

Abstract
A multiprotein, high molecular weight complex active in both U-insertion and U-deletion as judged by a pre-cleaved RNA editing assay was isolated from mitochondrial extracts of Leishmania tarentolae by the tandem affinity purification (TAP) procedure, using three different TAP-tagged proteins of the complex. This editing- or E-complex consists of at least three protein-containing components interacting via RNA: the RNA ligase-containing L-complex, a 3′ TUTase (terminal uridylyltransferase) and two RNA-binding proteins, Ltp26 and Ltp28. Thirteen approximately stoichiometric components were identified by mass spectrometric analysis of the core L-complex: two RNA ligases; homologs of the four Trypanosoma brucei editing proteins; and seven novel polypeptides, among which were two with RNase III, one with an AP endo/exonuclease and one with nucleotidyltransferase motifs. Three proteins have no similarities beyond kinetoplastids.
Keywords: editosome/RNA editing/TAP/TUTase
Introduction
Uridine insertion/deletion RNA editing is a post-transcriptional RNA modification phenomenon that occurs in the mitochondrion of kinetoplastid protists (Estévez and Simpson, 1999). The mechanism involves the initial hybridization to an mRNA of a complementary guide RNA (gRNA) which guides a specific endonuclease cleavage at the first editing site (Blum et al., 1990). This is followed by either deletion of the unpaired uridines from the cleavage fragment or the 3′ addition to the mRNA 5′ cleavage fragment, hybridization of the added Us to the guiding nucleotides in the gRNA, and religation of the two mRNA cleavage fragments. Each gRNA specifies the 3′ to 5′ editing of a small number of sites and, in the case of a multiple gRNA-mediated editing domain, creates the anchor sequence for hybridization of the adjacent upstream gRNA, thus producing an overall 3′ to 5′ progression of editing. A minimal non-progressive editing activity at one or two sites has been demonstrated in vitro using crude or partially purified mitochondrial extract, and the reaction was shown to involve high molecular weight RNP complexes (Byrne et al., 1996; Cruz-Reyes and Sollner-Webb, 1996; Kable et al., 1996; Seiwert et al., 1996).
The mechanism described above was proposed >12 years ago (Blum et al., 1990), and was verified experimentally in 1996 for both Trypanosoma brucei and Leishmania tarentolae (Byrne et al., 1996; Cruz-Reyes and Sollner-Webb, 1996; Seiwert et al., 1996). However, progress in the identification of specific proteins involved in editing has been hampered by their low abundance and by the low efficiency of the in vitro editing assays. A seven polypeptide complex from T.brucei mitochondria that supported in vitro insertion and deletion editing was isolated by two chromatographic steps and was proposed to represent a core editing complex (Rusche et al., 1997). An ∼20 polypeptide complex with similar activities was isolated in another laboratory by a similar fractionation (Panigrahi et al., 2001a,b).
The genes for several of the major components of these complexes have been identified, but only a few proteins so far have been ascribed enzymatic functions. Two components of both the seven polypeptide complex and the 20 polypeptide complex were shown to represent the adenylatable mitochondrial RNA ligases, REL1 (also termed band 4 or DREL) and REL2 (also termed band 5 or IREL) (Sabatini and Hajduk, 1995; Panigrahi et al., 2001a; Rusché et al., 2001), and a conditional gene knockout showed that the REL1 ligase was required both for viability of bloodstream T.brucei and for RNA editing in vivo (Schnaufer et al., 2001). A dominant-negative mutant of the REL1 ligase was also shown to affect parasite growth when overexpressed in T.brucei procyclic cells (Huang et al., 2001). Three other T.brucei proteins, TbMP81 (also known as band II), TbMP63 (also known as band III) and TbMP42 (also known as band VI), with zinc finger motifs, and the related TbMP18 (also known as band VII) were identified from the ∼20 polypeptide complex (Panigrahi et al., 2001b) and from the seven polypeptide complex (Huang et al., 2002). Down-regulation of TbMP81 and TbMP63 expression by RNA interference (RNAi) inhibited RNA editing, supporting an involvement of these proteins in editing (Drozdz et al., 2002; Huang et al., 2002). However, in spite of the fact that terminal uridylyltransferase (TUTase), 3′ exonuclease and gRNA-mediated endonuclease enzymatic activities were detected in both purified preparations, none of the polypeptides in either complex has been shown definitely to represent any of these enzymes or to contain putative enzymatic motifs.
Here we describe the affinity isolation of essentially all protein components of the RNA ligase-containing L-complex from L.tarentolae and identification of their orthologs in the closely related Leishmania major genome. We also show that the L-complex interacts with the 121 kDa 3′ TUTase and with two RNA-binding proteins via an RNA component. The resulting multienzyme complex is active in pre-cleaved editing assays for both U-insertion and U-deletion.
Results
The RNA ligase-containing L-complex
In order to assess the amount and size of RNA ligase-containing complexes in mitochondria of L.tarentolae, mitochondrial extract was fractionated on a 10–30% glycerol gradient followed by incubation of each fraction with [α-32P]ATP and separation on a native gel. This analysis showed the presence of two distinct ATP-labeled bands of ∼750 and 1500 kDa, as reported previously (Peris et al., 1997) (Figure 1A, panel 1, labeled I-complex and L-complex, respectively). The L-complex sedimented in gradient fractions 9 and 10, which corresponded to an approximate sedimentation value of 25S. Similar results were obtained using mitochondria from procyclic T.brucei (Figure 1B, panel 1), as also reported previously (Peris et al., 1997). Analysis of the same gradient fractions in an SDS gel showed the presence of the 50 (LtREL1) and 45 kDa (LtREL2) adenylated bands co-sedimenting with the L-complex in gradient fractions 8–11 (Figure 1A, panel 2); the lack of correspondence between native and SDS gels in fractions 5–7 indicated that labeling of the I-complex with [α-32P]ATP was not due to the presence of RNA ligase and that the majority of the label was not covalently bound. This was confirmed by labeling the I-complex in the absence of Mg2+, a cofactor that is absolutely required for ligase adenylation (Figure 1A, panel 3, and B, panel 2), and showing that no labeled protein bands were detectable upon SDS gel electrophoresis (Figure 1A, panel 4). The nature of the I-complex is unclear, but there is no evidence to suggest an involvement in editing, whereas the L-complex is clearly involved in RNA editing due to the presence of the editing ligases.

Fig. 1. Identification of the L-complex in mitochondrial extracts of L.tarentolae and T.brucei. The L-complex and the I-complex can be distinguished by labeling with [α-32P]ATP in the presence and absence of Mg2+. Mitochondrial S100 extracts were separated on a 10–30% glycerol gradient in an SW41 rotor for 20 h at 30 000 r.p.m. (A) Leishmania tarentolae. Panel 1: gradient fractions were incubated with [α-32P]ATP in the presence of Mg2+ and separated on a 4–20% native gel. The locations of the L-complex and I-complex are indicated by arrows. Panel 2: same fractions as in 1, SDS gel. The adenylated LtREL1 and LtREL2 proteins are indicated. Panel 3: fractions incubated with [α-32P]ATP in the absence of Mg2+, 4–20% native gel. Panel 4: same fractions as in panel 3, SDS gel. (B) Trypanosoma brucei. Panel 1: same conditions as panel 1 in (A). Panel 2: same conditions as panel 3 in (A).
Interaction of 3′ TUTase and Ltp26 and Ltp28 RNA-binding proteins with the L-complex
Evidence for an interaction of the L-complex with the 121 kDa 3′ TUTase (Aphasizhev et al., 2002) and the Ltp26/Ltp28 RNA-binding proteins (Aphasizhev et al., 2003) is provided by the experiments in Figure 2. Mitochondrial extract from wild-type cells was fractionated on a glycerol gradient, and each fraction was adenylated to detect the L-complex (Figure 2A). As a control for non-specific adsorption, half of each fraction was incubated with IgG beads coated with antiserum against glutamate dehydrogenase, which is an abundant mitochondrial protein not involved in editing (Estévez et al., 1999) (Figure 2A, panel 1), and the other half with antigen-purified TUTase antibodies (Aphasizhev et al., 2002) (Figure 2A, panel 2). After removing the beads, supernatants were incubated with [α-32P]ATP and separated on native gels. As shown in Figure 2A, panel 2, the ATP-labeled L-complex in the native gel was almost completely immunodepleted by the anti-TUTase antiserum. Panel 3 in Figure 2A shows an SDS gel analysis of the washed IgG–Sepharose beads from the experiment in panel 2, and demonstrates that the LtREL1 and LtREL2 components of the L-complex co-immunoprecipitated with the TUTase. No adenylated proteins were detected to be precipitated by the control beads (not shown). Similar immunodepletion and co-immunoprecipitation results were obtained when the gradient fractions were immunoprecipitated with anti-Ltp26/Ltp28 antiserum, as shown in Figure 2B. This evidence suggests an interaction between the L-complex, the 3′ TUTase and Ltp26/Ltp28.

Fig. 2. Interactions of the L-complex with 3′ TUTase and Ltp26/Ltp28 RNA-binding proteins. (A) Immunodepletion and co-immunoprecipitation of the L-complex with anti-TUTase antibody. Mitochondrial extract from L.tarentolae was fractionated on a 10–30% glycerol gradient for 20 h at 30 000 r.p.m. in an SW41 rotor, and two rounds of immunodepletion with affinity-purified antiserum against glutamate or antigen-purified polyclonal antibodies against recombinant TUTase were performed on each fraction. Panel 1: GDH immunodepletion control, native gel of adenylated fractions. The adenylated L-complex and I-complex bands are indicated. Panel 2: TUTase immunodepletion, native gel of adenylated fractions. Panel 3: IgG–Sepharose beads from the TUTase immunoprecipitation in panel 2 were incubated with [α-32P]ATP, washed and analyzed on a 10–20% SDS gel. The adenylated RNA ligase proteins are indicated. (B) Immunodepletion and co-immunoprecipitation of L-complex with antibodies against Ltp26 and Ltp28 RNA-binding proteins. Panel 1: control, native gel of adenylated gradient fractions after incubation with IgG beads saturated with pre-immune serum. Panel 2: native gel of Ltp26/Ltp28-immunodepleted adenylated gradient fractions. Panel 3: SDS gel of adenylated gradient fractions from IgG beads from panel 2. (C) Formation of L-complex in T.brucei procyclic cells is not affected by inhibition of TUTase expression. RNAi was induced with tetracycline for 3 days in procyclic T.brucei to down-regulate TUTase expression as described previously (Aphasizhev et al., 2002). Mitochondrial extracts were fractionated on 10–30% glycerol gradients for 20 h at 30 000 r.p.m. in an SW41 rotor. Panel 1: gradient fractions from uninduced and RNAi-induced cells were assayed for TUTase activity by incorporation of a [α-32P]UTP into a synthetic RNA substrate. Panel 2: TUTase western of SDS gels of gradient fractions from uninduced (Tet–) and RNAi-induced (Tet+) cells. Panels 3 and 4: native gels of adenylated gradient fractions from uninduced and RNAi-induced cells. The adenylated L-complex and I-complex bands are indicated by arrows.
To determine whether the interaction with TUTase is required for formation of the L-complex, the presence of the adenylatable L-complex was examined in T.brucei procyclic cells in which TUTase expression has been down-regulated by RNAi (Aphasizhev et al., 2002). Trypanosoma brucei cells were used since RNAi is not yet applicable to Leishmania, but the organization of editing complexes in both species is likely to be very similar. Cells were collected after 72 h of RNAi, a time period that is sufficient to deplete TUTase but not to change the growth rate or phenotype. As shown in Figure 2C, the loss of ∼80% of total TUTase activity (panel 1) and TUTase protein (panel 2), including all detectable TUTase in the 25–30S region, had no detectable effect on the L-complex as identified by an adenylatable band in a native gel (panels 3 and 4). This suggests that the 121 kDa 3′ TUTase is not required for the formation of the L-complex.
The interaction of the L-complex with the 3′ TUTase is RNase sensitive
Treatment of the mitochondrial extract with RNase A prior to gradient fractionation caused a loss of TUTase protein that could be immunoprecipitated with anti-TUTase antiserum from the 25–30S region of the gradient (Figure 3A and B, panel 3), as well as a loss of the co-immunoprecipitation of the adenylated LtREL1 and LtREL2 proteins (Figure 3A and B, panel 4). There was no effect of the RNase treatment on the L-complex itself, as seen by native gel analysis, other than a small but reproducible enhancement of the adenylation signal and a decrease in sedimentation by one to two fractions. We speculate that the enhancement of the signal and the slight decrease in S value is due to the removal of a ligatable RNA component from the L-complex, but this remains to be investigated. Our previous finding that the L-complex and the ∼700 kDa TUT II form of TUTase could be separated on a glycerol gradient after poly(U)–Sepharose and anion-exchange chromatography, conditions that involve exposure to high salt concentration and removal of weakly bound RNA (Aphasizhev et al., 2002), is also consistent with an RNA involvement in TUTase– L-complex interactions.

Fig. 3. Interaction of L-complex with the 121 kDa 3′ TUTase is disrupted by RNase A treatment. Mitochondrial extracts were incubated without (A) or with (B) RNase A (0.1 mg/ml) and fractionated on 10–30% glycerol gradients in an SW 28 rotor for 40 h at 24 000 r.p.m. Panel 1: 8–16% Tris-glycine native gel of adenylated gradient fractions. Panel 2: SDS gel of adenylated gradient fractions from panel 1. Panel 3: western blotting detection of TUTase immunoprecipitated with anti-TUTase antibody and recovered from IgG beads. Panel 4: SDS gel of adenylated LtREL1 and LtREL2 proteins co-immunoprecipitated with anti-TUTase antibody and recovered from IgG beads. (C) The L-complex band from (A) was excised and the protein components eluted and separated in an SDS gel, which was silver stained. Lane 1, stained gel; lane 2, autoradiograph of adenylated LtREL1 and LtREL2; lane 3, western blot using anti-TbREL1 monoclonal P3C1-G2 antibody; lane 4, western blotting with anti-TUTase antibody. (D) The L-complex band from the RNase-treated extract in (B) was eluted as in (C). Lane 1, stained gel; lane 2, autoradiograph of adenylated LtREL1 and LtREL2; lane 3, western blot using anti-TbREL1 monoclonal P3C1-G2 antibody; lane 4, western blotting with anti-TUTase antibody.
We show elsewhere that the interaction of the Ltp26/Ltp28 100 kDa complex with the L-complex is also RNase sensitive (Aphasizhev et al., 2003).
Polypeptide profile of gel-isolated L-complex
The native gel band of the adenylated L-complex from the experiment in Figure 3A was eluted and the component polypeptides separated in an SDS gel. The protein profile (Figure 3C, lane 1) was relatively simple and, as shown below, is almost identical to that obtained from the tandem affinity purification (TAP)-purified L-complex. The prominent 95 kDa band is unique to this preparation, but the relative intensity could indicate a contaminant. The 50 and 45 kDa bands were identified as LtREL1 and LtREL2 by adenylation (Figure 3C, lane 2) and western analysis (Figure 3C, lane 3). A 121 kDa 3′ TUTase could be identified by western analysis, but it did not correspond to a stoichiometric stained band (Figure 3C, lane 4).
The adenylated L-complex band from the experiment in Figure 3B, in which the extract was pre-treated with RNase A, was also eluted and the component polypeptides separated. As shown in Figure 3D, a protein pattern identical to that in Figure 3C was obtained of the L-complex from extract not pre-treated with RNase. However, the amount of the 121 kDa 3′ TUTase decreased below the level of detection by western analysis.
LtREL1–TAP affinity isolation of L-complex
The native gel isolation of the L-complex did not provide sufficient material for protein identification. Therefore, an affinity purification strategy was employed. The LtREL1 protein was fused at the C-terminus to the TAP double epitope tag (Rigaut et al., 1999) which consists of a calmodulin-binding peptide (CBP) and two protein A peptides in tandem, and inserted into the pX expression vector (LeBowitz et al., 1990) for transfection into L.tarentolae cells. Using PAP reagent, which is specific for protein A, LtREL1–TAP was detected only in the total cell (Figure 4A, lane 2) and mitochondrial fractions (lane 4) and not in the cytosolic fraction (lane 3) or in untransfected cells (lane 1), demonstrating that the fusion protein was expressed and targeted to the mitochondrion.
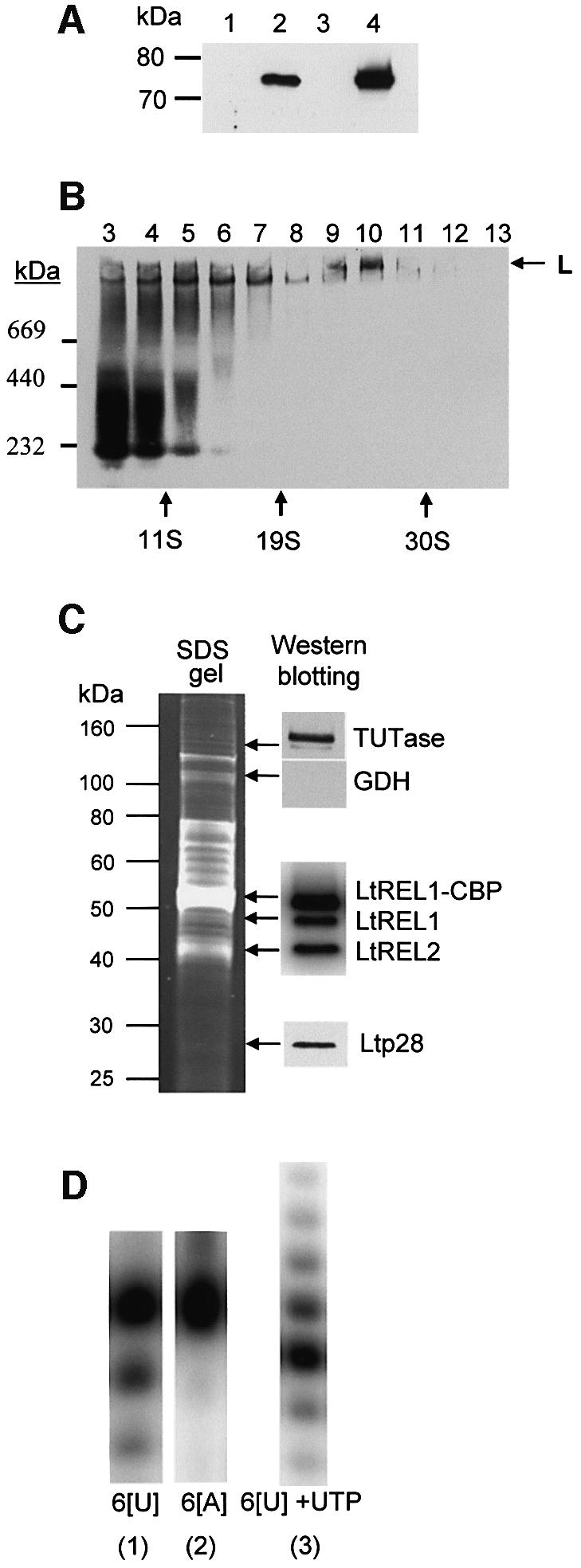
Fig. 4. Expression of LtREL1–TAP fusion protein and affinity isolation of the L-complex. (A) Mitochondrial localization of LtREL1–TAP fusion protein. Total protein (10 µg) from wild-type cells (lane 1), transfected (lane 2) cells, the cytoplasmic fraction of transfected cells (lane 3), and 2 µg of protein from purified mitochondria of transfected cells (lane 4) were analyzed with the PAP reagent for the presence of protein A as a part of the TAP fusion protein. (B) The REL1–TAP fusion protein is incorporated into the L-complex. Mitochondrial S100 extract from transfected cells was fractionated on a 10–30% glycerol gradient in an SW41 rotor for 20 h at 30 000 r.p.m. Each fraction was separated on a 4–12% native gel, and the protein A fusion protein was detected with the PAP reagent. (C) Protein composition of the TAP-purified L-complex. The fraction eluted from the calmodulin column was analyzed for the presence of both LtREL1 and LtREL2 ligases by adenylation, and for TUTase and p28 by western analysis with the respective polyclonal antibodies. (D) Enzymatic activities of the TAP-purified complex. The same fraction as in (C) was analyzed for the presence of 3′–5′ U-specific exonuclease activity by incubating the calmodulin fraction with 5′-labeled synthetic RNA oligonucleotides with six uridine (6[U]) or adenosine (6[A]) residues at the 3′ end. 3′ TUTase activity was detected by including 100 µM UTP. Products were separated on 15% polyacrylamide–urea.
The mitochondrial extract from transgenic cells was fractionated in a glycerol gradient, and each fraction was electrophoresed in a native gel which was blotted and treated with the PAP reagent. Figure 4B shows the incorporation of the LtREL1–TAP into a high molecular weight complex (indicated by an arrow) sedimenting in fractions 9 and 10 at ∼25S. The majority of the material detectable with PAP sedimented in fractions 3–7 and represented unincorporated fusion protein and/or breakdown. Adenylation of the gradient fractions showed that the LtREL1–TAP fusion protein did not self-adenylate, probably due to interference of the C-terminal protein A tag with activity (not shown). We show below that removal of the C-terminal protein A tag restores enzymatic activity to the tagged protein.
The tagged LtREL1 and associated components were isolated directly from mitochondrial extracts of transgenic cells by successive binding to IgG–Sepharose, cleavage of the protein A tag with TEV protease, and calmodulin affinity chromatography. A number of proteins co-eluted from the calmodulin column, as shown by the gel stained with Sypro Ruby in Figure 4C. The presence of adenylatable endogenous LtREL1 and LtREL2 and LtREL1–CBP is shown in the insert on the right, and the presence of TUTase and the Ltp28 protein is shown by western analysis using specific antisera. The material that was eluted from the calmodulin column showed a U-specific 3′–5′ exonuclease activity (Aphasizhev and Simpson, 2001) (Figure 4D, panels 1 and 2). This activity was not the reversal of 3′ TUTase since the presence of UTP in the reaction produced addition of a ladder of Us to the 3′ end (Figure 4D, panel 3).
The eluted material was subjected to glycerol gradient sedimentation to separate unincorporated LtREL1–CBP protein from the L-complex. As shown in the stained gel of the gradient fractions in Figure 5A, panel 1, a group of ∼13–15 major polypeptide bands in close to stoichiometric amounts co-sedimented in fractions 7–8 at ∼20S. The polypeptide profile is similar if not identical to that obtained from the gel-eluted L-complex native gel band in Figure 3C above, except for a 95 kDa band in the latter which could represent a gel contaminant due to its relatively high abundance. The bands, which were operationally labeled LC-1 to LC-11 (for L-complex 1–11), were subjected to LC MS/MS analysis.

Fig. 5. Characterization of LtREL1–TAP associated proteins. (A) The TAP-purified fraction was separated further on a 10–30% glycerol gradient and each fraction was subjected to adenylation with [α-32P]ATP and analyzed by native and SDS gel electrophoresis. Panel 1: protein profile on an 8–16% SDS gel, Sypro Ruby staining. Panel 2: western analysis of RNA ligase proteins with polyclonal antibodies against both LtREL1 and LtREL2, SDS gel. Panel 3: adenylated proteins, SDS gel. The LtREL1–CBP and the two endogenous ligases are indicated. Panel 4: detection of adenylated L-complex sedimenting at ∼20S (arrow labeled L) and an ∼200 kDa LtREL1-containing subcomplex in fractions 2 and 3 (arrows on the left), native gel. (B) Comparison of major polypeptides from LtREL2–TAP and LmLC-4–TAP complex with those from the LtREL1–TAP complex. Left panel: REL1, gradient-purified LtREL1–TAP complex; REL2, gradient-purified LtREL2–TAP complex; M, molecular weight markers. Right panel: M, molecular weight markers; REL1, gradient-purified LtREL1–TAP complex; LC-4, LmLC-4–TAP complex. Lane 4 is the material after the calmodulin elution and lane 3 is the concentrated 20–25S gradient fractions. Sypro Ruby-stained, SDS gel. (C) Identification of individual bands of L-complex by mass spectroscopy and database analysis. The lane shown is the Sypro Ruby-stained fraction 8 from (A). Novel proteins corresponding to L.major unidentified open reading frames are indicated on the left, and proteins homologous to known T.brucei proteins on the right. See Table I for details of proteins.
The presence of adenylatable LtREL1–CBP, LtREL1 and LtREL2 proteins in the gradient peak of the calmodulin-eluted material is shown in Figure 5A, panels 2 and 3. A distinct high molecular weight band was observed by native gel analysis of the adenylated material in fractions 7 and 8 (Figure 5A, panel 4). The S value of the L-complex decreased from 25S in the mitochondrial extract (fraction 10, Figure 4B) to 20S after TAP purification (fractions 7 and 8, Figure 5A), most probably due to removal of unstably interacting components. Also, the 5–10S region of the gradient (fractions 2–3) unexpectedly contained not only tagged LtREL1–CBP but also two other proteins (LC-3 and LC-4) in addition. Consistent with the presence of a stable subcomplex containing LtREL1 and the LC-3 and LC-4 polypeptides, an adenylated band of ∼200 kDa as well as a minor band of monomeric REL1–CBP could be detected in the same fractions using a native gel (Figure 5A, panel 4, arrow).
Identification of proteins in the L-complex
Since L.major and L.tarentolae protein sequences generally share extensive amino acid identity, it was possible to use peptide sequences obtained by mass spectrometric analysis of gel-isolated bands for identification of the genes encoding the major polypeptides in the L-complex using the L.major genome database. A total of 58 peptide sequences were obtained from 13 bands, of which 52 were assigned to 13 L.major proteins with high confidence (Table I). Identification of band 10, for which three peptide sequences were obtained, awaits further progress in the L.major genome project. The location of the LtREL1–CBP ligase is indicated in Figure 5C by an open arrow. The endogenous LtREL1 and LtREL2 RNA ligases were components of bands 7a and 9, respectively. Four proteins in bands 1, 4, 7b and 11 were homologs of the TbMP81, TbMP63, TbMP42 and TbMP18 proteins, respectively, obtained from a T.brucei editing complex (Panigrahi et al., 2001b). Seven novel proteins were components of bands 2, 3, 5, 6a, 6b, 7c and 8. Two of these contained RNase III motifs and one contained an AP exonuclease/endonuclease motif. These proteins represent possible candidates for the gRNA-dependent endonuclease, and also for the 3′–5′ exonuclease shown above to be present in the LtREL1–TAP complex. The stained gel lane from fraction 8 in Figure 5A is shown in Figure 5C, with the identified proteins indicated.
Table I. Major components of the core L-complex.
| Protein | Mol. wt (kDa) | Mol. wt (kDa) | No. of assigned | T.brucei | Functional motifs |
|---|---|---|---|---|---|
| |
SDS gel |
pre-protein |
peptides |
homolog |
|
| LmLC-1 | 139 | 113 | 5 | TbMP81a | Znf C2H2 |
| LmLC-2 | 109 | 108 | 4 | Exo_Endo_Phos (DNA) | |
| LmLC-3 | 75 | 72.3 | 5 | None | |
| LmLC-4 | 71 | 62.3 | 6 | TbMP63a | Two Znf C2H2 |
| LmLC-5 | 65 | 75.6 | 8 | None | |
| LmLC-6a | 60.5 | 65.7 | 6 | RNase_3_2 | |
| LmLC-6b | 60.5 | 57.7 | 2 | Nucleotidyltransferase, PAP associated | |
| LmLC-7a | ∼50 | 54.2 | 4 | TbREL1b | RNA ligase |
| (LtREL1) | |||||
| LmLC-7b | ∼50 | 46.1 | 1 | TbMP42a | Two Znf C2H2 |
| LmLC-7c | ∼50 | Fragment | 1 | None | |
| LmLC-8 | 45.4 | 48.5 | 3 | RNase_3_2 | |
| LmLC-9 | 42.2 | 47.6 | 4 | TbREL2b | RNA ligase |
| (LtREL2) | |||||
| LmLC-11 | 29.8 | 20.8 | 3 | TbMP18a | None |
The L.tarentolae LC-4 homolog of TbMP63 has been cloned and sequenced, and cloning of the other homologs is in progress. The L.tarentolae and L.major LC-4 proteins showed 74.5% identities and 79.5% conservative matches, whereas the L.tarentolae LC-4 and T.brucei TbMP63 proteins showed only 35.5% identities and 47.3% conservative amino acid replacements.
Although the 121 kDa 3′ TUTase and the Ltp26/Ltp28 proteins were detected in the LtREL1–TAP purified material by western analysis (see Figure 4C above), stained polypeptide bands of the appropriate sizes were not present in the TAP purified material, and no peptides from these proteins appeared in the mass spectrometric analyses. This suggests that the interaction of the 3′ TUTase and Ltp26/Ltp28 with the L-complex is not stable enough to withstand the extensive TAP purification procedures. Consistent with this interpretation is the fact that a higher relative amount of TUTase and Ltp26/Ltp28 was present directly after the IgG–Sepharose binding step than after the calmodulin column (not shown).
LtREL2–TAP and LmLC-4 affinity isolation of the editing complex
To determine if there was a single complex containing both LtREL1 and LtREL2, the TAP isolation procedure was performed using the LtREL2 protein fused to the TAP tag. The fusion protein was expressed and was incorporated into the L-complex (not shown). Purification of the complex proved relatively inefficient due to an apparent lack of exposure of the C-terminal CBP epitope in the folded protein. Nevertheless, an amount of protein was obtained from the 20S region of a glycerol gradient which was sufficient to be able to compare the protein profile with that of the LtREL1–TAP L-complex. As shown in Figure 5B, left panel, all polypeptides except for the 109 kDa LC-2 are present in the LtREL2–TAP complex. This protein, however, is non-stoichiometric in the LtREL1–TAP material, suggesting it is less stably bound to the complex.
As another test for the homogeneity of the L-complex and as a demonstration that a heterologous tagged protein from L.major could be incorporated into the L.tarentolae L-complex, the LmLC-4 protein was fused to the TAP tag and expressed in L.tarentolae. As in the case of the LtREL2–TAP, purification of the complex through the calmodulin binding step proved relatively inefficient, although expression and mitochondrial targeting were comparable with those of the LtREL1–TAP protein (data not shown). A comparison of the polypeptide profiles of the LtREL1–TAP purified L-complex and the LmLC-4–TAP purified material from the 20–25S region and also from the material layered on the gradient is shown in Figure 5B, right panel. As in the case of the LtREL2–TAP complex, the profile appears identical to that of the LtREL1–TAP complex.
Identification of a second 3′ TUTase in the L-complex
Two tryptic peptides from the band 6 region matched with a novel 57 kDa protein recently deposited in the L.major database, which has a significant level of amino acid similarity with the previously described L.tarentolae 121 kDa 3′ TUTase. The predicted protein has the nucleotidyltransferase and poly(A)-polymerase-associated motifs found in the 121 kDa TUTase (Figure 6). This protein also has the large insertion between the second and third aspartate residues of the catalytic triad, a characteristic feature of the L.tarentolae and T.brucei TUTase sequences. All other nucleotidyltransferases lack this insertion, which is within the catalytic domain (Keller and Martin, 2002). We have no direct evidence for 3′ TUTase activity, but it is likely that this protein represents a member of the TUTase family. In addition, database searching led to the identification of several similar TUTase sequences in T.brucei, suggesting that 3′ RNA uridylyltransferases may represent a multigene family in trypanosomes (data not shown). The Stuart lab has also detected a 57 kDa TUTase-related polypeptide in an editing complex from T.brucei (K.Stuart, personal communication). Since the band 6 region has at least two overlapping bands, the stoichiometry of the 57 kDa protein cannot be determined visually. However, the fact that two of eight tryptic peptides were derived from this protein (Table I) suggests that the protein is approximately stoichiometric with the other L-complex proteins and that the interaction with other proteins is sufficiently stable to survive the TAP isolation procedure.

Fig. 6. Partial alignment of the 57 kDa putative 3′ TUTase with the 121 kDa 3′ TUTase from L.major. The three aspartates of the nucleotidyltransferase catalytic triad are indicated by arrows, and the TUTase-specific insertion between D2 and D3 is indicated by boxing. Alignment performed by AlignX in Vector NTI.
The LtREL1–TAP complex has in vitro pre-cleaved U-insertion/deletion editing activity
The LtREL1–TAP complex that had undergone a final glycerol gradient fractionation step was analyzed for in vitro editing activity using a pre-cleaved RNA editing substrate (Igo et al., 2000). Little if any U-insertion activity was observed using intact pre-edited mRNA substrates (data not shown), suggesting an absence or inactivation of gRNA-mediated endonuclease activity in this material. The RNA substrates used for the U-insertion and U-deletion assays are shown schematically in Figure 7.

Fig. 7. In vitro gRNA-directed U-insertion and U-deletion activities of LtREL1–TAP purified material. (A) U-insertion in vitro activity of the LtREL1–TAP purified material. Fraction 8 from Figure 5 was incubated with pre-annealed RNA substrates for the pre-cleaved insertion assay in the presence of 200 µM UTP and 20 µM ATP, and products were separated on a 15% acrylamide–urea gel. The bridged RNA substrates are shown schematically above. Guiding nucleotides are indicated in bold. ‘0’ indicates no guiding nucleotide. ‘CCC’ indicates three non-guiding C nucleotides. The locations of the ligated edited products are indicated, as are the 5′ fragments with 1–3 Us added. Control, no protein added. (B) U-deletion in vitro activity. The same fraction as in (A) was incubated with RNA substrates for pre-cleaved deletion assay. The bridged substrate which should guide the deletion of two Us is shown schematically.
A 3′ exonuclease activity can be seen in the samples with only a 5′ fragment or with 5′ + 3′ fragments, but this activity was suppressed when the complementary gRNA was annealed (Figure 7A). A gRNA-directed U-insertion editing activity is evidenced by the appearance of ligated edited products dependent on the presence of the 5′ and 3′ fragments bridged by a complementary gRNA containing guiding purine nucleotides in the gap region. In addition to a prominent +1 edited product, there is a major +3 product mediated by the +3 gRNA, a major +2 product mediated by a +2 gRNA, and a major +1 product mediated by a +1 gRNA. The efficiency of the pre-cleaved assay varied dependent on the substrate, but generally 4–25% of input 5′ fragment could be converted into correctly edited product. The intensity of n guided insertion was at least 10-fold higher than n + 1 background signal. The gRNA without guiding nucleotides produced mainly the self-ligated 5′ and 3′ fragments in spite of the presence of a substantial +1 5′ fragment. A gRNA with CCC non-guiding nucleotides yielded an 8-fold lower amount of correctly extended 5′ fragment and half as much of the ligated product with three inserted uridines than AGA-containing gRNA. This demonstrates a requirement for base pairing of the guiding nucleotides with the inserted Us for precise nucleotide addition and, to a lesser extent, ligation. The presence of a minor background ladder of unguided U-insertions is characteristic of the Leishmania system (Byrne et al., 1996), but there is a definite stimulation of the correct ligated product in each case.
A pre-cleaved U-deletion activity was also detected. The 5′ fragment with two more uridines at the 3′ end was used with the same set of substrates as in Figure 7A. A gRNA containing no guiding nucleotides directed the deletion of two overhanging Us and the ligation of the edited products (Figure 7B) with 10% efficiency.
Discussion
We have isolated and characterized a high molecular weight, multienzyme complex active in both pre-cleaved U-insertion and U-deletion RNA editing in vitro. We label this the ‘E-complex’ since it shows in vitro editing activity. This complex consists of at least three different protein-containing components linked by RNA. The most stable is the ∼15 polypeptide core L-complex, which contains in approximately stoichiometric amounts the two RNA ligases, three zinc finger-containing proteins and one protein with strong C-terminal homology to these zinc finger proteins that are also present in an editing complex isolated from T.brucei. The L-complex also contains several novel proteins which include two putative RNase III-like enzymes, one protein with an exonuclease/endonuclease motif, one protein with a nucleotidyltransferase motif and three proteins with no recognizable motifs. To isolate this complex, an LtREL1–TAP fusion protein was overexpressed and incorporated into the L-complex in vivo. The tagged LtREL1 and interacting proteins were isolated by the TAP affinity procedure. The TAP purification, although performed under relatively low salt conditions, resulted in isolation of a 20S ligase-containing particle rather than the ∼25S particle observed in mitochondrial extract from wild-type cells, indicating a possible loss of interacting components. It is interesting that the LtREL1–TAP and LtREL2–TAP fusion proteins were not capable of self-adenylation but, after removal of the protein A tag, the CBP-tagged proteins could be adenylated. It should be noted that glycerol gradient fractionation separated the L-complex from an ∼200 kDa complex of LtREL1 with the LC-3 and LC-4 proteins, which may represent immediate interacting partners within the L-complex. The amount of free LtREL1 protein appears to be insignificant and is likely to be due to degradation of unincorporated protein.
Since an almost identical protein profile was obtained using an overexpressed LtREL2–TAP fusion protein or the heterologous L.major LmLC-4–TAP fusion protein, we conclude that there is a single L-complex containing both RNA ligases. Furthermore, the presence of both the CBP-tagged proteins and the endogenous proteins indicates that the L-complex has at least two copies of each enzyme, and may have an overall dimeric or tetrameric organization. Functional separation of U-insertion and U-deletion pathways has been suggested in T.brucei due to different nucleotide requirements in editing (Cruz-Reyes et al., 1998, 2002; Huang et al., 2001) and due to differential inhibition of U-insertions and U-deletions after RNAi of TbMP81 (Drozdz et al., 2002) and REL1 proteins (Huang et al., 2001). It has been proposed (Huang et al., 2001; Cruz-Reyes et al., 2002) that REL1 is responsible for ligation of the products of the U-deletion reaction and REL2 for the products of the U-addition reaction, although the latter is dispensable for editing in vivo (Drozdz et al., 2002). Our data provide evidence that there is indeed only a single complex that contains both RNA ligases. The functional separation of two editing pathways therefore occurs within one particle. Consistent with this interpretation is the preliminary evidence that LtREL1 and two interacting proteins, LC-3 and LC-4, may represent a subdomain of the L-complex.
The other two components of the E-complex are the 121 kDa 3′ TUTase (Aphasizhev et al., 2002) and the Ltp26/Ltp28 RNA-binding proteins (Allen et al., 1998; Lambert et al., 1999; Muller et al., 2001; Aphasizhev et al., 2003). We showed previously that the 3′ TUTase is present in two configurations, one of which, TUT I, consists of a trimer or tetramer and the other, TUT II, an ∼700 kDa complex that has not yet been well characterized (Aphasizhev et al., 2002). The TUT II TUTase sediments in the same region of the gradient as the L-complex, and therefore most probably is the form that binds to the L-complex. The evidence for an interaction of the L-complex and TUT II TUTase included co-immunoprecipitation from mitochondrial extracts of wild-type cells and co-purification together with the TAP-tagged L-complex from transfected cells. We show elsewhere that the Ltp26 and Ltp28 proteins are present as a stable 100 kDa heterotetramer, and therefore this is most probably the form that interacts with the L-complex (Aphasizhev et al., 2003).
The interaction of the L-complex with the 121 kDa TUTase was found to be dependent on RNA. We also showed previously that ∼40% of the total gRNA can be immunoprecipitated with anti-TUTase antibody and that gRNA can be cross-linked specifically to TUTase (Aphasizhev et al., 2002); we also showed that the Ltp26/Ltp28 100 kDa complex has a high affinity for RNA (Aphasizhev et al., 2003). RNase digestion of the mitochondrial extract prior to gradient fractionation prevented the TUT II TUTase and the Ltp26/Ltp28 (Aphasizhev et al., 2003) from interacting with the L-complex, suggesting that the interactions require RNA.
TUTase activity is required not only for insertion of Us at editing sites (Blum et al., 1990), but also for the formation of the 3′ oligo(U) tails of the gRNAs (Blum and Simpson, 1990), rRNAs (Adler et al., 1991) and the 3′ oligo(A + U) tails of mRNAs (Campbell et al., 1989). The presence of a 57 kDa putative second 3′ TUTase, which is related but not identical to the 121 kDa TUTase, as an apparent stoichiometric component of the L-complex, raises several questions as to the precise roles of the two enzymes. Our original scenario was that the 121 kDa TUTase was involved directly in all of these U-addition reactions. This was based on the fact that biochemical fractionation of TUTase activity from L.tarentolae mitochondrial extract, using several different substrate RNAs, coincided with purification of the 121 kDa TUTase in all separation steps (Aphasizhev et al., 2002). In addition, down-regulation of expression of the 110 kDa TUTase ortholog in T.brucei by RNAi also down-regulated RNA editing, including editing of the CO2 pre-mRNA which does not require a gRNA in trans and hence does not require a gRNA 3′ oligo(U) tail (Aphasizhev et al., 2002), but had no effect on formation of the L-complex (see Figure 2C). This evidence would suggest that this enzyme is involved in the insertion of Us at editing sites but does not remove the possibility that the 57 kDa TUTase is also involved. Furthermore, it was shown previously that immunoprecipitation of the 121 kDa enzyme from L.tarentolae mitochondrial extract depleted almost all detectable TUTase activity (Aphasizhev et al., 2002), but this result would not eliminate the possibility that there was a co-precipitation of the 57 kDa TUTase.
It is entirely possible that the 121 kDa TUTase and perhaps an associated as yet unidentified U-specific 3′–5′ exonuclease (Aphasizhev and Simpson, 2001) are involved solely in creation and regulation of the oligo(U) tail of gRNA and perhaps also mRNA and rRNA. In this scenario, the 57 kDa TUTase would be involved directly with U-insertions at editing sites, and a transient RNA-dependent interaction of the L-complex with the 121 kDa TUTase and the RNA-binding proteins would be required for regulation of this reaction. This would also suggest that the oligo(U) tail of gRNA is absolutely required for in vivo editing. Further work is required to ascertain the precise roles of the 121 kDa TUTase and the 57 kDa putative TUTase in the editing reaction.
We propose as a working hypothesis that the active RNP E-complex is formed by an initial interaction of the TUT II TUTase with the Ltp26/Ltp28 100 kDa complex, perhaps through bound RNA (gRNA?), and is followed by annealing of the gRNA to the cognate pre-edited mRNA and recruitment of the L-complex and possibly additional auxiliary factors. Since the level of gRNA-dependent endonuclease activity in the Leishmania in vitro system is low (Byrne et al., 1996), the location of this enzyme is unclear. However, the LC-2 protein with an AP endonuclease/exonuclease motif is a candidate for the endonuclease, and this protein appears to be interacting less stably with the L-complex than the other proteins. The entire RNP complex is competent to carry out both the U-insertion and the U-deletion editing reactions, at least at a single site. Since processive editing involving multiple overlapping gRNAs as occurs in vivo does not occur, it is likely that additional components must be recruited, such as, for example, an RNA helicase and other RNA-binding proteins.
Many questions remain, such as the regulation of the E-complex in terms of the known differences between U-insertion and U-deletion editing. This model almost certainly represents an oversimplification, and the actual reaction may be much more complex and involve the dynamic appearance of RNP complexes of varying composition, but it represents a starting point for the detailed molecular dissection of the editing machinery.
Materials and methods
Plasmid construction and cell transfection
Database searching using the T.brucei REL1 and REL2 sequences yielded the full-length L.major REL1 sequence and an L.major REL2 fragment sequence. Oligonucleotides from conserved regions of L.major REL1 and REL2 were used to amplify fragments of the L.tarentolae orthologs (REL1 5′ primer, 5′-GAGAAGGTGCACGGCACAAACTT-3′; REL1 3′ primer, 5′-GGCCGATCTTCGACAGCACGT-3′; REL2 5′ primer, 5′-ATCCTCATACCTGATCTCGC-3′; REL2 3′ primer, GTGTCTTACAACGAGGCCCT-3′). A 4 kb XbaI–KpnI L.tarentolae genomic fragment containing the entire LtREL1 gene was cloned and sequenced, and a 2.8 kb L.tarentolae genomic fragment containing the entire LtREL2 gene was cloned and sequenced. For details of TAP plasmid construction, see the Supplementary data available at The EMBO Journal Online.
TAP purification
Mitochondria were isolated as described (Braly et al., 1974) from late log phase cell cultures at 108 cells/ml in BHI medium with 10 µg/ml hemin and 100 µg/ml geneticin. The TAP purification protocol was adapted from published methods (Puig et al., 2001). See Supplementary data for experimental details.
In-gel tryptic digestion and mass spectrometry
Protein bands excised from SDS–acrylamide gels were crushed, washed in 25 mM ammonium bicarbonate/50% acetonitrile, dried, reduced, derivatized with iodoacetamide, and digested with trypsin by standard methods. Experimental details on the mass spectrometric analysis are in the Supplementary data.
Glycerol gradient fractionation, adenylation of ligases and native gel electrophoresis
Mitochondrial extract from wild-type L.tarentolae cells was prepared as described above and fractionated on a 10–30% glycerol gradient containing 25 mM HEPES pH 7.5, 60 mM KCl, 10 mM MgCl2 and 1 mM CHAPS. Sedimentation values were calculated using aldolase (7.6S), catalase (11.5S), thyroglobulin (19.3S) and 30S Escherichia coli ribosome subunits. For detection of Mg2+-independent [α-32P]ATP incorporation, MgCl2 was present during extract preparation and substituted by 0.1 mM EDTA in the glycerol gradient. Self-adenylation of fractions was carried out with 5 µCi of [α-32P]ATP per 10 µl of gradient fraction for 15 min at 27°C. The reaction was diluted 2-fold with water and electrophoresed on an 8–16, 4–12 or 4–20% native Tris-glycine gel for 16 h at 4°C. For analysis under denaturing conditions, the reaction was stopped with SDS loading buffer, separated on 10–20% denaturing gradient gels and blotted onto a nitrocellulose membrane.
Immunoprecipitation
Mouse polyclonal antibody against TUTase was affinity purified on immobilized antigen columns by standard techniques. A 100 µg aliquot of mouse antibody, 100 µl of rabbit antiserum against the p26/p28 gRNA-binding proteins (Aphasizhev et al., 2003) or 100 µl of rabbit anti-glutamate dehydrogenase serum was bound to 100 µl of GammaBind protein G plus Sepharose (AP Biotech) in 0.5 ml of 1× phosphate-buffered saline (PBS), 5 mg/ml bovine serum albumin (BSA) for 1 h at 4°C. Immunodepletions (two rounds) were carried out in 300–400 µl of glycerol gradient fractions with 10–20 µl of beads for 2 h at 4°C with constant mixing. Supernatants were incubated with [α-32P]ATP and analyzed on native gels. The beads were pelleted by brief centrifugation, washed three times with 1 ml of Tris-buffered saline (TBS) plus 0.3% Triton X-100 and re-suspended in 50 µl of 1× SDS loading buffer. For adenylation, the washed beads were incubated with 50 µCi/ml [α-32P]ATP for 30 min at 27°C in glycerol gradient buffer and washed with 1 ml of TBS/0.3% Triton X-100. An equal volume of 2× SDS loading buffer was added to the beads in order to stop the reaction and strip the bound proteins for gel analysis. See Supplementary data for details of western blotting.
RNA substrates and in vitro pre-cleaved editing assay
RNA substrates were chemically synthesized (Oligos Etc., Inc., Xeragon) and gel purified: 6-[U], 5′-GCUAUGUCUGCUAACUUGUUUUUU-3′; 6-[A], 5′-GCUAUGUCUGCUAACUUGAAAAAA-3′; 5′ fragment, 5′-GCACUACACGAUAAAUAUAAAAAG-3′; 5′-UU fragment, 5′-GC ACUACACGAUAAAUAUAAAAAGUU-3′; 3′ fragment, 5′-AACAU UAUGCUUCUUCGddC-3′; AGA gRNA, 5′-AGAAGCAUAAUGUU AGACUUUUUAUAUUUAUCGUGUAGUCdG-3′; AG gRNA, 5′-A AGAAGCAUAAUGUUAGCUUUUUAUAUUUAUCGUGUAGUCG-3′; A gRNA, 5′-AAGAAGCAUAAUGUUACUUUUUAUAUUUAUCGUG UAGUCdG-3′; 0 gRNA, 5′-AAGAAGCAUAAUGUUCUUUUUAUAU UUAUCGUGUAGUCddC-3′; and CCC gRNA, 5′-AAGAAGCAUAA UGUUCCCUUUUUAUAUUUAUCGUGUAGUCdG-3′. RNAs were 5′-phosphorylated with T4 polynucleotide kinase (BRL) and [γ-32P]ATP. The 5′ fragment, 3′ fragment and the gRNA were annealed by heating and slow cooling. The 20 µl reaction contained 0.2 pmol of 5′ end, 2 pmol of 3′end and 1 pmol of gRNA in 20 mM HEPES pH 7.5, 10 mM magnesium acetate, 1 mM dithiothreitol (DTT), 200 µM UTP, 20 µM ATP and 2–4 µl of glycerol fraction. After incubation for 2 h at 27°C, products were analyzed on a 15% sequencing gel.
DDBJ/EMBL/GenBank accession numbers
The sequences have been deposited in the DDBJ/EMBL/GenBank database with the accession Nos AY148476 for LtREL1, AY148475 for LtREL2 and AY190132 for LtLC-4.
Supplementary data
Supplementary data are available at The EMBO Journal Online.
Acknowledgments
Acknowledgements
We thank all members of the Simpson laboratory for advice and suggestions, Ken Stuart for providing the P3C1-G2 monoclonal antibody, and Sharleen Zhou for performing the tryptic digestions of the gel slices. This work was partially supported by National Institutes of Health Grant AI-09102 (to L.S.).
References
- Adler B.K., Harris,M.E., Bertrand,K.I. and Hajduk,S.L. (1991) Modification of Trypanosoma brucei mitochondrial rRNA by posttranscriptional 3′ polyuridine tail formation. Mol. Cell. Biol., 11, 5878–5884. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Allen T.E., Heidmann,S., Reed,R., Myler,P.J., Goringer,H.U. and Stuart,K.D. (1998) Association of guide RNA binding protein gBP21 with active RNA editing complexes in Trypanosoma brucei. Mol. Cell. Biol., 18, 6014–6022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aphasizhev R. and Simpson,L. (2001) Isolation and characterization of a U-specific 3′–5′ exonuclease from mitochondria of Leishmania tarentolae. J. Biol. Chem., 276, 21280–21284. [DOI] [PubMed] [Google Scholar]
- Aphasizhev R., Sbicego,S., Peris,M., Jang,S.H., Aphasizheva,I., Simpson,A.M., Rivlin,A. and Simpson,L. (2002) Trypanosome mitochondrial 3′ terminal uridylyl transferase (TUTase): the key enzyme in U-insertion/deletion RNA editing. Cell, 108, 637–648. [DOI] [PubMed] [Google Scholar]
- Aphasizhev R., Aphasizheva,I., Nelson,R.E. and Simpson,L. (2003) A 100 kDa complex of two RNA-binding proteins from mitochondria of Leishmania tarentolae catalyzes RNA annealing and interacts with several editing components. RNA, 9, 62–76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blum B. and Simpson,L. (1990) Guide RNAs in kinetoplastid mitochondria have a nonencoded 3′ oligo-(U) tail involved in recognition of the pre-edited region. Cell, 62, 391–397. [DOI] [PubMed] [Google Scholar]
- Blum B., Bakalara,N. and Simpson,L. (1990) A model for RNA editing in kinetoplastid mitochondria: ‘guide’ RNA molecules transcribed from maxicircle DNA provide the edited information. Cell, 60, 189–198. [DOI] [PubMed] [Google Scholar]
- Braly P., Simpson,L. and Kretzer,F. (1974) Isolation of kinetoplast–mitochondrial complexes from Leishmania tarentolae. J. Protozool., 21, 782–790. [DOI] [PubMed] [Google Scholar]
- Byrne E.M., Connell,G.J. and Simpson,L. (1996) Guide RNA-directed uridine insertion RNA editing in vitro. EMBO J., 15, 6758–6765. [PMC free article] [PubMed] [Google Scholar]
- Campbell D.A., Spithill,T.W., Samaras,N., Simpson,A. and Simpson,L. (1989) Sequence of a cDNA for the ND1 gene from Leishmania major: potential uridine addition in the polyadenosine tail. Mol. Biochem. Parasitol., 36, 197–200. [DOI] [PubMed] [Google Scholar]
- Cruz-Reyes J. and Sollner-Webb,B. (1996) Trypanosome U-deletional RNA editing involves guide RNA-directed endonuclease cleavage, terminal U exonuclease and RNA ligase activities. Proc. Natl Acad. Sci. USA, 93, 8901–8906. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cruz-Reyes J., Rusche,L., Piller,K.J. and Sollner-Webb,B. (1998) T.brucei RNA editing: adenosine nucleotides inversely affect U-deletion and U-insertion reactions at mRNA cleavage. Mol. Cell, 1, 401–409. [DOI] [PubMed] [Google Scholar]
- Cruz-Reyes J., Zhelonkina,A.G., Huang,C.E. and Sollner-Webb,B. (2002) Distinct functions of two RNA ligases in active Trypanosoma brucei RNA editing complexes. Mol. Cell. Biol., 22, 4652–4660. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Drozdz M., Palazzo,S.S., Salavati,R., O’Rear,J., Clayton,C. and Stuart,K. (2002) TbMP81 is required for RNA editing in Trypanosoma brucei. EMBO J., 21, 1791–1799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Estévez A.M. and Simpson,L. (1999) Uridine insertion/deletion RNA editing in trypanosome mitochondria—a review. Gene, 240, 247–260. [DOI] [PubMed] [Google Scholar]
- Estévez A.M., Kierszenbaum,F., Wirtz,E., Bringaud,F., Grunstein,J. and Simpson,L. (1999) Knockout of the glutamate dehydrogenase gene in bloodstream Trypanosoma brucei in culture has no effect on editing of mitochondrial mRNAs. Mol. Biochem. Parasitol., 100, 5–17. [DOI] [PubMed] [Google Scholar]
- Huang C.E., Cruz-Reyes,J., Zhelonkina,A.G., O’Hearn,S., Wirtz,E. and Sollner-Webb,B. (2001) Roles for ligases in the RNA editing complex of Trypanosoma brucei: band IV is needed for U-deletion and RNA repair. EMBO J., 20, 4694–4703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang C.E., O’Hearn,S.F. and Sollner-Webb,B. (2002) Assembly and function of the RNA editing complex in Trypanosoma brucei requires band III protein. Mol. Cell. Biol., 22, 3194–3203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Igo R.P., Palazzo,S.S., Burgess,M.L., Panigrahi,A.K. and Stuart,K. (2000) Uridylate addition and RNA ligation contribute to the specificity of kinetoplastid insertion RNA editing. Mol. Cell. Biol., 20, 8447–8457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kable M.L., Seiwert,S.D., Heidmann,S. and Stuart,K. (1996) RNA editing: a mechanism for gRNA-specified uridylate insertion into precursor mRNA. Science, 273, 1189–1195. [DOI] [PubMed] [Google Scholar]
- Keller W. and Martin,G. (2002) Gene regulation: reviving the message. Nature, 419, 267–268. [DOI] [PubMed] [Google Scholar]
- Lambert L., Muller,U.F., Souza,A.E. and Goringer,H.U. (1999) The involvement of gRNA-binding protein gBP21 in RNA editing—an in vitro and in vivo analysis. Nucleic Acids Res., 27, 1429–1436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- LeBowitz J.H., Coburn,C.M., McMahon-Pratt,D. and Beverley,S.M. (1990) Development of a stable Leishmania expression vector and application to the study of parasite surface antigen genes. Proc. Natl Acad. Sci. USA, 87, 9736–9740. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Muller U.F., Lambert,L. and Goringer,H.U. (2001) Annealing of RNA editing substrates facilitated by guide RNA-binding protein gBP21. EMBO J., 20, 1394–1404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Panigrahi A.K. et al. (2001a) Association of two novel proteins, TbMP52 and TbMP48, with the Trypanosoma brucei RNA editing complex. Mol. Cell Biol., 21, 380–389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Panigrahi A.K. et al. (2001b) Four related proteins of the Trypanosoma brucei RNA editing complex. Mol. Cell. Biol., 21, 6833–6840. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peris M., Simpson,A.M., Grunstein,J., Liliental,J.E., Frech,G.C. and Simpson,L. (1997) Native gel analysis of ribonucleoprotein complexes from a Leishmania tarentolae mitochondrial extract. Mol. Biochem. Parasitol., 85, 9–24. [DOI] [PubMed] [Google Scholar]
- Puig O., Caspary,F., Rigaut,G., Rutz,B., Bouveret,E., Bragado-Nilsson,E., Wilm,M. and Seraphin,B. (2001) The tandem affinity purification (TAP) method: a general procedure of protein complex purification. Methods, 24, 218–229. [DOI] [PubMed] [Google Scholar]
- Rigaut G., Shevchenko,A., Rutz,B., Wilm,M., Mann,M. and Seraphin,B. (1999) A generic protein purification method for protein complex characterization and proteome exploration. Nat. Biotechnol., 17, 1030–1032. [DOI] [PubMed] [Google Scholar]
- Rusche L.N., Cruz-Reyes,J., Piller,K.J. and Sollner-Webb,B. (1997) Purification of a functional enzymatic editing complex from Trypanosoma brucei mitochondria. EMBO J., 16, 4069–4081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rusché L.N., Huang,C.E., Piller,K.J., Hemann,M., Wirtz,E. and Sollner-Webb,B. (2001) The two RNA ligases of the Trypanosoma brucei RNA editing complex: cloning the essential band IV gene and identifying the band V gene. Mol. Cell. Biol., 21, 979–989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sabatini R. and Hajduk,S.L. (1995) RNA ligase and its involvement in guide RNA/mRNA chimera formation. J. Biol. Chem., 270, 7233–7240. [DOI] [PubMed] [Google Scholar]
- Schnaufer A., Panigrahi,A.K., Panicucci,B., Igo,R.P., Salavati,R. and Stuart,K. (2001) An RNA ligase essential for RNA editing and survival of the bloodstream form of Trypanosoma brucei. Science, 291, 2159–2161. [DOI] [PubMed] [Google Scholar]
- Seiwert S.D., Heidmann,S. and Stuart,K. (1996) Direct visualization of uridylate deletion in vitro suggests a mechanism for kinetoplastid RNA editing. Cell, 84, 831–841. [DOI] [PubMed] [Google Scholar]