

Abstract
Transcriptional coactivators such as p300 act as crucial elements in the eukaryotic gene regulation network. These proteins bind to various transcription factors which recruit them to specific gene regions whose chromatin structure subsequently is remodeled. Previously, we have shown that C/EBPβ recruits p300 by interacting with the E1A-binding site of the coactivator. We now show that C/EBPβ not only binds to p300 but also triggers massive phosphorylation of p300. This novel activity of C/EBPβ is dependent on the E1A-binding region of p300 as well as on several subdomains of C/EBPβ, all of which are involved in the p300–C/EBPβ interaction. We have identified several sites of C/EBPβ-inducible phosphorylation within the C-terminal domain of p300. Mutation of these sites substantially impairs the activity of p300 as a coactivator of C/EBPβ. Interestingly, phosphorylation of p300 is also triggered by other C/EBP family members as well as by various other transcription factors that interact with the E1A-binding domain of p300, suggesting that this novel phosphorylation mechanism may be of general relevance.
Keywords: CEBP/coactivator/p300/phosphorylation
Introduction
In eukaryotic cells, assembly of the transcriptional machinery on the DNA is hampered by the tight packing of DNA into chromatin (for reviews see Grunstein, 1997; Wade and Wolffe, 1997; Struhl, 1998). There is a large body of evidence indicating that one major mechanism to overcome the repressive effects of chromatin involves the coordinate action of transcription factors and so-called coactivators, such as p300 or the CREB-binding protein (CBP), leading to a localized remodeling of the chromatin structure and permitting the transcriptional machinery access to the DNA (Janknecht and Hunter, 1996; Shikama et al., 1997; Giordano and Avantaggiati, 1999; Goodman and Smolik, 2000). One important aspect of chromatin remodeling that has been studied intensely is the acetylation of histones by histone acetyltransferases (HATs). The coactivators p300 and CBP, which were identified originally on the basis of their interaction with the adenovirus E1A protein and the transcription factor CREB (Chrivia et al., 1993; Eckner et al., 1994), have intrinsic HAT activity (Bannister and Kouzarides, 1996; Ogryzko et al., 1996); in addition, they recruit other cofactors possessing HAT activity via protein–protein interactions, such as P/CAF, ACTR, SRC-1 and p/CIP (Chakravarti et al., 1996; Yang et al., 1996; Chen et al., 1997; Spencer et al., 1997; Torchia et al., 1997). The importance of p300 and CBP for the regulation of transcription is supported by their ability to interact with and potentiate the activity of a large number of transcription factors, including nuclear hormone receptors, CREB, Myb, Jun, Fos, MyoD, p53, CCAAT box/enhancer-binding protein β (C/EBPβ) and others (Arias et al., 1994; Kwok et al., 1994; Chakravarti et al., 1996; Janknecht and Nordheim, 1996; Oelgeschlager et al., 1996; Yuan et al., 1996; Gu and Roeder, 1997; Mink et al., 1997). It is generally believed that the interaction of p300 and CBP with these factors provides a means to localize the coactivators to specific DNA regions, resulting in chromatin remodeling and, ultimately, the activation of specific genes.
The C/EBPs are members of the basic region/leucine zipper class of transcription factors and have been implicated in a variety of processes, including the differentiation of certain cell types, such as liver and fat cells, granulocytes and macrophages (Darlington et al., 1998; Lekstrom-Himes and Xanthopoulos, 1998; Yamanaka et al., 1998). C/EBP family members are highly expressed during the differentiation of these cell types and, in concert with other transcription factors, play key roles in the regulation of cell-type specific gene expression in these lineages (Cao et al., 1991; Natsuka et al., 1992; Scott et al., 1992; Katz et al., 1993; Yeh et al., 1995). In the myelomonocytic lineage, C/EBPβ cooperates with Myb to generate a combinatorial molecular switch that is responsible for the activation of several myeloid-specific genes, such as mim-1, the lysozyme gene and the C/EBPβ gene itself (Burk et al., 1993; Ness et al., 1993; Mink et al., 1996, 1999). Both Myb and C/EBPβ interact with p300/CBP, making this coactivator an important component of this differentiation switch.
We and others have shown before that C/EBPβ recruits p300/CBP by interacting with the E1A-binding domain of the coactivator (Oelgeschlager et al., 1996; Mink et al., 1997). We have now analyzed the cooperation of C/EBPβ and p300 in more detail. Our results reveal a novel aspect of the mechanism by which C/EBPβ recruits p300 and provide further insight into the interplay between transcription factors and coactivators. Our data show that docking of p300 to C/EBPβ not only is involved in translocating the coactivator to specific gene regions but, in addition, triggers massive phosphorylation of the C-terminal domain of p300 and thereby modulates its activity as a coactivator. We also show that this activity of C/EBPβ is shared by a number of other transcription factors interacting with p300.
Results
C/EBPβ induces modified forms of p300
Previously, we have shown that C/EBPβ binds the coactivator p300 resulting in stimulation of C/EBPβ activity (Mink et al., 1997). During this work, we noted that the mobility of p300 in an SDS–polyacryamide gel was affected by C/EBPβ. As shown in Figure 1, in cells expressing C/EBPβ, a fraction of p300 was converted to a slower migrating form. In addition, the total amount of p300 was slightly increased in the presence of C/EBPβ. Because of the large size of the protein, the different forms of p300 were difficult to resolve. To provide a more convenient assay for the C/EBPβ-induced mobility shift, we expressed a partially deleted form of p300 with or without C/EBPβ and analyzed its mobility. As shown in Figure 2A, the mobility shift was much more obvious when a C-terminal fragment of p300, p300/1751–2370, was used instead of the full-length protein. The shifted band appeared fuzzy, suggesting that C/EBPβ induces a variety of forms of p300 migrating with slightly different mobilities. Again, the total amount of p300/1751–2370 was increased in the presence of C/EBPβ.
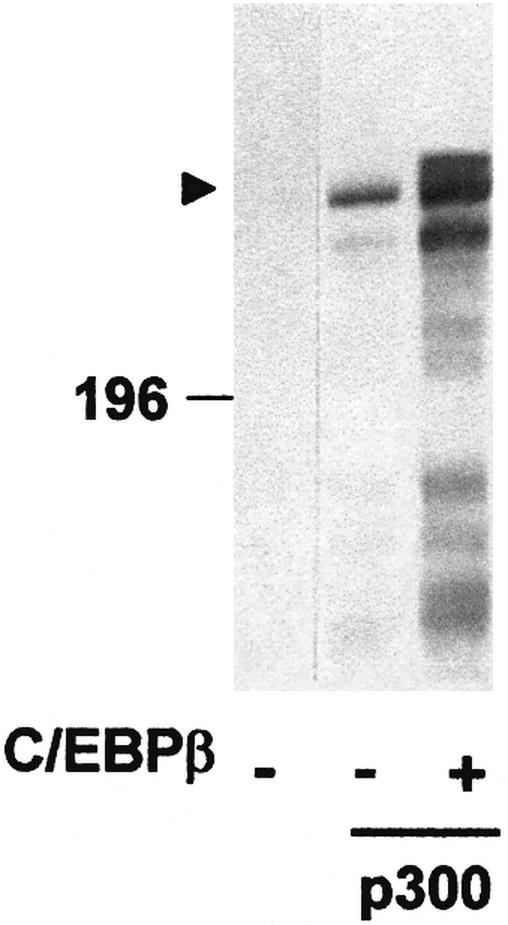
Fig. 1. Appearance of a slow migrating form of p300 in the presence of C/EBPβ. QT6 cells were transfected with expression vectors for full-length p300 (10 µg) and C/EBPβ (1 µg), as indicated below the lanes. Controls were transfected with the corresponding empty expression vectors. Cells were analyzed 24 h after transfection by SDS–PAGE and western blotting using p300-specific antibodies. The position of a molecular weight marker (in kDa) is indicated. Full-length p300 is marked by an arrowhead.

Fig. 2. Detection of slow-migrating forms of the C-terminal domain of p300 in the presence of C/EBPβ. The schematic structures of full-length p300 and truncated derivatives are shown at the top. QT6 cells were transfected with expression vectors for p300 constructs (10 µg each) and C/EBPβ (1 µg), as indicated. Controls were transfected with empty expression vectors. (A and B) After 24 h, cell extracts were analyzed by SDS–PAGE and western blotting with p300-specific antibodies. (C and D) Cell extracts prepared 24 h after transfection were fractionated by SDS–PAGE (lanes 1 and 2) or immunoprecipitated with C/EBPβ-specific antiserum (lanes 3 and 4), followed by SDS–PAGE. All lanes were then analyzed by western blotting using p300-specific antibodies. In (B), the intense bands at the bottom of lanes 3 and 4 are due to the antibodies used for immunoprecipitation. Molecular weight markers (in kDa) are indicated.
To determine whether the mobility shift was dependent on the interaction with C/EBPβ, different parts of p300 were co-expressed with C/EBPβ. As illustrated further in Figure 2A, neither p300/1515–1998 nor p300/1853–2370 were affected by C/EBPβ. As shown before, p300/1853–2370 lacks the C/EBP-binding site, whereas p300/1515–1998 interacts with C/EBPβ (Mink et al., 1997). Together, these observations suggest that binding to C/EBPβ is necessary for the induction of a mobility shift but that sequences C-terminal of the C/EBP-binding site of p300 are also required. Deletion of sequences from the C-terminus of p300 resulted in less dramatic shifts (Figure 2B), indicating that the C/EBPβ-dependent mobility shift is not due to a unique site in p300 but that several sites located in the C-terminal domain of p300 contribute to the shift.
To investigate whether the slow migrating forms of p300 were bound to C/EBPβ, we performed in vivo co-immunoprecipitation experiments. Figure 2C and D shows that the slow migrating forms of p300/1751–2370 and p300/1751–2218 could be co-precipitated with C/EBPβ-specific antibodies. In contrast, the fast migrating forms of p300/1751–2370 and p300/1751–2218 were not precipitated (compare lanes 2 and 4 of Figure 2C and D). This suggests that the fast migrating forms of p300 interact with C/EBPβ more weakly than the slow migrating forms (or not at all).
The ability to induce a mobility shift of p300 is shared by different C/EBP family members and depends on the interaction of p300 and C/EBP
To explore further the significance of the mobility shift induced by C/EBPβ, we wished to know whether the ability to trigger this shift is a unique property of C/EBPβ or whether it is shared by other C/EBP family members. p300/1751–2370 was expressed together with C/EBPα, C/EBPβ or C/EBPδ followed by analysis of the mobility of p300. As illustrated in Figure 3, all C/EBP family members tested were able to induce slow migrating forms of p300. Thus, the mechanism revealed by our studies is shared by several C/EBP family members.
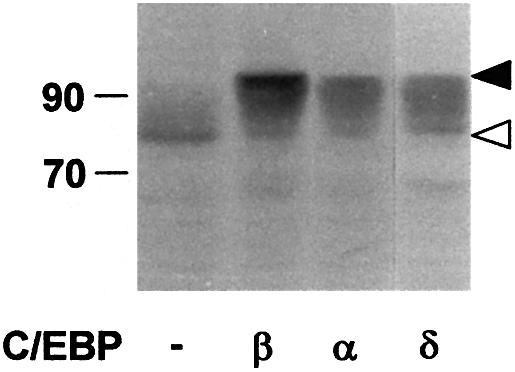
Fig. 3. Induction of slow migrating forms of p300 by different C/EBP family members. QT6 cells were transfected with expression vectors for p300/1751–2370 (10 µg) and different C/EBP family members (1 µg each), as indicated. After 24 h, cells were analyzed by SDS–PAGE and western blotting using p300-specific antibodies. Slow and fast migrating forms of p300/1751–2370 are marked by black and white arrows. Molecular weight markers (in kDa) are indicated.
Comparison of the N-terminal domains of different C/EBPs has revealed several highly conserved regions whose integrity is critical for C/EBP-mediated transactivation (Katz et al., 1993). As shown in the case of C/EBPα, mutations that disrupt these regions diminish the transactivation potential of the protein (Nerlov and Ziff, 1995). We suspected that these conserved regions might be responsible for inducing the mobility shift of p300. To address this possibility, we used a set of previously described point mutations of C/EBPα, referred to as Y67A, FL77,78AA and Y67A, FL77,78AA (Nerlov and Ziff, 1995). Figure 4A shows that mutants Y67A and FL77,78AA both have some effect on the mobility shift of p300/1751–2370. Combination of both mutants, however, strongly reduced the mobility shift. This shows that two highly conserved regions within the transactivation domain of C/EBPα cooperate to induce the mobility shift of p300. Since the mobility shift was not completely abolished in the double mutant, it is possible that yet another region of C/EBPα contributes slightly to the mobility shift of p300.

Fig. 4. Mutation of the transactivation domain of C/EBPα prevents appearance of slow migrating forms of p300. (A) QT6 cells were transfected with expression vectors for p300/1751–2370 (10 µg) and wild-type or mutant C/EBPα (1 µg each), as indicated. After 24 h, cells were analyzed by SDS–PAGE and western blotting with antibodies against p300 (top) or C/EBPα (bottom). (B) QT6 cells were transfected with the Gal4-responsive reporter pG5E4-38Luc (3 µg), the β-galactosidase plasmid pCMVβ (0.2 µg) and expression vectors for different Gal4–C/EBPα fusion proteins (0.02 µg). Western blotting showed that these proteins were expressed in similar amounts (data not shown). In addition, an expression vector for a hybrid protein consisting of the VP16 transactivation domain fused to p300 amino acids 1737–1983 (pCDNA3-SN1G/3-VP16, 2 µg) was included (white bars). As control (black bars), an expression vector encoding only the VP16 transactivation domain was used (pCDNA3-VP16, 2 µg). The columns show the average luciferase activity determined 24 h after transfection and normalized with respect to the β-galactosidase activity. Thin lines show standard deviations.
To investigate whether these domains of C/EBPα are required for interaction with p300, we used a two-hybrid assay described previously (Mink et al., 1997). Gal4–C/EBPα fusion proteins containing either wild-type or mutant C/EBPα sequences were co-expressed with a p300–VP16 protein encompassing the C/EBP-binding site of p300. As shown in Figure 4B, expression of the p300–VP16 fusion protein strongly increased the activity of a Gal4-dependent reporter gene in the presence of Gal4–C/EBPα, but not in its absence, indicating that both proteins interact. The results obtained with the mutant proteins agree with the results of the shift assay: mutants Y67A and FL77,78AA showed partially diminished interaction in the two-hybrid assay, whereas the mutant Y67A, FL77,78AA, which had the strongest effect on the ability to induce a mobility shift of p300, had lost the ability to interact with p300. Thus, this experiment shows that two of the conserved domains of C/EBPα are involved in interacting with p300 and that the same regions are required for the observed mobility shift of p300.
In the light of these findings, we wished to know whether in the case of C/EBPβ the same conserved regions were responsible for inducing the mobility shift of p300. We generated various mutants of C/EBPβ and analyzed their effects on the mobility of p300 (Figure 5). Mutant C/EBPβ-mut3a corresponds to the FL77,78AA mutant of C/EBPα, and mutant C/EBPβ-mut3d is analogous to the Y67A,FL77,78AA mutant of C/EBPα. As shown in Figure 5B, in the presence of C/EBPβ-wt, the forms of p300 with the lowest mobility were the most abundant. In contrast, in the presence of C/EBPβ mutants 3a and 3d, p300 forms with intermediate mobility increased in abundance, indicating that both mutants were less active in inducing a mobility shift of p300. However, it also appeared that mutant 3d had a less strong effect than the corresponding C/EBPα mutant, suggesting that additional domains of C/EBPβ are involved in altering the mobility of p300. Further analysis showed that these additional sequences reside at the N-terminus of C/EBPβ and within its DNA-binding domain. The relevant mutants and their effects on the mobility of p300 are illustrated in Figure 5B. Deletion of 33 amino acids from the N-terminus of C/EBPβ had a similar effect on the mobility of p300 to that of mutants 3a and 3d. The combination of the N-terminal deletion with mutants 3a or 3d led to a much stronger effect on the ability of C/EBPβ to cause a mobility shift of p300. However, even after combining these different mutations, C/EBPβ retained some activity to shift the mobility of p300 (see lanes 6 and 7 of Figure 5B). Surprisingly, the region responsible for this residual activity is the DNA-binding domain. Replacement of this domain of C/EBPβ with the DNA-binding domain of C/EBPα (in the presence of the other mutations described above) resulted in a protein that was completely unable to shift the mobility of p300.

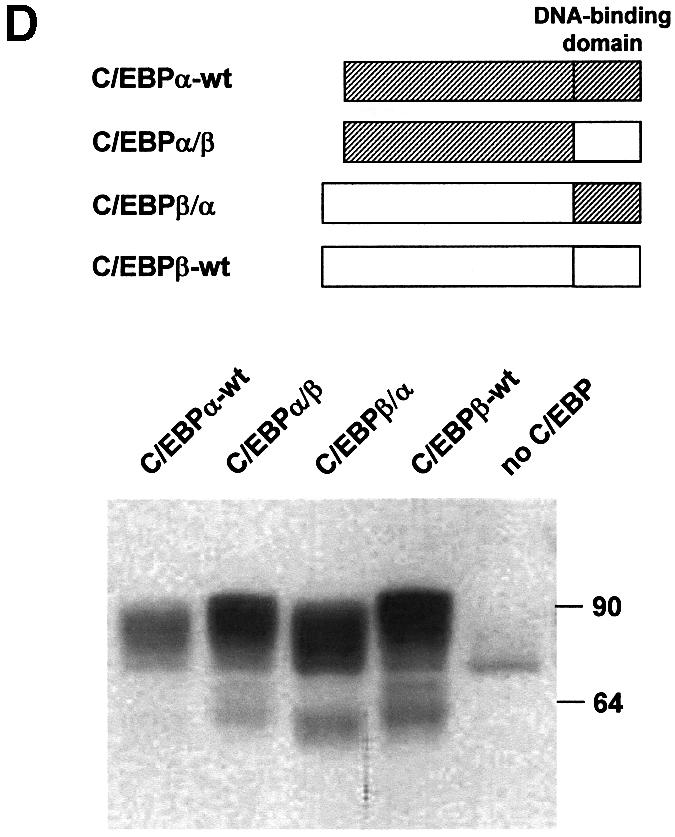
Fig. 5. Induction of the p300 mobility shift depends on multiple domains of C/EBPβ. (A) Schematic illustration of wild-type and mutant C/EBPβ proteins. (B and C) QT6 cells were transfected with expression vectors for p300/1751–2370 (10 µg) and wild-type or mutant C/EBPβ (1 µg each), as indicated. After 24 h, cells were analyzed by SDS–PAGE and western blotting with antibodies against p300 (top) or C/EBPβ (bottom). (D) Recombinants of chicken C/EBPα and β were analyzed as described above. Bands in the lower part of the gel represent degradation products.
That the C/EBPβ DNA-binding domain is involved in the mobility shift of p300 was also apparent with recombinants of C/EBPα and β, whose DNA-binding domains had been exchanged (Figure 5D). A recombinant of C/EBPα with the DNA-binding domain of C/EBPβ (C/EBPα/β) caused a slightly stronger mobility shift of p300 than C/EBPα, whereas replacement of the DNA-binding domain of C/EBPβ (C/EBPβ/α) with that of C/EBPα resulted in a reduced mobility shift compared with C/EBPβ-wt (Figure 5D).
To investigate whether these different domains of C/EBPβ all contribute to the interaction with p300, we constructed various Gal4 fusion proteins and tested them in two-hybrid experiments. Figure 6 illustrates that there was a good correlation between the ability of the different mutants to stimulate the Gal4-dependent reporter gene in the presence of p300–VP16 and their effects on the mobility of p300. We therefore concluded that multiple regions of C/EBPβ are involved in the interaction with p300 as well as in altering the mobility of p300. Our results indicate that C/EBPα and C/EBPβ differ significantly with respect to their interaction with p300. That C/EBPβ causes a more drastic mobility shift of p300 than C/EBPα is consistent with the finding that apparently more regions of C/EBPβ than of C/EBPα interact with p300.

Fig. 6. p300 interacts with multiple domains of C/EBPβ. QT6 cells were transfected with the Gal4-responsive reporter pG5E4-38Luc (3 µg), the β-gal actosidase plasmid pCMVβ (0.2 µg) and expression vectors for the different Gal4–C/EBPβ fusion proteins (0.02 µg). Western blotting showed that these proteins were expressed in similar amounts (data not shown). In addition, an expression vector for a hybrid protein consisting of the VP16 transactivation domain fused to p300 amino acids 1737–1983 (pCDNA3-SN1G/3-VP16, 2 µg) was included (white bars). As control (black bars), an expression vector encoding only the VP16 transactivation domain was used (pCDNA3-VP16, 2 µg). Columns show the average luciferase activity determined 24 h after transfection and normalized with respect to the co-transfected β-galactosidase activity.
Finally, we investigated whether C/EBPβ and p300 interact directly or whether the interaction is mediated by another protein. To address this question, we subjected bacterially expressed C/EBPβ to a pull-down assay using different GST–p300 fusion proteins bound to glutathione– Sepharose. As shown in Figure 7, bacterial C/EBPβ bound to GST–p300(1752–1998) and GST–p300(1710–1891), but not to GST itself or a GST–p300(1883–1927) fusion protein. This experiment demonstrates that the interaction of p300 and C/EBPβ does not depend on other eukaryotic proteins and, thus, probably is due to direct contacts between both proteins. Furthermore, this experiment localizes the sequence of p300 involved in C/EBPβ binding to amino acids 1752–1891.

Fig. 7. Interaction of bacterially expressed C/EBPβ and p300. (A) Schematic illustration of GST–p300 fusion proteins. (B) Similar amounts of the indicated GST fusion proteins were bound to glutathione–Sepharose, incubated with bacterially expressed C/EBPβ and subjected to a GST pull-down assay. Input (2% of total amount, lane 1) and bound proteins (lanes 2–5) were analyzed by 10% SDS–PAGE and western blotting using C/EBPβ-specific antibodies.
Slow migrating forms of p300 appear during myeloid differentiation of promyelocytic HL60 cells
In the experiments described above, we have examined the effect of C/EBPβ on the mobility of p300 by using transiently transfected cells expressing high levels of the respective proteins. It was therefore of interest to see whether slow migrating forms of p300 also occur under physiological conditions. To address this question, we used the promyelocytic HL60 cell line whose differen tiation to macrophages can be induced by the tumor promoter tetradecanoylphorbol-13-acetate (TPA) and is accompanied by an increase in the expression of several C/EBP family members, including C/EBPβ (Natsuka et al., 1992; Scott et al., 1992). Macrophage differentiation of HL60 cells was induced by treating the cells with TPA for 2 days, and p300 was visualized by SDS–PAGE and western blotting. As shown in Figure 8, slower migrating forms of p300, whose mobility was identical to that of the slow migrating p300 observed after co-expression of C/EBPβ, accumulated in the differentiated cells, suggesting that such slow migrating forms also appear in untransfected cells.
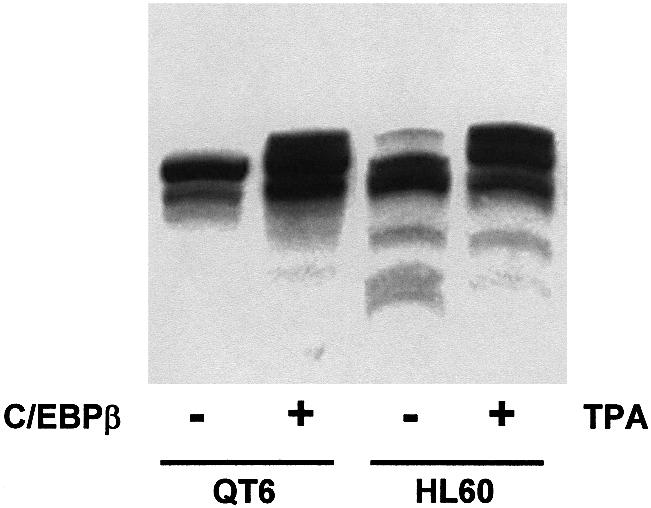
Fig. 8. Appearance of slow migrating forms of p300 in HL60 cells treated with PMA. Cell extracts from untreated HL60 cells (lane 3) or from HL60 cells treated for 2 days with 100 nM TPA (lane 4) were analyzed by SDS–PAGE and western blotting using p300-specific antibodies. Extracts from QT6 cells transfected with the expression vector for p300 (lane 1) or with expression vectors for p300 and C/EBPβ (lane 2) were analyzed in parallel.
C/EBPβ induces phosphorylation of p300
To address the question of whether the C/EBPβ-induced mobility shift of p300 is due to phosphorylation, we first determined whether p300 was affected by phosphatase treatment. As shown in Figure 9A and B, the slow migrating forms of full-length p300 or p300/1751–2370 were no longer detected after incubation with phosphatase, providing initial evidence that C/EBPβ induces phosphorylation of p300.

Fig. 9. C/EBPβ induces phosphorylation of p300. (A and B) QT6 cells were transfected with expression vectors for full-length p300 (10 µg), p300/1751–2370 (10 µg), C/EBPβ (1 µg) or empty expression vectors, as indicated. After 24 h, cell extracts were immunoprecipitated with p300-specific antibodies. Immunoprecipitates were incubated with shrimp alkaline phosphatase (AP, lanes marked +), mock incubated without phosphatase (AP, lanes marked –) or incubated with heat- inactivated phosphatase (AP, lanes marked h), followed by SDS–PAGE and western blotting with p300-specific antibodies. The last two lanes of (B) show total cell extract from transfected cells. The intense band at the bottom of the first four lanes of (B) is due to the antibodies used for immunoprecipitation. Molecular weight markers (in kDa) are indicated. Slow and fast migrating forms of p300/1751–2370 are marked by black and white arrows. (C) QT6 cells were transfected with expression vectors for p300/1751–2370 (10 µg) and C/EBPβ (1 µg), as indicated. After 24 h, cells were labeled with [32P]orthophosphate and proteins were immunoprecipitated with C/EBPβ- or p300-specific antibodies, followed by SDS–PAGE and autoradiography. The band marked by a black arrow corresponds to the slow migrating form of p300/1751–2370.
To confirm this conclusion, cells expressing p300/1751–2370 alone or p300/1751–2370 and C/EBPβ together were radiolabeled with [32P]orthophosphate, followed by immunoprecipitation with C/EBPβ- or p300-specific antibodies. This experiment demonstrated that the C-terminal fragment of p300 was phosphate labeled in the presence of C/EBPβ, but not in its absence (Figure 9C). The phosphate-labeled band (marked by a black arrow in Figure 9C) had the same mobility as the slow migrating form of p300/1751–2370 detected by western blotting, confirming that the modification of p300 induced by C/EBPβ is a phosphorylation. Virtually identical amounts of phosphate-labeled p300/1751–2370 were precipitated by p300- and by C/EBPβ-specific antibodies, indicating that most of the phosphorylated p300/1751–2370 is bound to C/EBPβ.
Identification of C/EBPβ-inducible phosphorylation sites of p300
To explore the relevance of the C/EBPβ-induced phosphorylation of p300, we set out to map relevant phosphorylation sites. The C-terminal domain of p300 contains many potential phosphorylation sites for proline-directed protein kinases, as well as sites for protein kinases A and C (Blom et al., 1999). We mutated a substantial number of these sites, using the C/EBPβ-induced mobility shift as an assay. Some of our data are shown in Figure 10A. p300/1751–2159, containing the mutation STTT1849,1851, 1854,1857AAAA (mutA), showed a mobility shift that was significantly smaller than the shift of the unmutated protein. Each of the relevant single mutations on their own had only minor effects (data not shown). In addition, we found that p300/1751–2370 containing the mutation S2279A (mutB) also had an effect on the C/EBPβ-induced mobility shift. Together, our data show that C/EBPβ induces phosphorylation of p300 at amino acids S1849, T1851, T1854, T1857 and T2279. However, these sites comprise only a subset of the phosphorylation sites that are affected by C/EBPβ because full-length p300 mutated at these positions still showed a significant mobility shift in the presence of C/EBPβ (see below). As pointed out before, the magnitude of the C/EBPβ-induced mobility shift also argues that C/EBPβ induces phosphorylation of p300 at multiple sites.

Fig. 10. Identification of C/EBPβ-inducible phosphorylation sites of p300. (A) Relevant amino acid sequences of wild-type p300 and mutants A and B are shown at the top. p300/1751–2159, p300/1751–2159 carrying mutation A, p300/1751–2370 and p300/1751–2370 carrying mutation B (5 µg each) were co-transfected into QT6 cells with or without C/EBPβ expression vector (1 µg), as indicated. After 24 h, cells were analyzed by SDS–PAGE and western blotting with p300-specific antibodies. (B) QT6 cells were transfected with the C/EBPβ-responsive reporter p-240Luc (3 µg), the β-galactosidase plasmid pCMVβ (0.2 µg) and 5 µg each of expression vectors for full-length p300 containing wild-type sequences (p300-wt) or carrying mutations A and B (p300-mutAB). Controls were transfected with empty expression vectors. After 24 h, cells were analyzed for luciferase and β-galactosidase activities. The bars show the average luciferase activity normalized with respect to the β-galactosidase activity. Thin lines show standard deviations. Aliquots of transfected cells were also analyzed by western blotting with p300-specific antibodies. The relevant part of the blot is shown at the bottom.
To determine whether mutation of the phosphorylation sites affects the ability of p300 to act as a coactivator for C/EBPβ, we transferred these mutations into full-length p300 and performed transactivation experiments with the C/EBPβ-responsive promoter of the chicken mim-1 gene (Figure 10B). Consistent with previous work (Mink et al., 1997), wild-type p300 strongly increased the ability of C/EBPβ to activate this reporter gene. In contrast, p300-mutAB was substantially less active in stimulating C/EBPβ-dependent transactivation, suggesting that C/EBPβ-induced phosphorylation of p300 is required for full activity of p300 as a coactivator of C/EBPβ. Analysis of the mobility of mutant p300 showed that it was still significantly affected by C/EBPβ. Thus, in addition to the sites identified here, there must be further sites of C/EBPβ-inducible phosphorylation.
The mobility of p300 is affacted by several other transcription factors
To determine whether or not the ability to trigger phosphorylation of p300 is a unique property of the C/EBP family, we analyzed the effects of other transcription factors known to interact with the E1A-binding domain of p300/CBP (such as c-Jun, c-Ets-1, c-Ets-2 and Pu.1) on the mobility of p300/1751–2370. As a control, we also included transcription factors that do not interact with the C-terminal domain of p300, such as ATF2 and AML1. Figure 11 shows that all C/EBP family members as well as c-Jun, c-Ets-1, c-Ets-2 and Pu.1 shifted the mobility of p300, while ATF2 and AML1 failed to do so. Thus, the modification described here is not unique to the C/EBPs but may have a more general relevance.
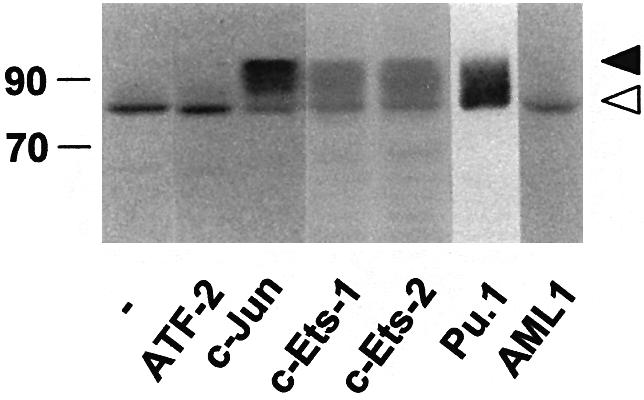
Fig. 11. Induction of slow migrating forms of p300 by different transcription factors. QT6 cells were transfected with expression vectors for p300/1751–2370 (10 µg) and different transcription factors (1 µg each), as indicated. After 24 h, cells were analyzed by SDS–PAGE and western blotting using p300-specific antibodies. Slow and fast migrating forms of p300/1751–2370 are marked by black and white arrows. Molecular weight markers (in kDa) are indicated.
C/EBPβ induces a shift in the subnuclear localization of p300
Finally, we were interested to know if C/EBPβ also affects other properties of p300 that might provide an explanation for how C/EBPβ induces phosphorylation of p300, and for the increased activity of phosphorylated p300. As a first step, we analyzed the subnuclear localization of p300 in the presence or absence of C/EBPβ by immunofluorescence microscopy. As illustrated in Figure 12A–F, p300 showed a speckled distribution in the absence of C/EBPβ and was more evenly distributed throughout the nucleus if C/EBPβ was present. In the presence of C/EBPβ, the distribution of p300 was very similar to that of C/EBPβ. The distribution of C/EBPβ was not influenced by p300 (not shown). Thus, it appears that C/EBPβ causes a change of the subnuclear localization of p300. Figure 12G–L shows the same experiment using the chicken fibroblast cell line DF-1 instead of the quail fibroblast line QT6. DF-1 cells have a flatter morphology than QT6 cells and, therefore, permit better resolution of nuclear structures. Again, it is evident that the distribution of p300 is altered in the presence of C/EBPβ from a speckled to a more even distribution and that p300 co-localizes with C/EBPβ in doubly transfected cells.

Fig. 12. Effect of C/EBPβ on the subnuclear localization of p300. QT6 cells (A–F) or DF-1 cells (G–L) were transfected with expression vector for HA-tagged full-length p300 alone (A, B, G and H) or together with C/EBPβ expression vector (C–F and I–L). After 24 h, cells were analyzed by fluorescence microscopy using HA-specific (A–D and G–J) or C/EBPβ-specific (E, F, K and L) antibodies coupled to different fluorophores. (A), (B), (G) and (H) show individual nuclei of different cells. Each pair of (C) and (E), (D and (F), (I) and (K), and (J) and (L) shows the same nucleus stained for p300 and C/EBPβ.
In addition to analyzing the effect of C/EBPβ on the subcellular distribution of p300, we also investigated whether phosphorylation of p300 affects its interaction with the p300-associated factor P/CAF. These experiments did not provide any evidence for an altered interaction between these proteins (data not shown).
Discussion
According to current view, coactivators such as p300 or CBP interact with various transcription factors and thereby are recruited to specific genes where they participate in further protein–protein interactions and affect the local chromatin structure leading, ultimately, to the activation of specific genes (Janknecht and Hunter, 1996; Shikama et al., 1997; Goodman and Smolik, 2000). In this simple view, transcription factors provide a sequence-specific DNA-binding activity to the coactivator, thereby enabling the coactivator to gain access to specific genes. The data presented here suggest that recruitment of a coactivator by a transcription factor is a more complex process. It appears that docking of p300 to a transcription factor such as C/EBPβ not only provides a mechanism for translocating p300 to specific gene regions but, in addition, initiates a process, in the course of which the phosphorylation state and the activity of p300 are modulated. This idea is supported by several key observations. First, our studies demonstrate that C/EBPβ triggers the phosphorylation of full-length p300 as well as of the isolated C-terminal domain of p300. We have shown conclusively that the shift of the mobility of p300 induced by C/EBPβ is due to phosphorylation. Phosphorylated p300 migrates as a multitude of bands differing slightly in electrophoretic mobility. This is particularly apparent with the isolated C-terminal fragment of p300 and suggests that C/EBPβ induces phosphorylation at multiple sites within this domain. This conclusion is supported by mutagenesis studies which have led to the identification of five different sites of C/EBPβ-inducible phosphorylation in p300. Secondly, our studies demonstrate that mutation of the sites of C/EBPβ-inducible phosphorylation diminishes the activity of p300 as a coactivator of C/EBPβ, indicating that the C/EBPβ-induced phosphorylation of p300 is relevant for its function. Since C/EBPβ still affects the mobility of the most extensive p300 phosphorylation site mutant, it is clear that we have not yet identified all sites of C/EBPβ-inducible phosphorylation, providing an explanation for why this mutant is still partially active. Analysis of the HAT activity of p300 in the presence or absence of C/EBPβ has not shown significant differences (C.Schwartz, unpublished data); hence, it is unlikely that the C/EBPβ-induced phosphorylation affects this activity of p300. Thirdly, we have found that the ability of C/EBPβ to trigger phosphorylation of p300 is shared by other C/EBP family members as well as by other unrelated transcription factors, raising the interesting possibility that the mechanism that causes phosphorylation of p300 in response to C/EBPβ may have general relevance for the function of p300 as a coactivator. Together, these observations provide clear evidence that the recruitment of p300 is more complex than simple binding of the coactivator to the transcription factor, and suggest a novel mechanism for the cooperation between coactivators and transcription factors.
Analysis of the sequences of C/EBPα and C/EBPβ necessary for inducing phosphorylation of p300 has shown that although C/EBPα and β are highly conserved, both proteins differ substantially with respect to how they cooperate with p300. In the case of C/EBPα, two conserved domains of the transactivation region mediate phosphorylation of p300. The mutations that abrogate the ability of C/EBPα to induce phosphorylation of p300 also destroy the ability of C/EBPα to interact with p300 and, as shown before (Nerlov and Ziff, 1995), lead to the loss of most of the transactivation potential of C/EBPα. This is consistent with the idea that p300 plays a crucial role as a coactivator of C/EBPα. In the case of C/EBPβ, mutation of the same regions of the transactivation domain had only a minor effect on its ability to trigger phosphorylation of p300. We found that two additional domains of C/EBPβ are involved, namely the N-terminal domain (for which there is no counterpart in C/EBPα) and the DNA-binding domain. The N-terminus of C/EBPβ has already been shown to be crucial for C/EBPβ function as it mediates interaction with the SWI–SNF complex (Kowenz-Leutz and Leutz, 1999). In addition, due to alternative use of translational initiation sites, it is present in some C/EBPβ isoforms but not in others, thereby increasing the functional diversity of C/EBPβ isoforms (Calkhoven et al., 2000). Our results suggest that the functional differences between these isoforms may be mediated not only by their differential interaction with SWI–SNF but also by differences in their ability to cooperate with p300. The amino acid sequences of the DNA-binding domains of C/EBPα and β are very similar; it is therefore surprising that these domains show clear functional differences with respect to the interaction with p300. Our results provide clear evidence that the C/EBP DNA-binding domain is implicated not only in DNA binding but also in other functions, such as protein–protein interactions. The C/EBP DNA-binding domain has been shown before to interact directly with Myb transcription factors (Mink et al., 1996; Tahirov et al., 2002); however, this is the first time that differential interaction of a protein with the DNA-binding domains of two highly related C/EBP family members has been clearly demonstrated. Functional differences between the DNA-binding domains of C/EBPα and β have been noted before. The DNA-binding domain of C/EBPβ, but not that of C/EBPα, has been implicated in calcium-regulated transcriptional stimulation of certain genes (Wegner et al., 1992).
We do not yet know how C/EBPβ causes phosphorylation of p300 or which protein kinase is involved. C/EBPβ could act by increasing the expression of certain genes whose activation, in turn, is responsible for the phosphorylation of p300. For example, C/EBPβ could stimulate expression of a protein kinase that uses p300 as substrate. We do not think that this is a likely explanation, but favor the idea that phosphorylation of p300 is a consequence of the interaction between p300 and C/EBPβ. First, p300 and C/EBPβ appear to be direct interaction partners, as evidenced by the use of bacterially expressed proteins. Secondly, phosphorylation of p300 depends on the binding site for C/EBP in p300 as well as on the binding sites for p300 in C/EBPβ and C/EBPα. Furthermore, virtually all of the phosphorylated p300 is bound to C/EBPβ. These findings are difficult to reconcile with an indirect mechanism, but strongly suggest that phosphorylation of p300 is a direct consequence of the interaction of C/EBPβ and p300. An indirect mechanism is also unlikely because a number of unrelated transcription factors, which presumably activate different sets of target genes, cause phosphorylation of p300. There are a number of possible mechanisms that might explain how interaction with C/EBPβ could trigger phosphorylation of p300. Interaction with C/EBPβ might cause a conformational change in p300 or sequester C/EBPβ-bound p300 to a particular compartment where it is a better substrate for a protein kinase, resulting in phosphorylation of C/EBPβ-bound p300. Alternatively, C/EBPβ itself might be associated with a protein kinase that phosphorylates p300 once it interacts with C/EBPβ. Although we have no definitive evidence for or against any of these mechanisms, the observation that the localization of p300 in the nucleus is affected by C/EBPβ is consistent with the idea that a C/EBPβ-induced redistribution of p300 might play a role in the phosphorylation of p300. A C/EBPβ-induced redistribution of p300 within the nucleus might also be relevant for the increased transcriptional activity of phosphorylated p300.
All phosphorylation sites that we have identified so far contain SP or TP motifs, suggesting that the C/EBPβ-induced phosphorylation of p300 is mediated by a proline-directed kinase. It has been reported that p300 (or CBP) is a substrate for mitogen-activated protein (MAP) kinase or cyclin-dependent kinases (Janknecht and Nordheim, 1996; Ait-Si Ali et al., 1998, 1999), making these kinases possible candidates for the p300 kinase responsible for the phosphorylations described here. The C-terminal domain of p300 also contains potential phosphorylation motifs for protein kinases A and C (Blom et al., 1999); however, mutation of these motifs did not affect the C/EBPβ-induced mobility shift (data not shown).
Irrespective of the identity of the kinase(s) involved, the C/EBPβ-induced phosphorylation has interesting functional consequences. First, the complexity of the interplay between p300 and C/EBPβ revealed by our studies could provide additional levels of regulation which could be used for controlling the activity of p300. These regulatory mechanisms, presumably, would not affect all p300 molecules present in a cell in the same way, but would allow subsets of p300, interacting with particular transcription factors and targeted to specific genes, to be regulated independently. Secondly, since p300 phosphorylation increases the activity of p300 as a coactivator (as shown by the analysis of phosphorylation site mutants), p300 not recruited by C/EBPβ will remain less active. This ‘activation on demand’ could provide a mechanism to permit free p300 (i.e. p300 molecules not recruited by C/EBPβ to specific genes) to rest in a less active state, thereby preventing squelching of components of the transcriptional machinery by free p300, which might occur if p300 was constitutively active. Since, in addition to C/EBPβ, several other transcription factors which are known to interact with the C-terminal part of p300 trigger phosphorylation of p300, the underlying processes appear not only to be relevant for C/EBPβ but may be important for the function of many other transcription factors.
Materials and methods
Expression vectors
An expression vector for p300 (pCMV-p300CHA) was obtained from R.Eckner (Eckner et al., 1994). Expression vectors for truncated forms of p300, p300/1751–2370, p300/1853–2370 and p300/1515–1998 or a VP16–p300/1751–1998 fusion protein have been described (Mink et al., 1997). Expression vectors p300/1751–2299, p300/1751–2218 and p300/1751–2159 are derived from p300/1751–2370 and were generated by truncating the p300 coding region at restriction sites located at the respective positions. p300 point mutations were generated by PCR using appropriate primers. All mutants were verified by sequencing. Expression vectors for full-length chicken C/EBPβ, pCRNC-CCR and pCDNA3-CCR (Burk et al., 1993; Mink et al., 1997), chicken C/EBPα (Mink et al., 1996), mouse C/EBPδ (Cao et al., 1991), chicken c-Ets-1 and c-Ets-2, and mouse Pu.1 (Burk and Klempnauer, 1999) have been described. Expression vectors for human ATF2, human c-Jun and human AML-1 were generated by cloning the respective coding regions into pCDNA3 (Invitrogen). Expression vectors for point mutants of C/EBPα were obtained from C.Nerlov (Nerlov and Ziff, 1995). Point mutants of C/EBPβ were generated by PCR using appropriate primers and verified by sequencing. In the mutant mutA, S1848, T1851, T1854 and T1857 were replaced by alanines. In the mutant mutB, S2279 was replaced by alanine. In the N-terminal deletion mutant C/EBPβ-ΔN33, the first 33 amino acids are missing. The chicken C/EBPα- and β-binding recombinant constructs pCDNA3-C/EBPα/β and pCDNA3-C/EBPβ/α have been described (Mink et al., 1999). Expression vectors encoding Gal4 fusion proteins of wild-type or mutant C/EBPα and C/EBPβ were obtained by cloning the coding region of the Gal4 DNA-binding domain upstream of the C/EBP coding sequence.
Transfections
The Gal4-responsive reporter plasmid pG5E4-38Luc has been described (Mink et al., 1997). pCMVβ was from Clontech. Transfection of the QT6 cell line and the analysis of reporter gene expression have been described (Burk et al., 1993). The amounts of DNA used for transfection of cells in a 10 cm culture dish are indicated in the figure legends.
GST pull-down assay
A bacterial expression vector for full-length C/EBPβ has been described (Mink et al., 1996). Recombinant C/EBPβ was purified as the insoluble protein and solubilized by treatment with 6 M urea followed by 50-fold dilution into binding buffer [10 mM Tris–HCl pH 8, 50 mM NaCl, 1 mM EDTA, 1 mM dithiothreitol (DTT)]. Expression vectors GST–p300 (1752–1998), GST–p300(1883–1927) and GST–p300(1710–1891) encode the indicated amino acids of p300 fused to GST. Equal amounts of these proteins were bound to glutathione–Sepharose, followed by incubation with C/EBPβ overnight in binding buffer. Beads were washed three times in 50 mM Tris–HCl pH 7.5, 120 mM NaCl, 1 mM EDTA, 6 mM EGTA, 0.1% NP-40. Bound proteins were analyzed by SDS–PAGE and western blotting with C/EBPβ-specific antibodies.
Differentiation of HL60 cells
Macrophage differentiation of the promyelocytic HL60 cell line cells was induced by treatment with 100 nM TPA for 2 days. The differentiation of the cells was confirmed by microscopic inspection.
Antibodies, western blotting and immunoprecipitation
Cells were lysed in ELB buffer [50 mM Tris–HCl pH 7.5, 120 mM NaCl, 20 mM NaF, 1 mM benzamidine, 1 mM EDTA, 6 mM EGTA, 15 mM sodium pyrophosphate, 1 mM phenylmethylsulfonyl fluoride (PMSF), 0.1% NP-40] followed by centrifugation for 30 min at 14 000 g. Lysates were analyzed by SDS–PAGE and western blotting, or precipitated with antibodies against C/EBPβ (Mink et al., 1996) or p300 (UBI), followed by SDS–PAGE and western blotting. For phosphatase treatment, Sepharose beads carrying immunoprecipitated proteins were washed three times with ELB buffer lacking NaF and sodium pyrophosphate, washed once in phosphatase buffer (50 mM Tris–HCl pH 8.5, 5 mM MgCl2), suspended in 30 µl of phosphatase buffer containing 1 U of shrimp alkaline phosphatase (Boehringer) and incubated for 20 min at 37°C. Controls were performed using phosphatase heat treated for 15 min at 65°C.
Radiolabeling of cells
Cells were labeled with [32P]orthophosphate by first pre-incubating them in phosphate-free Dulbecco’s modified Eagle’s medium (DMEM; ICN) for 30 min and then incubating them for 5 h with 200 µCi/ml [32P]orthophosphate (Amersham; HCl- and carrier-free). For immunoprecipitation (all steps on ice), cells were lysed in RIPA buffer (50 mM Tris–HCl pH 7.4, 150 mM NaCl, 1 mM EDTA, 0.5% NP-40, 0.5% sodium deoxycholate, 0.1% SDS, 5 mM DTT, 1 mM NaF, 15% glycerol). The lysate was clarified by centrifugation, and p300-specific antibodies were added, followed by incubation overnight. Protein A–Sepharose and rabbit anti-mouse immunoglobulins were then added and incubated for an additional hour. The Sepharose was washed three times with RIPA buffer and once with phosphate-buffered saline (PBS), and bound proteins were detected by SDS–PAGE and autoradiography.
Immunofluorescence microscopy
QT6 quail fibroblasts or DF-1 chicken fibroblasts (Himly et al., 1998) were plated on glass coverslips and transfected as described above. After 24 h, cells were fixed with 3.5% paraformaldehyde, permeabilized by 0.2% NP-40 and incubated with the hemagglutinin (HA)-specific mouse monoclonal antibody HA.11 (BabCO) or with rabbit anti C/EBPβ antiserum (Mink et al., 1996). Secondary antibodies were rhodamine-conjugated anti-mouse and fluorescein-conjugated anti-rabbit immunoglobulins. Cells were analyzed with a confocal laser scanning microscope.
Acknowledgments
Acknowledgements
We thank U.Hommes for excellent technical assistance, and R.Eckner and C.Nerlov for plasmids and antibodies. This work was supported by grants from the DFG (KL461/8-1), the BMBF (0311707) and the Fonds der chemischen Industrie. K.B. is supported by the NRW Graduate School of Chemistry.
References
- Ait-Si-Ali S. et al. (1998) Histone acetyltransferase activity of CBP is controlled by cycle-dependent kinases and oncoprotein E1A. Nature, 396, 184–186. [DOI] [PubMed] [Google Scholar]
- Ait-Si-Ali S., Carlisi,D., Ramirez,S., Upegui-Gonzalez,L.C., Duquet,A., Robin,P., Rudkin,B., Harel-Bellan,A. and Trouche,D. (1999) Phosphorylation by p44 MAP kinase/ERK1 stimulates CBP histone acetyltransferase activity in vitro. Biochem. Biophys. Res. Commun., 262, 157–162. [DOI] [PubMed] [Google Scholar]
- Arias J., Alberts,A.S., Brindle,P., Claret,F.X., Smeal,T., Karin,M., Feramisco,J. and Montminy,M. (1994) Activation of cAMP and mitogen responsive genes relies on a common nuclear factor. Nature, 370, 226–229. [DOI] [PubMed] [Google Scholar]
- Bannister A.J. and Kouzarides,T. (1996) The CBP co-activator is a histone acetyltransferase. Nature, 384, 641–643. [DOI] [PubMed] [Google Scholar]
- Blom N., Gammeltoft,S. and Brunak,S. (1999) Sequence and structure-based prediction of eucaryotic protein phosphorylation sites. J. Mol. Biol., 294, 1351–1362. [DOI] [PubMed] [Google Scholar]
- Burk O. and Klempnauer,K.-H. (1999) Myb and Ets transcription factors cooperate at the myb-inducible promoter of the tom-1 gene. Biochim. Biophys. Acta, 1446, 243–252. [DOI] [PubMed] [Google Scholar]
- Burk O., Mink,S., Ringwald,M. and Klempnauer,K.-H. (1993) Synergistic activation of the chicken mim-1 gene by v-myb and C/EBP transcription factors. EMBO J., 12, 2027–2038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Calkhoven C.F., Muller,C. and Leutz,A. (2000) Translational control of C/EBPα and C/EBPβ isoform expression. Genes Dev., 14, 1920–1932. [PMC free article] [PubMed] [Google Scholar]
- Cao Z. Umek,R.M. and McKnight,S.L. (1991) Regulated expression of three C/EBP isoforms during adipose conversion of 3T3-L1 cells. Genes Dev., 5, 1538–1552. [DOI] [PubMed] [Google Scholar]
- Chakravarti D., LaMorte,V.J., Nelson,M.C., Nakajima,T., Schulman,I.G., Juguilon,H., Montminy,M. and Evans,R. (1996) Role of CBP/P300 in nuclear receptor signalling. Nature, 383, 99–103. [DOI] [PubMed] [Google Scholar]
- Chen H., Lin,R.J., Schiltz,R.L., Chakravarti,D., Nash,A., Nagy,L., Privalsky,M.L., Nakatani,Y. and Evans,R.M. (1997) Nuclear receptor coactivator ACTR is a novel histone acetyltransferase and forms a multimeric activation complex with P/CAF and CBP/p300. Cell, 90, 569–580. [DOI] [PubMed] [Google Scholar]
- Chrivia J.C., Kwok,R.P.S., Lamb,N., Hagiwara,M., Montminy,M.R. and Goodman,R.H. (1993) Phosphorylated CREB binds specifically to the nuclear protein CBP. Nature, 365, 855–859. [DOI] [PubMed] [Google Scholar]
- Darlington G.J., Ross,S.E. and MacDougald,O.A. (1998) The role of C/EBP genes in adipocyte differentiation. J. Biol. Chem., 273, 30057–30060. [DOI] [PubMed] [Google Scholar]
- Eckner R., Ewen,M.E., Newsome,D., Gerdes,M., DeCaprio,J.A., Bentley-Lawrence,J. and Livingston,D.M. (1994) Molecular cloning and functional analysis of the adenovirus E1A-associated 300-kD protein (p300) reveals a protein with properties of a transcriptional adaptor. Genes Dev., 8, 869–884. [DOI] [PubMed] [Google Scholar]
- Giordano A. and Avantaggiati,M.L. (1999) p300 and CBP: partners for life and death. J. Cell Physiol., 181, 218–230. [DOI] [PubMed] [Google Scholar]
- Goodman R.H. and Smolik,S. (2000) CBP/p300 in cell growth, transformation and development. Genes Dev., 14, 1553–1577. [PubMed] [Google Scholar]
- Grunstein M. (1997) Histone acetylation in chromatin structure and transcription. Nature, 389, 349–352. [DOI] [PubMed] [Google Scholar]
- Gu W. and Roeder,R.G. (1997) Activation of p53 sequence-specific DNA binding by acetylation of the p53 C-terminal domain. Cell, 90, 595–606. [DOI] [PubMed] [Google Scholar]
- Himly M., Foster,D.N., Bottoli,I., Iacovoni,J.S. and Vogt,P.K. (1998) The DF-1 chicken fibroblast cell line: transformation induced by diverse oncogenes and cell death resulting from infection by avian leukosis viruses. Virology, 248, 295–304. [DOI] [PubMed] [Google Scholar]
- Janknecht R. and Hunter,T. (1996) Transcriptional control: versatile molecular glue. Curr. Biol., 6, 951–954. [DOI] [PubMed] [Google Scholar]
- Janknecht R. and Nordheim,A. (1996) Regulation of the c-fos promoter by the ternary complex factor Sap-1a and its coactivator CBP. Oncogene, 12, 1961–1969. [PubMed] [Google Scholar]
- Katz S., Kowenz,L.E., Müller,C., Meese,K., Ness,S.A. and Leutz,A. (1993) The NF-M transcription factor is related to C/EBPβ and plays a role in signal transduction, differentiation and leukemogenesis of avian myelomonocytic cells. EMBO J., 12, 1321–1332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kowenz-Leutz E. and Leutz,A. (1999) A C/EBPβ isoform recruits the SWI/SNF complex to activate myeloid genes. Mol. Cell., 4, 735–743. [DOI] [PubMed] [Google Scholar]
- Kwok R.P.S., Lundblad,J.R., Chrivia,J.C., Richards,J.P., Bächinger,H.P., Brennan,R.G., Roberts,S.G.E., Green,M.R. and Goodman,R.H. (1994) Nuclear protein CBP is a coactivator for the transcription factor CREB. Nature, 370, 223–226. [DOI] [PubMed] [Google Scholar]
- Lekstrom-Himes J. and Xanthopoulos,K.G. (1998) Biological role of the CCAAT/enhancer-binding protein family of transcription factors. J. Biol. Chem., 273, 28545–28548. [DOI] [PubMed] [Google Scholar]
- Mink S., Kerber,U. and Klempnauer,K.-H. (1996) Interaction of C/EBPβ and v-Myb is required for synergistic activation of the mim-1 gene. Mol. Cell. Biol., 16, 1316–1325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mink S., Haenig,B. and Klempnauer,K.-H. (1997) Interaction and functional collaboration of p300 and C/EBPβ. Mol. Cell. Biol., 17, 6609–6617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mink S., Jaswal,S., Burk,O. and Klempnauer,K.-H. (1999) The v-Myb oncoprotein activates C/EBPβ expression by stimulating an autoregulatory loop at the C/EBPβa promoter. Biochim. Biophys. Acta, 1447, 175–184. [DOI] [PubMed] [Google Scholar]
- Natsuka S., Akira,S., Nishio,Y., Hashimoto,S., Sugita,T., Isshiki,H. and Kishimoto,T. (1992) Macrophage differentiation-specific expression of NF-IL6, a transcription factor for interleukin-6. Blood, 79, 460–466. [PubMed] [Google Scholar]
- Nerlov C. and Ziff,E.B. (1995) CCAAT/enhancer binding protein-α amino acid motifs with dual TBP and TFIIB binding ability co-operate to activate transcription in both yeast and mammalian cells. EMBO J., 14, 4318–4328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ness S.A., Kowentz-Leutz,E., Casini,T., Graf,T. and Leutz,A. (1993) Myb and NF-M: combinatorial activators of myeloid genes in heterologous cell types. Genes Dev., 7, 749–759. [DOI] [PubMed] [Google Scholar]
- Oelgeschlager M., Janknecht,R., Krieg,J., Schreek,S. and Lüscher,B. (1996) Interaction of the co-activator CBP with Myb proteins: effects on Myb-specific transactivation and on the cooperativity with NF-M. EMBO J., 15, 2771–2780 [PMC free article] [PubMed] [Google Scholar]
- Ogryzko V.V., Schiltz,R.L., Russnova,V., Howard,B. and Nakatani,Y. (1996) The transcriptional coactivators p300 and CBP are histone acetyltransferases. Cell, 87, 953–959. [DOI] [PubMed] [Google Scholar]
- Scott L.M., Civin,C.I., Rorth,P. and Friedman,A.D. (1992) A novel temporal expression pattern of three C/EBP family members in differentiating myelomonocytic cells. Blood, 80, 1725–1735. [PubMed] [Google Scholar]
- Shikama N., Lyon,J and La Thangue,N.B. (1997) The p300/CBP family: integrating signals with transcription factors and chromatin. Trends Cell Biol., 7, 230–236. [DOI] [PubMed] [Google Scholar]
- Spencer T.E. et al. (1997) Steroid receptor coactivator-1 is a histone acetyltransferase. Nature, 389, 194–198. [DOI] [PubMed] [Google Scholar]
- Struhl K. (1998) Histone acetylation and transcriptional regulatory mechanisms. Genes Dev., 12, 599–606. [DOI] [PubMed] [Google Scholar]
- Tahirov T.H. et al. (2002) Mechanism of c-Myb-C/EBPβ cooperation from separated sites on a promoter. Cell, 108, 57–70. [DOI] [PubMed] [Google Scholar]
- Torchia J., Rose,D.W., Inostroza,J., Kamei,Y., Westin,S., Glass,C.K. and Rosenfeld,M.G. (1997) The transcriptional co-activator p/CIP binds CBP and mediates nuclear-receptor function. Nature, 387, 677–684. [DOI] [PubMed] [Google Scholar]
- Wade P. and Wolffe,A. (1997) Histone acetyltransferases in control. Curr. Biol., 7, R82–R84. [DOI] [PubMed] [Google Scholar]
- Yamanaka R., Lekstrom-Himes,J., Barlow,C., Wynshaw-Boris,A. and Xanthopoulos,K.G. (1998) CCAAT/enhancer binding proteins are critical components of the transcriptional regulation of hematopoiesis. Int. J. Mol. Med., 1, 213–221. [DOI] [PubMed] [Google Scholar]
- Wegner M., Cao,Z. and Rosenfeld,M.G. (1992) Calcium-regulated phosphorylation within the leucine zipper of C/EBPβ. Science, 256, 370–373. [DOI] [PubMed] [Google Scholar]
- Yang X.J., Ogryzko,V.V., Nishikawa,J., Howard,B.H. and Nakatani,Y. (1996) A p300/CBP-associated factor that competes with the adenoviral oncoprotein E1A. Nature, 382, 319–324. [DOI] [PubMed] [Google Scholar]
- Yeh W.-C., Cao,Z., Classon,M. and McKnight,S.L. (1995) Cascade regulation of terminal adipocyte differentiation by three members of the C/EBP family of leucine zipper proteins. Genes Dev., 9, 168–181. [DOI] [PubMed] [Google Scholar]
- Yuan W., Condorelli,G., Caruso,M., Felsani,A. and Giordano,A. (1996) Human p300 protein is a coactivator for the transcription factor MyoD. J. Biol. Chem., 271, 9009–9013. [DOI] [PubMed] [Google Scholar]