuronal calcium signaling. Neuron 21: 13-26. [DOI] [PubMed] [Google Scholar]
- Bezprozvanny I, Watras J, Ehrlich BE (1991) Bell-shaped calcium-response curves of Ins(1,4,5)P3- and calcium-gated channels from endoplasmic reticulum of cerebellum. Nature 351: 751-754. [DOI] [PubMed] [Google Scholar]
- Boland LM, Morrill JA, Bean BP (1994) ω-Conotoxin block of N-type calcium channels in frog and rat sympathetic neurons. J Neurosci 14: 5011-5027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bolton TB, Imaizumi Y (1996) Spontaneous transient outward currents in smooth muscle cells. Cell Calcium 20: 141-152. [DOI] [PubMed] [Google Scholar]
- Bond CT, Maylie J, Adelman JP (1999) Small-conductance calcium-activated potassium channels. Ann NY Acad Sci 868: 370-378. [DOI] [PubMed] [Google Scholar]
- Brailoiu E, Patel S, Dun NJ (2003) Modulation of spontaneous transmitter release from the frog neuromuscular junction by interacting intracellular Ca(2+) stores: critical role for nicotinic acid-adenine dinucleotide phosphate (NAADP). Biochem J 373: 313-318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brown DA, Constanti A, Adams PR (1983) Ca-activated potassium current in vertebrate sympathetic neurons. Cell Calcium 4: 407-420. [DOI] [PubMed] [Google Scholar]
- Cancela JM, Churchill GC, Galione A (1999) Coordination of agonist-induced Ca2+-signalling patterns by NAADP in pancreatic acinar cells. Nature 398: 74-76. [DOI] [PubMed] [Google Scholar]
- Cardozo DL, Bean BP (1995) Voltage-dependent calcium channels in rat midbrain dopamine neurons: modulation by dopamine and GABAB receptors. J Neurophysiol 74: 1137-1148. [DOI] [PubMed] [Google Scholar]
- Carter AG, Regehr WG (2002) Quantal events shape cerebellar interneuron firing. Nat Neurosci 5: 1309-1318. [DOI] [PubMed] [Google Scholar]
- Castellanos FX, Tannock R (2002) Neuroscience of attention-deficit/hyperactivity disorder: the search for endophenotypes. Nat Rev Neurosci 3: 617-628. [DOI] [PubMed] [Google Scholar]
- Chuhma N, Zhang H, Masson J, Zhuang X, Sulzer D, Hen R, Rayport S (2004) Dopamine neurons mediate a fast excitatory signal via their glutamatergic synapses. J Neurosci 24: 972-981. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clements JD, Bekkers JM (1997) Detection of spontaneous synaptic events with an optimally scaled template. Biophys J 73: 220-229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ehrlich BE, Kaftan E, Bezprozvannaya S, Bezprozvanny I (1994) The pharmacology of intracellular Ca(2+)-release channels. Trends Pharmacol Sci 15: 145-149. [DOI] [PubMed] [Google Scholar]
- Fiorillo CD, Williams JT (1998) Glutamate mediates an inhibitory postsynaptic potential in dopamine neurons. Nature 394: 78-82. [DOI] [PubMed] [Google Scholar]
- Fletcher GH, Chiappinelli VA (1992) Spontaneous miniature hyperpolarizations of presynaptic nerve terminals in the chick ciliary ganglion. Brain Res 579: 165-168. [DOI] [PubMed] [Google Scholar]
- Galione A (1994) Cyclic ADP-ribose, the ADP-ribosyl cyclase pathway and calcium signalling. Mol Cell Endocrinol 98: 125-131. [DOI] [PubMed] [Google Scholar]
- Galione A, Churchill GC (2002) Interactions between calcium release pathways: multiple messengers and multiple stores. Cell Calcium 32: 343-354. [DOI] [PubMed] [Google Scholar]
- Ghosh TK, Eis PS, Mullaney JM, Ebert CL, Gill DL (1988) Competitive, reversible, and potent antagonism of inositol 1,4,5-trisphosphate-activated calcium release by heparin. J Biol Chem 263: 11075-11079. [PubMed] [Google Scholar]
- Gomez TM, Spitzer NC (1999) In vivo regulation of axon extension and pathfinding by growth-cone calcium transients. Nature 397: 350-355. [DOI] [PubMed] [Google Scholar]
- Gonon FG (1988) Nonlinear relationship between impulse flow and dopamine released by rat midbrain dopaminergic neurons as studied by in vivo electrochemistry. Neuroscience 24: 19-28. [DOI] [PubMed] [Google Scholar]
- Hausser M, Clark BA (1997) Tonic synaptic inhibition modulates neuronal output pattern and spatiotemporal synaptic integration. Neuron 19: 665-678. [DOI] [PubMed] [Google Scholar]
- Kinsbourne M (1973) Minimal brain dysfunction as a neurodevelopmental lag. Ann NY Acad Sci 205: 268-273. [DOI] [PubMed] [Google Scholar]
- Kolta MG, Scalzo FM, Ali SF, Holson RR (1990) Ontogeny of the enhanced behavioral response to amphetamine in amphetamine-pretreated rats. Psychopharmacology (Berl) 100: 377-382. [DOI] [PubMed] [Google Scholar]
- Kononenko NI, Dudek FE (2004) Mechanism of irregular firing of suprachiasmatic nucleus neurons in rat hypothalamic slices. J Neurophysiol 91: 267-273. [DOI] [PubMed] [Google Scholar]
- Lee HC, Aarhus R (1995) A derivative of NADP mobilizes calcium stores insensitive to inositol trisphosphate and cyclic ADP-ribose. J Biol Chem 270: 2152-2157. [DOI] [PubMed] [Google Scholar]
- Lee JH, Gomora JC, Cribbs LL, Perez-Reyes E (1999) Nickel block of three cloned T-type calcium channels: low concentrations selectively block alpha1H. Biophys J 77: 3034-3042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Loizou LA (1972) The postnatal ontogeny of monoamine-containing neurones in the central nervous system of the albino rat. Brain Res 40: 395-418. [DOI] [PubMed] [Google Scholar]
- Martin RL, Lee JH, Cribbs LL, Perez-Reyes E, Hanck DA (2000) Mibefradil block of cloned T-type calcium channels. J Pharmacol Exp Ther 295: 302-308. [PubMed] [Google Scholar]
- Mathers DA, Barker JL (1984) Spontaneous voltage and current fluctuations in tissue cultured mouse dorsal root ganglion cells. Brain Res 293: 35-47. [DOI] [PubMed] [Google Scholar]
- Merriam LA, Scornik FS, Parsons RL (1999) Ca(2+)-induced Ca(2+) release activates spontaneous miniature outward currents (SMOCs) in parasympathetic cardiac neurons. J Neurophysiol 82: 540-550. [DOI] [PubMed] [Google Scholar]
- Meszaros LG, Bak J, Chu A (1993) Cyclic ADP-ribose as an endogenous regulator of the non-skeletal type ryanodine receptor Ca2+ channel. Nature 364: 76-79. [DOI] [PubMed] [Google Scholar]
- Montague PR, McClure SM, Baldwin PR, Phillips PE, Budygin EA, Stuber GD, Kilpatrick MR, Wightman RM (2004) Dynamic gain control of dopamine delivery in freely moving animals. J Neurosci 24: 1754-1759. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morikawa H, Imani F, Khodakhah K, Williams JT (2000) Inositol 1,4,5-triphosphate-evoked responses in midbrain dopamine neurons. J Neurosci 20: RC103(1-5). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morikawa H, Khodakhah K, Williams JT (2003) Two intracellular pathways mediate metabotropic glutamate receptor-induced Ca2+ mobilization in dopamine neurons. J Neurosci 23: 149-157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Newcomb R, Szoke B, Palma A, Wang G, Chen X, Hopkins W, Cong R, Miller J, Urge L, Tarczy-Hornoch K, Loo JA, Dooley DJ, Nadasdi L, Tsien RW, Lemos J, Miljanich G (1998) Selective peptide antagonist of the class E calcium channel from the venom of the tarantula Hysterocrates gigas Biochemistry 37: 15353-15362. [DOI] [PubMed] [Google Scholar]
- Paladini CA, Fiorillo CD, Morikawa H, Williams JT (2001) Amphetamine selectively blocks inhibitory glutamate transmission in dopamine neurons. Nat Neurosci 4: 275-281. [DOI] [PubMed] [Google Scholar]
- Parsons RL, Barstow KL, Scornik FS (2002) Spontaneous miniature hyperpolarizations affect threshold for action potential generation in mudpuppy cardiac neurons. J Neurophysiol 88: 1119-1127. [DOI] [PubMed] [Google Scholar]
- Patel S, Churchill GC, Sharp T, Galione A (2000) Widespread distribution of binding sites for the novel Ca2+-mobilizing messenger, nicotinic acid adenine dinucleotide phosphate, in the brain. J Biol Chem 275: 36495-36497. [DOI] [PubMed] [Google Scholar]
- Patel S, Churchill GC, Galione A (2001) Coordination of Ca2+ signalling by NAADP. Trends Biochem Sci 26: 482-489. [DOI] [PubMed] [Google Scholar]
- Perez-Reyes E (2003) Molecular physiology of low-voltage-activated t-type calcium channels. Physiol Rev 83: 117-161. [DOI] [PubMed] [Google Scholar]
- Petersen OH, Cancela JM (1999) New Ca2+-releasing messengers: are they important in the nervous system? Trends Neurosci 22: 488-495. [DOI] [PubMed] [Google Scholar]
- Pitts DK, Freeman AS, Chiodo LA (1990) Dopamine neuron ontogeny: electrophysiological studies. Synapse 6: 309-320. [DOI] [PubMed] [Google Scholar]
- Porter VA, Bonev AD, Knot HJ, Heppner TJ, Stevenson AS, Kleppisch T, Lederer WJ, Nelson MT (1998) Frequency modulation of Ca2+ sparks is involved in regulation of arterial diameter by cyclic nucleotides. Am J Physiol 274: C1346-C1355. [DOI] [PubMed] [Google Scholar]
- Randall A, Tsien RW (1995) Pharmacological dissection of multiple types of Ca2+ channel currents in rat cerebellar granule neurons. J Neurosci 15: 2995-3012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Randall AD, Tsien RW (1997) Contrasting biophysical and pharmacological properties of T-type and R-type calcium channels. Neuropharmacology 36: 879-893. [DOI] [PubMed] [Google Scholar]
- Satin LS, Adams PR (1987) Spontaneous miniature outward currents in cultured bullfrog neurons. Brain Res 401: 331-339. [DOI] [PubMed] [Google Scholar]
- Seidler NW, Jona I, Vegh M, Martonosi A (1989) Cyclopiazonic acid is a specific inhibitor of the Ca2+-ATPase of sarcoplasmic reticulum. J Biol Chem 264: 17816-17823. [PubMed] [Google Scholar]
- Seutin V, Massotte L, Scuvee-Moreau J, Dresse A (1998) Spontaneous apamin-sensitive hyperpolarizations in dopaminergic neurons of neonatal rats. J Neurophysiol 80: 3361-3364. [DOI] [PubMed] [Google Scholar]
- Seutin V, Mkahli F, Massotte L, Dresse A (2000) Calcium release from internal stores is required for the generation of spontaneous hyperpolarizations in dopaminergic neurons of neonatal rats. J Neurophysiol 83: 192-197. [DOI] [PubMed] [Google Scholar]
- Smith JS, Imagawa T, Ma J, Fill M, Campbell KP, Coronado R (1988) Purified ryanodine receptor from rabbit skeletal muscle is the calcium-release channel of sarcoplasmic reticulum. J Gen Physiol 92: 1-26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tang F, Dent EW, Kalil K (2003) Spontaneous calcium transients in developing cortical neurons regulate axon outgrowth. J Neurosci 23: 927-936. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tepper JM, Trent F, Nakamura S (1990) Postnatal development of the electrical activity of rat nigrostriatal dopaminergic neurons. Brain Res Dev Brain Res 54: 21-33. [DOI] [PubMed] [Google Scholar]
- Trent F, Nakamura S, Tepper JM (1991) Amphetamine exerts anomalous effects on dopaminergic neurons in neonatal rats in vivo. Eur J Pharmacol 204: 265-272. [DOI] [PubMed] [Google Scholar]
- Verkhratsky A, Shmigol A (1996) Calcium-induced calcium release in neurones. Cell Calcium 19: 1-14. [DOI] [PubMed] [Google Scholar]
- Voorn P, Kalsbeek A, Jorritsma-Byham B, Groenewegen HJ (1988) The preand postnatal development of the dopaminergic cell groups in the ventral mesencephalon and the dopaminergic innervation of the striatum of the rat. Neuroscience 25: 857-887. [DOI] [PubMed] [Google Scholar]
- Walseth TF, Lee HC (1993) Synthesis and characterization of antagonists of cyclic-ADP-ribose-induced Ca2+ release. Biochim Biophys Acta 1178: 235-242. [DOI] [PubMed] [Google Scholar]
- Wang L, Pitts DK (1994) Postnatal development of mesoaccumbens dopamine neurons in the rat: electrophysiological studies. Brain Res Dev Brain Res 79: 19-28. [DOI] [PubMed] [Google Scholar]
- Wilson CJ, Callaway JC (2000) Coupled oscillator model of the dopaminergic neuron of the substantia nigra. J Neurophysiol 83: 3084-3100. [DOI] [PubMed] [Google Scholar]
- Wolfart J, Roeper J (2002) Selective coupling of T-type calcium channels to SK potassium channels prevents intrinsic bursting in dopaminergic midbrain neurons. J Neurosci 22: 3404-3413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Womack MD, Khodakhah K (2004) Dendritic control of spontaneous bursting in cerebellar Purkinje cells. J Neurosci 24: 3511-3521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xia XM, Fakler B, Rivard A, Wayman G, Johnson-Pais T, Keen JE, Ishii T, Hirschberg B, Bond CT, Lutsenko S, Maylie J, Adelman JP (1998) Mechanism of calcium gating in small-conductance calcium-activated potassium channels. Nature 395: 503-507. [DOI] [PubMed] [Google Scholar]
- Zamponi GW, Bourinet E, Snutch TP (1996) Nickel block of a family of neuronal calcium channels: subtype- and subunit-dependent action at multiple sites. J Membr Biol 151: 77-90. [DOI] [PubMed] [Google Scholar]
- Zucchi R, Ronca-Testoni S (1997) The sarcoplasmic reticulum Ca2+ channel/ryanodine receptor: modulation by endogenous effectors, drugs and disease states. Pharmacol Rev 49: 1-51. [PubMed] [Google Scholar]