

Abstract
Herpes simplex virus-1 (HSV-1) virions are large, complex enveloped particles containing a proteinaceous tegument layer connected to an icosahedral capsid. The major capsid protein, VP5 (149 kDa), makes up both types of capsomere, pentons and hexons. Limited trypsin digestion of VP5 identified a single stable 65 kDa fragment which represents a proposed protein folding nucleus. We report the 2.9 Å crystal structure of this fragment and its modeling into an 8.5 Å resolution electron cryomicroscopy map of the HSV-1 capsid. The structure, the first for any capsid protein from Herpesviridae, revealed a novel fold, placing herpesviruses outside any of the structurally linked viral groupings. Alterations in the geometrical arrangements of the VP5 subunits in the capsomeres exposes different residues, resulting in the differential association of the tegument and VP26 with the pentons and hexons, respectively. The rearrangements of VP5 subunits required to form both pentavalent and hexavalent capsomeres result in structures that exhibit very different electrostatic properties. These differences may mediate the binding and release of other structural proteins during capsid maturation.
Keywords: assembly/capsid protein/crystal structure/folding nucleus/herpesviruses
Introduction
Herpesviridae is a large family of viruses including several members that are pathogenic to humans, causing a variety of disorders ranging from cold sores and chicken pox to less frequent conditions such as blindness and cancers (Roizman and Sears, 1996). Herpes simplex virus-1 (HSV-1), the prototypical member of this family, is a ubiquitous human pathogen with a seroprevalence of >80% in some populations (Sharp, 2002).
The herpesvirus virion is one of the largest and most complex viruses studied structurally. It has a diameter of ∼2000 Å and consists of four distinct structural layers: a glycoprotein-containing envelope, a proteinaceous tegument layer, a capsid (1250 Å diameter) and a DNA core (Steven and Spear, 1997).
The capsid is a well-defined icosahedron that has proven amenable to study by electron cryomicroscopy (Zhou et al., 2000). The capsid shell is made of four distinct proteins that lie on a T = 16 icosahedral lattice (Caspar and Klug, 1962). The major capsid protein, VP5 (149 kDa), makes up both capsomere structures, the pentons and hexons, which contain five and six VP5 monomers, respectively. In the capsid, pentons are located at the icosahedral 5-fold vertices, while the hexons form the faces and edges. In addition to the obvious differences in their rotational symmetries, the hexons also differ from the pentons through the presence of six copies of VP26 (12 kDa). These are attached to the upper portion of the VP5 molecules where they form a continuous ring around the top of each hexon (Zhou et al., 1995). Connecting the capsomeres is a heterotrimeric complex known as the triplex, which consists of two copies of VP23 (34 kDa) and one copy of VP19C (50 kDa). The triplex has been compared with an external scaffold in that it aids in morphogenesis, although it differs from the classical definition of a scaffold since it remains in the mature capsid (Spencer et al., 1998). An asymmetric unit contains 16 VP5s (one penton subunit, 2.5 hexons) and five and one-third triplexes, giving a total molecular weight of 3.2 MDa.
The tegument layer contains proteins that are important for various aspects of the virus life cycle. In many cases, the functions of the tegument proteins are poorly understood. However, it is believed that they have key roles in the early events of infection, as well as in transport of the capsid through the cell, DNA insertion into the nucleus, and envelope formation (Lemaster and Roizman, 1980; Post et al., 1981; Batterson et al., 1983; Fenwick and Everett, 1990; Coulter et al., 1993; Zhang and McKnight, 1993). While the HSV-1 capsid structure has been studied to a relatively high resolution by electron cryomicroscopy, structural analysis on the tegument as a whole has proven difficult, due to its size, complexity and lack of symmetry. Structural studies on the entire virion using electron cryomicroscopy have shown very little ordered density in the regions of the envelope and tegument. However, ordered tegument density has been observed on and around the penton, suggesting some icosahedral symmetry where the tegument is in contact with the nucleocapsid (Zhou et al., 1999). This density has been suggested to be the protein VP1-3, an extremely large protein (336 kDa) thought to be involved in nucleocapsid attachment to the nuclear pore and subsequent DNA release into the nucleoplasm (Knipe et al., 1981; Batterson et al., 1983; Ojala et al., 2000). In addition, null mutants of VP1-3 accumulate newly assembled, unenveloped, DNA-filled capsids in the cytoplasm of infected cells (Desai, 2000), suggesting that VP1-3 is important at various stages of the viral life cycle.
HSV-1 VP5 is large for a capsid protein (1374 amino acids) and can be considered as having three domains: floor, middle and upper. The floor domains form a thin (25 Å), largely continuous layer, or shell, and are the only parts of VP5 that interact directly to form intercapsomeric connections. They also interact with the internal scaffolding protein during capsid assembly (Zhou et al., 1999). The remainder of the VP5 molecule extends radially outward from the capsid floor to produce the characteristic towers of the hexons and pentons. The middle domains are involved in binding to the triplexes that lie between and link adjacent capsomeres (Zhou et al., 2000). The upper domains form the tops of the penton and hexon towers and are the binding sites for the small capsid protein VP26 in the hexons and for tegument proteins in the pentons.
In a first step towards understanding the structural basis of assembly of the HSV-1 capsid at the atomic level, we report the crystal structure of a 604 residue, protease-resistant fragment of VP5. A combination of X-ray crystallographic, electron cryomicroscopy and biochemical information (Spencer et al., 1997) identified this fragment as the upper domain of VP5 (VP5ud). Fitting the upper domain structure into the 8.5 Å electron cryomicroscopy reconstruction of the HSV-1 capsid (Zhou et al., 2000) uncovered details of the interactions between these regions of VP5 within the hexons and pentons. In addition, their relationship to VP26 and the tegument could be inferred, shedding light on the structure–function relationships within this region of the capsid. The upper domain structure itself has a novel fold, and the implications of this structure for capsid assembly, the evolutionary relationships among viruses and the folding of large proteins in general are discussed.
Results and discussion
Identification of a protease-stable fragment
The VP5 monomer undergoes extensive rearrangements during capsid assembly (Newcomb et al., 1996; Trus et al., 1996), therefore implying that the folding pattern of the molecule must exhibit some degree of flexibility. In accordance with this, biophysical studies on the VP5 monomer suggest that some portions of the protein lack definitive tertiary structure (D.McClelland, unpublished results). With the high protein concentrations required for crystallography, VP5 was polydispersed, suggesting that it lacks a defined oligomeric state in solution as determined by dynamic light scattering (data not shown). However, upon limited trypsin digestion of VP5, much of the sequence (∼55%) was digested into small fragments, leaving a single stable 65 kDa fragment, corresponding to the middle 604 amino acids of the protein sequence (residues 451–1054), hereafter referred to as VP5ud. The rapid and extensive digestion of the remaining sequences suggests they are flexible and readily exposed to the protease. The VP5ud exists as a monomer in solution; however, it dimerizes following addition of sodium acetate, the precipitant agent used to grow the VP5ud crystals.
Novel viral domain fold
The structure of the VP5ud was determined by multiwavelength anomalous dispersion (MAD) phasing and refined to 2.9 Å resolution (Table I). The VP5ud has a pyramid-like structure with a height of 80 Å and a rectangular base of 56 × 55 Å (Figure 1). The base consists mostly of loop structure and harbors both the N- and C-termini of the fragment. Overall, the structure is predominantly α-helical. A cluster of nine α-helices forms one face of the VP5ud (Figure 1B). The largest component of this cluster is a 24 residue, 37 Å long helix that traverses the entire length of this face (Figure 1B). This helix appears to be important in VP5ud–VP5ud interactions within the capsomeres. The other half of the domain is composed largely of loops, with a few β-sheet strands and the remaining α-helices. A set of three antiparallel β-strands (Figure 1B) makes up the largest array of β-sheets in the domain. The apical region of the VP5ud consists of a polyproline loop important in VP5ud interactions with VP26 and the tegument. An antibody epitope (Spencer et al., 1997) was mapped to this region (residues 862–880) and is highlighted in Figure 1B.
Table I. Crystallographic analysis statistics.
| Data collection |
|
|
|
|
|
|
|
| Crystal |
Wavelength (Å) |
dmin |
No. of measurements |
Unique reflections |
Completeness (%) |
<I>/<σ> |
Rsyma,b (%) |
| MAD phasing data | SeMet λ1 (0.97898) | 3.5 | 689 657 | 29 841 | 99.7 (98.4) | 70 (6.2) | 8.9 (23.3) |
| SeMet λ2 (0.97885) | 3.2 | 575 686 | 38 704 | 98.3 (90.5) | 7.1 (3) | 8.8 (19.9) | |
| SeMet λ3 (0.96363) | 3.6 | 639 010 | 27 543 | 99.8 (98.9) | 7.0 (6.9) | 9.4 (24.7) | |
| Refinement data |
SeMet λ4 (0.95679) |
2.9 |
308 549 |
47 194 |
91.2 (80.4) |
7.0 (3.3) |
10.1 (20.5) |
| Phasing |
|
|
|
|
|
|
|
| Observed diffraction ratiosc | |||||||
| Resolution range 50>d>3.6 (Å) | λ1 | λ2 | λ3 | ||||
| λ1 | 0.0839 | 0.0459 | 0.0741 | ||||
| λ2 | 0.0328 | 0.0611 | |||||
| λ3 | 0.0238 | ||||||
| Figure of merit (FOM) |
0.57 |
|
|
|
|
|
|
| Refinement |
|
|
|
|
|
|
|
| Resolution range (Å) | Rvalued (%) | Rfreee (%) | Bond length deviation (Å) | Bond angle deviation (°) | Bonded main chain atom B-factor r.m.s.d. (Å2) | Bonded side chain atom B-factor r.m.s.d. (Å2) | |
| 50–2.9 | 25.4 | 28.9 | 0.013 | 1.8 | 11.4 | 19.3 |
aValues in parentheses are for the outer resolution shell.
bRsym = ΣhΣi|Il(h) – <I(h)|/ΣhΣiIl(h).
cValues are <(Δ|F|)2>1/2/<|F|2>1/2.
dRvalue = Σ(|Fobs| – k|Fcalc|)/Σ|Fobs|.
eRfree is obtained for a test set of reflections, consisting of a randomly selected 10% of the data and not used during refinement.

Fig. 1. The 2.9 Å structure of the upper domain of VP5 (VP5ud, residues 451–1054). (A) Stereoview of the VP5ud color-ramped from the N- (blue) to the C-terminus (red). Every 80 residues and the ends of breaks are numbered and identified by small filled circles. (B) Ribbon representation of the overall fold of the VP5ud. The secondary structural elements in the structure are colored: α-helices in red, β-strands in blue and loops in green. Key elements are labeled in the structure.
The VP5ud has a novel α + β protein folding motif. A search for overall structural similarities against the DALI (Holm and Sander, 1995) database failed to reveal any significant matches. Many structures of a variety of viruses and viral capsid proteins have been determined by X-ray crystallography, revealing surprising structural similarities between capsid proteins of several apparently unrelated virus types. The most common viral fold, the eight-stranded β-barrel ‘jelly roll’ motif, has been seen in RNA viruses from plant, insect and animal origins (Rossmann and Johnson, 1989; Harrison et al., 1996), in double-stranded DNA (dsDNA) viruses such as adenovirus (Roberts et al., 1986), PRD1 (Benson et al., 1999) and SV40 (Stehle et al., 1996), and in dsRNA viruses such as blue tongue virus (Grimes et al., 1998) and orthoreovirus (Reinisch et al., 2000). While the jelly roll motif is the most common fold found in viral capsid proteins, some viruses, including hepatitis B virus and HK 97 (Böttcher et al., 1997; Conway et al., 1997; Wikoff et al., 2000), have capsid proteins that exhibit completely unrelated folds. Although it remains to be seen whether the remainder of VP5 contains known folds, we have shown here that the VP5ud from HSV-1 exhibits a fold that is unlike those seen in any other virus to date, and therefore places herpesviruses outside of any of the structurally linked groupings that have been identified so far.
Fitting the crystal structure into the electron cryomicroscopy reconstruction of the HSV-1 capsid
The model of the VP5ud was fit unambiguously into the 8.5 Å resolution electron cryomicroscopy reconstruction of the HSV-1 capsid (Figure 2) (Zhou et al., 2000), thereby confirming that the protease-resistant, 65 kDa fragment comprises the upper domain of VP5. The VP5ud represents 45% of the entire VP5 monomer and 33% of the entire capsid shell (Figure 2B). The fitting was carried out on 6-fold averaged peripentonal, center and edge hexons, with similar results for all three, indicating very similar conformations within the VP5uds of the hexon subunits (data not shown). The major secondary structure elements of the VP5ud X-ray structure are all contained within the electron cryomicroscopy density envelope of the hexon and, in some cases, especially for most of the α-helices, are well defined (Figure 2C).

Fig. 2. Computational fitting of the VP5ud into the hexon and penton subunits. (A) Part of the 8.5 Å electron cryomicroscopy reconstruction of the HSV-1 B capsid. The penton capsomere is displayed in red, the hexon in blue and the triplex in green. The asymmetric unit of the HSV-1 nucleocapsid is enclosed by the white line. P, C and E represent the three types of hexons, the peripentonal, center and edge, respectively. (B) The crystal structure of VP5ud fitted into each of the positions it would be expected to occupy in the electron cryomicroscopy reconstruction of the HSV-1 capsid. The Cα backbones of the VP5uds are color coded yellow, red, green and blue depending on which capsomere (penton, central hexon, edge hexon or peripentonal hexon, respectively) they comprise. (C) Side view of the Cα trace of the VP5ud (magenta) modeled into the 8.5 Å electron cryomicroscopy density of a single hexon subunit. The blue density at the apical end of the VP5ud represents VP26. VP26 was delineated by calculating a difference map between the fitted VP5ud and a computationally isolated VP5 monomer from a hexon. (D) Side view of the Cα trace of the VP5ud (magenta) modeled into the 8.5 Å electron cryomicroscopy density of a single penton subunit, viewed from the same direction as in (A).
The X-ray structure was not fitted as easily into the penton subunit as into the hexon subunit, due primarily to the poorer quality of the penton density in the electron cryomicroscopy map. The reason for this is not clear, but it may be due to the presence of a portal complex at one 5-fold vertex (Newcomb et al., 2001) or may simply reflect greater flexibility within the penton channel (Newcomb and Brown, 1991). Nevertheless, in contrast to the hexon subunit where the density is smooth and continuous, the electron cryomicroscopy density of the penton subunit is broken and discontinuous in several places. As a consequence, the individual secondary structure elements such as the α-helices are not as readily identifiable as is the case in the hexon. The VP5ud was initially fitted to a low resolution, 17 Å map with Foldhunter (Jiang et al., 2001), to generate an envelope fit with the correct position and orientation of the VP5ud structure. Using this orientation, the fitting of the VP5ud was refined in reference to the 8.5 Å map (Figure 2D). Despite the poorer quality of the penton density, the crystal structure fits the envelope of the electron cryomicroscopy density reasonably well.
Comparison of hexons and pentons
The ability of VP5 to assemble into both the pentons and hexons allows it to satisfy the geometrical constraints imposed in forming a closed shell capsid with icosahedral symmetry. The hexons and pentons are cylindrical structures that occupy roughly the same volume despite differences in the number of their subunits. Spreading a smaller number of subunits over a similar volume causes changes in the nature of the contacts between the VP5uds. The consequences of this are seen in the arrangement of the subunits, which appear skewed in the penton, with their major axis pointing towards the neighboring subunit rather than towards the center of the channel, as is the case in the hexon (Figure 3). To accommodate the altered arrangement, individual residues at the interfaces between the VP5ud subunits form different contacts within the two types of capsomere.
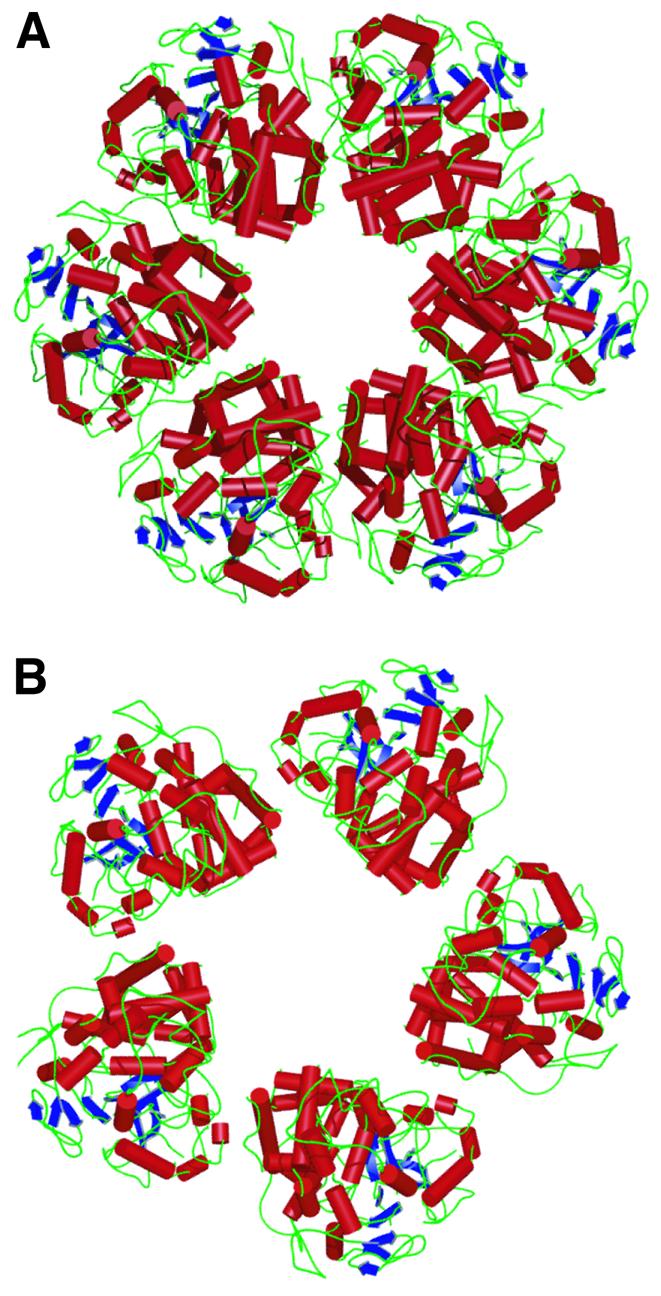
Fig. 3. Comparison of the hexon and penton structures. (A) Top view showing the ribbon representation of the VP5ud structures in the hexon. The ribbon diagrams are a representation of the VP5uds as they lie in the C hexon. (B) Top view as in (A), showing the ribbon representation of the VP5ud structures in the penton.
VP5ud interactions within the hexons and pentons
Because the VP5ud was crystallized as an isolated fragment, there is uncertainty over the exact residues involved in the interactions between neighboring subunits in the penton and hexon. Nevertheless, inferences can be made based on the quantitative fitting of the crystal structure to the electron cryomicroscopy map and an analysis of the residues involved in contacts within the crystal packing. The calculated buried surface area between VP5uds in the hexon is 2100 Å2. The two binding surfaces have been defined arbitrarily as the helix face and the loop face (Figure 4). Residues were identified as possible candidates for interaction if they are located within a 4.0 Å radius of each other. All the potentially interacting residues were charged or polar, implying that the upper domain interactions are mediated primarily by electrostatics rather than by hydrophobic packing. The interacting residues on the helix face were clustered primarily in and around the major helix (Figure 4A). Many of the residues identified as forming interactions in the hexon are also responsible for interactions found within the crystal lattice. For example, Arg691, located at the top of the helix, was found as a residue potentially involved in a salt link with Glu620 in the neighboring VP5ud. In the crystal packing, Arg691 is involved in a salt link with Glu852 in the neighboring dimer along the c-axis, therefore exhibiting a propensity to form intersubunit contacts. In addition, a loop on the helix face, consisting of residues 790–803, was another likely candidate for participation in subunit–subunit interactions. Arg790 is involved in a crystal contact with the neighboring dimer along the c-axis, and therefore also exhibits a propensity to interact with other VP5ud subunits. The loop face consists of loops and short helices located throughout the interface (Figure 4A). In the crystal, two VP5uds interact via the same loops that are present in the loop face of the hexon VP5ud. However, the interaction in the crystal was between two loop faces, rather than between a loop face and a helix face, as is the case in the hexon. This conservation of the loop face interface between the modeled hexon structure and the crystal packing supports its proposed role in maintaining the hexon structure in the upper domain region of the VP5 subunits.

Fig. 4. VP5ud interactions within the hexon and penton. (A) Ribbon representation of two VP5uds as they interact in the hexon. The face of the major helix (red) interacts with one loop face (blue). The secondary structure elements are colored if any of their constituent residues were flagged as being within 4.0 Å of another residue on the adjacent VP5ud. (B) Ribbon representation of two VP5uds as they interact in the penton. One helix face (red) interacts with one loop face (blue). The secondary structure elements are colored if any of their constituent residues were flagged as being within 4.0 Å of another residue on the adjacent VP5ud.
With its extensive interactions and large buried surface area, the upper domain region of the hexon appears to be a rigid and stable structure. In contrast, the interactions between VP5uds in the penton are much more limited, with a calculated buried surface area between VP5uds of only 650 Å2. Nevertheless, similar, though less extensive, regions of the proteins are involved, with the areas of interaction on the helix face confined to the top loop and upper regions of the major helix (Figure 4B). On the loop face, they consist of residues located on only two loops, 943–947 and 981–985 (Figure 4B). The interacting residues that are conserved between both capsomere structures appear to have different binding partners. For instance, Arg691 appears to be salt linked to Glu620 in the hexon and to Asp946 in the penton, while Glu684 is putatively salt linked to Arg660 in the hexon and His944 in the penton. Arg790, which is salt linked to Asp946 in the hexon, was not found to be an interacting residue in the penton.
Chemical properties of hexon and penton channels
Examination of the electrostatic surface potential of VP5ud reveals that the charge is distributed unevenly. Thus, the helix-rich surface, which points into the hexon channel (Figure 3A), is very acidic (Figure 5A), while the basic patch seen in the molecule on the left of Figure 5B is buried in the interface between neighboring VP5uds. This results in a strongly acidic hexon channel (Figure 5A and B). Due to the skewed arrangement of the VP5uds in the penton, the acidic surface is turned towards the neighboring VP5ud subunit, while the basic patch that was hidden by the VP5ud interaction in the hexon is exposed on the wall of the penton channel (Figure 5D). The result is a penton channel with a more basic character (Figure 5C and D). Therefore, the subunit rearrangements between hexons and pentons that allow VP5 to form capsomeres with different structural properties also result in structures that appear to have completely different chemical properties.

Fig. 5. Electrostatics of the hexons and pentons. (A) Top view as in Figure 3A, showing the electrostatic surface potential of the VP5ud subunits in the hexon. The arrow represents a rotation of the hexon channel by 90°. (B) The internal electrostatics of the hexon channel from the perspective of the center of the channel outward. The hexon has been cut in half with only three of the six VP5uds being displayed. (C) Top view as in (A), showing the electrostatic surface potential of the VP5ud subunits in the penton. The arrow represents a rotation of the hexon channel by 90°. (D) The internal electrostatics of the penton channel from the perspective of the center of the channel outward. Only three of the five VP5uds are being displayed.
Whether the differing properties of the channels have a functional significance is not yet clear. Channels through the shell do provide potential routes into and out of the capsid. Treatment of DNA-containing capsids with 0.5 M guanidine–HCl allows the DNA to escape through the pentagonal vertices (Newcomb and Brown, 1994). However, as the HSV-1 DNA is now thought to be packaged through a portal complex occupying a unique position at one vertex of the capsid (Newcomb et al., 2001), it is less likely that either the penton or hexon channels have a role in DNA entry or exit. However, during DNA packaging, the internal scaffolding protein of HSV-1 must be displaced, and the relatively large penton channel may provide the exit route. The acidic nature of the internal scaffold protein would be more compatible with exiting through the less charged and more basic penton channel than via the highly acidic hexon channel.
Tegument and VP26 binding
Another consequence of the altered subunit arrangement in the capsomeres is the differential association of proteins with the hexons and pentons. The smallest capsid protein, VP26, is part of the hexon, where six copies form a continuous ring around the top of the VP5ud, but is absent from the equivalent position on the penton (Zhou et al., 1995). This contrasts with the tegument binding seen in virions, which is restricted to the pentons, and does not involve the hexons (Zhou et al., 1999). Studies have shown that these mutually exclusive distributions are not a result of competition for binding sites but appear to reflect differences in the properties at the top of each capsomere (Wingfield et al., 1997; Chen et al., 2001). The results presented here do indeed show marked differences between the hexons and pentons in this region. As with the channel, the altered packing of the VP5ud subunits exposes different sets of residues at the outer surfaces of the two types of capsomeres, giving them dissimilar properties. Superimposition of the electron cryomicroscopy densities of the VP26 and tegument proteins onto the crystal-based capsomere structures revealed their binding footprints and highlighted the capsomere-specific nature of the interactions (Figure 6).

Fig. 6. VP26 and tegument binding to the VP5ud. (A) Top view of the electrostatic surface potential of two hexon VP5uds bound to two VP26 molecules (blue-gray semi-transparent electron density). VP26 was delineated by calculating a difference map between the fitted VP5ud and a computationally isolated VP5 monomer from a hexon. (B) Top view of the entire cryo-EM map of the penton channel with the Cα trace of the five fitted VP5uds (magenta). The red density located at the top of the penton channel is the icosahedrally ordered tegument putatively identified as VP1-3. (C) Top view of the electrostatic surface potential of two penton VP5uds bound to the tegument (blue-gray semi-transparent electron density).
The highly basic nature of VP26 (pI of 11.1) makes it likely for charge–charge interactions to occur between basic residues on VP26 and acidic residues on the VP5ud. Although the bulk of VP26 is in fact in contact with a relatively uncharged area on the outside of the hexon (Figure 6A), the VP26-binding site does extend into the charged regions near the interface between the subunits, with a portion reaching the vicinity of the extremely acidic polyproline loop that forms the apex of the VP5ud. This loop is an attractive candidate for participating in the binding of VP26 as it contains three glutamic acid residues at positions 846, 851 and 852, the last of which makes a salt bridge with Arg691 in the crystal packing.
The tegument footprint covers some of the same regions on the penton as that covered by VP26 on the hexon but, instead of forming a continuous ring, the binding is confined to the interfaces between the subunits (Figure 6B). In contrast to the hexon VP5ud interface that has a large buried surface area of 2100 Å2, the buried surface area between VP5uds in the penton is only 650 Å2. As a result, it appears that there are portions of the tegument density that are ‘threaded’ in the much larger space between the VP5ud penton subunits and are involved in simultaneous interactions with two subunits (Figure 6C). Interactions across such junctions will be particularly sensitive to the relationship between the residues on either side and, since their pattern is not preserved in the hexon, binding at this location presumably accounts for the very high specificity of tegument attachment to the penton. Similar considerations are likely to account for the hexon-only binding of VP26.
Folding nucleus function of VP5ud
VP5ud by itself is as large as many intact viral capsid proteins such as the 56 kDa, T4 hexon protein, gp23. This poses interesting questions as to the importance of this domain to the structure and function of VP5. A notable aspect of the comparison between the VP5ud crystal and electron cryomicroscopy structures is their very close structural correspondence. In addition, its resistance to proteolytic digestion suggests that VP5ud represents a very compact stable structure. Such a structure could serve as a folding nucleus for the VP5 monomer, forming a rigid core around which the flexible middle and floor domains are able to undertake the conformational changes required for capsid assembly. A similar phenomenon is seen in the outer scaffold protein D from φX174, where a rigid core structure is surrounded by flexible regions, capable of existing in different conformations depending on their local environments (Dokland et al., 1997). This protein folding strategy would provide VP5 with the flexibility needed to switch between the different conformations required for viral assembly while still maintaining some structural rigidity. Furthermore, accurate folding of large proteins is a demanding and complicated process and, in many cases, must be carefully controlled. For example, in the case of the adenovirus hexon protein, a viral-encoded chaperone binds to the nascent polypeptide while it is still on the ribosomes, and is only released when the protein adopts its final conformation in the hexon trimer (Cepko and Sharp, 1983). There is no evidence to suggest that any viral or cellular chaperone is required for correct folding of VP5, and it is possible that the VP5ud provides an equivalent function, acting as a surrogate internal chaperone to hold the rest of the protein in an appropriate state until it is able to participate in capsid assembly.
Materials and methods
Upper domain identification, expression, purification and crystallization
Full-length VP5 was purified as described previously (Newcomb et al., 1999). The protein solution was concentrated to 1 mg/ml and digested with trypsin at 2% of total protein by weight. Proteolysis was performed at room temperature for 3 h and then terminated by addition of excess aminoethylbenzenesulfonyl fluoride (AEBSF). N-terminal sequencing and MALDI-TOF mass spectrometric analysis identified a stable domain consisting of residues 451–1054.
The DNA sequence corresponding to the upper domain of full-length VP5 was amplified by PCR and cloned into the pET32 expression vector. Protein was expressed in Escherichia coli BL21 (DE3) cells at 16°C for 24 h following induction with 0.3 mM isopropyl-β-d-thiogalactopyranoside (IPTG). Cells were lysed with BPER (Pierce), and the protein purified to near homogeneity using Ni2+-chelating Sepharose (Pharmacia). The protein was eluted with 250 mM imidazole in buffer B (250 mM NaCl, 20 mM Tris pH 7.4). After overnight dialysis at 4°C against enterokinase cleavage buffer [50 mM NaCl, 2 mM CaCl2, 5 mM dithiothreitol (DTT), 20 mM Tris pH 7.4], proteolysis to remove the thioredoxin tag was performed at 37°C for 6–8 h at a protein concentration of 2 mg/ml with 2 U/ml of protease. An additional overnight incubation at 4°C was usually needed to complete the cutting reaction. The cleaved product was purified on a Pharmacia S75 gel filtration column equilibrated with buffer C (250 mM NaCl, 10 mM Tris pH 8.0 and 10 mM DTT). Selected fractions were pooled and concentrated to 20 mg/ml in buffer C. The protein was crystallized at 4°C using the vapor diffusion method, with the drop consisting of a 3:1 mixture of the stock protein solution and the reservoir solution of 0.8–1 M sodium acetate and 100 mM imidizole pH 6.0–6.8. Prior to data collection, the crystals were placed in increasing percentages of PEG 400 in the crystallization solution to a final concentration of 30% PEG 400, and flash-frozen in liquid nitrogen.
Structure determination
A three-wavelength data set from a crystal of selenomethione-substituted protein was collected on beamline BM-19 at the SBC-CAT at the Advanced Photon Source (APS) at the Argonne National Laboratory. Data were processed with HKL2000 and merged with SCALEPACK (Otwinowski and Minor, 1997). The crystals have two molecules in the asymmetric unit and belong to the tetragonal space group P41212 with unit cell dimensions a = b = 99.07 Å, c = 454.68 Å, and α = β = γ = 90°. The positions of 26 of the 34 SeMet sites in the two molecules in the asymmetric unit were determined, heavy atom parameters refined, and MAD phases calculated to 3.6 Å resolution using the suite of programs in CNS (Brünger et al., 1998). The calculated electron density map was solvent flattened, 2-fold non-crystallographic symmetry averaged, and phase extended to 3.2 Å resolution. This initial map was used to build ∼80% of the total residues using O (Jones et al., 1991). The model was refined in CNS interspersed with model building. A total of 1076 out of 1208 residues (89.1%) (542/604 in the A molecule and 534/604 in the B molecule) were ordered within the asymmetric unit. In both molecules, the residues 451–483, 523–539, 922–924 and 1046–1054 were disordered. In addition, residues 918–920 and 945–950 were disordered in the B molecule. The r.m.s. deviation in the α-carbon positions between the A and B molecules is 0.153 Å. The current model, refined at 2.9 Å resolution against a data set collected at beamline 19ID at the SBC-CAT and processed and scaled with d*TREK (Pflugrath, 1999), has an Rcryst of 25.4% and an Rfree of 28.9%. The model has good geometry, with all of the backbone dihedral angles in the most favored or allowed regions. Coordinates have been deposited under PDB accession code 1N07.
Fitting into the electron cryomicroscopy reconstruction
A single hexon subunit was extracted from an averaged C hexon of the 8.5 Å electron cryomicroscopy structure of the capsid, as described previously (Zhou et al., 2000). To fit the structure of the VP5 fragment to the hexon subunit, the PDB file of the fragment was converted to an electron density map (size = 1283) at the corresponding resolution (8.5 Å) and step size (1.4 Å/pixel) as the capsid map using pdb2mrc (Ludtke et al., 1999). The low resolution fragment was then localized to the single hexon subunit using Foldhunter (Jiang et al., 2001) with a 10° angular search and the ‘smart’ option. As the hand of the reconstruction had not been determined previously, both hands of the extracted VP5 monomer, created using mirror, were used (Ludtke et al., 1999). The original PDB file for the VP5 fragment was then transformed to the Foldhunter-derived coordinates using the Molecular Modeling Toolkit (MMTK).
To localize the VP5 fragment in the intact hexon, the single hexon subunit (padded to 2563) was fit to the average C hexon using Foldhunter (angular search of 15°). From these coordinates, the transformed PDB file was again transformed using MMTK to the new Foldhunter-derived coordinates. Localization of the VP5 fragment within an intact penton subunit and penton was carried out in the same way. The correlation coefficient for the fit of the X-ray structure into the electron cryomicroscopy density of the VP5ud is 0.90 for the hexon and 0.88 for the penton.
Illustrations
Ribbon diagrams were computed using MOLSCRIPT (Kraulis, 1991) and RASTER3D (Merritt and Bacon, 1997) (Figures 1, 3 and 4) or PyMol (http://www.pymol.org) as a renderer. Ribbon diagrams fitted into the electron cryomicroscopy map were calculated using RIBBONS (Carson, 1997) and rendered using Iris Explorer (NAG, Downers Grove, IL) with custom-designed modules (Figures 2 and 6B). Electrostatic potential maps were calculated using Delphi, visualized using GRASP (Nicholls et al., 1993) and rendered using the POV4GRASP utility (http://pov4grasp.free.fr) (Figures 5, and 6A and C). All representations of the VP5ud were made using the A molecule.
Acknowledgments
Acknowledgements
We thank Drs H.Jayaram, Z.H.Zhou, B.V.V.Prasad, J.Pflugrath, F.Tsai, S.Lee and D.McNab for technical assistance and useful discussions, and the staff at the APS SBC-CAT for help with data collection. This research was supported by grants from the Human Frontier Science Program, the National Institutes of Health (P415502250, R01AI3860 to W.C.), the R.Welch Foundation (Q1242 to W.C. and Q0581 to F.A.Q.) and Howard Hughes Medical Institute, with which F.A.Q. is an Investigator. B.R.B. was supported in part by an NIH Training Grant (GM08280) to the Houston Area Molecular Biophysics Program. M.L.B. was supported in part by BRASS and the W.M.Keck Center for Computational Biology through a training grant from the National Library of Medicine (2T15LM07093).
References
- Batterson W., Furlong,D. and Roizman,B. (1983) Molecular genetics of herpes simplex virus. VIII. Further characterization of a temperature-sensitive mutant defective in release of viral DNA and in other stages of the viral reproductive cycle. J. Virol., 45, 397–407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Benson S.D., Bamford,J.K., Bamford,D.H. and Burnett,R.M. (1999) Viral evolution revealed by bacteriophages PRD1 and human adenovirus coat protein structures. Cell, 98, 825–833. [DOI] [PubMed] [Google Scholar]
- Böttcher B., Wynne,S.A. and Crowther,R.A. (1997) Determination of the fold of the core protein of hepatitis B virus by electron cryomicroscopy. Nature, 386, 88–91. [DOI] [PubMed] [Google Scholar]
- Brünger A.T. et al. (1998) Crystallography and NMR system: a new software suite for macromolecular structure determination. Acta Crystallogr. D, 54, 905–921. [DOI] [PubMed] [Google Scholar]
- Carson M. (1997) Ribbons. Methods Enzymol., 277, 493–505. [PubMed] [Google Scholar]
- Caspar D.L. and Klug,A. (1962) Physical principles in the construction of regular viruses. Cold Spring Harbor Symp. Quant. Biol., 48, 1–24. [DOI] [PubMed] [Google Scholar]
- Cepko C.L. and Sharp,P.A. (1983) Analysis of Ad5 hexon and 100K ts mutants using conformation-specific monoclonal antibodies. Virology, 129, 137–154. [DOI] [PubMed] [Google Scholar]
- Chen D.H., Jakana,J., McNab,D., Mitchell,J., Zhou,Z.H., Dougherty,M., Chiu,W. and Rixon,F.J. (2001) The pattern of tegument–capsid interaction in the herpes simplex virus type 1 virion is not influenced by the small hexon-associated protein VP26. J. Virol., 75, 11863–11867. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Conway J.F., Cheng,N., Zlotnick,A., Wingfield,P.T., Stahl,S.J. and Steven,A.C. (1997) Visualization of a 4-helix bundle in the hepatitis B virus capsid by cryo-electron microscopy. Nature, 386, 91–94. [DOI] [PubMed] [Google Scholar]
- Coulter L.J., Moss,H.W., Lang,J. and McGeoch,D.J. (1993) A mutant of herpes simplex virus type 1 in which the UL13 protein kinase gene is disrupted. J. Gen. Virol., 74, 387–395. [DOI] [PubMed] [Google Scholar]
- Desai P.J. (2000) A null mutation in the UL36 gene of herpes simplex virus type 1 results in accumulation of unveloped DNA-filled capsids in the cytoplasm of infected cells. J. Virol., 74, 11608–11618. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dokland T., McKenna,R., Ilag,L.L., Bowman,B.R., Incardona,N.L., Fane,B.A. and Rossmann,M.G. (1997) Structure of a viral procapsid with molecular scaffolding. Nature, 389, 308–313. [DOI] [PubMed] [Google Scholar]
- Fenwick M.L. and Everett,R.D. (1990) Inactivation of the shutoff gene (UL41) of herpes simplex virus types 1 and 2. J. Gen. Virol., 71, 2961–2967. [DOI] [PubMed] [Google Scholar]
- Grimes J.M., Burroughs,J.N., Gouet,P., Diprose,J.M., Malby,R., Zientara,S., Mertens,P.P. and Stuart,D.I. (1998) The atomic structure of the bluetongue virus core. Nature, 395, 470–478. [DOI] [PubMed] [Google Scholar]
- Harrison S.C., Skehel,J.J. and Wiley,D.C. (1996) Virus structure. In Fields,B.N., Knipe,D.M., Howley,P.M., Chanock,R.M., Melnick,J.L., Monath,T.P., Roizman,B. and Strauss,S.E. (eds), Field’s Virology. Lippincott-Raven, Philadelphia, PA, pp. 59–99.
- Holm L. and Sander,C. (1995) Dali: a network tool for protein structure comparison. Trends Biochem. Sci., 20, 478–480. [DOI] [PubMed] [Google Scholar]
- Jiang W., Baker,M.L., Ludtke,S.J. and Chiu,W. (2001) Bridging the information gap: computational tools for intermediate resolution structure interpretation. J. Mol. Biol., 308, 1033–1044. [DOI] [PubMed] [Google Scholar]
- Jones T.A., Zou,J.Y., Cowan,S.W. and Kjeldgaard,M. (1991) Improved methods for binding protein models in electron density maps and the location of errors in these models. Acta Crystallogr. A, 47, 110–119. [DOI] [PubMed] [Google Scholar]
- Knipe D M., Batterson,W., Nosal,C., Roizman,B. and Buchan A. (1981) Molecular genetics of herpes simplex virus. VI. Characterization of a temperature-sensitive mutant defective in the expression of all early viral gene products. J. Virol., 38, 539–547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kraulis P. (1991) MOLSCRIPT: a program to produce both detailed and schematic plots of protein structures. J. Appl. Crystallogr., 24, 946–950. [Google Scholar]
- Lemaster S. and Roizman,B. (1980) Herpes simplex virus phosphoproteins. II. Characterization of the virion protein kinase and of the polypeptides phosphorylated in the virion. J. Virol., 35, 798–811. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ludtke S.J., Baldwin,P.R. and Chiu,W. (1999) EMAN: semiautomated software for high resolution single-particle reconstructions. J. Struct. Biol., 128, 82–97. [DOI] [PubMed] [Google Scholar]
- Merritt E.A. and Bacon,D.J. (1997) Raster3D version 2.0: a program for photorealistic molecular graphics. Methods Enzymol., 277, 505–524. [DOI] [PubMed] [Google Scholar]
- Newcomb W.W. and Brown,J.C. (1991) Structure of the herpes simplex virus capsid: effects of extraction with guanidine hydrochloride and partial reconstitution of extracted capsids. J. Virol., 65, 613–620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Newcomb W.W. and Brown,J.C. (1994) Induced extrusion of DNA from the capsid of herpes simplex virus type 1. J. Virol., 68, 433–440 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Newcomb W.W., Homa F.L., Thomsen,D.R., Booy,F.P., Trus,B.L., Steven,A.C., Spencer,J.V. and Brown,J.C. (1996) Assembly of the herpes simplex virus capsid: characterization of intermediates observed during cell-free capsid formation. J. Mol. Biol., 263, 432–446. [DOI] [PubMed] [Google Scholar]
- Newcomb W.W., Homa,F.L., Thomsen D.R., Trus B.L., Cheng,N., Steven,A.C., Booy F.P. and Brown,J.C. (1999) Assembly of the herpes simplex virus procapsid from purified components and identification of small complexes of the major capsid and scaffolding proteins. J. Virol., 73, 4239–4350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Newcomb W.W., Juhas,R.M., Thomsen,D.R., Homa,F.L., Burch,A.D., Weller,S.K. and Brown,J.C. (2001) The UL6 gene product forms the portal for entry of DNA into the herpes simplex virus capsid. J. Virol., 75, 10923–10932. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nicholls A., Bharadwaj,R. and Honig,B. (1993) GRASP: graphical representation and analysis of surface properties. Biophys. J., 64, 166–170. [Google Scholar]
- Ojala P.M., Sodeik,B., Ebersold,M.W., Kutay,U. and Helenius,A. (2000) Herpes simplex virus type 1 entry into host cells: reconstitution of capsid binding and uncoating at the nuclear pore complex in vitro. Mol. Cell Biol., 20, 4922–4931. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Otwinowski Z. and Minor,W. (1997) Processing of X-ray diffraction data collected in oscillation mode. Methods Enzymol., 276, 307–326. [DOI] [PubMed] [Google Scholar]
- Pflugrath J.W. (1999) The finer things in X-ray diffraction data collection. Acta Crystallogr. D, 55, 1718–1725. [DOI] [PubMed] [Google Scholar]
- Post L.E., Mackem,S. and Roizman,B. (1981) Regulation of α genes of herpes simplex virus: expression of chimeric genes produced by fusion of thymidine kinase with α gene promoters. Cell, 24, 555–565. [DOI] [PubMed] [Google Scholar]
- Reinisch K.M., Nibert,M.L. and Harrison,S.C. (2000) Structure of the reovirus core at 3.6 Å resolution. Nature, 404, 960–967. [DOI] [PubMed] [Google Scholar]
- Roberts M.M., White,J.L., Grutter,M.G. and Burnett,R.M. (1986) Three dimensional structure of the adenovirus major coat protein hexon. Science, 232, 1148–1151. [DOI] [PubMed] [Google Scholar]
- Roizman B.N. and Sears,A.E. (1996) Herpes simplex viruses and their replication. In Fields,B.N., Knipe,D.M., Howley,P.M., Chanock,R.M., Melnick,J.L., Monath,T.P., Roizman,B. and Strauss,S.E. (eds), Field’s Virology. Lippincott-Raven, Philadelphia, PA, pp. 2231–2295.
- Rossmann M.G. and Johnson,J.E. (1989) Icosahedral RNA virus structure. Annu. Rev. Biochem., 58, 533–573. [DOI] [PubMed] [Google Scholar]
- Sharp P.M. (2002) Origins of human virus diversity. Cell, 108, 305–312. [DOI] [PubMed] [Google Scholar]
- Spencer J.V., Trus,B.L., Booy,F.P., Steven,A.C., Newcomb,W.W. and Brown,J.C. (1997) Structure of the herpes simplex virus capsid: peptide A862–H880 of the major capsid protein is displayed on the rim of the capsomer protrusions. Virology, 228, 229–235. [DOI] [PubMed] [Google Scholar]
- Spencer J.V., Newcomb,W.W., Thomsen,D.R., Homa,F.L. and Brown,J.C. (1998) Assembly of the herpes simplex virus capsid: preformed triplexes bind to the nascent capsid. J. Virol., 72, 3944–3951. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stehle T., Gamblin,S.J., Yan,Y. and Harrison S.C. (1996) The structure of simian virus 40 refined at 3.1 Å. Structure, 4, 165–182. [DOI] [PubMed] [Google Scholar]
- Steven A.C. and Spear,P.G. (1997) Herpesvirus capsid assembly and envelopment. In Chiu,W., Burnett,R.M. and Garcea,R.L. (eds), Structural Biology of Viruses. Oxford University Press, New York, NY, pp. 312–351.
- Trus B.L., Booy,F.P., Newcomb,W.W., Brown,J.C., Homa,F.L., Thomsen,D.R. and Steven,A.C. (1996) The herpes simplex virus procapsid: structure, conformational changes upon maturation and roles of the triplex proteins VP19c and VP23 in assembly. J. Mol. Biol., 263, 447–462. [DOI] [PubMed] [Google Scholar]
- Wikoff W.R., Liljas,L., Duda,R.L., Tsuruta,H., Hendrix,R.H. and Johnson,J.E. (2000) Topologically linked rings in the bacteriophage HK97 capsid. Science, 289, 2129–2133. [DOI] [PubMed] [Google Scholar]
- Wingfield P.T., Stahl,S.J., Thomsen,D.R., Homa,F.L., Booy,F.P. Trus,B.L. and Steven,A.C. (1997) Hexon-only binding of VP26 reflects differences between the hexon and penton conformations of VP5, the major capsid protein from herpes simplex virus. J. Virol., 71, 8955–8961. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Y. and McKnight,J.L. (1993) Herpes simplex virus type 1 UL46 and UL47 deletion mutants lack VP11 and VP12 or VP13 and VP14, respectively and exhibit altered viral thymidine kinase expression. J. Virol., 67, 1482–1492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou Z.H., He,J., Jakana,J., Tatman,J.D., Rixon,F.J. and Chiu,W. (1995) Assembly of VP26 in herpes simplex virus-1 inferred from structures of wild-type and recombinant capsids. Nat. Struct. Biol., 2, 1026–1030. [DOI] [PubMed] [Google Scholar]
- Zhou Z.H., Chen D.H., Jakana,J., Rixon,F.J. and Chiu,W. (1999) Visualization of tegument–capsid interactions and DNA in intact herpes simplex virus type 1 virions. J. Virol., 73, 3210–3218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou Z.H., Dougherty,M., Jakana,J., He,J., Rixon,F.J. and Chiu,W. (2000) Seeing the herpesvirus capsid at 8.5 Å. Science, 288, 877–880. [DOI] [PubMed] [Google Scholar]