

Abstract
Ethanol stimulates the firing activity of midbrain dopamine (DA) neurons, leading to enhanced dopaminergic transmission in the mesolimbic system. This effect is thought to underlie the behavioral reinforcement of alcohol intake. Ethanol has been shown to directly enhance the intrinsic pacemaker activity of DA neurons, yet the cellular mechanism mediating this excitation remains poorly understood. The hyperpolarization-activated cation current, Ih, is known to contribute to the pacemaker firing of DA neurons. To determine the role of Ih in ethanol excitation of DA neurons, we performed patch-clamp recordings in acutely prepared mouse midbrain slices. Superfusion of ethanol increased the spontaneous firing frequency of DA neurons in a reversible fashion. Treatment with ZD7288, a blocker of Ih, irreversibly depressed basal firing frequency and significantly attenuated the stimulatory effect of ethanol on firing. Furthermore, ethanol reversibly augmented Ih amplitude and accelerated its activation kinetics. This effect of ethanol was accompanied by a shift in the voltage dependence of Ih activation to more depolarized potentials and an increase in the maximum Ih conductance. Cyclic AMP mediated the depolarizing shift in Ih activation but not the increase in the maximum conductance. Finally, repeated ethanol treatment in vivo induced downregulation of Ih density in DA neurons and an accompanying reduction in the magnitude of ethanol stimulation of firing. These results suggest an important role of Ih in the reinforcing actions of ethanol and in the neuroadaptations underlying escalation of alcohol consumption associated with alcoholism.
INTRODUCTION
The dopaminergic projection from the ventral tegmental area (VTA) to the nucleus accumbens (NAc) and other limbic structures, termed the mesolimbic dopamine (DA) system, is critically involved in reward processing, behavioral reinforcement, and addictive behaviors. Several lines of evidence show that this system plays an important role in the reinforcing/rewarding actions of ethanol. Administration of DA antagonists, either systemically or directly into the NAc, suppresses ethanol-reinforced behavioral responses, such as ethanol drinking, self-administration, and preference (George et al. 1995; Kaczmarek and Kiefer 2000; Myers and Robinson 1999; Samson et al. 1993; Weiss et al. 1990), although neurotoxin-induced DA neuron lesion studies have produced some equivocal results (Ikemoto et al. 1997; Rassnick et al. 1993). The brain DA system has also been implicated in ethanol-induced euphoria and behavioral stimulation in humans (Ahlenius et al. 1973). Consistent with the role played by the mesolimbic DA system in the behavioral actions of ethanol, active and passive administration of ethanol elevates extracellular DA levels in the NAc (Di Chiara and Imperato 1988; Weiss et al. 1993), most likely through its effect at the VTA DA cell body region rather than the NAc terminal region (Budygin et al. 2001; Yim and Gonzales 2000). Indeed, rats self-administer ethanol directly into the VTA (Gatto et al. 1994; Rodd et al. 2004) and ethanol facilitates action potential firing of DA neurons both in vivo (Gessa et al. 1985; Mereu et al. 1984) and in vitro (Brodie and Appel 1998; Brodie et al. 1990). It has been shown that this stimulatory effect of ethanol results, at least partly, from its direct action on the intrinsic excitability of DA neurons (Brodie et al. 1999).
DA neurons display a slowly activating, inward cation current, termed Ih, in response to membrane hyperpolarization (Harris and Constanti 1995; Neuhoff et al. 2002). Ih, originally described in cardiac pacemaker cells (Brown and Difrancesco 1980), has been shown to contribute to the autonomous pacemaker activity of a variety of neurons (Chan et al. 2004; Luthi and McCormick 1998; Maccaferri and McBain 1996). DA neurons are also capable of spontaneous, low-frequency pacemaker discharge (1-5 Hz) independent of synaptic input drive (Grace 1991; Kitai et al. 1999). Recent studies have shown that Ih is actively engaged in controlling the pacemaker frequency in a subset of DA neurons (Neuhoff et al. 2002; Seutin et al. 2001). A previous intracellular recording study has also demonstrated that ethanol can enhance Ih in DA neurons (Brodie and Appel 1998). However, the contribution of Ih to ethanol-induced modulation of DA neuron excitability is not known.
In the present investigation, we show that pharmacological blockade of Ih attenuates ethanol enhancement of DA neuron pacemaker firing. Using whole cell voltage-clamp recordings, we found that ethanol facilitates the voltage gating of Ih in a cAMP-dependent fashion and also increases the maximum Ih conductance independent of cAMP. Furthermore, we present data demonstrating downregulation of Ih density in DA neurons after repeated ethanol exposure in vivo, suggesting that Ih plasticity may play an important role in the neuroadaptations underlying alcoholism.
METHODS
Slices and solutions
Horizontal midbrain slices (200-220 μm) containing both the VTA and the substantia nigra pars compacta (SNc) were prepared from C57BL/6J mice [postnatal day 21 (P21) to P35]. Animals were anesthetized with halothane or isoflurane and then killed by cervical dislocation or decapitation in accordance with a protocol approved by the University of Texas Institutional Animal Care and Use Committee. Slices were cut using a vibratome (VT1000S; Leica, Nussloch, Germany) in ice-cold (4°C) physiological saline containing (in mM) 126 NaCl, 2.5 KCl, 1.2 NaH2PO4, 1.2 MgCl2, 2.4 CaCl2, 11 glucose, and 21.4 NaHCO3, saturated with 95% O2-5% CO2 (pH 7.4, 300 mOsm/kg), and then incubated in the same solution warmed to 35°C for ≥1 h before being used for electrophysiological recordings. MK-801 (50-100 μM) was added to the cutting and incubation solutions to block N-methyl-d-aspartate (NMDA)-mediated excitotoxicity. For recording, slices were placed in a recording chamber superfused with physiological saline (35°C) at 2.5-3 ml/min. Unless noted otherwise, pipette solutions used for whole cell and cell-attached recordings contained (in mM) 115 K-methylsulfate, 20 KCl, 1 MgCl2, 10 HEPES, 5 EGTA, 2 Mg-ATP, 0.2 Na-GTP, and 10 Na-phosphocreatine (pH 7.3, 280 mOsm/kg).
Electrophysiological recordings
Cells were visualized using a ×40 water-immersion objective on an upright microscope with IR/DIC optics (BX51WI; Olympus, Melville, NY). All recordings were performed in DA neurons identified by spontaneous pacemaker firing (1-5 Hz) and the presence of large Ih (>200 pA), evoked by a 1.5-s hyperpolarizing step from -55 to -105 mV. Whole cell voltage-clamp recordings were made at a holding potential of -55 mV unless stated otherwise. A MultiClamp 700A amplifier (Axon Instruments, Foster City, CA) was used to record the data, which were filtered at 1 kHz, digitized at 2 kHz, and collected on a personal computer using Axograph 4.9 (Axon Instruments).
Action potential firing was monitored using either perforated-patch or cell-attached recording configurations because the spontaneous firing of DA neurons is significantly distorted with a standard whole cell recording configuration (Morikawa et al. 2003). For perforated-patch recordings, the pipette was tip-filled with a solution containing (in mM) 135 KCl and 10 HEPES (pH 7.3, 295 mOsm/kg) and then back-filled with the same solution added with gramicidin (50-250 μg/ml). The typical series resistance obtained 15-30 min after the formation of a gigaseal was 20-40 MΩ. Perforated-patch and cell-attached recordings gave equivalent results; hence, the data were pooled from both recording conditions.
Some of the firing recordings were done in the presence of 6,7-dinitroquinoxaline-2,3-dione (DNQX) (10 μM), picrotoxin (100 μM), and strychnine (1 μM) to block AMPA, GABAA, and glycine-mediated synaptic inputs. The NMDA-mediated input was routinely blocked by pretreatment with MK-801 (50-100 μM), as described in the preceding text.
Drugs
Drugs were applied by superfusion to the slice, except for cAMP analogues, Rp-cAMPS and Sp-cAMPS, which were infused into the cytosol through the whole cell pipette. Superfusion of forskolin (10 μM), an activator of adenylyl cyclase, produced variable effects on Ih and hence was not used in the present study. This was most likely due to the well-documented facilitation of neurotransmitter release by forskolin (Chavis et al. 1998) because a variety of neurotransmitters can modulate Ih via activation of G-protein-coupled receptors in DA neurons (Cathala and Paupardin-Tritsch 1997, 1999; Jiang et al. 1993; Liu et al. 2003). DNQX, MK-801, and ZD7288 were purchased from Tocris Cookson (Ellisville, MO). All other chemicals were obtained from Sigma/RBI (St. Louis, MO).
Repeated ethanol treatment
Mice received once daily intraperitoneal injections of either normal saline or 2 g/kg ethanol (20% wt/vol in saline) for 5 consecutive days. Midbrain slices were prepared 24 h after the final injection and were used for electrophysiological recordings. No more than two data were obtained from a single animal in this series of experiments. Repeated ethanol treatment had no significant effect on the cell capacitance in both the VTA and the SNc (data not shown), suggesting that there was no significant change in the size of DA neurons.
Data analysis
Ih was evoked once per minute by a 1.5-s hyperpolarizing step to -105 mV from -55 mV. Ih amplitude was defined as the difference in the current values measured at the onset, after the capacitative transient has subsided, and the end of the voltage step. The effect of ethanol on Ih was determined with respect to the average of control and washout values. The activation time constant of Ih was determined by fitting the activating phase of the current trace with a single exponential function over the initial 500 ms of the hyperpolarizing step. This gave much more consistent values in each recorded cell than fitting the entire activating phase during the 1.5-s hyperpolarizing step with a double-exponential function.
For the construction of Ih activation curves, 1.5-s hyperpolarizing steps to various potentials (-55 to -125 mV) were applied from a holding potential of -45 mV and tail currents were measured at -105 mV. These experiments were done in TEA-Cl (10 mM), with equimolar reduction of NaCl, to block noninactivating voltage-dependent K+ conductances. Tail current amplitudes at -105 mV, after subtraction of the current following no hyperpolarizing step, were plotted as a function of test potentials. The obtained curve was fitted with a Boltzmann function: I = Imax/[1 + exp(V - V1/2)/s], where Imax is the maximal tail current amplitude, V is the test potential, V1/2 is the half-activation potential, and s is the slope factor. Imax was normalized to the cell capacitance to estimate Ih density in each cell. When comparing saline- and ethanol-treated mice for Ih density and V1/2, these values were routinely obtained 10 min after establishing the whole cell configuration.
Data are expressed as means ± SE. Statistical significance was determined by paired or unpaired Student's t-test. The difference was considered significant at P < 0.05.
RESULTS
Ethanol stimulates DA neuron firing via Ih
The spontaneous firing of DA neurons was monitored with perforated-patch or cell-attached current-clamp recordings in midbrain slices from C57BL/6J mice. DA neurons displayed spontaneous pacemaker-like firing at 1-5 Hz, as reported previously (Grace 1991). Superfusion of ethanol (100 mM) reversibly increased the firing frequency by 18.4 ± 1.7% (from 2.27 ± 0.10 to 2.66 ± 0.11 Hz, n = 33, P < 0.0001; Fig. 1, A and B). Eleven of these 33 recordings were done in the presence of antagonists of ionotropic neurotransmitter receptors (see methods). The effect of ethanol was not significantly different in the absence and presence of these antagonists (19.4 ± 2.4%, n = 22 vs. 16.3 ± 2.2%, n = 11, P = 0.41; Fig. 1C). Ethanol produced nearly identical increases in the firing frequency in the VTA and the SNc (18.5 ± 2.6%, n = 16 vs. 18.2 ± 2.3%, n = 17, P = 0.93; Fig. 1D). Ethanol (50 mM) increased the firing frequency by 10.2 ± 2.0% (from 2.38 ± 0.21 to 2.62 ± 0.23 Hz, n = 8, P < 0.01; Fig. 1B), whereas 25 mM ethanol produced a small increase (approximately 5%) in one of five cells tested.
fig. 1.

Ethanol facilitates dopamine (DA) neuron firing. A: time graph illustrating the effect of ethanol (100 mM) on DA neuron firing. This experiment was done with a perforated-patch configuration. Representative traces of action potential firing in control (C) and in ethanol (E) are shown in the inset. B: summary bar graph showing the effects of 50 and 100 mM ethanol. C: comparison of the effects of ethanol in the absence [Antg (-)] and presence [Antg (+)] of major neurotransmitter receptors. D: comparison of the effects of ethanol in the VTA and the SNc.
To examine the role played by Ih in ethanol stimulation of firing, we used ZD7288, an Ih blocker (Harris and Constanti 1995). Using whole cell voltage-clamp recordings, we confirmed that ZD7288 (30 μM) completely and irreversibly abolished Ih in ∼10 min (n = 5; Fig. 2A). ZD7288 produced no significant changes in the holding current and the membrane input resistance at a holding potential of -55 mV (data not shown). Because prolonged ZD7288 treatment (>20 min) can produce effects independent of Ih blockade (Chevaleyre and Castillo 2002), we tested the effect of 10-min superfusion of ZD7288 (30 μM) on firing. ZD7288 irreversibly decreased the firing frequency with a time course similar to its effect on Ih (Fig. 2B). The reduction in the firing frequency produced by ZD7288 was of similar magnitude in the VTA and the SNc (28.2 ± 4.1%, n = 7 vs. 33.2 ± 7.9%, n = 7, P = 0.58; Fig. 2C). Furthermore, ZD7288 treatment dramatically reduced the stimulatory effect of ethanol on firing from 17.6 ± 2.9 to 0.5 ± 3.9% (n = 7, P < 0.01; Fig. 2, B and D). Ethanol produced a small inhibition of firing after ZD7288 treatment in two of seven cells, suggesting the presence of an inhibitory action of ethanol when Ih is blocked. On the other hand, injection of a hyperpolarizing current (-20 to -60 pA), which decreased the firing frequency by 30-40% (33.3 ± 3.8%, n = 3), did not significantly affect the magnitude of ethanol response (from 22.6 ± 5.8 to 28.8 ± 9.1%, P = 0.20; Fig. 2D, right), showing that a mere reduction in the firing frequency does not attenuate the effect of ethanol. In 11 cells where both ethanol and ZD7288 were tested, the magnitude of ethanol-induced enhancement of firing was positively correlated with that of ZD7288-induced suppression (r = 0.74; Fig. 2E). Altogether, these data suggest that Ih contributes to the stimulation of pacemaker activity by ethanol.
fig. 2.
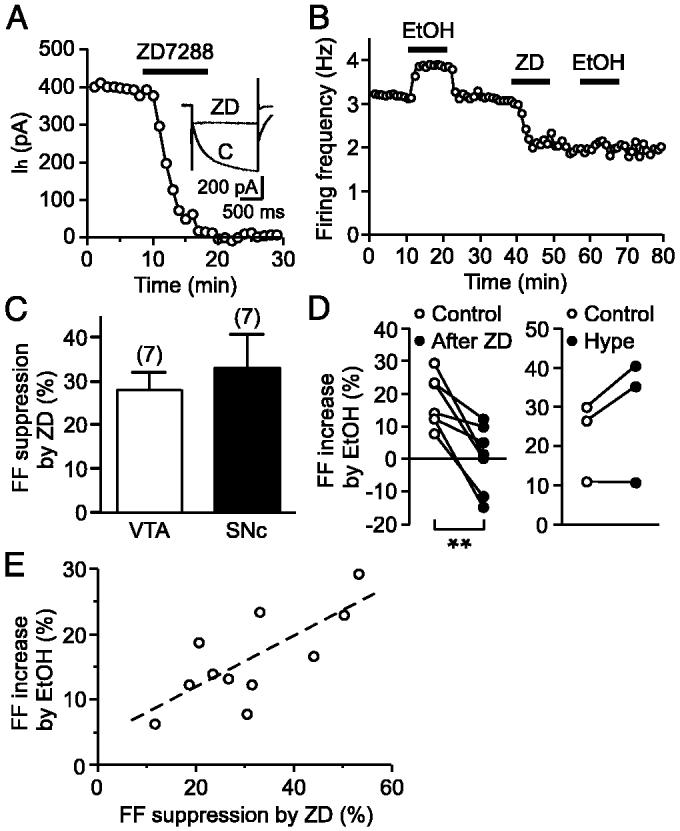
Ih is involved in ethanol stimulation of firing. A: time graph showing the effect of ZD7288 (30 μM) on Ih. Representative traces of Ih in control (C) and in ZD7288 (ZD) are shown in the inset. B: ZD7288 (30 μM) irreversibly decreased the firing frequency and blocked the effect of ethanol (100 mM). This experiment was performed with a cell-attached configuration. C: comparison of the effects of ZD7288 on the firing frequency in the VTA and the SNc. D: magnitude of ethanol-induced increase in the firing frequency (FF) before and after ZD7288 treatment (left) and hyperpolarizing current injection (right) is plotted in each cell. E: magnitude of ethanol-induced increase in FF is plotted vs. the magnitude of ZD7288-induced suppression. - - -, linear fit to the data. **P < 0.01.
Ethanol enhances Ih via two mechanisms
We next directly examined the effect of ethanol on Ih using whole cell voltage-clamp recordings. Ih was evoked once a minute by a 1.5-s hyperpolarizing voltage step to -105 from -55 mV. Superfusion of ethanol (100 mM) reversibly increased Ih amplitude by 26.2 ± 4.2% (from 501 ± 61 to 621 ± 69 pA, n = 11, P < 0.001; Fig. 3, A and B). Ethanol (50 mM) also enhanced Ih by 11.7 ± 1.7% (from 436 ± 68 to 484 ± 72 pA, n = 5, P < 0.01; Fig. 3B), although 20-25 mM ethanol did not have a measurable effect. Ethanol enhancement of Ih was accompanied by an acceleration of the activation kinetics (Fig. 3A1). Thus ethanol (100 mM) reduced the activation time constant of Ih from 384 ± 53 to 267 ± 25 ms (n = 11, P < 0.01; Fig. 3C). It has been reported that neurotransmitters can produce an apparent inhibition of Ih in DA neurons secondary to a reduction in the membrane input resistance, which would worsen space clamp of the recorded cell in voltage-clamp recordings (Watts et al. 1996). Hence, it is possible that ethanol-induced augmentation of Ih was due to an increase in the membrane input resistance and improved space clamp. However, ethanol (100 mM) actually reduced the membrane input resistance from 344 ± 50 to 254 ± 44 MΩ (n = 11, P < 0.0001). Ethanol also caused variable changes in the holding current, ranging from inward to outward, which were not further investigated in the present study.
fig. 3.

Ethanol augments Ih in DA neurons. A, 1 and 2: representative traces (A1) and time graph (A2) depicting the effect of ethanol (100 mM) on Ih. The gray lines represent single-exponential fit to the activating phase of the current during the initial 500 ms of the voltage step. B: summary bar graph showing the effects of 50 and 100 mM ethanol on Ih. C: activation time constant of Ih is plotted before and after ethanol application in individual cells. **P < 0.01.
To gain a mechanistic insight into the ethanol action, we further investigated the effect of ethanol on the voltage dependence of Ih activation. Tail current amplitudes after a series of hyperpolarizing voltage steps were measured to construct an Ih activation curve. Ethanol (100 mM) shifted the Ih activation curve to more depolarized potentials and also produced an increase in the maximal current (Fig. 4). In 10 cells tested, the half-activation potential (V1/2) was shifted from -95.2 ± 1.9 to -91.5 ± 2.0 mV (P < 0.001; Fig. 5, A and E) and the maximal Ih density, obtained by normalizing the maximal current to the cell capacitance, was increased from 12.9 ± 1.2 to 14.0 ± 1.2 pA/pF (P < 0.01; Fig. 5, A and G).
fig. 4.

The effect of ethanol on Ih activation curve. A: representative traces of Ih evoked by a series of voltage steps as indicated. Traces before and during superfusion of ethanol (100 mM) are overlaid. Tail current amplitudes at -105 mV were measured at the time indicated by the arrow. B: tail current amplitudes, after subtraction of the current amplitude after no hyperpolarizing voltage step, are plotted versus the preceding test potentials. The dashed lines represent fit to a Boltzmann function. The data are from the same experiment as in A.
fig. 5.

Ethanol enhancement of Ih has cAMP-dependent and -independent components. A-C: summary of the effects of ethanol on Ih activation curves in cells recorded with a control internal solution (n = 10; A), Rp-cAMPS (100 μM, n = 7; B), and Sp-cAMPS (100 μM, n = 7; C). In each cell, the current amplitudes were normalized to the control maximal amplitude in the absence of ethanol, which was estimated from the Boltzmann fit as shown in Fig. 4. - - -, Boltzmann fit to the averaged data. D-G: summary bar graphs showing the V1/2 value of Ih activation (D), ethanol-induced shift in V1/2 (E), the maximal Ih density (F), and ethanol-induced increase in maximal Ih (G) in cells recorded with a control solution (Con), Rp-cAMPS (Rp), and Sp-cAMPS (Sp). The maximal Ih amplitude, estimated from the Boltzmann fit, was divided by the cell capacitance to obtain the maximal Ih density in each cell. *P < 0.05, **P < 0.01, ***P < 0.001 vs. control.
Direct binding of cAMP, without the involvement of protein kinase A, can facilitate the voltage gating of Ih, thereby producing a depolarizing shift in the voltage dependence of activation and accelerating the activation kinetics (Chen et al. 2001b; Wainger et al. 2001). Indeed, cAMP-mediated facilitation of Ih has been reported in DA neurons (Cathala and Paupardin-Tritsch 1997; Franz et al. 2000; Liu et al. 2003). Furthermore, ethanol can increase intracellular cAMP levels in certain cells (Asher et al. 2002; Hoffman and Tabakoff 1990). Therefore it is possible that cAMP is involved in the ethanol action on Ih. To test this possibility, Rp-cAMPS (100 μM) and Sp-cAMPS (100 μM), an inhibitor and an activator of cAMP-dependent processes, respectively, were infused into the cell through the whole cell pipette. In seven cells tested with intracellular Rp-cAMPS, ethanol (100 mM) failed to cause a significant change in the V1/2 value of Ih activation (from -95.7 ± 3.0 to -95.5 ± 2.9 mV, P = 0.75; Fig. 5B). Accordingly, the magnitude of the depolarizing shift in V1/2 was significantly smaller in Rp-cAMPS than in control (0.2 ± 0.6 mV, n = 7 in Rp-cAMPS vs. 3.7 ± 0.7 mV, n = 10 in control, P < 0.01; Fig. 5E). In contrast, ethanol was still able to produce a significant increase the maximal Ih in the presence of Rp-cAMPS (from 11.4 ± 1.3 to 12.0 ± 1.2 pA/pF, P < 0.05; Fig. 5B). Furthermore, the magnitude of ethanol-induced increase in maximal Ih was comparable in Rp-cAMPS and control (6.0 ± 2.3% in Rp-cAMPS vs. 8.3 ± 2.2% in control, P = 0.49; Fig. 5G). The basal V1/2 value and the maximal Ih density in Rp-cAMPS were not significantly different from those values in control (P = 0.90 for V1/2 and P = 0.39 for maximal Ih density; Fig. 5, D and F); hence Ih is not modulated by tonic levels of cAMP. On the other hand, the basal V1/2 value was more depolarized in seven cells dialyzed with Sp-cAMPS (P < 0.05 vs. control; Fig. 5D), showing that Ih gating can be facilitated by cAMP in DA neurons. Sp-cAMPS prevented the depolarizing shift in Ih activation produced by ethanol (Fig. 5C). On the contrary, ethanol induced a small but significant hyperpolarizing shift in V1/2 in the presence of Sp-cAMPS (from -88.8 ± 1.9 to -91.1 ± 1.6 mV, n = 7, P < 0.05; Fig. 5E). The reason for this hyperpolarizing shift in V1/2 caused by ethanol in Sp-cAMPS is not known. Sp-cAMPS did not affect the maximal Ih density (P = 0.50 vs. control; Fig. 5F) or the magnitude of ethanol effect on maximal Ih (P = 0.99 vs. control; Fig. 5G). Altogether these results demonstrate that ethanol enhances Ih via two mechanisms: a cAMP-dependent mechanism causing a depolarizing shift in the voltage dependence of activation and a cAMP-independent mechanism resulting in augmentation of the maximal Ih conductance.
Ih downregulation after repeated ethanol exposure
Repeated exposure to ethanol produces various compensatory changes in the mesolimbic system to counter the stimulatory action of ethanol (Weiss and Porrino 2002). Recent studies indicate that Ih undergoes activity-dependent plasticity under various neuropathological conditions (Chaplan et al. 2003; Chen et al. 2001a; Shah et al. 2004). Therefore we asked if repeated ethanol exposure in vivo could induce plastic changes in the expression and properties of Ih in DA neurons. Mice received daily injections of either saline or ethanol (2 g/kg ip), to produce blood ethanol concentrations of 40-50 mM (Grisel et al. 2002), for 5 days, and recordings were made 1 day after the final injection. Although the neuroadaptive consequences in the brain can be different after active versus passive administration of drugs of abuse (Jacobs et al. 2003), the passive ethanol treatment protocol used in this study has been shown to cause facilitation of ethanol drinking in C57BL/6J mice (Lessov et al. 2001)
We first obtained the maximum Ih density and V1/2 of Ih activation in each cell using the tail current analysis illustrated in Fig. 4. Ih density was significantly reduced after repeated ethanol treatment in both the VTA and the SNc (Fig. 6A). Overall, Ih density was 11.4 ± 0.7 pA/pF (n = 20) and 8.8 ± 0.4 pA/pF (n = 19) in saline- and ethanol-treated mice, respectively (P < 0.01). A previous study using neonatal mice (P12-P15) has reported that DA neurons in the SNc generally have higher density of Ih than in the VTA (Neuhoff et al. 2002); however, we found no significant difference between the VTA and the SNc in this study using older mice (P21-P35). The V1/2 value (Fig. 6B) and the activation kinetics of Ih (Fig. 6C) were not significantly different between saline- and ethanol-treated mice in both the VTA and the SNc, implying that the gating properties of Ih were not affected by repeated ethanol exposure. These results suggest that repeated ethanol treatment induces downregulation of functional Ih expression in DA neurons. The membrane input resistance at a holding potential of -55 mV was similar in saline- and ethanol-treated mice (Fig. 6D), consistent with the Ih activation curve exhibiting no activation at -55 mV (Fig. 5A).
fig. 6.

Ih plasticity after repeated ethanol treatment in vivo. A-C: summary bar graphs depicting Ih density (A), V1/2 of Ih activation (B), and Ih activation time constant (C) in the VTA and the SNc for saline- and ethanol-treated mice. D: summary bar graph showing the input resistance in saline- and ethanol-treated mice. *P < 0.05 vs. saline.
To determine the functional consequence of Ih downregulation after repeated ethanol exposure, we next compared the firing activity in saline- and ethanol-treated mice. The basal firing frequency was not significantly different in these two types of mice (2.18 ± 0.10 Hz, n = 27 in saline-treated mice vs. 2.31 ± 0.09 Hz, n = 29 in ethanol-treated mice, P = 0.35; Fig. 7A). However, the magnitude of decrease in the firing frequency produced by ZD7288 (30 μM) was significantly reduced after repeated ethanol treatment in both the VTA and the SNc (Fig. 7B). Overall, the magnitude of ZD7288-induced suppression of firing was 32.6 ± 2.7% (n = 10) and 17.8 ± 2.5% (n = 12) in saline- and ethanol-treated mice (P < 0.001). Thus the contribution of Ih to the pacemaker activity was diminished after repeated ethanol exposure. Furthermore, the magnitude of increase in the firing frequency caused by acute ethanol challenge (100 mM) was also significantly smaller in ethanol-treated mice (11.3 ± 2.0%, n = 13) than in saline-treated mice (19.0 ± 2.7%, n = 10, P < 0.05; Fig. 7C), in agreement with the involvement of Ih in the stimulatory effect of ethanol. Ethanol-induced enhancement of Ih was similar in both types of animals (Fig. 7D), demonstrating that the ethanol sensitivity of Ih itself was not altered. Taken together, repeated ethanol exposure in vivo resulted in tolerance to ethanol stimulation of DA neuron activity, most likely due to down-regulation of Ih.
fig. 7.

The effects of ZD7288 and ethanol after repeated ethanol treatment in vivo. Summary bar graphs showing the basal firing frequency (FF; A), ZD7288-induced suppression of FF (B), ethanol-induced increase in FF (C), and ethanol-induced increase in Ih (D) for saline- and ethanol-treated mice. The data are shown separately for the VTA and the SNc in B. *P < 0.05 vs. saline.
DISCUSSION
Ethanol modulates the excitability of neurons by acting on a multitude of ion channels (Harris 1999). In this study, we have identified Ih, a canonical pacemaker current, as an ethanol target in mediating the stimulation of DA neuron firing. Furthermore, repeated ethanol exposure produced down-regulation of functional Ih in DA neurons. The resulting reduction in the stimulatory effect of ethanol may contribute to the progressive increase in alcohol consumption in alcoholic individuals (Schuckit 1994).
Ih and pacemaker activity
ZD7288 (30 μM) suppressed the firing of DA neurons in both the VTA and the SNc, consistent with the involvement of Ih in the regulation of pacemaker activity. This observation is in contrast with a recent study by Neuhoff et al. (2002) demonstrating that Ih is engaged in the pacemaker frequency control of a subpopulation of DA neurons only in the SNc. In that study, ZD7288 selectively inhibited pacemaker activity of SNc DA neurons that do not express the calcium-binding protein calbindin, whereas ZD7288 had no effect in calbindin-positive SNc DA neurons or in any of VTA DA neurons. In line with this, calbindin-negative SNc DA neurons had a higher density of Ih compared with other subpopulations of DA neurons, whereas we detected similar Ih densities in the VTA and the SNc. One difference between the two studies is the age of animals used, i.e., P21-P35 in this study versus P12-P15 in the study of Neuhoff et al., although both used the same strain of mice (C57BL/6J). In this regard, developmental increase in functional Ih expression between P1 and P20 has been reported in hippocampal pyramidal neurons (Vasilyev and Barish 2002). Therefore it is conceivable that maturation of Ih expression during development could affect their impact on DA neuron pacemaker activity. In support of the prominent role of Ih in mature animals, it has been shown that ZD7288 can suppress the spontaneous firing of a majority of VTA DA neurons in adult rats (Seutin et al. 2001). It should also be noted that calbindin-positive VTA DA neurons, which have minimal expression of Ih, displayed a highly irregular firing pattern in the study of Neuhoff et al. It is likely that these VTA neurons were not included in our study because we recorded only from those neurons with regular pacemaker activity.
Acute ethanol effect
An initial study using brain slices demonstrated that ethanol increased the firing frequency of DA neurons in a concentration-dependent fashion at 20-320 mM with an EC50 of ∼100 mM (Brodie et al. 1990), the concentration mainly used in the present investigation. Ethanol facilitation of DA neuron firing was not significantly affected by blockade of major synaptic inputs, including GABAergic inputs, consistent with the direct stimulatory action of ethanol (Brodie et al. 1999). An in vivo study has shown that ethanol suppresses the firing of GABAergic neurons in the VTA (Stobbs et al. 2004), an effect that may lead to disinhibition of DA neurons. GABAergic tone on DA neurons may not be large enough in a brain slice preparation to detect this component of ethanol action.
Inhibition of Ih by ZD7288 treatment caused marked attenuation of the stimulatory effect of ethanol. Ethanol was also able to facilitate Ih, further supporting the role of Ih as an ethanol target. In slow pacemaking neurons like DA neurons with an interspike interval of 0.2-1 s, Ih is thought to be activated by the large afterhyperpolarization (AHP) after each action potential, thus curtailing the AHP. Consistent with this scenario, augmentation of Ih by ethanol reduced the size and duration of the AHP (Fig. 1A, inset), whereas blocking Ih with ZD7288 had an opposite effect (data not shown).
Four genes encoding Ih, termed HCN1-4, have been identified, each having distinct biophysical properties and expression profiles (Kaupp and Seifert 2001). The major type expressed in the VTA appears to be HCN2 (Notomi and Shigemoto 2004), which has an activation time constant of 200-500 ms and is highly sensitive to cAMP-induced facilitation of the voltage gating. In line with this, the activation time constant of Ih was ∼400 ms in this study. Furthermore, ethanol facilitation of Ih had a cAMP-dependent component, most likely via stimulation of adenylyl cyclase and subsequent increase in cytoplasmic cAMP levels (Yoshimura and Tabakoff 1995), mediating a depolarizing shift in the voltage dependence of activation. Ethanol also augmented the maximal Ih amplitude in a cAMP-independent fashion. It is possible that ethanol can directly bind to the Ih channel protein itself, as has been shown for other proteins (Harris 1999), and increase the single-channel conductance and/or maximal open probability of individual Ih channels.
Ethanol stimulation of firing was not entirely eliminated after ZD7288 treatment in three of seven cells (Fig. 2D), suggesting that Ih may not be the only target mediating the effect of ethanol. Our preliminary data show that ethanol can inhibit rapidly inactivating A-type K+ currents in DA neurons (unpublished observation). A-type K+ channels are abundantly expressed in DA neurons and play an opposing role to Ih in controlling the pacemaker frequency (Liss et al. 2001), raising the possibility that these channels may be another target of ethanol. Appel et al. (2003) reported that ethanol excitation of DA neurons was blocked by quinidine, a blocker of multiple types of K+ channels, but not by ZD7288, in rats. Therefore ethanol excitation of DA neurons may involve different mechanisms in different species of animals.
Repeated ethanol exposure
Ih density was reduced by ∼25% in DA neurons after repeated ethanol treatment in vivo (2 g/kg, once daily for 5 days). The reduction in functional Ih resulted in a smaller contribution of Ih to pacemaker activity, as shown by a significantly smaller inhibition of firing by ZD7288. The magnitude of ethanol stimulation of firing was also reduced after repeated ethanol treatment, although ethanol facilitated Ih to a similar degree in saline- and ethanol-treated mice. Thus reduced expression of functional Ih is likely responsible for this tolerance to the excitatory effect of ethanol. The ethanol tolerance that we observed in this study is in contrast to the development of sensitization reported previously using more vigorous and prolonged ethanol treatment protocol (3.5 g/kg, twice daily for ≥21 days) (Brodie 2002). It would be interesting to examine if differential plasticity of Ih underlies the difference in these two studies.
We confirmed that 100 nM ZD7288, which inhibits Ih amplitude by ∼25% (Harris and Constanti 1995), was able to depress the firing frequency by ∼10% in naïve mice (n = 5), indicating that a small reduction in functional Ih can affect DA neuron firing. However, the basal firing frequency was not decreased after repeated ethanol exposure. This may be a consequence of plastic changes in other ion channels that counterbalance the reduction in Ih. In this regard, it has been reported that A-type K+ currents described in the preceding text are co-regulated with Ih in pyloric neurons, resulting in homeostatic regulation of their pacemaker activity (MacLean et al. 2003).
Functional significance
Mereu et al. (1984) reported that intravenous injections of ethanol (0.5-2 g/kg) dose-dependently enhanced the firing of SNc DA neurons by 40-80% in awake rats. Although the actual ethanol level was not measured in the study of Mereu et al., blood and brain ethanol concentrations have been shown to reach 60-70 mM after an intravenous injection of 1 g/kg ethanol (Robinson et al. 2002). In the present investigation using a brain slice preparation, ethanol at 50-100 mM produced smaller increases (10-20%) in the DA neuron firing frequency. The firing of DA neurons in vivo is diverted from a simple pacemaker pattern due to the influence of active synaptic inputs. Thus it is likely that dual actions of ethanol on the intrinsic pacemaker activity and on synaptic inputs work in concert to attain its full stimulatory effect on DA neuron firing in vivo. It is well documented that Ih can dampen the impact of synaptic inputs by reducing the membrane input resistance (Magee 1998). Although Ih does not contribute to the input resistance of DA neurons held at -55 mV, it is certainly involved in the dynamic regulation of the input resistance when DA neurons are tonically firing. The predominant input to DA neurons is GABAergic, which exerts powerful inhibitory control of firing (Tepper et al. 1998). These GABAergic inputs are thought to be suppressed by acute ethanol exposure in vivo (Mereu and Gessa 1985; Stobbs et al. 2004). Ethanol-induced enhancement of Ih may further reduce the inhibitory influence of GABAergic inputs and facilitate the disinhibition of DA neurons. On the other hand, an increase in presynaptic GABA release has been reported after withdrawal from ethanol treatment (Melis et al. 2002). The reduction in Ih density observed in this study may augment the postsynaptic impact of these facilitated GABAergic inputs, thus contributing to the marked reduction in DA neuron activity during ethanol withdrawal (Diana et al. 1993). Therefore Ih may play a role not only in the acute reinforcing action of ethanol but also in the hypodopaminergic state underlying the emotional/motivational component of ethanol withdrawal symptoms.
ACKNOWLEDGMENTS
We thank Drs. R. Adron Harris and R. Dayne Mayfield for comments on the earlier version of the manuscript and Dr. Yuri Blednov for providing excellent animal care.
Footnotes
The costs of publication of this article were defrayed in part by the payment of page charges. The article must therefore be hereby marked “advertisement” in accordance with 18 U.S.C. Section 1734 solely to indicate this fact.
GRANTS
This work was supported by a grant from the Alcoholic Beverage Medical Research Foundation and a National Institute on Drug Abuse Grant DA-015687. M. T. Harnett was supported by a National Science Foundation Graduate Research Fellowship.
REFERENCES
- Ahlenius S, Carlsson A, Engel J, Svensson T, Sodersten P. Antagonism by alpha methyltyrosine of the ethanol-induced stimulation and euphoria in man. Clin Pharmacol Ther. 1973;14:586–591. doi: 10.1002/cpt1973144part1586. [DOI] [PubMed] [Google Scholar]
- Appel SB, Liu Z, McElvain MA, Brodie MS. Ethanol excitation of dopaminergic ventral tegmental area neurons is blocked by quinidine. J Pharmacol Exp Ther. 2003;306:437–446. doi: 10.1124/jpet.103.050963. [DOI] [PubMed] [Google Scholar]
- Asher O, Cunningham TD, Yao L, Gordon AS, Diamond I. Ethanol stimulates cAMP-responsive element (CRE)-mediated transcription via CRE-binding protein and cAMP-dependent protein kinase. J Pharmacol Exp Ther. 2002;301:66–70. doi: 10.1124/jpet.301.1.66. [DOI] [PubMed] [Google Scholar]
- Brodie MS. Increased ethanol excitation of dopaminergic neurons of the ventral tegmental area after chronic ethanol treatment. Alcohol Clin Exp Res. 2002;26:1024–1030. doi: 10.1097/01.ALC.0000021336.33310.6B. [DOI] [PubMed] [Google Scholar]
- Brodie MS, Appel SB. The effects of ethanol on dopaminergic neurons of the ventral tegmental area studied with intracellular recording in brain slices. Alcohol Clin Exp Res. 1998;22:236–244. [PubMed] [Google Scholar]
- Brodie MS, Pesold C, Appel SB. Ethanol directly excites dopaminergic ventral tegmental area reward neurons. Alcohol Clin Exp Res. 1999;23:1848–1852. [PubMed] [Google Scholar]
- Brodie MS, Shefner SA, Dunwiddie TV. Ethanol increases the firing rate of dopamine neurons of the rat ventral tegmental area in vitro. Brain Res. 1990;508:65–69. doi: 10.1016/0006-8993(90)91118-z. [DOI] [PubMed] [Google Scholar]
- Brown H, Difrancesco D. Voltage-clamp investigations of membrane currents underlying pace-maker activity in rabbit sino-atrial node. J Physiol. 1980;308:331–351. doi: 10.1113/jphysiol.1980.sp013474. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Budygin EA, Phillips PE, Wightman RM, Jones SR. Terminal effects of ethanol on dopamine dynamics in rat nucleus accumbens: an in vitro voltammetric study. Synapse. 2001;42:77–79. doi: 10.1002/syn.1101. [DOI] [PubMed] [Google Scholar]
- Cathala L, Paupardin-Tritsch D. Neurotensin inhibition of the hyperpolarization-activated cation current (Ih) in the rat substantia nigra pars compacta implicates the protein kinase C pathway. J Physiol. 1997;503:87–97. doi: 10.1111/j.1469-7793.1997.087bi.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cathala L, Paupardin-Tritsch D. Effect of catecholamines on the hyperpolarization-activated cationic Ih and the inwardly rectifying potassium I(Kir) currents in the rat substantia nigra pars compacta. Eur J Neurosci. 1999;11:398–406. doi: 10.1046/j.1460-9568.1999.00452.x. [DOI] [PubMed] [Google Scholar]
- Chan CS, Shigemoto R, Mercer JN, Surmeier DJ. HCN2 and HCN1 channels govern the regularity of autonomous pacemaking and synaptic resetting in globus pallidus neurons. J Neurosci. 2004;24:9921–9932. doi: 10.1523/JNEUROSCI.2162-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chaplan SR, Guo HQ, Lee DH, Luo L, Liu C, Kuei C, Velumian AA, Butler MP, Brown SM, Dubin AE. Neuronal hyperpolarization-activated pacemaker channels drive neuropathic pain. J Neurosci. 2003;23:1169–1178. doi: 10.1523/JNEUROSCI.23-04-01169.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chavis P, Mollard P, Bockaert J, Manzoni O. Visualization of cyclic AMP-regulated presynaptic activity at cerebellar granule cells. Neuron. 1998;20:773–781. doi: 10.1016/s0896-6273(00)81015-6. [DOI] [PubMed] [Google Scholar]
- Chen K, Aradi I, Thon N, Eghbal-Ahmadi M, Baram TZ, Soltesz I. Persistently modified h-channels after complex febrile seizures convert the seizure-induced enhancement of inhibition to hyperexcitability. Nat Med. 2001a;7:331–337. doi: 10.1038/85480. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen S, Wang J, Siegelbaum SA. Properties of hyperpolarization-activated pacemaker current defined by coassembly of HCN1 and HCN2 subunits and basal modulation by cyclic nucleotide. J Gen Physiol. 2001b;117:491–504. doi: 10.1085/jgp.117.5.491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chevaleyre V, Castillo PE. Assessing the role of Ih channels in synaptic transmission and mossy fiber LTP. Proc Natl Acad Sci USA. 2002;99:9538–9543. doi: 10.1073/pnas.142213199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Di Chiara G, Imperato A. Drugs abused by humans preferentially increase synaptic dopamine concentrations in the mesolimbic system of freely moving rats. Proc Natl Acad Sci USA. 1988;85:5274–5278. doi: 10.1073/pnas.85.14.5274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Diana M, Pistis M, Carboni S, Gessa GL, Rossetti ZL. Profound decrement of mesolimbic dopaminergic neuronal activity during ethanol withdrawal syndrome in rats: electrophysiological and biochemical evidence. Proc Natl Acad Sci USA. 1993;90:7966–7969. doi: 10.1073/pnas.90.17.7966. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Franz O, Liss B, Neu A, Roeper J. Single-cell mRNA expression of HCN1 correlates with a fast gating phenotype of hyperpolarization-activated cyclic nucleotide-gated ion channels (Ih) in central neurons. Eur J Neurosci. 2000;12:2685–2693. doi: 10.1046/j.1460-9568.2000.00151.x. [DOI] [PubMed] [Google Scholar]
- Gatto GJ, McBride WJ, Murphy JM, Lumeng L, Li TK. Ethanol self-infusion into the ventral tegmental area by alcohol-preferring rats. Alcohol. 1994;11:557–564. doi: 10.1016/0741-8329(94)90083-3. [DOI] [PubMed] [Google Scholar]
- George SR, Fan T, Ng GY, Jung SY, O'Dowd BF, Naranjo CA. Low endogenous dopamine function in brain predisposes to high alcohol preference and consumption: reversal by increasing synaptic dopamine. J Pharmacol Exp Ther. 1995;273:373–379. [PubMed] [Google Scholar]
- Gessa GL, Muntoni F, Collu M, Vargiu L, Mereu G. Low doses of ethanol activate dopaminergic neurons in the ventral tegmental area. Brain Res. 1985;348:201–203. doi: 10.1016/0006-8993(85)90381-6. [DOI] [PubMed] [Google Scholar]
- Grace AA. Regulation of spontaneous activity and oscillatory spike firing in rat midbrain dopamine neurons recorded in vitro. Synapse. 1991;7:221–234. doi: 10.1002/syn.890070307. [DOI] [PubMed] [Google Scholar]
- Grisel JE, Metten P, Wenger CD, Merrill CM, Crabbe JC. Mapping of quantitative trait loci underlying ethanol metabolism in BXD recombinant inbred mouse strains. Alcohol Clin Exp Res. 2002;26:610–616. [PubMed] [Google Scholar]
- Harris NC, Constanti A. Mechanism of block by ZD 7288 of the hyperpolarization-activated inward rectifying current in guinea pig substantia nigra neurons in vitro. J Neurophysiol. 1995;74:2366–2378. doi: 10.1152/jn.1995.74.6.2366. [DOI] [PubMed] [Google Scholar]
- Harris RA. Ethanol actions on multiple ion channels: which are important? Alcohol Clin Exp Res. 1999;23:1563–1570. [PubMed] [Google Scholar]
- Hoffman PL, Tabakoff B. Ethanol and guanine nucleotide binding proteins: a selective interaction. Faseb J. 1990;4:2612–2622. doi: 10.1096/fasebj.4.9.2161371. [DOI] [PubMed] [Google Scholar]
- Ikemoto S, McBride WJ, Murphy JM, Lumeng L, Li TK. 6-OHDA-lesions of the nucleus accumbens disrupt the acquisition but not the maintenance of ethanol consumption in the alcohol-preferring P line of rats. Alcohol Clin Exp Res. 1997;21:1042–1046. [PubMed] [Google Scholar]
- Jacobs EH, Smit AB, de Vries TJ, Schoffelmeer AN. Neuroadaptive effects of active versus passive drug administration in addiction research. Trends Pharmacol Sci. 2003;24:566–573. doi: 10.1016/j.tips.2003.09.006. [DOI] [PubMed] [Google Scholar]
- Jiang ZG, Pessia M, North RA. Dopamine and baclofen inhibit the hyperpolarization-activated cation current in rat ventral tegmental neurones. J Physiol. 1993;462:753–764. doi: 10.1113/jphysiol.1993.sp019580. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaczmarek HJ, Kiefer SW. Microinjections of dopaminergic agents in the nucleus accumbens affect ethanol consumption but not palatability. Pharmacol Biochem Behav. 2000;66:307–312. doi: 10.1016/s0091-3057(00)00182-9. [DOI] [PubMed] [Google Scholar]
- Kaupp UB, Seifert R. Molecular diversity of pacemaker ion channels. Annu Rev Physiol. 2001;63:235–257. doi: 10.1146/annurev.physiol.63.1.235. [DOI] [PubMed] [Google Scholar]
- Kitai ST, Shepard PD, Callaway JC, Scroggs R. Afferent modulation of dopamine neuron firing patterns. Curr Opin Neurobiol. 1999;9:690–697. doi: 10.1016/s0959-4388(99)00040-9. [DOI] [PubMed] [Google Scholar]
- Lessov CN, Palmer AA, Quick EA, Phillips TJ. Voluntary ethanol drinking in C57BL/6J and DBA/2J mice before and after sensitization to the locomotor stimulant effects of ethanol. Psychopharmacology. 2001;155:91–99. doi: 10.1007/s002130100699. [DOI] [PubMed] [Google Scholar]
- Liss B, Franz O, Sewing S, Bruns R, Neuhoff H, Roeper J. Tuning pacemaker frequency of individual dopaminergic neurons by Kv4.3L and KChip3.1 transcription. Embo J. 2001;20:5715–5724. doi: 10.1093/emboj/20.20.5715. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu Z, Bunney EB, Appel SB, Brodie MS. Serotonin reduces the hyperpolarization-activated current (Ih) in ventral tegmental area dopamine neurons: involvement of 5-HT2 receptors and protein kinase C. J Neurophysiol. 2003;90:3201–3212. doi: 10.1152/jn.00281.2003. [DOI] [PubMed] [Google Scholar]
- Luthi A, McCormick DA. Periodicity of thalamic synchronized oscillations: the role of Ca2+-mediated upregulation of Ih. Neuron. 1998;20:553–563. doi: 10.1016/s0896-6273(00)80994-0. [DOI] [PubMed] [Google Scholar]
- Maccaferri G, McBain CJ. The hyperpolarization-activated current (Ih) and its contribution to pacemaker activity in rat CA1 hippocampal stratum oriens-alveus interneurons. J Physiol. 1996;497:119–130. doi: 10.1113/jphysiol.1996.sp021754. [DOI] [PMC free article] [PubMed] [Google Scholar]
- MacLean JN, Zhang Y, Johnson BR, Harris-Warrick RM. Activity-independent homeostasis in rhythmically active neurons. Neuron. 2003;37:109–120. doi: 10.1016/s0896-6273(02)01104-2. [DOI] [PubMed] [Google Scholar]
- Magee JC. Dendritic hyperpolarization-activated currents modify the integrative properties of hippocampal CA1 pyramidal neurons. J Neurosci. 1998;18:7613–7624. doi: 10.1523/JNEUROSCI.18-19-07613.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Melis M, Camarini R, Ungless MA, Bonci A. Long-lasting potentiation of GABAergic synapses in dopamine neurons after a single in vivo ethanol exposure. J Neurosci. 2002;22:2074–2082. doi: 10.1523/JNEUROSCI.22-06-02074.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mereu G, Fadda F, Gessa GL. Ethanol stimulates the firing rate of nigral dopaminergic neurons in unanesthetized rats. Brain Res. 1984;292:63–69. doi: 10.1016/0006-8993(84)90890-4. [DOI] [PubMed] [Google Scholar]
- Mereu G, Gessa GL. Low doses of ethanol inhibit the firing of neurons in the substantia nigra, pars reticulata: a GABAergic effect? Brain Res. 1985;360:325–330. doi: 10.1016/0006-8993(85)91249-1. [DOI] [PubMed] [Google Scholar]
- Morikawa H, Khodakhah K, Williams JT. Two intracellular pathways mediate metabotropic glutamate receptor-induced Ca2+ mobilization in dopamine neurons. J Neurosci. 2003;23:149–157. doi: 10.1523/JNEUROSCI.23-01-00149.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Myers RD, Robinson DE. Mmu and D2 receptor antisense oligonucleotides injected in nucleus accumbens suppress high alcohol intake in genetic drinking HEP rats. Alcohol. 1999;18:225–233. doi: 10.1016/s0741-8329(99)00015-4. [DOI] [PubMed] [Google Scholar]
- Neuhoff H, Neu A, Liss B, Roeper J. I(h) channels contribute to the different functional properties of identified dopaminergic subpopulations in the midbrain. J Neurosci. 2002;22:1290–1302. doi: 10.1523/JNEUROSCI.22-04-01290.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Notomi T, Shigemoto R. Immunohistochemical localization of Ih channel subunits, HCN1-4, in the rat brain. J Comp Neurol. 2004;471:241–276. doi: 10.1002/cne.11039. [DOI] [PubMed] [Google Scholar]
- Rassnick S, Stinus L, Koob GF. The effects of 6-hydroxydopamine lesions of the nucleus accumbens and the mesolimbic dopamine system on oral self-administration of ethanol in the rat. Brain Res. 1993;623:16–24. doi: 10.1016/0006-8993(93)90004-7. [DOI] [PubMed] [Google Scholar]
- Robinson DL, Brunner LJ, Gonzales RA. Effect of gender and estrous cycle on the pharmacokinetics of ethanol in the rat brain. Alcohol Clin Exp Res. 2002;26:165–172. [PubMed] [Google Scholar]
- Rodd ZA, Melendez RI, Bell RL, Kuc KA, Zhang Y, Murphy JM, McBride WJ. Intracranial self-administration of ethanol within the ventral tegmental area of male Wistar rats: evidence for involvement of dopamine neurons. J Neurosci. 2004;24:1050–1057. doi: 10.1523/JNEUROSCI.1319-03.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Samson HH, Hodge CW, Tolliver GA, Haraguchi M. Effect of dopamine agonists and antagonists on ethanol-reinforced behavior: the involvement of the nucleus accumbens. Brain Res Bull. 1993;30:133–141. doi: 10.1016/0361-9230(93)90049-h. [DOI] [PubMed] [Google Scholar]
- Schuckit MA. Low level of response to alcohol as a predictor of future alcoholism. Am J Psychiatry. 1994;151:184–189. doi: 10.1176/ajp.151.2.184. [DOI] [PubMed] [Google Scholar]
- Seutin V, Massotte L, Renette MF, Dresse A. Evidence for a modulatory role of Ih on the firing of a subgroup of midbrain dopamine neurons. Neuroreport. 2001;12:255–258. doi: 10.1097/00001756-200102120-00015. [DOI] [PubMed] [Google Scholar]
- Shah MM, Anderson AE, Leung V, Lin X, Johnston D. Seizure-induced plasticity of h channels in entorhinal cortical layer III pyramidal neurons. Neuron. 2004;44:495–508. doi: 10.1016/j.neuron.2004.10.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stobbs SH, Ohran AJ, Lassen MB, Allison DW, Brown JE, Steffensen SC. Ethanol suppression of ventral tegmental area GABA neuron electrical transmission involves N-methyl-d-aspartate receptors. J Pharmacol Exp Ther. 2004;311:282–289. doi: 10.1124/jpet.104.071860. [DOI] [PubMed] [Google Scholar]
- Tepper JM, Paladini CA, Celada P. GABAergic control of the firing pattern of substantia nigra dopaminergic neurons. Adv Pharmacol. 1998;42:694–699. doi: 10.1016/s1054-3589(08)60843-1. [DOI] [PubMed] [Google Scholar]
- Vasilyev DV, Barish ME. Postnatal development of the hyperpolarization-activated excitatory current Ih in mouse hippocampal pyramidal neurons. J Neurosci. 2002;22:8992–9004. doi: 10.1523/JNEUROSCI.22-20-08992.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wainger BJ, DeGennaro M, Santoro B, Siegelbaum SA, Tibbs GR. Molecular mechanism of cAMP modulation of HCN pacemaker channels. Nature. 2001;411:805–810. doi: 10.1038/35081088. [DOI] [PubMed] [Google Scholar]
- Watts AE, Williams JT, Henderson G. Baclofen inhibition of the hyperpolarization-activated cation current, Ih, in rat substantia nigra zona compacta neurons may be secondary to potassium current activation. J Neurophysiol. 1996;76:2262–2270. doi: 10.1152/jn.1996.76.4.2262. [DOI] [PubMed] [Google Scholar]
- Weiss F, Lorang MT, Bloom FE, Koob GF. Oral alcohol self-administration stimulates dopamine release in the rat nucleus accumbens: genetic and motivational determinants. J Pharmacol Exp Ther. 1993;267:250–258. [PubMed] [Google Scholar]
- Weiss F, Mitchiner M, Bloom FE, Koob GF. Free-choice responding for ethanol versus water in alcohol preferring (P) and unselected Wistar rats is differentially modified by naloxone, bromocriptine, and methysergide. Psychopharmacology. 1990;101:178–186. doi: 10.1007/BF02244123. [DOI] [PubMed] [Google Scholar]
- Weiss F, Porrino LJ. Behavioral neurobiology of alcohol addiction: recent advances and challenges. J Neurosci. 2002;22:3332–3337. doi: 10.1523/JNEUROSCI.22-09-03332.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yim HJ, Gonzales RA. Ethanol-induced increases in dopamine extracellular concentration in rat nucleus accumbens are accounted for by increased release and not uptake inhibition. Alcohol. 2000;22:107–115. doi: 10.1016/s0741-8329(00)00121-x. [DOI] [PubMed] [Google Scholar]
- Yoshimura M, Tabakoff B. Selective effects of ethanol on the generation of cAMP by particular members of the adenylyl cyclase family. Alcohol Clin Exp Res. 1995;19:1435–1440. doi: 10.1111/j.1530-0277.1995.tb01004.x. [DOI] [PubMed] [Google Scholar]