

Abstract
Transcriptional activation is associated commonly with recruitment of histone acetylases, while repression involves histone deacetylases (HDACs). Here, we provide evidence to suggest that STAT5 activates gene expression by recruiting HDAC. The interleukin-3 (IL-3)-dependent expression of the Id-1 gene, encoding a helix–loop–helix (HLH) transcriptional inhibitor, is activated by both C/EBPβ and STAT5 transcription factors bound to its pro-B-cell enhancer (PBE), but is inhibited by HDAC inhibitors in Ba/F3 cells. STAT5 interacts with HDAC1 in the PBE region, resulting in deacetylation of histones, as well as C/EBPβ, whose acetylation diminishes its DNA-binding activity. Consistently, expression of an acetylation-resistant mutant of C/EBPβ results in IL-3-independent expression of the Id-1 gene. Thus, we propose a novel mechanism by which STAT5 mediates the deacetylation of C/EBPβ, allowing transcriptional activation.
Keywords: acetylation/cytokine/helix–loop–helix/STAT/transcription
Introduction
The Id-1 gene encodes a dominant-negative inhibitor of basic helix–loop–helix (bHLH) transcription factors such as E2A, HEB and E2-2 (Norton et al., 1998; Massari and Murre, 2000). Ablation of the function of these bHLH proteins arrests B- and T-cell development at early progenitor stages (Engel and Murre, 2001). Although the bHLH proteins are expressed constitutively, Id-1 expression is regulated during lymphocyte development, which in turn controls bHLH function and lymphocyte differentiation (Sun, 1994; Heemskerk et al., 1997; Kim et al., 1999). Id-1 is highly expressed in hematopoietic stem cells and down-regulated during B-cell differentiation in the bone marrow (Jaleco et al., 1999; Igarashi et al., 2002), a process probably controlled by growth factors and cytokines critical for lymphocyte development. Therefore, knowledge of how the Id1 gene is regulated and what the signal transduction pathways are leading to its regulation will facilitate an understanding of lymphocyte development.
Regulation of specific gene expression is achieved largely by controlling production of transcription factors responsible for the expression of target genes, alteration of the activities of transcription factors through post-translational modifications and localized remodeling of chromatin structures. Recently, histone acetylation has attracted tremendous attention. Acetylation of histone proteins, particularly H3 and H4, is thought to lead to relaxed chromatin structure and, therefore, an enhanced rate of transcription. Acetylation reactions are catalyzed by various families of coactivators with histone acetyl transferase activities, represented by PCAF, TAF250 and p300/CBP (H.Chen et al., 2001; Marmorstein and Roth, 2001). On the other hand, deacetylation of histone proteins by histone deacetylases (HDACs) is believed to cause chromosomal condensation and gene repression (Struhl, 1998; Tyler and Kadonaga, 1999).
In addition to modification of histone proteins, acetyl ation has been shown to impact on the activities of transcription factors (Kouzarides, 2000). Acetylation of components in the basal transcriptional machinery, such as TFIIEβ and TFIIF, may impact directly on transcription of genes controlled by PCAF, p300 and TAF250 (Imhof et al., 1997). Acetylation of p53, EKLF, GATA1 and MyoD increases in DNA binding or transactivating abilities (Gu and Roeder, 1997; Boyes et al., 1998; Zhang and Bieker, 1998; Hung et al., 1999; Sartorelli et al., 1999; Yamagata et al., 2000). The acetylated p65 subunit of NF-κB has lower affinity for IκB (L.Chen et al., 2001). Despite these different underlying mechanisms, a common outcome of acetylation is to favor transcription controlled by acetyl ated factors. However, it is also possible that acetylation might negatively influence the activities of transcription factors. For instance, acetylation has been shown to disrupt interaction between coactivators and ACTR or TCF (Waltzer and Bienz, 1998; Chen et al., 1999). Recently, it has been found that β-catenin is acetylated at a single lysine residue frequently mutated in thyroid anaplastic cancer. An unacetylatable form of β-catenin activates c-myc gene expression more efficiently than the wild-type protein (Wolf et al., 2002). In this report, we provide evidence that expression of the Id-1 gene depends on HDAC activities through a molecular mechanism involving the deacetylation of C/EBPβ, a transcription factor essential for Id-1 expression.
Previous studies of Id-1 gene regulation led to identification of a pro-B-cell-specific enhancer (PBE), located 3 kb downstream of the gene (Saisanit and Sun, 1995; Saisanit and Sun, 1997). Within this 460 bp PBE enhancer, a C/EBPβ-binding site was found to be important to the enhancer activity, which can be abolished by an inhibitor of C/EBPβ, CHOP (Saisanit and Sun, 1997). We have now demonstrated that HDAC activities are required for STAT5-mediated Id-1 gene expression in the presence of interleukin-3 (IL-3). Consistent with this observation, an acetylation-resistant mutant of C/EBPβ enables Id-1 expression in the absence of IL-3. It thus appears that HDAC activities recruited by STAT5 proteins may be necessary for the deacetylation of C/EBPβ and its transcriptional activation of the Id-1 gene.
Results
Three C/EBPβ-binding sites in PBE are required for Id-1 expression
We have shown previously that a C/EBPβ-binding site is important for the enhancer activity of PBE (Saisanit and Sun, 1995, 1997). Further examination of the PBE sequence revealed two additional C/EBPβ-binding sites with considerably divergent sequences (Figure 1A). These sites, named β1–β3, are clustered within an 80 bp region. Electrophoretic mobility shift assays (EMSAs) with each site as a probe revealed that nuclear extracts from Ba/F3 cells formed three complexes, C1–C3, which were supershifted with antibodies against C/EBPβ (Figure 1B). The C1 complex co-migrates with a non-specific complex, which can be separated from the C1 complex upon prolonged electrophoresis (data not shown).

Fig. 1. The C/EBPβ-binding site is required for Id-1 expression. (A) Three C/EBPβ-binding sites are diagrammed as filled boxes, and their sequences shown on top. Mutations are indicated with lower case letters. The position of PBE (gray box) relative to Id-1 exons (hatched boxes) is illustrated. (B) EMSA with each of the three C/EBPβ-binding sites as probes. A 70 bp probe containing the β1, β2 or β3 site was incubated with Ba/F3 nuclear extracts with or without antibodies against C/EBPβ. The binding reactions were analyzed on a 6% poly acrylamide gel in Tris–glycine buffer. C/EBPβ-containing complexes are labeled as C1–C3. Supershifted complexes are marked with an open arrow. Additional bands are non-specific complexes found in all cell types. (C) Mutational analysis in transient transfection assays using Ba/F3 cells. Constructs containing the 460 bp PBE placed upstream of a luciferase reporter gene driven by a c-fos minimal promoter are diagrammed. Filled and open boxes represent wild-type and mutant C/EBPβ sites, respectively. Luciferase activities were normalized against the β-galactosidase activity. The data are presented as activities relative to that of the PBE-luc construct and are averages of at least three independent experiments with standard deviations. (D) Mutational analysis in stable Ba/F3 cell lines. The ΔId-1 gene is diagrammed as in (A). Three independent cell lines stably transfected with the indicated constructs were analyzed using RPAs, and transcripts were detected with the probes as labeled.
The functional significance of the C/EBPβ-binding sites for PBE activity was tested by mutational analyses using a luciferase reporter gene. Transient transfection of the reporter gene driven by wild-type PBE into Ba/F3 cells usually resulted in >100-fold increases in luciferase activity compared with the construct without the enhancer. Mutations of 2 bp in each of the C/EBPβ sites caused ∼70% reduction in the enhancer activity, suggesting that all three sites are necessary for the enhancer activity. Mutations in all three binding sites almost completely abolished the enhancer activity (Figure 1C).
To examine further the importance of these three sites in directing Id-1 expression, stable Ba/F3 cell lines were generated by using an Id-1-minigene, ΔId-1 (Saisanit and Sun, 1995). The ΔId-1 gene, controlled by its 6 kb of 5′- and 7 kb of 3′-flanking sequences, contains a 200 bp deletion in its exon 1 so that its transcript can be distinguished from the endogenous Id-1 transcript. Ribonuclease protection assays (RPAs) revealed that the ΔId-1 gene controlled by wild-type PBE (15K/wt) was expressed at high levels in multiple cell lines, but ΔId-1 expression was dramatically inhibited by mutations in the three C/EBPβ-binding sites (Figure 1D). Therefore, data obtained from both transient and stable transfection assays strongly suggest that the three C/EBPβ-binding sites are essential for Id-1 expression in Ba/F3 cells.
Combinations of C/EBPβ dimeric proteins are bound to its binding sites in PBE
The C/EBPβ gene encodes three polypeptides through differential usage of AUG codons within the same transcript (Descombes and Schibler, 1991). The 31 and 29 kDa polypeptides contain an N-terminal transcriptional activation domain (Williams et al., 1995), generally referred to as LAP (liver-enriched transcriptional activator protein). The 16 kDa form of C/EBPβ lacks the activation domain, and is called LIP (liver-enriched transcriptional inhibitory protein). These leucine zipper-containing proteins bind to DNA as dimers. To determine the identities of the C1–C3 complexes found in Ba/F3 cells, we compared the mobility of these complexes with those formed with LAP and/or LIP proteins expressed in NIH-3T3 cells (Figure 2A). Although NIH-3T3 cells produced LAP and LIP proteins, which exist as LAP–LAP and LAP–LIP dimers, expression of LAP or LIP led to increased levels of LAP–LAP or LIP–LIP dimers. The LAP construct also makes a small amount of LIP because of internal translation initiation, which formed LAP–LIP dimers. LIP-expressing cells had an increased amount of LAP–LIP dimers due to interaction with endogenous LAP. Expression of both LAP and LIP generated LAP and LIP homo- and heterodimers. The C1–C3 complexes in Ba/F3 cells co-migrated with complexes of LAP–LIP, LIP–LIP and LAP–LAP dimers, respectively, suggesting that these complexes are likely to be combinations of different products of the C/EBPβ gene. Intriguingly, western blot analysis showed that Ba/F3 cells produced a larger amount of LAP than LIP (Figure 2A), but the amount of DNA-binding complexes formed with LAP–LAP dimers (the C3 complex) was much less than those with LAP–LIP and LIP–LIP dimers. In contrast, similar amounts of LAP and LIP proteins co-expressed in NIH-3T3 fibroblasts readily formed the LAP–LAP complex (Figure 2A). These results thus suggested that the LAP protein might be modified post-translationally specifically in Ba/F3 cells such that its DNA-binding activity was inhibited.

Fig. 2. Identities of the C1–C3 complexes. (A) EMSA was performed with nuclear extracts from NIH-3T3 cells transfected with the indicated constructs. ‘C’ stands for extracts transfected with an empty vector, pcDNA3. The positions of the indicated homo- and heterodimers of LAP and LIP are labeled on the left, while C1–C3 complexes in Ba/F3 cells are marked on the right. Supershifted complexes with antibodies against C/EBPβ are indicated by an open arrow. Nuclear extracts used in lanes 4 and 5 were analyzed using a western blot with antibodies against the C-terminus of C/EBPβ. The doublets represent the 29 and 31 kDa LAP. (B) ChIP assays with antibodies against C/EBPβ and a negative control, Skp2. A fragment containing all three C/EBP-binding sites was amplified. (C) Transient co-transfection assay of Ba/F3 cells with the indicated constructs; normalized luciferase activities are shown relative to that of PBE-luc.
To demonstrate that C/EBPβ binds to PBE in vivo, we performed chromatin immunoprecipitation (ChIP) using cross-linked protein–DNA complexes prepared from Ba/F3 cells cultured with or without IL-3. The immunoprecipitates obtained with antibodies against the C-terminus of C/EBPβ and a non-specific control protein were used as templates to amplify a 170 bp DNA fragment containing all three C/EBP sites (Figure 2B). Indeed, anti-C/EBPβ but not control antibodies brought down the PBE fragment in Ba/F3 cells cultured with and without IL-3. Because the antibodies recognize both LAP and LIP, this assay could not distinguish the different dimers of C/EBPβ bound to the enhancer region. However, the above results suggest that C/EBPβ does bind to the three sites found in PBE.
We then tested if C/EBPβ could activate PBE-mediated transcription. The PBE-luc reporter construct was co-transfected into Ba/F3 cells with plasmids expressing LAP, LIP or LGL, which is a forced dimer between LAP and LIP connected through a glycine hinge. LAP increased the reporter activity by 8-fold, but LIP and LGL had no effect on reporter expression (Figure 2C), suggesting that LAP–LAP dimers, but not LIP–LIP or LAP–LIP dimers, are able to activate transcription through PBE. Furthermore, expression of a potent inhibitor of C/EBPβ, CHOP (Ron and Habener, 1992), resulted in a 10-fold reduction of the reporter activity (Figure 2C), indicating that C/EBPβ proteins, specifically LAP–LAP dimers, are probably responsible for the basal activity of PBE-luc in Ba/F3 cells.
IL-3-dependent Id-1 expression correlates with STAT5 activation
Ba/F3 is an IL-3-dependent cell line (Kinashi et al., 1988), and thus serves as a good model system to study the signaling pathways that influence Id-1 gene expression. Stimulation through the IL-3 receptor leads to activation of STAT5 (Reddy et al., 2000). It has been shown previously that a dominant-negative STAT5 mutant can inhibit Id-1 expression in Ba/F3 cells (Mui et al., 1996). Consistently, we found that upon IL-3 deprivation, levels of Id-1 mRNA decreased immediately, along with levels of phospho-STAT5 proteins, suggesting an involvement of STAT5 in Id-1 gene regulation (Figure 3A). Furthermore, Id-1 expression upon IL-3 re-stimulation occurred in the presence of cycloheximide and hence the absence of new protein synthesis (Figure 3B), perhaps through activation of pre-existing STAT5 transcription factors.
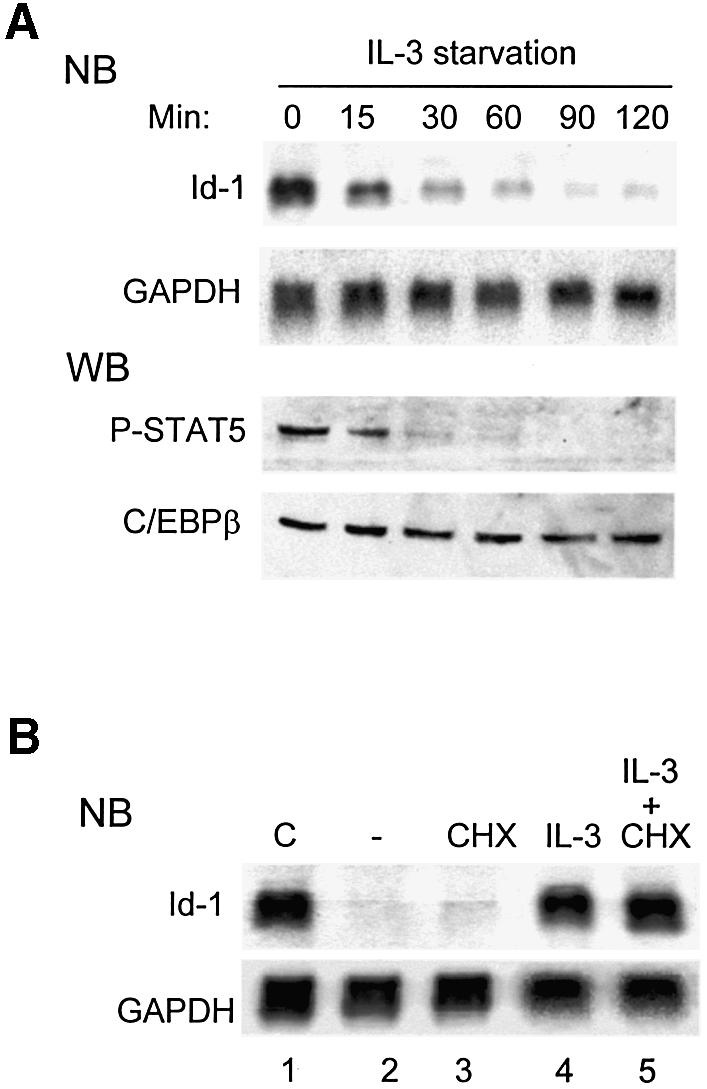
Fig. 3. STAT5 mediates IL-3-dependent Id1 expression. (A) Northern and western blots (NB and WB) for the levels of Id-1 mRNA and phospho-STAT5 protein in Ba/F3 cells cultured without IL-3 for the indicated times. The levels of GAPDH mRNA and C/EBPβ protein were used as internal controls. Twenty percent of WEHI-3 conditioned medium was used as a source of IL-3 in all experiments. (B) Northern blots for Id-1 and GAPDH mRNA levels in Ba/F3 cells: lane 1, continuously cultured in IL-3; lane 2, deprived of IL-3 for 5 h; lane 3, as lane 2 plus treatment with CHX at 50 µg/ml for the last 80 min; lane 4, re-stimulated with IL-3 for 60 min after a 4 h starvation; and lane 5, as lane 4 plus treatment with CHX at 50 µg/ml for the last 80 min.
STAT5 is bound to PBE
Although both C/EBPβ and STAT5 are involved in the regulation of Id-1 expression, mutations that destroy the three C/EBPβ-binding sites almost completely abolish Id-1 expression (Figure 1C and D). This thus raises the question of how STAT5 contributes to the regulation of Id-1 expression. We hypothesized that STAT5 could potentiate the transcriptional activity of C/EBPβ. If so, STAT5 would probably bind to sequences near the C/EBPβ sites. To locate STAT5-binding sites, we searched a 1.4 kb sequence surrounding PBE, and found three putative sites (S1–S3) within PBE (Figure 4A). EMSA was performed using the S1 site as a probe, which is identical to the STAT5 consensus binding sequence, TTCNNNGAA (Wakao et al., 1994). A specific binding complex was supershifted by antibodies against STAT5, and competed off by oligonucleotides containing a known STAT5 site or the S1 site (Figure 4A). The S2 site, whose sequence resembles the consensus sequence poorly, did not compete for binding. The S3 site only competed at a high concentration. Therefore, the S1 site appeared to have the highest affinity for STAT5, as judged by EMSA. However, it is not clear whether the S2 and S3 sites, being close to each other, can be bound by STAT5 in vivo.

Fig. 4. STAT5 binds to PBE. (A) Identification of STAT5-binding sites. Three potential sites shown in triangles, named S1–S3, were found within PBE (thin line). The three C/EBPβ-binding sites are marked as in Figure 1. EMSA was performed using end-labeled 30 bp oligonucleotides containing the S1 site. STAT5 binding and supershifted complexes are indicated by filled and open arrows. Unlabeled 30 bp oligonucleotides containing the S1, S2 or S3 site, plus a known STAT5 site, were used as competitors at the indicated molar excess to the probe. (B) ChIP assays with anti-STAT5a antibodies. Fragments containing the S1 site (S1), S2 plus S3 sites (S23) and C/EBP sites (C/EBP), as diagrammed in (A), were amplified using DNA templates before (input) or after immunoprecipitation (IP) with anti-STAT5 and control antibodies. (C) Effect of deletion of STAT sites on PBE activity. Left: Ba/F3 cells were transfected with the indicated constructs along with the CMV-LacZ construct. Luciferase activities are normalized with that of β-galactosidase and are shown as averages of activities relative to that of PBE-luc obtained from at least three independent experiments. Right: C/EBP-dependent activity is the ratio of relative activities between enhancers with wild-type and mutant C/EBP sites (open and closed ovals).
To determine whether STAT5 binds to the PBE region in vivo, ChIP assays were performed with antibodies against STAT5 (Figure 4B). The amount of DNA brought down in the precipitates was determined by PCR using primers specific to regions containing the S1 or the S2 and S3 sites (S23). A larger amount of S1- and S23-containing DNA was recovered from the anti-STAT5 precipitates of cells cultured with IL-3 than those without IL-3, or those from the precipitates with control antibodies. As a negative control, an intervening region between the S1 and S23 sites, containing the C/EBP sites (Figure 4A), was not precipitated specifically by anti-STAT5 antibodies, suggesting that the S1- and S23-containing fragments were brought down as separate fragments due to their independent binding to STAT5. Taken together, these results indicate that STAT5 indeed binds to the DNA sequences in PBE in Ba/F3 cells. Interestingly, EMSA did not reveal high affinities of the S2 and S3 sites for STAT5; these sites together could bind to STAT5 in vivo, perhaps through cooperation.
If STAT5 can potentiate the transcriptional activity of C/EBPβ, deletion of these STAT sites would have a negative impact on C/EBP-mediated transcription. Indeed, we found that deletion of all three STAT sites in the ΔPBE construct led to a 55% reduction of overall PBE activity even though all three C/EBP sites are intact (Figure 4C). Furthermore, C/EBP-dependent activities can be determined by comparing the activity of PBE or ΔPBE with that of its cognate construct lacking C/EBP sites. Such activities for PBE and ΔPBE were therefore estimated to be 6.25- and 1.5-fold, which meant that deletion of the STAT sites caused a 76% reduction of the C/EBP-dependent activity in ΔPBE (Figure 4C). This would suggest that the STAT5 sites contribute to the enhancer activity of PBE, most probably by influencing the function of C/EBPβ. The increased activity of ΔPBE-luc-β123 compared with PBE-luc-β123 is probably due to removal of unknown inhibitory sequences in PBE.
Histone deacetylase activities are involved in activation of Id-1 transcription
To explore the possibility that Id-1 expression is regulated by changes in chromatin structure, we examined histone acetylation in the Id-1 locus using ChIP assays with antibodies against both acetylated histones H3 and H4 (Figure 5A). Regions near the STAT sites (regions B and C) were analyzed along with a region in the promoter (region A) (Figure 5A). Surprisingly, levels of acetylated histones H3 and H4 bound within PBE were significantly and reproducibly lower in cells cultured with IL-3 than those without IL-3. In contrast, the amounts of acetylated histone H3 and H4 in the promoter region (region A) did not decrease under the same condition, suggesting that histone deacetylation occurred specifically in the PBE region (Figure 5A). Furthermore, the reductions in acetylated histone H3 and H4 bound to PBE were not due to decreased histone occupancy because similar or slightly larger amounts of histone H3 and H2B proteins were found in the presence of IL-3 (Figure 5A).

Fig. 5. Deacetylase activities are necessary for PBE-mediated transcription of the Id-1 gene. (A) ChIP assays of the Id-1 locus depicted as in Figure 1. PCR fragments corresponding to various regions in the locus are shown as short black bars and labeled A–C. Each fragment was amplified from immunoprecipitates of Ba/F3 cells cultured in medium plus or minus IL-3. Fragment B was also amplified using DNA preparations prior to IP as input controls. Data shown are representative of three independent experiments with the indicated antibodies. (B) Northern blot analyses of Id-1 and GAPDH levels in 4 h IL-3-deprived Ba/F3 cells, which were re-stimulated with (lanes 5–7) or without (lanes 2–4) IL-3 for 1 h. TSA (lane 3, 4, 6 and 7) was added 1 h before and during re-stimulation at the indicated concentrations. Lane 1 contains RNA from Ba/F3 cells continuously cultured with IL-3. The percentage Id-1 re-stimulation (lanes 5–7) was calculated by comparing Id-1 levels normalized with levels of GAPDH and shown in the bar graph. (C) Northern blots to measure the levels of Id-2 and GAPDH mRNA. Treatments in lanes 1–6 were identical to those in lanes 2–7 of (B). (D) Transient transfection assays for expression of the indicated reporter genes under the following conditions: (1) continuous culture in IL-3; (2) IL-3 deprivation for 16 h; (3) IL-3 deprivation for 10 h followed by 6 h re-stimulation; (4) the same as (3) plus TSA treatment 1 h before and during IL-3 re-stimulation. SEAP activities accumulated in media during the last 6 h of the treatment were normalized against β-galactosidase activities. Data are presented as activities relative to SEAP activities under condition 1.
To test whether histone deacetylation is of any functional significance, IL-3-dependent expression of Id-1 was measured in cells treated with a deacetylase inhibitor, trichostatin A (TSA). Pre-treatment of IL-3-deprived cells with TSA for 1 h inhibited Id-1 expression by 60% upon IL-3 re-stimulation (Figure 5B). As a negative control, expression of an IL-3-independent gene, Id-2, was not inhibited by TSA, suggesting that inhibition of Id-1 expression is not due to any potential deleterious effects on Ba/F3 cells (Figure 5C). Therefore, IL-3-stimulated Id-1 expression appears, at least in part, to depend on a deacetylase activity, most probably operating at the PBE region.
We next tested if histone deacetylation is important for PBE activity by using a secreted alkaline phosphatase (SEAP) reporter gene in transient transfection assays. The SEAP reporter allows us to assay for transcriptional activities within defined time periods after different treatments (Figure 5D). Removal of IL-3 for 16 h diminished SEAP expression by at least 70%, indicating that PBE could mediate IL-3-stimulated expression of SEAP. More importantly, treatment with TSA inhibited PBE-SEAP expression upon IL-3 re-stimulation, suggesting that deacetylase activities played an important role in PBE-activated gene expression even in the context of a heterologous promoter and a reporter gene. As a control, IL-3 removal and TSA treatment had little effect on expression of the CMV-SEAP construct. Because the IL-3 response and deacetylase dependence of PBE can be recapitulated when taken out of its natural context, it is unlikely that histone deacetylation or nucleosomal positioning of the enhancer is crucial for Id-1 expression in vivo. Histone deacetylation observed in the PBE region might be a by-product of HDACs recruited to the vicinity. Instead, deacetylase activities could be important for activities of other transcription factors involved in Id-1 expression.
STAT5 recruits histone deacetylase 1
Since IL-3-dependent Id-1 expression required protein deacetylation, we tested whether STAT5 could recruit deacetylases by performing co-IP assays. Immuno precipitates obtained with antibodies against STAT5a were western blotted with antibodies against HDAC1. Anti-STAT5a antibodies brought down not only STAT5a but also HDAC1 from total extracts of Ba/F3 cells cultured with IL-3, but not those without IL-3 (Figure 6A). Control mouse IgG precipitated neither STAT5a nor HDAC1. Co-precipitation of STAT5a and HDAC1 was also observed in 293T cells transfected with both STAT5a and HDAC1 expression constructs, but not either alone (Figure 6B). Consistently, we have shown using a ChIP assay that an increased amount of HDAC1 was present near the S1 STAT5 site in the PBE (region B), but not in the promoter (region A), in cells cultured with IL-3 compared with those without IL-3 (Figure 6C). These results thus suggest that STAT5, by recruiting HDAC1 to PBE, is responsible for the deacetylase activity associated with PBE.

Fig. 6. Interaction between STAT5 and HDAC1. (A) Total cell extracts derived from Ba/F3 cells cultured with or without (+ or –) IL-3 were precipitated with the indicated antibodies. Immunoprecipitates and inputs were analyzed using western blots with antibodies against HDAC1 or STAT5a. (B) 293T cells were transfected with the indicated constructs, and nuclear extracts were analyzed after IP with antibodies against HDAC1 and western blotting with anti-STAT5 antibodies. Inputs were analyzed using western blots with the indicated antibodies. (C) ChIP assay was performed using antibodies against HDAC1 and Ba/F3 cells cultured with or without IL-3. PCR products for regions A and B were as described in Figure 5A.
Acetylation of C/EBPβ inhibits its DNA-binding activity
What are the intended targets, if not histones, of HDAC1 recruited by STAT5? Since C/EBPβ is crucial for Id-1 expression, it becomes a logical candidate. From studies on acetylation of p53 and GATA1 (Gu and Roeder, 1997; Boyes et al., 1998), an acetylation motif of K/RXKK has been derived, where the acetyl group is usually added to the two adjacent lysine residues (Figure 7A). A similar motif, KAKK, was found in a region N-terminal to the DNA-binding domain of C/EBPβ, which is evolutionarily conserved (Figure 7A). To determine whether C/EBPβ can be acetylated, partially purified recombinant LAP proteins were used in an in vitro acetylation reaction with [14C]acetyl-CoA. Immunoprecipitates containing CBP overexpressed in 293T cells or partially purified GST–PCAF fusion proteins were used as the sources of acetylases. Indeed, LAP was acetylated in vitro by both CBP and PCAF (Figure 7B). Furthermore, mutations of the acetylation site (Lys215 and Lys216 to arginine) greatly reduced the acetylation in LAP (Figure 7A and B). These results thus suggest that C/EBPβ can be a target for acetylation.

Fig. 7. Acetylation of C/EBPβ. (A) Sequence alignment of a potential acetylation site in C/EBPβ and other proteins as indicated. The three conserved lysines in the motif are boxed. Acetylation motifs are underlined, with acetylated lysine in bold. (B) In vitro acetylation. Wild-type and mutant LAP were incubated with the indicated acetyltransferases and [14C]acetyl-CoA, and labeled proteins were analyzed by SDS–PAGE followed by autoradiography. Input proteins (3%) were analyzed by western blots. The mutant LAP protein contains an HA tag, thus migrating more slowly than LAP. (C) Acetylation inhibits the DNA-binding activity of LAP–LAP dimers. Pull-down assays were performed with agarose beads conjugated with C/EBPβ-binding sites and in vitro acetylated LAP proteins. Equal portions of the bound and unbound fractions were analyzed using SDS–PAGE and quantified with a phosphoimager. The amounts of proteins in these fractions were also analyzed by western blots and quantified with a LumiImager. The amount of acetylated LAP was normalized against that of total LAP in each fraction. The ratio between the proportion of acetylated LAP in the unbound and bound fractions is shown below. (D) EMSA using nuclear extracts from Ba/F3 cells cultured with (+) or without (–) IL-3. Different forms of C/EBPβ dimers were identified by supershifting with anti-C/EBPβ antibodies. Oct-1-binding activities were used as controls for the nuclear extracts.
We then examined the effect of acetylation on the DNA-binding activity of C/EBPβ. Because in vitro acetylation reactions are incomplete, the DNA-binding activity of acetylated proteins could not be determined using EMSA without separating the acetylated from unacetylated protein. We thus designed a pull-down assay. LAP was labeled with [14C]acetyl-CoA and incubated with agarose beads coupled with oligonucleotides containing a C/EBPβ-binding site. Bound and unbound fractions of LAP were analyzed by SDS–PAGE, and the proportion of 14C-labeled LAP present in each fraction was used as an indication of the DNA-binding activity (Figure 7C). The amounts of LAP acetylated by CBP and PCAF in the bound fraction were found to be lower than those in the unbound fraction by 3.7- and 2.6-fold, respectively, suggesting that acetylation of LAP could impair its DNA-binding activity. It is thus conceivable that deacetylation of LAP is necessary for LAP–LAP dimers to bind to DNA, and STAT5 may facilitate the deacetylation by recruiting HDAC1 to the vicinity of PBE. If so, the DNA-binding activity of LAP–LAP dimers may be influenced by the function of STAT5 in an IL-3-dependent manner. Indeed, EMSA revealed that the amount of LAP–LAP binding complexes (C3) was moderately but significantly reduced in Ba/F3 cells deprived of IL-3 for 4 h compared with those with IL-3 (Figure 7D). In contrast, the amount of LIP–LIP dimers increased upon IL-3 deprivation. Interestingly, although the acetylation site is present in both LAP and LIP, only the LAP–LAP dimers appeared to be affected. This is probably due to the presence of a bulky N-terminus of the LAP protein, as proposed by Williams et al. (1995), which makes its DNA binding or dimeriz ation more susceptible to acetylation.
Acetylation of C/EBPβ occurs in vivo and IL-3-dependent deacetylation is necessary for Id-1 expression
To determine if acetylation of C/EBPβ occurs in vivo, we performed IP experiments using a monoclonal antibody against acetyl-lysine and nuclear extracts from Ba/F3 and PD31 cells. The immunoprecipitates were then western blotted with a monoclonal antibody against the C-terminus of C/EBPβ, which detects both LAP and LIP. As shown in Figure 8A, Ba/F3 cells expressed a substantial amount of C/EBP, while PD31 cells had a barely detectable amount. The anti-acetyl-lysine precipitates from Ba/F3 cells, but not PD31 cells, contained both LAP and LIP. However, the immunoprecipitates of a control antibody, anti-E47, did not include either LAP or LIP. These results thus suggest that C/EBPβ proteins indeed are acetylated in Ba/F3 cells.

Fig. 8. An acetylation mutant of C/EBPβ enables IL-3-independent expression of Id-1. (A) In vivo acetylation assay. Immunoprecipitates of an anti-acetyl-lysine antibody (KAc) and a control antibody (anti-E47) from nuclear extracts of Ba/F3 (B) or PD31 (P) cells were analyzed using western blots with a monoclonal antibody against the C-terminus of C/EBPβ. Inputs contain 2% of nuclear extracts used for IP. (B) Real-time PCR assays for Id-1 expression in Ba/F3 cells expressing HA-tagged wild-type or K2 mutant C/EBPβ. The experimental procedure is as outlined. Data shown are averages of three real-time PCR assays with standard deviations, and expressed as levels relative to that at time zero. Protein expression at each time point was analyzed using western blots with an antibody against C/EBPβ. The endogenous (endo) and exogenous (exo) LAP proteins are indicated by arrows. The level of TFIIH serves as a loading control. (C) Acetylation of wild-type and K2 mutant protein. Ba/F3 cells infected with retrovectors expressing HA-tagged wild-type and K2 mutant were used to immunoprecipitate the proteins with an anti-HA antibody. The immunoprecipitates were probed with anti-acetyl-lysine and anti-HA antibodies.
If the IL-3 dependence of Id-1 expression is due to the requirement for the deacetylation of C/EBPβ mediated by STAT5, introduction of an acetylation-resistant mutant of C/EBPβ into Ba/F3 cells should enable Id-1 gene expression in the absence of IL-3. Therefore, we infected Ba/F3 cells with retroviruses expressing hemagglutinin (HA)-tagged LAP or LAP-K2, which was shown to resist acetylation in vitro (Figure 7B) but to have DNA-binding activity comparable with wild-type LAP (data not shown). Since the retroviral vector also expresses green fluorescent protein (GFP), infected cells were sorted for their fluorescence and used to measured Id-1 expression by real-time PCR (Figure 8B). Upon IL-3 deprivation, Id-1 expression decreased dramatically in vector- or LAP-infected cells after 1 and 2 h. In contrast, LAP-K2-infected cells retained high levels of Id-1 expression at both time points. As a control, we measured the amounts of wild-type or K2 mutant protein expressed at each time point and found that both proteins were expressed at levels much higher than the endogenous LAP protein, whereas amounts of wild-type and K2 mutant proteins differ by <30% as determined using a LumiImager (Figure 8B). Furthermore, we determined the acetylation status of wild-type and K2 mutant proteins. Immunoprecipitates against the HA tag on these proteins were western blotted with monoclonal antibodies against acetylated lysine and the HA tag. Although the amounts of wild-type and K2 proteins precipitated were similar, the wild-type but not K2 protein reacted to the anti-acetyl-lysine antibody, suggesting that the K2 mutant is not acetylated in vivo (Figure 8C). Therefore, this result would suggest that the LAP-K2 mutant is capable of activating Id-1 gene expression in the absence of IL-3 because it is independent of deacetylation mediated by STAT5/HDAC.
Discussion
In this report, we have provided several lines of direct and indirect evidence to suggest that expression of the Id-1 gene in Ba/F3 cells requires deacetylation of the C/EBPβ transcription factor in an IL-3-dependent manner. This conclusion is supported by several key observations: (i) C/EBPβ binds to three sites in PBE, which is essential for Id-1 expression; (ii) HDAC1 is recruited to PBE, and HDAC inhibitors diminish IL-3-stimulated Id-1 expression; (iii) Id-1 expression depends on STAT5 activation, which leads to STAT5 binding to sites adjacent to C/EBPβ sites in PBE and its recruitment of HDAC1; (iv) C/EBPβ can be acetylated in vitro and is acetylated in vivo; and (v) acetylation of C/EBPβ diminishes its DNA-binding activity and an acetylation-resistant mutant of LAP rescues Id-1 expression upon IL-3 deprivation.
Based on these results, we propose that C/EBPβ proteins are normally present in acetylated states in Ba/F3 cells, which may be incapable of binding DNA and activating transcription. During IL-3 stimulation, activated STAT5 binds to its sites adjacent to the C/EBPβ sites in PBE and recruits HDAC1. Consequently, HDAC causes deacetylation of LAP proteins bound either as heterodimers with LIP or as homodimers with low affinity. Deacetylated LAP–LAP dimers can then bind with high affinity and activate transcription. This hypothesis is strongly supported by the result that expression of an acetylation-resistant mutant renders IL-3-independent expression of Id-1 in Ba/F3 cells (Figure 8).
Our current data showed that acetylation causes a moderate reduction in the DNA-binding activity of LAP–LAP dimers in vitro (∼3-fold). This observation is consistent with the finding that the abundance of LAP–LAP binding complexes (C3 complex) is disproportionally low in Ba/F3 cells as compared with NIH-3T3 cells (Figure 2A). Perhaps the C3 complexes detected in Ba/F3 cells consist primarily of deacetylated LAP protein bound adjacent to STAT5 in PBE, whereas the overall pool of LAP protein in the cells remains acetylated even in the presence of IL-3. Therefore, IL-3 deprivation in Ba/F3 cells further decreases the amount of C3 complexes, crucial for Id-1 transcription (Figure 7D). However, it remains to be determined if acetylation of C/EBPβ could also interfere with its interaction with putative coactivators. Once deacetylated, LAP–LAP or LAP–LIP dimers could recruit the necessary coactivators to stimulate transcription of the Id-1 gene. It is also possible that acetylated C/EBPβ could bind to corepressors, which would be released by deacetylation of C/EBPβ. Further more, a C/EBPβ-independent model could be invoked, which would suggest that STAT5 may recruit HDAC1 to deacetylate histone proteins, which may cause chromatin condensation, leading to exclusion of any potential repressors which might bind in the PBE region.
Because Id-1 gene expression is controlled primarily by its enhancer, PBE, located 3 kb downstream of its coding sequence, recruitment of HDAC and histone deacetylation in the enhancer might not have as much of a negative effect on transcription as one might expect. We have shown that the acetylation status of histones bound in the promoter region of Id-1 is unchanged in response to IL-3 deprivation. Perhaps IL-3-dependent transcription of the Id-1 gene is insensitive to alterations in histone acetylation or chromatin structure. It is possible that the Id-1 locus is already accessible in Ba/F3 cells but requires transcription factors to facilitate the initiation of its transcription.
Our studies have established a connection between HDACs and the Jak–STAT signal transduction pathway utilized by a variety of cytokines and growth factors (Bromberg and Darnell, 2000). STAT5 is activated by Jaks in response to stimulation through a large number of cytokines (Lin and Leonard, 2000). In addition to Id-1, we found that the IL-3-dependent expression of two other STAT5 target genes, CIS and Pim-1, was also inhibited by TSA (data not shown). Expression of IL-2-stimulated genes has also been shown to be suppressed upon treatment with deacetylase inhibitors (Koyama et al., 2000), possibly through a similar mechanism involving STAT5 and HDACs. However, it is important to note that the requirement for HDACs for transcriptional activation is not restricted to STAT5-activated genes. Expression of the genes encoding IL-8, IL-10 and CD154 can also be inhibited by TSA (Huang et al., 1997; Mishra et al., 2001). In lower eukaryotes, yeast and Drosophila, ablation of deacetylase function can cause suppression as well as induction of gene expression (Vidal and Gaber, 1991; Van Lint et al., 1996). Recently, the Hos2 family of HDACs has been shown to activate transcription by deacetylating the lysines in the H3 and H4 histone tails (Wang et al., 2002). Therefore, deacetylases, besides their well-documented role as transcription repressors, may also function as positive regulators of gene expression.
Moreover, it will be interesting to understand how different cytokines control activation and repression of the Id-1 gene during lymphocyte development since Id-1 is a potent inhibitor of bHLH proteins essential for lymphocyte development (Sun, 1994; Heemskerk et al., 1997; Kim et al., 1999). The cytokines may utilize STAT proteins to influence the acetylation status of C/EBPβ proteins if different STATs have different abilities in recruiting acetylases and deacetylases, and thus exert distinct biological effects on lymphocyte development.
Materials and methods
Plasmids and transfection
Constructions of the PBE-luc (pfLUC/B1) and ΔId-1 minigene have been described previously (Saisanit and Sun, 1995). Point mutations in the three C/EBPβ-binding sites in the above two constructs were generated using a two-step PCR protocol as described previously (Vitola et al., 1996). The ΔPBE-luc construct was created by PCR amplification of a 258 bp fragment with HindIII and SalI sites added at the ends for cloning into the pfluc vector (Saisanit and Sun, 1995). The PBE-SEAP constructs was generated by replacing the luciferase coding sequence with the SEAP sequence in the PBE-luc construct. CMV-SEAP was created by inserting the SEAP sequence into pcDNA3. The LGL construct was generated by fusing LAP with LIP through a linker encoding a stretch of glycines. Transient transfection of the reporter constructs along with the CMV-lacZ internal control was performed using the DEAE–dextran method. Reporter activities were measured using the luciferase assay (Promega, WI), Galaco-light Plus (Tropix, MA) and Great EscAPe SEAP (Clontech, CA) kits with a luminometer. Stable cell lines were established as described previously (Saisanit and Sun, 1995).
ChIP assays
ChIP assays were performed using ChIP assay kits for acetylhistones H3 and H4 (Upstate Biotechnology, NY) according to the manufacturer’s instructions for ChIP. Similar ChIP assays were also carried out using antibodies against H3 and H2B histone, as well as HDAC1 (Upstate Biotechnology, NY). Anti-STAT5a and anti-C/EBPβ were from Zymed and Santa Cruz Biotechnology, CA, respectively. DNA fragments brought down by IP were detected by PCR for ∼30 cycles. The primer pairs used for PCR detecting regions A, B, C and S23 in the Id-1 locus were as follows: Id1-A, CCCAAAGCTAGCCACTTCCCCGTTC and GTTCAAAAGCAACCAATAGGCTGC; Id1-B, GCTTCAAACTTCC AGAGTAC and AGCCAGGCAGAATCTGAGATC; Id1-C, ACAGG TGGGGGTTGGGGGGAGCAG and GCGTCGACGAGAGCCAGAC AGAAGC; and S23, CTGATCGGAAGTCTGCTGCTTCTGTC and GCTTAGGCTGAAGCTCAGAGCATATC. The primers used for S1 and C/EBP fragments were the Id1-A and B pairs, respectively.
Co-IP assays
Ba/F3 cells cultured with or without IL-3 were lysed on ice for 30 min in 150 mM NaCl, 1 mM EDTA, 20 mM Tris–HCl pH 8.0, 0.5% NP-40 and 10% glycerol. A 600 µg aliquot of total cell lysates was incubated with 3 µg of anti-STAT5a antibodies or control IgG for 2 h at 4°C. A 20 µl aliquot of 50% protein A–agarose slurry was added and incubated for another hour, followed by washes with 0.1% NP-40 in PBS. Bound proteins were western blotted with rabbit anti-HDAC1 and anti-Stat5a antibodies. A similar procedure was performed using nuclear extracts from transfected 293T cells.
Acetylation and pull-down assays
LAP was subcloned into T7 expression vector pRSET and expressed in Escherichia coli after IPTG induction. The K2 mutant was generated by PCR-assisted mutagenesis, and an HA tag was fused to its C-terminus. Recombinant LAP and K2 proteins were partially purified using a DEAE–Sepharose column. In vitro acetylation of LAP was carried out at 30°C for 1 h in a 30 µl reaction containing 50 mM Tris pH 8.0, 2 µg of LAP, 1 µl of [14C]acetyl-CoA (52 mCi/mmol, Amersham Pharmacia Biotech, NJ), 10% glycerol, 1 mM DTT, 1 mM PMSF and CBP immunoprecipitates from 293T cells transfected with a CMV–CBP construct or 0.8 µg of GST–PCAF (Upstate Biotechnology, NY). Pull-down assays were performed by incubating acetylated LAP and agarose beads conjugated with C/EBP-binding sites (Santa Cruz Biotechnology, CA) in a reaction containing 25 mM NaCl, 0.5 mM Tris pH 7.5, 0.25 mM EDTA, 1 mM DTT and 5% glycerol at room temperature for 30 min. The supernatant was collected and beads were then washed four times with cold PBS plus 0.1% NP-40. Proteins in the supernatant (unbound) and beads (bound) were analyzed by SDS–PAGE and quantified on a phosphoimager. The total amount of LAP was determined by western blotting with anti-C/EBPβ antibodies and quantified using a LumiImager.
To detect acetylation of C/EBPβ in vivo, Ba/F3 or PD31 cells were treated with 0.5 µM TSA and 5 mM nicotinamide for 6 h before preparation of nuclear extracts as described previously, except that all buffers contained 0.25 µM TSA and 2.5 mM nicotinamide (Luo et al., 2001; Kim et al., 2002). IP was carried out by incubating 400 µg of nuclear extracts with 6 µg of monoclonal antibodies (anti-acetyl-lysine from Upstate Biotechnology, NY or anti-HA from Roche Molecular Biochemicals, Indianapolis, IN) overnight in a buffer containing 40 mM Na2HPO4, 10 mM NaH2PO4, 250 mM NaCl, 0.5 mM PMSF, 1 mM DTT, 0.25 mM TSA, 2.5 mM nicotamide, 2 µg/ml leupeptin and 2 µg/ml pepstatin. The immunoprecipitates were western blotted with the desired monoclonal antibodies.
Retroviral infection and real-time PCR
Retroviral constructs were created by inserting cDNAs encoding LAP and LAP-K2 carrying an HA tag into the MIGR retrovector, which also expresses EGFP (Pui et al., 1999). Retroviruses were produced by transient transfection of Phoenix-E cells and collection 1 day later of the culture medium for 24 h. Ba/F3 cells were infected by centrifugation for 1.5 h at 1800 r.p.m. followed by 12 h incubation. Cells were then incubated in fresh medium containing IL-3 for 8 h. GFP-positive cells, sorted on a cell sorter, were cultured for 6 h before switching to an IL-3-free medium for various times.
Real-time PCR was performed using an ABI PRISM 770 Sequence Detector according to the manufacturer’s instruction (Applied Biosystems, CA). The 5′ and 3′ primers were CCCTGCCC CAGAACCG and ACTCCGAGTTCAGCTCCAGC. The fluorogenic probe used was FAM-AAAGTGAGCAAGGTGGAGATCCTGCA-TAMRA. For internal control of the amount of cDNA, expression of GAPDH was examined by using TaqMan Rodent GAPDH control reagents. One cycle of denaturation (95°C for 10 min) was performed, followed by 45 cycles of amplification (95°C for 15 s, 56°C for 1 min). Threshold cycle values (Ct) for Id-1 mRNA were normalized with those of GAPDH.
Acknowledgments
Acknowledgements
We thank Drs David Levy, Naoko Tanese, Hua Lu and Wei Gu for advice, Drs Linda Thompson, Carol Webb and Paul Kincade for critical reading of the manuscript, Dr Yang Xiao for technical assistance, and Kerry Humphrey for secretarial help. This work was supported by grants from the NIH (AI33597 and CA77553 to X.-H.S.).
References
- Boyes J., Byfield,P., Nakatani,Y. and Ogryzko,V. (1998) Regulation of activity of the transcription factor GATA-1 by acetylation. Nature, 396, 594–598. [DOI] [PubMed] [Google Scholar]
- Bromberg J. and Darnell,J.E.,Jr (2000) The role of STATs in transcriptional control and their impact on cellular function. Oncogene, 19, 2468–2473. [DOI] [PubMed] [Google Scholar]
- Chen H., Lin,R.J., Xie,W., Wilpitz,D. and Evans,R.M. (1999) Regulation of hormone-induced histone hyperacetylation and gene activation via acetylation of an acetylase. Cell, 98, 675–686. [DOI] [PubMed] [Google Scholar]
- Chen H., Tini,M. and Evans,R.M. (2001) HATs on and beyond chromatin. Curr. Opin. Cell Biol., 13, 218–224. [DOI] [PubMed] [Google Scholar]
- Chen L., Fischle,W., Verdin,E. and Greene,W.C. (2001) Duration of nuclear NF-κB action regulated by reversible acetylation. Science, 293, 1653–1657. [DOI] [PubMed] [Google Scholar]
- Descombes P. and Schibler,U. (1991) A liver-enriched transcription activator protein, LAP and a transcription inhibitory protein, LIP, are translated from the same mRNA. Cell, 67, 569–579. [DOI] [PubMed] [Google Scholar]
- Engel I. and Murre,C. (2001) The function of E- and Id proteins in lymphocyte development. Nat. Rev. Immunol., 1, 193–199. [DOI] [PubMed] [Google Scholar]
- Gu W. and Roeder,R.G. (1997) Activation of p53 sequence-specific DNA binding by acetylation of the p53 C-terminal domain. Cell, 90, 595–606. [DOI] [PubMed] [Google Scholar]
- Heemskerk M.H., Blom,B., Oda,K., Stegmann,A.P., Bakker,A.Q., Weijer,K., Res,P.C. and Spits,H. (1997) Inhibition of T cell and promotion of natural killer cell development by the dominant negative helix loop helix factor Id3. J. Exp. Med., 186, 1597–1602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang N., Katz,J.P., Martin,D.R. and Wu,G.D. (1997) Inhibition of IL-8 gene expression in Caco-2 cells by compounds which induce histone hyperacetylation. Cytokine, 9, 27–36. [DOI] [PubMed] [Google Scholar]
- Hung H.L., Lau,J., Kim,A.Y., Weiss,M.J. and Blobel,G.A. (1999) CREB-binding protein acetylates hematopoietic transcription factor GATA-1 at functionally important sites. Mol. Cell. Biol., 19, 3496–3505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Igarashi H., Gregory,S.C., Yokota,T., Sakaguchi,N. and Kincade,P.W. (2002) Transcription from the RAG1 locus marks the earliest lymphocyte progenitors in bone marrow. Immunity, 17, 117–130. [DOI] [PubMed] [Google Scholar]
- Imhof A., Yang,X.J., Ogryzko,V.V., Nakatani,Y., Wolffe,A.P. and Ge,H. (1997) Acetylation of general transcription factors by histone acetyltransferases. Curr. Biol., 7, 689–692. [DOI] [PubMed] [Google Scholar]
- Jaleco A.C., Stegmann,A.P., Heemskerk,M.H., Couwenberg,F., Bakker,A.Q., Weijer,K. and Spits,H. (1999) Genetic modification of human B-cell development: B-cell development is inhibited by the dominant negative helix loop helix factor Id3. Blood, 94, 2637–2646. [PubMed] [Google Scholar]
- Kim D., Peng,X.C. and Sun,X.H. (1999) Massive apoptosis of thymocytes in T-cell-deficient Id1 transgenic mice. Mol. Cell. Biol., 19, 8240–8253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim D., Xu,M., Nie,L., Peng,X.C., Jimi,E., Voll,R.E., Nguyen,T., Ghosh,S. and Sun,X.H. (2002) Helix–loop–helix proteins regulate pre-TCR and TCR signaling through modulation of Rel/NF-κB activities. Immunity, 16, 9–21. [DOI] [PubMed] [Google Scholar]
- Kinashi T., Inaba,K., Tsubata,T., Tashiro,K., Palacios,R. and Honjo,T. (1988) Differentiation of an interleukin 3-dependent precursor B-cell clone into immunoglobulin-producing cells in vitro. Proc. Natl Acad. Sci. USA, 85, 4473–4477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kouzarides T. (2000) Acetylation: a regulatory modification to rival phosphorylation? EMBO J., 19, 1176–1179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koyama Y., Adachi,M., Sekiya,M., Takekawa,M. and Imai,K. (2000) Histone deacetylase inhibitors suppress IL-2-mediated gene expression prior to induction of apoptosis. Blood, 96, 1490–1495. [PubMed] [Google Scholar]
- Lin J.X. and Leonard,W.J. (2000) The role of Stat5a and Stat5b in signaling by IL-2 family cytokines. Oncogene, 19, 2566–2576. [DOI] [PubMed] [Google Scholar]
- Luo J., Nikolaev,A.Y., Imai,S., Chen,D., Su,F., Shiloh,A., Guarente,L. and Gu,W. (2001) Negative control of p53 by Sir2α promotes cell survival under stress. Cell, 107, 137–148. [DOI] [PubMed] [Google Scholar]
- Marmorstein R. and Roth,S.Y. (2001) Histone acetyltransferases: function, structure and catalysis. Curr. Opin. Genet. Dev., 11, 155–161. [DOI] [PubMed] [Google Scholar]
- Massari M.E. and Murre,C. (2000) Helix–loop–helix proteins: regulators of transcription in eucaryotic organisms. Mol. Cell. Biol., 20, 429–440. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mishra N., Brown,D.R., Olorenshaw,I.M. and Kammer,G.M. (2001) Trichostatin A reverses skewed expression of CD154, interleukin-10 and interferon-γ gene and protein expression in lupus T cells. Proc. Natl Acad. Sci. USA, 98, 2628–2633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mui A.L., Wakao,H., Kinoshita,T., Kitamura,T. and Miyajima,A. (1996) Suppression of interleukin-3-induced gene expression by a C-terminal truncated Stat5: role of Stat5 in proliferation. EMBO J., 15, 2425–2433. [PMC free article] [PubMed] [Google Scholar]
- Norton J.D., Deed,R.W., Craggs,G. and Sablitzky,F. (1998) Id helix–loop–helix proteins in cell growth and differentiation. Trends Cell Biol., 8, 58–65. [PubMed] [Google Scholar]
- Pui J.C. et al. (1999) Notch1 expression in early lymphopoiesis influences B versus T lineage determination. Immunity, 11, 299–308. [DOI] [PubMed] [Google Scholar]
- Reddy E.P., Korapati,A., Chaturvedi,P. and Rane,S. (2000) IL-3 signaling and the role of Src kinases, JAKs and STATs: a covert liaison unveiled. Oncogene, 19, 2532–2547. [DOI] [PubMed] [Google Scholar]
- Ron D. and Habener,J.F. (1992) CHOP, a novel developmentally regulated nuclear protein that dimerizes with transcription factors C/EBP and LAP and functions as a dominant-negative inhibitor of gene transcription. Genes Dev., 6, 439–453. [DOI] [PubMed] [Google Scholar]
- Saisanit S. and Sun,X.H. (1995) A novel enhancer, the pro-B enhancer, regulates Id1 gene expression in progenitor B cells. Mol. Cell. Biol., 15, 1513–1521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saisanit S. and Sun,X.H. (1997) Regulation of the pro-B-cell-specific enhancer of the Id1 gene involves the C/EBP family of proteins. Mol. Cell. Biol., 17, 844–850. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sartorelli V., Puri,P.L., Hamamori,Y., Ogryzko,V., Chung,G., Nakatani,Y., Wang,J.Y. and Kedes,L. (1999) Acetylation of MyoD directed by PCAF is necessary for the execution of the muscle program. Mol. Cell, 4, 725–734. [DOI] [PubMed] [Google Scholar]
- Struhl K. (1998) Histone acetylation and transcriptional regulatory mechanisms. Genes Dev., 12, 599–606. [DOI] [PubMed] [Google Scholar]
- Sun X.-H. (1994) Constitutive expression of the Id1 gene impairs mouse B cell development. Cell, 79, 893–900. [DOI] [PubMed] [Google Scholar]
- Tyler J.K. and Kadonaga,J.T. (1999) The ‘dark side’ of chromatin remodeling: repressive effects on transcription. Cell, 99, 443–446. [DOI] [PubMed] [Google Scholar]
- Van Lint C., Emiliani,S. and Verdin,E. (1996) The expression of a small fraction of cellular genes is changed in response to histone hyperacetylation. Gene Expr., 5, 245–253. [PMC free article] [PubMed] [Google Scholar]
- Vidal M. and Gaber,R.F. (1991) RPD3 encodes a second factor required to achieve maximum positive and negative transcriptional states in Saccharomyces cerevisiae. Mol. Cell. Biol., 11, 6317–6327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vitola S.J., Wang,A. and Sun,X.H. (1996) Substitution of basic amino acids in the basic region stabilizes DNA binding by E12 homodimers. Nucleic Acids Res., 24, 1921–1927. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wakao H., Gouilleux,F. and Groner,B. (1994) Mammary gland factor (MGF) is a novel member of the cytokine regulated transcription factor gene family and confers the prolactin response. EMBO J., 13, 2182–2191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Waltzer L. and Bienz,M. (1998) Drosophila CBP represses the transcription factor TCF to antagonize Wingless signalling. Nature, 395, 521–525. [DOI] [PubMed] [Google Scholar]
- Wang A., Kurdistani,S.K. and Grunstein,M. (2002) Requirement of Hos2 histone deacetylase for gene activity in yeast. Science, 298, 1412–1414. [DOI] [PubMed] [Google Scholar]
- Williams S.C., Baer,M., Dillner,A.J. and Johnson,P.F. (1995) CRP2 (C/EBPβ) contains a bipartite regulatory domain that controls transcriptional activation, DNA binding and cell specificity. EMBO J., 14, 3170–3183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wolf D., Rodova,M., Miska,E.A., Calvet,J.P. and Kouzarides,T. (2002) Acetylation of β-catenin by CBP. J. Biol. Chem., 277, 25562–25567. [DOI] [PubMed] [Google Scholar]
- Yamagata T., Mitani,K., Oda,H., Suzuki,T., Honda,H., Asai,T., Maki,K., Nakamoto,T. and Hirai,H. (2000) Acetylation of GATA-3 affects T-cell survival and homing to secondary lymphoid organs. EMBO J., 19, 4676–4687. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang W. and Bieker,J.J. (1998) Acetylation and modulation of erythroid Kruppel-like factor (EKLF) activity by interaction with histone acetyltransferases. Proc. Natl Acad. Sci. USA, 95, 9855–9860. [DOI] [PMC free article] [PubMed] [Google Scholar]