

Abstract
Passage through the restriction point late in G1 normally commits cells to replicate their DNA. Here we show that the previously reported cell cycle block mediated by the human cytomegalovirus (HCMV) immediate early 2 (IE2) protein uncouples this association. First, IE2 expression leads to elevated levels of cyclin E-associated kinase activity via transcriptional activation of the cyclin E gene. This contributes to post-restriction point characteristics of IE2-expressing cells. Then these cells fail to undergo substantial DNA replication although they have entered S phase, and the induction of DNA replication observed after overexpression of cyclin E or D can be antagonized by IE2 without impinging on cyclin-associated kinase activities. These data suggest that IE2 secures restriction-point transition of cells before it stops them from replicating their genome. Our results fit well with HCMV physiology and support the view that IE2 is part of a viral activity which, on the one hand, promotes cell cycle-dependent expression of cellular replication factors but, on the other hand, disallows competitive cellular DNA synthesis.
Keywords: cell cycle/DNA replication/HCMV/IE2
Introduction
Cell cycle progression in human cells is driven by the periodic activity of cyclin-dependent kinases (cdk). Mitogen-dependent expression and activation of cyclin D– cdk4/6 complexes leads to progression through G1 phase, mainly by phosphorylation of the retinoblastoma protein (Rb), thereby alleviating its repressor function on E2F-controlled gene transcription (Sherr, 1995; Harbour and Dean, 2000). As a consequence, later in G1 cyclin E becomes expressed. A positive feedback loop via cyclin E–cdk2-mediated Rb hyperphosphorylation (Ohtani et al., 1995; Geng et al., 1996) has been suggested to produce the rapid rise of cyclin E–cdk2 activity, reaching its maximum at the G1–S boundary (Dulic et al., 1992; Koff et al., 1992). This is believed to constitute the molecular basis for the loss of mitogen dependency after passage through the restriction point (R point) (Pardee, 1974; Bartek et al., 1996; Planas-Silva and Weinberg, 1997). Once cells have passed the R point they are committed to enter S phase and to complete the cell division cycle governed by coordinated and sequential induction of cyclin A- and cyclin B-dependent kinase activities. However, if critical cell cycle events have not been completed properly, surveillance mechanisms called checkpoints are activated and prevent cells from entering the next phase of the cell cycle (Hartwell and Weinert, 1989).
Cell cycle arrest generally is achieved by negatively regulating cdk activity. A wide variety of external and internal antiproliferative signals during cell growth and differentiation results in downregulation of cyclin expression, in maintenance of inhibitory phosphorylations on cdk, or in upregulation of cdk inhibitory proteins (cki), which is the most widely used mechanism to block cell cycle progression before the R point in G1 (Sherr and Roberts, 1995). Whereas the CIP/KIP family of cki (Hengst and Reed, 1998) directly targets a broad spectrum of cyclin–cdk complexes, the INK4 family (Carnero and Hannon, 1998), including the bona fide tumour suppressor p16INK4a, inhibits only cdk4/6 activity directly but causes an additional indirect inhibition of cdk2 activity by redistribution of CIP/KIP proteins (Jiang et al., 1998; McConnell et al., 1999; Mitra et al., 1999). Whilst the establishment of long term arrest works by transcriptional regulation of cki, the rapid initiation of cell cycle arrest usually employs mechanisms acting on the post-translational level of cdk regulation (Agami and Bernards, 2000; Badie et al., 2000; Mailand et al., 2000).
Viruses modify the host cell cycle in many ways to optimize their own replication. Replication of small DNA viruses, for instance, depends on ongoing cellular DNA replication; therefore, infection with these tumorigenic viruses leads to S phase induction at least in part via inactivation of Rb (Vousden, 1995). In contrast, replication of large DNA viruses like herpesviruses which encode their own replication apparatus (Anders and McCue, 1996) appears to be favoured in the absence of host cell DNA synthesis. They are able to replicate in G0–G1-arrested cells (Shadan et al., 1994) and have been found to actively block cell cycle progression predominantly in G1 immediately after infection (Bresnahan et al., 1996; Dittmer and Mocarski, 1997; Ehmann et al., 2000; Song et al., 2000). With human cytomegalovirus (HCMV) infection, this demonstrates a rather unique cell cycle arrest: it is preceded by a virally mediated mitogenic response when quiescent cells are infected (Boldogh et al., 1990) and takes place in the presence of upregulated cyclin E–cdk2 activity (Jault et al., 1995; Bresnahan et al., 1996) and hyperphosphorylated Rb (Jault et al., 1995). In addition, HCMV induces expression of proliferation markers such as topoisomerase II and proliferating cell nuclear antigen (PCNA) (Benson and Huang, 1990; Dittmer and Mocarski, 1997), and of enzymes involved in nucleotide metabolism leading to a 20–30 fold increase of dNTP pools (Biron et al., 1986). Cyclin B1 is also expressed in HCMV-arrested cells (Jault et al., 1995; Dittmer and Mocarski, 1997) and is associated with high kinase activities (Jault et al., 1995). Remarkably, both HCMV-induced G1 arrest and cdk2 activity appear to be prerequisites for the viral replication cycle (Bresnahan et al., 1997; Salvant et al., 1998). Thus, HCMV seems to have evolved mechanisms that support viral but disallow cellular DNA replication. We have recently identified the immediate early 2 (IE2) protein of HCMV as a candidate regulatory factor to mediate the cell cycle arrest function of the virus (Wiebusch and Hagemeier, 1999). Transiently expressed IE2 protein can block cell cycle progression, leading to an accumulation of cells with a G1 DNA content when measured by propidium iodide (PI) staining (Wiebusch and Hagemeier, 1999). Although IE2 is known to be a strong transcriptional activator of numerous viral and host cell genes (Stenberg, 1996), the cell cycle arrest mechanism employed by IE2 appears to occur at a post-transcriptional level (Wiebusch and Hagemeier, 1999).
The work presented here was directed towards characterizing the IE2-mediated cell cycle arrest in order to delineate viral activities that contribute to the complex cell cycle phenotype of HCMV-infected cells. We show that IE2 is a rather unique protein in that it allows cells to pass the restriction point and to enter S phase, but then interferes with S phase progression, thereby stopping cells from replicating their genome. In contrast to cells arrested at the restriction point, these cells lack a downregulation of cdk activity, have hyperphosphorylated Rb and show E2F-dependent gene expression. In addition to these unusual cell cycle arrest properties per se, we demonstrate further that IE2 transcriptionally activates the cyclin E gene, leading to an additional increase of cyclin E-associated kinase activity in IE2-arrested cells. This defines a second and independent cell cycle activity of IE2 and both of these activities are also hallmarks of the HCMV-induced cell cycle arrest. Importantly, we show that both cell cycle activities of IE2 can function independently at the same time, since the induction of S phase after overexpression of cyclin E and D can be antagonized by IE2 without impairing their associated kinase activities.
Results
Experimental design
The functional characterization of a single viral protein outside the complex situation of viral infection necessitates the establishment of an appropriate expression system for this protein. We chose U373 cells since they can routinely be infected with HCMV to >90% and are fully permissive for viral infection (Jault et al., 1994). U373 cells can be growth factor-depleted and, after serum re-addition, a subpopulation of cells synchronously enters S phase and are fully in G2–M by 25 h (Figure 1A, top panel). Cyclin E-, A- and B-associated kinase activities in these cells could be detected with the expected kinetics (Figure 1B, lanes 1–8). When U373 cells are infected with HCMV, cell cycle parameters behave very similarly to those found in infected fibroblasts. First, infected serum-starved U373 cells do not progress through S phase and this occurs whether cells were supplemented at the time of infection with serum containing or serum missing media (Figure 1A, middle and bottom panel). Secondly, cdk activities were deregulated such that cyclin E- and B-associated kinase activities were upregulated, whereas cyclin A kinase activity was downregulated (Figure 1B, lanes 9–15). Since these findings together are perfectly consistent with results in fibroblasts (Jault et al., 1995; Bresnahan et al., 1996), U373 cells appear to qualify as a model cell system for the analysis of cell cycle functions of isolated viral factors.

Fig. 1. Cell cycle arrest and deregulation of cdk activities in HCMV-infected U373 cells. Cells were serum starved for 48 h. At the end of the starvation period (0 h) cells were serum restimulated (+ serum), HCMV infected (+ HCMV), or both (+serum, + HCMV). After restimulation/infection, cells were harvested at the indicated time points and aliquots were processed for flow cytometry and cyclin kinase assays. (A) DNA histograms in which the relative DNA content (measured by PI staining) was plotted against cell number. (B) The indicated cyclin complexes were immunoprecipitated (IP) from whole-cell extracts and the associated kinase activities measured with histone H1 (H1) as substrate.
In order to specifically characterize at a molecular level IE2-arrested cells we built upon the same experimental system already employed to demonstrate the cell cycle arrest function of IE2 (Wiebusch and Hagemeier, 1999). This system relies on the transient co-expression of IE2 and CD20. Importantly, IE2 expression levels were found to be comparable in infected and transfected U373 cells, suggesting that this system resembles near physiological conditions in this respect (Wiebusch and Hagemeier, 1999). Expression of CD20 enabled us not only to select cells for DNA content analysis by flow cytometry, as previously reported by several laboratories, but also to successfully use this system to physically separate cells by anti-CD20-directed magnetic cell sorting (MACS), thus enabling us to biochemically analyse IE2-arrested cells. We routinely analysed cells expressing either the full-length 86 kDa IE2 protein, which arrests cells with a G1 DNA content and functions as a powerful transcriptional activator, or an N-terminally truncated form of IE2 (IE2mut), which has lost the ability to activate transcription but retains the cell cycle arrest function (Figure 2A; Wiebusch and Hagemeier, 1999). In addition, control transfected cells and cells expressing p16INK4a were also included in the experiments. The quality of cell sorting was controlled for at three levels: (i) anti-CD20– fluorescein isothiocyanate (FITC) staining to show that the separated cell population corresponded to the gated CD20-positive population of non-sorted cells (Figure 2B); (ii) immunoblotting to demonstrate that IE2 becomes highly enriched via CD20-dependent MACS (Figure 2C); (iii) DNA staining to demonstrate that cell cycle distribution of CD20-positive cells is not being influenced by the sorting procedure per se (Figure 2D). Fluorescence-activated cell sorter (FACS) analysis of PI-stained IE2-expressing cells demonstrated that the DNA distribution pattern of non-sorted cells (Figure 2D, ‘before sorting’) and sorted cells (‘after sorting’) corresponded and was consistent with previously published results (Wiebusch and Hagemeier, 1999).

Fig. 2. Physical separation of IE2-expressing cells. U373 cells were transfected with the empty expression vector pSG5-3HA (control) or plasmids expressing p16INK4a (p16), HA-tagged full-length IE2 (IE2) or a HA-tagged IE2 deletion mutant (IE2mut). A CD20 expression vector was included in all transfections in a 1:4 molar ratio. Cells were harvested 48 h after transfection and stained with FITC-anti-CD20 antibody. Following this, cells were separated in fractions enriched for or depleted of CD20-positive cells by anti-CD20-directed MACS. (A) Schematic of IE2 and IE2mut. The amino acids (AA) present in each construct are indicated. AD, transcriptional activation domain; NLS, nuclear localization signal. (B) CD20 expression (measured as FITC fluorescence) of transfected cell populations before or after sorting was plotted against cell size (measured as forward scatter). (C) Equal amounts of extracts from sorted cells were separated by SDS–PAGE and analysed by immunoblotting with an anti-HA antibody. (D) DNA histograms show CD20-positive cell populations in which the relative DNA content (measured by PI staining) was plotted against cell number. The left panel is generated from gated [gate is shown in (B)] but unsorted cells, the right panel from sorted cells [the ‘enriched population’ in (B)]. Data shown in (B), (C) and (D) are from a single experiment representative of multiple cell separations with similar results.
Unlike p16INK4a-arrested cells, IE2-arrested cells have elevated levels of cyclin gene expression
We first set out to determine whether IE2 might alter cyclin gene expression in order to interfere with cell cycle progression. In this respect we were particularly interested in analysing expression from cyclin D, E, A and B genes and in directly comparing the expression levels with those from cells that were either cycling or arrested in G1 by p16INK4a. In comparison with cycling cells (Figure 3, lanes 2 and 11) p16INK4a-arrested cells contained low or undetectable levels of cyclin E (lane 3), A and B mRNAs (lane 12). In contrast expression of cyclin H (lane 3), which is not strictly cell cycle regulated, or expression of cyclin D1 and D2 (lane 12), which is upstream of p16INK4a function was not affected. Despite a similar FACS profile (Figure 2D; Wiebusch and Hagemeier, 1999), IE2-arrested cells were found to contain considerably higher levels of cyclin E, A and B mRNAs compared with p16INK4a-expressing cells (Figure 3, lanes 4 and 13). In this respect IE2-arrested cells more closely resembled cycling rather than G1 cells. Notably, cyclin E mRNA expression was even found to be higher in IE2-arrested versus cycling cells (compare lanes 2 and 4). The significance of this moderate increase is supported by the corresponding increases in cyclin E protein levels and the associated kinase activity (see below). This maximal expression of cyclin E mRNA (and to a lesser extent of cyclin A but not cyclin B mRNAs) appears to be due to IE2-mediated transcriptional activation since IE2mut-expressing cells contained lower levels of this mRNA (lane 5).

Fig. 3. Analysis of cyclin transcription in IE2-expressing cells. U373 cells were transfected and sorted as described in Figure 2. After isolation of RNAs from sorted cells mRNA expression levels of the indicated cyclins were determined by multiprobe ribonuclease protection assay. As loading controls, probes for L32 and GAPDH were included in each assay.
Cyclin expression levels amongst CD20-negative cells, i.e. the non-transfected populations that had been obtained from each flow-through fraction after MACS were found to be very similar (compare expressions of individual cyclin genes in Figure 3, lanes 6–9). This result serves as a further quality control for cell preparations and demonstrates that the differences observed in cyclin mRNA levels were indeed a specific consequence of IE2- or p16INK4a expression. Also, CD20-positive and CD20-negative control cells (lanes 2 and 6) had virtually identical cyclin mRNA expression levels, demonstrating that the transfection or purification procedures per se did not influence mRNA expression levels. Finally, L32 and GAPDH signals showed that comparable amounts of extracts were used in each lane.
Consistent with the analysis of cyclin mRNA expression, western blot analysis revealed comparatively high levels of cyclin E, A and B proteins in IE2-arrested cells (Figure 4A, lane 3). This was again in contrast to p16INK4a-arrested cells which contain nearly undetectable levels of cyclin A and B proteins (lane 2). Also, again in line with upregulated mRNA expression levels, cyclin E protein levels were markedly elevated in IE2-, but less in IE2mut-arrested cells (lanes 3 and 4). The relatively high amount of cyclin E protein in p16INK4a-arrested cells is well recognized and appears to be due to a lack of cdk2-dependent cyclin E degradation (see below; Won and Reed, 1996). Thus, the IE2-mediated cell cycle arrest is not due to a downregulation of cyclin gene expression. In contrast, mRNA and protein expression levels of cyclin genes, in particular cyclin E, were found to be unexpectedly high.
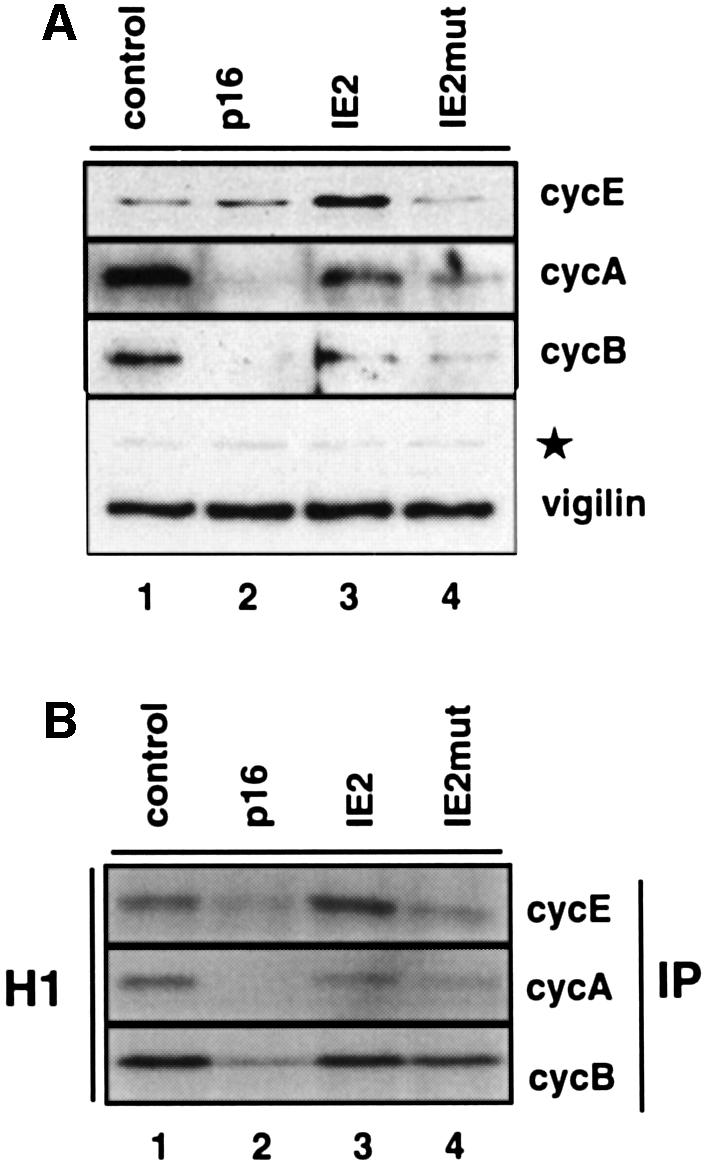
Fig. 4. Kinase activities of cyclin E, A and B complexes in IE2-expressing cells. U373 cells were transfected and sorted as described in Figure 2. (A) Equal amounts of whole-cell extracts from CD20-positive cells were separated by SDS–PAGE and analysed by immunoblotting with antibodies against cyclins E, A and B. A vigilin-specific antibody was used to control for equal protein loading. A non-specific background band is marked by a star. (B) The same extracts were used for immunoprecipitations (IP) with antibodies against the indicated cyclins followed by kinase assays using histone H1 (H1) as substrate.
The IE2-mediated cell cycle arrest occurs in the presence of high cdk activities
To address the question as to whether the expression of cyclins in IE2-arrested cells is paralleled by associated kinase activities, we immunoprecipitated cyclin-associated cdk from whole-cell extracts of CD20-positive cells and performed in vitro kinase assays with histone H1 as substrate. Surprisingly, during the IE2-induced cell cycle arrest we found that cyclin E-, A- and B-dependent kinase activities were not downregulated (Figure 4B, lane 3), which is in sharp contrast to the situation in p16INK4a-arrested cells (lane 2) where levels of kinase activities were found to be well below those of cycling cells (lane 1). In particular, cyclin E-associated kinase activity was found to be increased, which is consistent with the observed IE2-mediated transactivation of the cyclin E gene. In agreement with these results, immunoblot analysis of p21Cip and p27Kip showed no differences in the expression levels of these cdk inhibitors between IE2-arrested and control cells (data not shown). Also, consistent with mRNA and protein expression levels, cyclin E-, and to a lesser extent cyclin A-associated kinase activities were higher in IE2-arrested than IE2mut-arrested cells (Figure 4B, compare lanes 3 and 4). Since IE2mut blocks cell cycle progression as effectively as full-length IE2 (Figure 2; Wiebusch and Hagemeier, 1999) the data suggest that high cyclin E-associated kinase activity is not strictly associated with the ability of IE2 to arrest cell cycle progression, but rather is a consequence of the additional transcriptional capacity of IE2 that is lost in IE2mut. The data also indicate that the IE2-mediated cell cycle arrest functions despite high cdk activities, implying that IE2 does not need to downregulate cdk activities in order to stop cell cycle progression. In this respect it is also noteworthy that even in IE2mut-arrested cells cyclin-associated kinase activities were found to be elevated relative to p16INK4a-arrested cells (lane 4), suggesting fundamental differences in the strategies employed by IE2 and p16INK4a to stop cells from replicating their DNA.
IE2-arrested cells have post-restriction point characteristics
Cyclin-dependent kinase inhibitors are typically located at the end of signalling pathways whose goal it is to globally block cell cycle progression, including DNA replication and the cell cycle-dependent gene expression program directed towards preparing the later G1 cell for DNA synthesis. In contrast, cell cycle arrest after HCMV infection is believed to serve a different purpose. The virus apparently omits competitive cellular DNA synthesis, but at the same time needs to secure the expression of (at least some) cellular replication factors that, in addition to virally encoded proteins, are essential for viral replication. The unexpected observation that the G1 DNA content of IE2-arrested cells is associated with high cyclin-associated kinase activity in vitro fits in well with this consideration. However, if the in vitro measured increase in kinase activity should be meaningful in this respect then endogenous cdk targets should be phosphorylated in vivo and functional consequences of this phosphorylation should be detectable. In order to address this point we analysed the phosphorylation and functional state of the Rb protein in IE2-arrested cells. Rb is the best characterized bona fide target of G1–S cyclins, and the most important cell cycle function of Rb appears to be the negative regulation of a G1–S transcription program via repression of the E2F transcription factor family (Harbour and Dean, 2000).
In cycling cells several phosphorylated subforms of Rb could be observed (Figure 5A, lane 1) indicating cdk activity in vivo. As a consequence of downregulated cdk activity in p16INK4a-arrested cells only the un- or hypo-phosphorylated species could be detected (lane 2), which is consistent with published evidence (Alevizopoulos et al., 1997). In IE2-arrested cells, however, the entire spectrum of the Rb phosphorylation pattern was seen (lane 3), reflecting that of cycling (lane 1) but not p16INK4a-arrested cells. This result demonstrates that IE2 arrests cells in the presence of high cdk activities in vivo, and is consistent with in vitro measured cdk activities (Figure 4B). This result also shows that the cell cycle arrest function of IE2 is not due to a specific inhibition of Rb phosphorylation. This possibility had to be excluded since Rb and IE2 can directly interact (Hagemeier et al., 1994), which in theory could prevent Rb phosphorylation even in the presence of active cyclin–cdk complexes.
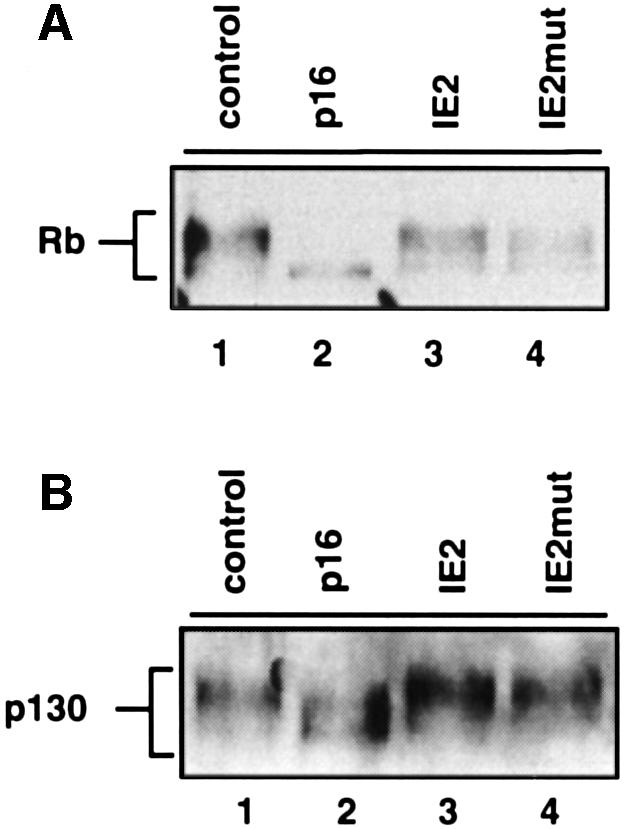
Fig. 5. Pocket proteins are hyperphosphorylated in IE2-expressing cells. U373 cells were transfected and sorted as described in Figure 2. Equal amounts of whole-cell extracts from CD20-positive cells were separated by SDS–PAGE and analysed by immunoblotting with antibodies against Rb (A) and p130 (B).
Also, in IE2mut-arrested cells all phosphorylated forms of Rb were present (Figure 5A, lane 4). This is consistent with the readily detectable cdk activity in IE2mut-arrested cells in vitro and demonstrates that, like IE2, IE2mut can arrest cell cycle progression in the presence of active cyclin–cdk complexes and hyperphosphorylated Rb in vivo. In agreement with the ability of IE2, but not IE2mut, to upregulate cyclin E mRNA and protein levels, compared with IE2-arrested cells the uppermost hyper-phosphorylated form of Rb in IE2mut-arrested cells is reduced with respect to the faster migrating, and hence less phosphorylated Rb species (Figure 5A, lane 4). Again, this is perfectly in line with the cyclin E-associated kinase activities of these cells measured in vitro (Figure 3). In addition, we have also looked at the phosphorylation state of a second independent cyclin–cdk target of the Rb family, p130, and in principle corresponding results were found (Figure 5B).
We next set out to analyse the functional consequences of hyper-phosphorylated Rb in IE2-arrested cells by examining expression levels of E2F target genes. First, we asked whether the hyper-phosphorylated forms of Rb detected in IE2-expressing cells had lost the ability to bind E2F-1 in a glutathione S-transferase (GST) pull-down assay in vitro. We found that, disregarding whether the extracts were prepared from cycling or IE2-arrested cells, GST–E2F-1 was only able to significantly interact with hypo-phosphorylated Rb (Figure 6A). This indicates that the phosphorylation of Rb in IE2-expressing cells is genuine in that it leads to a loss of E2F-1 binding. More specifically we could demonstrate that bona fide E2F target genes e2f-1 and p107 (Hurford et al., 1997) were de-repressed in IE2-arrested cells in vivo (Figure 6B, lane 3). This result suggests that Rb is indeed inactivated in IE2-arrested cells, with the functional consequence that (in principle) the E2F-dependent transcriptional program downstream of Rb is induced. Again, this is in contrast to p16INK4a-arrested cells which maintain Rb in its biologically active form repressing E2F-dependent gene transcription (see E2F-1 and p107 in lane 2). Genes whose mRNA expression is not considered to be cell cycle-regulated in an E2F-dependent manner, such as p130 and Rb, showed very similar mRNA expression levels in cycling as well as p16INK4a- and IE2-expressing cells, emphasizing that the observed differences in Rb-dependent gene expression were specific (lanes 1–3).
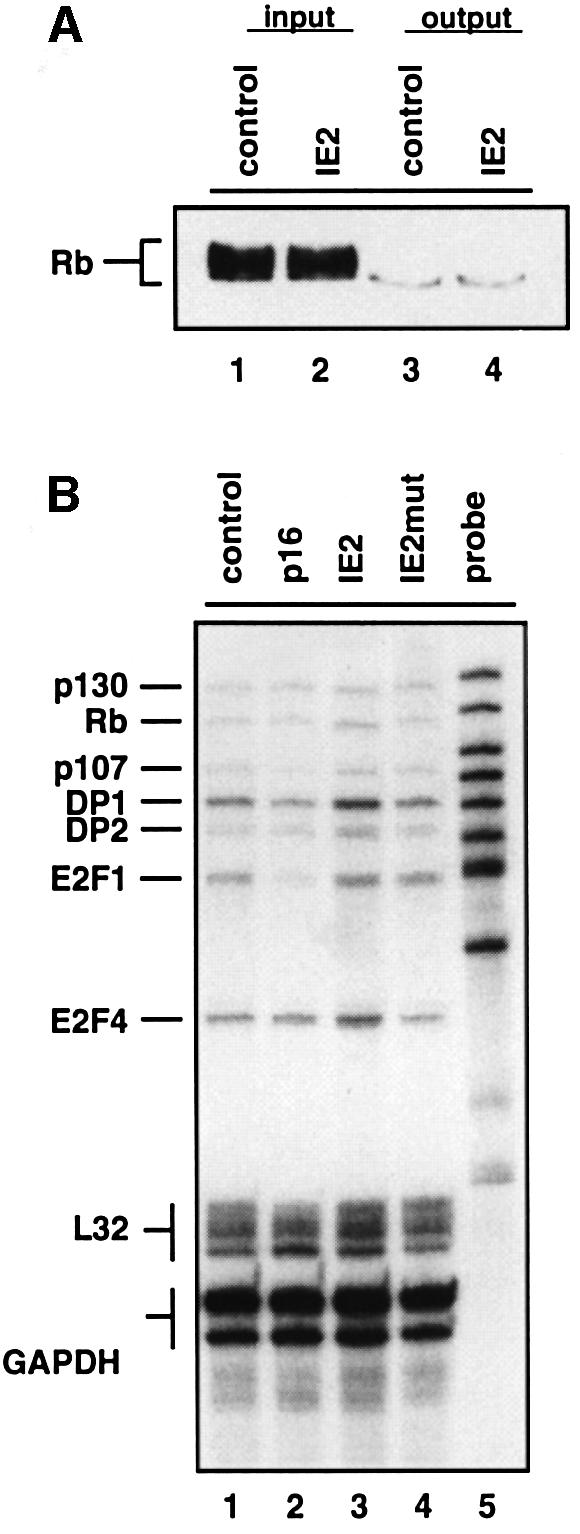
Fig. 6. IE2-mediated inactivation of transcriptional repression by E2F/Rb. U373 cells were transfected and sorted as described in Figure 2. (A) Whole-cell extracts from IE2-arrested (IE2) or cycling (control) cells were incubated with bacterially produced GST–E2F1 bound to glutathione–agarose beads. Input proteins and precipitates from the binding reaction were resolved by SDS–PAGE and analysed by Rb-specific immunoblotting. (B) RNAs of CD20-positive cells were analysed by multiprobe ribonuclease protection assay for mRNA expression levels of E2F and pocket protein family members as indicated. L32 and GAPDH mRNA expression was determined in the same assay for loading control.
The finding that cells expressing IE2mut showed the same level of E2F-dependent gene expression as cells expressing IE2 (see signals for e2f1 and p107 in lane 4) further suggests that elevated e2f-1 and p107 mRNA levels in these cells resulted from the observed increase in Rb phosphorylation rather than an IE2-mediated transactivation. In contrast, dp1 and e2f4 genes were slightly upregulated via IE2 transcriptional activity (lane 3). Since the gradual switch from the biologically active hypo-phosphorylated to the biologically inactive hyper-phosphorylated form of Rb is the most suitable molecular correlate available to mark the restriction point, our data suggest that IE2-arrested cells have passed through the restriction point.
IE2 blocks cell cycle progression at the beginning of S phase
A cell cycle block that does not allow DNA synthesis to precede despite restriction point transition and non-reduced levels of cdk activities is an unusual finding, and no proteins causing such a phenotype have been identified so far. Still, further support that this is indeed the case for IE2comes from analysing the pattern of cyclin-associated kinase activities in cells that have been arrested by the chemicals aphidicolin and hydroxyurea (HU), which interfere directly with DNA synthesis (Krakoff et al., 1968; Ikegami et al., 1978). We applied these drugs to CD20/control transfected cells in order to have the system most suitably arranged for correlating the results with IE2-transfected cells (Figure 4B). Also, using the drugs on transfected cells allowed us to benefit from the advantage that during transfection nuclear uptake of DNA is most effective in mitosis, when the nuclear membrane is disrupted. Consequently, the majority of transfected cells are entering the following G1 phase in a somewhat synchronous manner (Adams et al., 1997; Rodriguez and Flemington, 1999). Consistent with this notion, CD20-positive cells treated with aphidicolin or HU showed a cell cycle profile resembling a G1 arrest. (Figure 7A). However, compared with cycling cells, both aphidicolin- and HU-treated cells showed markedly increased levels of cyclin E, slightly reduced levels of cyclin A and only near-background levels of cyclin B-associated kinase activities (Figure 7B). This reflects what would be expected from cells that have been arrested at G1–S transition where cyclin E expression is known to peak (Dulic et al., 1992), cyclin A has only just begun to accumulate (Pines and Hunter, 1990) and cyclin B activity is still repressed by several levels of regulation. This pattern of cyclin-associated kinase activities resembles that of IE2-arrested cells but is in contrast to cells arrested before transition of the restriction point, e.g. by p16INK4a (Figure 4B). Therefore, this result supports the view that IE2 blocks cell cycle progression in a similar time window to DNA replication inhibitors. However, the high level of cyclin B-associated kinase activity in IE2-arrested cells (Figure 4B) contrasts the situation found after aphidicolin or HU treatment and suggests that this kinase activity may be specifically deregulated by IE2, an option that is consistent with elevated levels of cyclin B-associated kinase activity found in cells infected with HCMV (Figure 1A; Jault et al., 1995).

Fig. 7. IE2-arrested cells resemble cells at G1–S transition in their state of cyclin–cdk activity. U373 cells were transfected with the empty expression plasmid pSG5-3HA and pSG5CD20. After removal of the DNA precipitates, cells were treated for 32 h with aphidicolin (Aph) or hydroxyurea (HU) or left untreated (–). After harvesting, cells were sorted as detailed in Figure 2. (A) Cell cycle distributions of CD20-positive, PI-stained cells were analysed by flow cytometry. (B) Kinase activities associated with cyclins E, A and B in extracts of CD20-positive cells were assayed as detailed in the legend to Figure 4B.
A method based on DNA staining by PI is not suitable for distinguishing late G1 from early S phase cells since small amounts of newly replicated DNA cannot be detected. However, given the parallels in cdk activities between HU-, aphidicolin- and IE2-arrested cells it seemed important to answer exactly this question, i.e. whether IE2-expressing cells arrest in late G1 or early S phase after passing through the restriction point. In order to address this point we employed the experimental set up described above, but this time, in addition, scored for 5-bromo-2′-deoxyuridine (BrdU) incorporation of sorted cells, which allowed us to detect even small amounts of newly synthesized DNA. Indeed, the appearance of a distinct cell population with elevated levels of BrdU incorporation indicated that IE2-expressing cells had started DNA synthesis before being arrested (Figure 8). However, counterstaining with PI also demonstrated that these cells must be located very early in S phase since no significant additional PI staining could be detected in BrdU-positive cells. This picture differs from p16-arrested cells, which show no BrdU incorporation indicating a block in G1. It is, however, clearly reminiscent of aphidicolin- and particularly HU-arrested cells, which are known to be blocked at the very beginning of S phase (Figure 8).

Fig. 8. IE2-arrested cells locate at the beginning of S-phase rather than the end of G1. Cells were transfected with pSG5-3HA or the p16 and IE2 expression plasmids as described in Figure 2. The pSG5-3HA transfected cells were treated with aphidicolin (Aph) or hydroxyurea (HU) as detailed in Figure 7 or left untreated (control). In all transfections BrdU was included in the culture medium after removal of the DNA precipitates. Cells were harvested 32 h later, sorted for CD20 expression and analysed for BrdU incorporation and DNA content by staining with a FITC-labelled BrdU specific antibody and PI, followed by flow cytometry. Shown are density dot plots of CD20-positive cells.
IE2 uncouples DNA synthesis from cdk activation
The most direct interpretation of the above findings is that IE2 interferes with DNA replication thereby blocking cell cycle progression independently of downregulating cdk activity. One prediction from such a model is that IE2 should be able to sufficiently counteract DNA synthesis forced by overexpression of cyclin D or E without altering their associated kinase activities. To test directly this hypothesis we performed rescue experiments by transfecting U373 cells with cDNAs for cyclin E or D1 in the absence or presence of an IE2 expression vector. In the absence of IE2, cyclin E and to a lesser degree cyclin D1 overexpression lead to a significant increase of cells in S phase from 34% of control transfected cells to 47% of cyclin D1- and 63% of cyclin E-expressing cells (Figure 9A, top panels). This demonstrated that both cyclins can induce cells to enter S phase when overexpressed in our experimental system and is in line with previous findings (Resnitzky et al., 1994). Consistent with our prediction, IE2 co-expression was found to significantly antagonize cyclin-mediated DNA synthesis (Figure 9A, bottom panel). In the presence of IE2 the population of cyclin D1-expressing cells which had undergone clearly detectable DNA synthesis decreased from 47 to 25% and that of cyclin E-expressing cells from 63 to 30%. The fact that we did not observe a total rescue most likely reflects that subpopulations of cells would have been transfected with sufficient levels of cyclins but sub-functional levels of IE2 cDNAs. However, the data clearly show that IE2 can indeed antagonize cyclin-mediated cell cycle progression.

Fig. 9. Cdk-independent arrest of IE2-expressing cells. U373 cells were transiently transfected with pCMVneoBam (control) or vectors expressing cyclin D1 or cyclin E in the absence or presence of an IE2 expression vector. Forty-eight hours after transfection, cells were harvested and sorted for CD20 expression. (A) One aliquot of sorted cells was stained with PI and analysed by flow cytometry. Shown are DNA histograms of CD20-positive cells transfected as indicated. The percentages of cells in G1 are given. (B) A second aliquot of sorted cells was used for immunoblot analysis with an Rb-specific antibody.
Aliquots of cells taken for the above cell cycle analysis were then used to examine the state of Rb phosphorylation. As expected, cyclin D and E overexpression lead to a gradual shift towards higher phosphorylated forms of Rb as demonstrated by the loss of faster and the gain of slower migrating Rb species after cyclin D1 and cyclin E expression (Figure 9B, compare lanes 1, 3 and 5). Importantly, the dominant cell cycle arrest imposed by IE2 did not lead to a corresponding drop in cyclin-associated kinase activity as shown by the unchanged status of Rb phosphorylation in the presence or absence of IE2 (lanes 2, 4 and 6). Therefore, in IE2-expressing cells, cyclin-associated kinase complexes remained in a state of activity that was indistinguishable from cells not expressing IE2. Nevertheless, IE2-expressing cells were significantly impaired in replicating their DNA.
Together our results strongly suggest that IE2 interferes with S phase progression by uncoupling the process of DNA synthesis from the induction of cdk activities. For a cell cycle regulatory protein this is a novel and unusual mode of action but appears to be an advantageous strategy from a viral point of view.
Discussion
The central finding of this report is that the recently described cell cycle block induced by the HCMV IE2 protein (Wiebusch and Hagemeier, 1999) results in cells which have passed the restriction point and are arrested at the beginning of S phase in a cdk-independent manner. This suggests that IE2 can dissociate DNA synthesis from cdk activation.
Viral aspects of IE2 cell cycle functions
Importantly, the IE2-induced cell cycle arrest in many ways resembles characteristic features of the cell cycle block during HCMV infection. In Table I the status of cells arrested by IE2 and HCMV is compared. It is immediately apparent that the cell cycle state of HCMV-infected cells is paralleled by that of IE2-expressing cells. All the differences that can be observed concern only the strength of cyclin regulation. (i) The induction of cyclin E and B activities appears to be even stronger in infected (Figure 1) than in IE2-transfected cells (Figure 4). This is most likely due to additional viral factors which (as in the case of IE1) may even work synergistically with IE2. (ii) The downregulation of cyclin A activity during early viral infection (Figure 1) which occurs at the transcriptional level (Salvant et al., 1998) cannot be observed to the same extent in IE2-arrested cells. This activity is, therefore, most likely to be provided by other viral factors either synergistically with or independently from IE2. However, the question as to whether low cyclin A kinase levels contribute to the HCMV-induced cell cycle block or not has never been approached. It is, therefore, difficult to say how meaningful this difference is. In addition to the data shown here we have also observed that IE2-arrested cells have elevated levels of PCNA (data not shown), which is also a feature of HCMV-infected cells (Dittmer and Mocarski, 1997). In the absence of an IE2 mutant virus, our study, therefore, supports the view that the IE2 cell cycle arrest function is physiologically relevant and contributes to the seemingly contradictory cell cycle phenotype during HCMV infection.
Table I. Characteristics of HCMV- and IE2-induced cell cycle phenotypes.
| Cell cycle parameter | HCMV-arrested cells |
IE2-arrested cellsa |
||
|---|---|---|---|---|
| Compared with |
Compared with |
|||
| Quiescent | Cycling | p16-arrested | Cycling | |
| Rb phosphorylation | ++a,b | +a,b | ++ | 0 |
| cycE activity | ++a,b,c | ++a,b,c | ++ | + |
| cycA activity | 0a,b,c | –a,b,c | + | – |
| cycB activity | ++a,b | +a,b | ++ | 0 |
| E2F-induced transcription | ++d | +d | ++ | 0 |
| PCNA expression | ++e | ++ | ||
++, strongly increased; +, increased; 0, unchanged; -, reduced; –, strongly reduced.
The cell cycle state of cells arrested by HCMV infection is compared with proliferating cells (cycling) and with cells arrested by growth factor withdrawal or contact inhibition (quiescent). References b–e have used primary fibroblasts.
aThis study.
Our results show that, in principle, two separable cell cycle functions reside in IE2: the abilities to interfere with DNA replication and to activate cyclin E-associated kinase activity. In contrast to the cell cycle arrest function, the activation of cyclin E-associated kinase activity depends on the integrity of IE2 sequences required for transcriptional activation. Consistent with this we found that the increase in kinase activity was due to an increase in mRNA and protein expression from the cyclin E gene. These data suggest that cyclin E may be a target for transcriptional activation by IE2. This view is in agreement with an earlier report demonstrating that IE2 can directly transactivate the cyclin E promoter in a chloramphenicol acetyltransferase (CAT)-reporter assay (Bresnahan et al., 1998). In addition, IE2-independent mechanisms are likely to contribute to cyclin E activation during HCMV infection (McElroy et al., 2000), which would be consistent with the differences in cyclin E kinase activity observed in HCMV-infected versus IE2-transfected cells (see above). Our findings concerning the dual role of IE2 in cell cycle regulation are also reminiscent of studies with ts13 cells which show several cell cycle-specific defects in transcription and G1–S cell cycle progression due to a temperature-sensitive TAFII250 mutant (Wang and Tjian, 1994). Interestingly, IE2 expression in these cells can reverse the defect in the activation of the cyclin A promoter (Lukac et al., 1997), whereas it fails to rescue (and even enhances) the cell cycle block (Lukac and Alwine, 1999).
The identification of IE2 arresting cells in early S phase is based and was dependent on the BrdU incorporation assay (Figure 8). In contrast, a less sensitive staining method (PI staining) was not able to recognize these cells as S phase cells, but rather indicated these cells to be blocked with a G1-like DNA content (Figures 2D and 8; Wiebusch and Hagemeier, 1999). This explains why in our earlier report (when the BrdU incorporation assay was not yet available on sorted cells due to antibody limitations) the IE2-induced cell cycle arrest, monitored by PI staining only, was considered most likely to occur in G1 (Wiebusch and Hagemeier, 1999). Undoubtedly, in this report IE2 could be identified as arresting cells in early S phase. These findings are also consistent with an independent report on IE2-mediated cell cycle regulation which was published when this paper was under investigation (Murphy et al., 2000). Murphy et al. used an adenoviral expression system to also show that IE2-expressing cells arrest in S phase. However, since these authors did not address the biochemical state of IE2-arrested cells no further conclusions can be drawn at this stage.
A simple model of why the IE2-mediated cell cycle arrest would be advantageous in the presence of elevated cdk activities is to envisage that IE2 participates in two viral programs: one that blocks cellular DNA synthesis and one that secures the expression of cellular proteins essential for viral DNA replication. For instance, cytomegalovirus does not encode enzymes necessary for dNTP synthesis (Chee et al., 1990) but does depend on their function (Lembo et al., 1999; Gribaudo et al., 2000). Therefore, with the help of regulatory proteins like IE2, the virus may secure cell cycle-dependent expression of limiting replication factors by guiding cells into a post-restriction point state before competitive cellular DNA synthesis is inhibited. Naturally, such a model needs to include other viral regulators supporting the cell cycle arrest (Lu and Shenk, 1999; Hayashi et al., 2000) and the gene expression program of S phase (Margolis et al., 1995; Poma et al., 1996). The model is consistent with E2F-specific gene expression in IE2-arrested cells (Figures 4 and 5), the E2F-dependent activation of dihydrofolate reductase (Wade et al., 1992; Lembo et al., 1999) and thymidylate synthase (Gribaudo et al., 2000) in infected cells, and more generally with the dependency of HCMV replication on cdk activity (Bresnahan et al., 1997).
Cellular aspects of IE2 cell cycle functions
Apart from insights into viral strategies, an equally important aspect of this report deals with cellular regulatory mechanisms. The unusual combination of a DNA replication stop at the beginning of S phase and the presence of active cyclin-associated kinases is an unconventional finding and in clear contrast to cellular pathways which generally block cell cycle progression by downregulating cdk activity. This is not only true for cells arrested at the restriction point in G1 but also for cells that have already entered S phase and, for instance, as part of the DNA damage checkpoint, are stopped from continuing DNA replication. Even in these S phase cells cdk activity is targeted (e.g. resulting in hypophosphorylated Rb) rather than DNA replication directly in order to interfere with cell cycle progression (Knudsen et al., 2000). In contrast IE2 expression appears to selectively stop DNA synthesis without affecting cdk activity, i.e. IE2 in this point dissociates the regulation of DNA synthesis from the regulation of cdk activity.
The presence of elevated levels of cyclin E-associated kinase activity in IE2-arrested cells suggests that IE2 is most likely to interfere with a rate limiting (direct or indirect) downstream target of cyclin E or a process independent of cyclin E in order to block cell cycle progression. Given the central role of cyclin E–cdk2 in G1–S transition, surprisingly few bona fide substrates of this kinase are known. Rb, which has been shown to act both as an activator and substrate of cyclin E (Ohtani et al., 1995; Geng et al., 1996), is needed in its inactive, hyperphosphorylated form for progression through S phase (Chew et al., 1998; Knudsen et al., 1998). However, in IE2-expressing cells where Rb is clearly hyperphosphorylated and E2F-dependent gene expression is derepressed, cells are blocked at the beginning of S phase. This and recent reports suggesting that Rb may not be a rate-limiting target of cyclin E (Alevizopoulos et al., 1997; Lukas et al., 1997) make it unlikely that Rb serves as a target in the IE2-mediated cell cycle arrest. NPAT, a poorly characterized protein which when overexpressed accelerates G1–S progression (Zhao et al., 1998), has recently been found in a screen for cyclin E cdk targets. However, it is not known what role NPAT plays under physiological conditions in cell cycle regulation. Cyclin A, which has been suggested to serve as an indirect downstream target of cyclin E (Knudsen et al., 1999; Zhang et al., 2000), is not significantly downregulated in IE2-expressing cells. The fact that cyclin B protein can be detected in those cells provides further, indirect evidence for the presence of cyclin A-associated kinase activity, because it is the cyclin A–cdk2-mediated inactivation of the anaphase-promoting complex that leads to stabilization of cyclin B protein around G1–S transition (Lukas et al., 1999).
The finding that cyclin B in IE2-arrested cells was found to be associated with kinase activity needs further comment. As a consequence of cdk1 phosphorylation the cyclin B–cdk1 complex normally remains inactive until G2–M. This is reflected by low level cyclin B-associated cdk1 activity in aphidicolin- or HU-arrested cells (Lukas et al., 1999; Figure 7). This downregulation of cdk1 activity is part of the replication checkpoint which ensures that entry into mitosis does not occur before S phase is properly finished (Boddy and Russell, 1999). Surprisingly, and in contrast to aphidicolin- or HU-treated cells, IE2-arrested cells were found to contain elevated levels of cyclin B-associated kinase activity. Similarly, HCMV-arrested cells were found to upregulate cyclin B-associated kinase activity (Jault et al., 1995; Figure 1). This dissociation of cyclin B–cdk activation from cell cycle progression may indicate that the DNA replication checkpoint in infected cells is inactivated and that IE2 plays a role in mediating this effect.
Taken together, IE2 does not seem to globally interfere with cell cycle progression, suggesting that IE2 may have evolved mechanisms to more specifically stop cells from replicating their DNA, possibly by directly interfering with the process of cellular DNA synthesis. The finding that the state of cyclin E- and A-associated kinase activities in IE2-arrested cells resembles that of cells blocked by inhibitors of DNA replication (Figure 6) is consistent with this view. The possibility that IE2 might interfere with the assembly or activation of pre-replicative complexes (pre-RC), which occurs locally at origins of replication, seems unlikely since DNA replication appears to be interrupted by IE2 at an early stage rather than prohibited in the first instance. However, eukaryotic replication origins are activated in a characteristic temporal pattern during S phase (Fangman and Brewer, 1992; Ma et al., 1998). This appears, at least in part, to be accomplished by differential regulation of one or more pre-RC components as has been shown for the yeast Cdc45p (Aparicio et al., 1999; Zou and Stillman, 2000). Thus, it cannot be excluded that IE2 selectively allows the firing of early origins and inhibits the activation of later firing origins at the level of replication complex formation. Another possibility is that IE2 interferes with functions at replication forks. For instance, inhibition of the polymerase (pol) switch, i.e. the step from initiation of DNA synthesis by polα to the elongation of replication by polδ (Tsurimoto et al., 1990), could explain the limited BrdU incorporation that is observed as a consequence of the IE2-mediated arrest. Such an option would seem feasible since HCMV encodes its own DNA polymerase (Anders and McCue, 1996) and might, therefore, not depend on the cellular counterpart. In general, it seems an intriguing possibility for IE2 to exploit the cellular replication system locally leaving the more global aspects of G1–S transition untouched.
Finally, a set of 11 viral gene loci has been described to be necessary for efficient HCMV DNA replication in vivo (Pari and Anders, 1993; Anders and McCue, 1996). Interestingly, IE2 was found to be one of the encoded factors, pointing towards a more direct role of IE2 in HCMV DNA replication rather than only providing transactivating activity (Sarisky and Hayward, 1996). Given the (so far non-characterized) role of IE2 in viral DNA replication and the unusual cell cycle arrest function (Wiebusch and Hagemeier, 1999; this report) it seems possible that one aspect of IE2 function could be to diverge cellular replication factors to viral origins, thereby interfering with cellular DNA synthesis as a consequence of scavenging essential replication factors.
Disregarding the specific mechanism(s) employed by IE2 to deregulate the cell cycle for reasons discussed above, it seems advantageous for this HCMV protein to interfere with normal cell cycle progression independently of downregulating cdk activity. In doing so cdk-driven global aspects of the G1–S transition program could be maintained and at the same time uncoupled from cellular DNA replication. IE2 is the first protein for which such an activity has been described. Studying the unusual cell cycle arrest function of this viral protein should, therefore, not only be informative in understanding HCMV physiology, but also be helpful in analysing how the process of DNA replication is coupled to G1–S progression.
Materials and methods
Cell culture and virus infections
U373-MG cells were maintained in Dulbecco’s modified Eagle’s medium supplemented with 5% newborn and 5% fetal calf serum. For synchronizing in G0, cells were kept in serum-free medium for 48 h prior to infection. HCMV AD169 were used to infect these cells at a multiplicity of infection (MOI) of 10 PFU per cell, in the presence or absence of serum. In parallel, serum-starved cells were restimulated with normal, serum-containing culture medium.
Plasmids and transfections
The empty expression vector pSG5-3-hemagglutinin (HA) and the expression vectors for IE2, IE2mut, p16INK4a and CD20 [pSG5-3HA-IE2, pSG5-3HA-IE2(195–579), pXmycp16 and pSG5CD20] have been described previously (Wiebusch and Hagemeier, 1999). The cyclin D1 expression vector pRcCMV-CycD1 was kindly provided by Jiri Lukas (Danish Cancer Society, Copenhagen) and the cyclin E expression vector pCMX-CycE by Jon Pines (Wellcome/CRC Institute, Cambridge). U373 cells were transfected using the calcium phosphate precipitation method as described previously (Wiebusch and Hagemeier, 1999). Where indicated, 5 µg/ml aphidicolin or hydroxyurea at a final concentration of 1 mM were added to the pSG5-3HA/pSG5-CD20 transfected cells immediately after removal of the DNA precipitates.
Cell sorting and flow cytometry
Forty-eight hours post transfection, cells were harvested by trypsinization. Transfected cells were separated from untransfected cells by means of MACS technology (Miltenyi Biotec, Heidelberg) according to the manufacturer’s protocols. After labelling with a FITC-conjugated anti-CD20 antibody (2H7, Pharmingen), cells were incubated with MACS goat anti-mouse IgG microbeads and separated on a MACS large cell separation column. To increase purity, the eluate containing the CD20-positive cells was passed over a second large cell separation column with a flow resistor attached. The effluent was further depleted from CD20-positive cells on a LS+ separation column. To control for sorting efficiency and cell cycle distribution, aliquots of the sorted cells were routinely taken and analysed for CD20 expression and DNA content by flow cytometry as described previously (Wiebusch and Hagemeier, 1999).
Ribonuclease protection assays
Multi-probe ribonuclease protection assays were performed by using the Riboquant system (Pharmingen). Defined pools of [α-32P]UTP-labelled probes were synthesized by in vitro transcription from the template sets hCyc-2, hCyc-1 and hCC-2 (Pharmingen). Total RNAs from sorted cells were isolated using Tri-Reagent (Molecular Research Center, Inc.) following the manufacturer’s instructions. One microgram of each preparation was used as target RNA in the hybridization reaction. Afterwards the RNA samples were RNase treated, purified and subjected to denaturing polyacrylamide gel electrophoresis followed by autoradiography.
Immunoblotting and kinase assays
After sorting, cells were suspended in IPB [50 mM Tris–Cl pH 7.4, 150 mM NaCl, 10 mM MgCl2, 10 mM NaF, 0.5 mM Na3VO4, 0.5% Nonidet P-40, 10% glycerol, 1 mM dithiothreitol (DTT), 2 µg/ml aprotinin, 1 mM leupeptin, 1 mM phenylmethylsulfonylfluoride] and lysed by freeze–thaw. Extracts were clarified by centrifugation and then depleted from anti-CD20 antibodies by incubation with protein A–agarose beads. Protein concentrations were determined using the Bio-Rad DC protein assay. Immunoblot analyses were performed as described previously (Wiebusch and Hagemeier, 1999), using the following antibodies: rabbit polyclonal antibodies against Rb (C-15, Santa Cruz Biotechnology), p130 (C-20, Santa Cruz), cyclin A and cyclin B1 (a kind gift of Jon Pines); mouse monoclonal antibodies against the HA-tag (16B12, BAbCO) and cyclin E (HE12, kindly provided by Steve Reed, Scripps Research Institute, La Jolla). To control for equal loading, the upper parts of the blots were probed separately with an antiserum against the 155 kDa vigilin protein (Neu-Yilik et al., 1993), which is not cell cycle regulated.
For kinase assays 10–20 µg of protein extracts were incubated with 0.1 µg of antibody in a final volume of 50 µl for 1 h at 0°C. The following antibodies were used for precipitating cyclin–cdk complexes: H-432 against cyclin A (Santa Cruz), GNS1 against cyclin B (Santa Cruz) and HE172 against cyclin E (kindly provided by Steve Reed). After dilution to 1 ml with IPB, immunocomplexes were precipitated with 5 µl protein A– or protein G–agarose for 3–12 h at 4°C, washed four times with IPB and twice with 50 mM Tris–Cl pH 7.4, 10 mM MgCl2, 1 mM DTT. The immunoprecipitates were resuspended in 25 µl 50 mM Tris–Cl pH 7.4, 10 mM MgCl2, 1 mM DTT, 10 mM β-glycerophosphate, 50 µM ATP containing 5 µCi of [γ-32P]ATP and 200 ng histone H1 (kind gift of Sybille Mittnacht, ICRF, London), and incubated for 30 min at 30°C. Kinase reactions were analysed by 12% SDS–PAGE followed by autoradiography.
GST–E2F1 binding assay
GST–E2F1 was expressed in and purified from Escherichia coli as previously described (Hagemeier et al., 1993). GST–E2F1 (0.5 µg) bound to glutathione–agarose beads were incubated with 50 µg of protein extracts from sorted cells for 2 h at 4°C. After the binding reaction, agarose beads were washed four times with IPB and bound proteins were resolved by 7.5% SDS–PAGE. The Rb protein was then detected by immunoblotting as described above.
BrdU incorporation assay
Cells were transfected and sorted as described above, except that after removal of the DNA precipitates the culture medium was supplemented with 10 µM BrdU, and the anti-CD20 antibody used for sorting was not FITC-conjugated. After fixation in 70% ethanol, the sorted cells were washed in phosphate-buffered saline (PBS) and treated with 2 M HCl for 20 min at room temperature. Afterwards the cells were washed in PBS and PBS/0.5% bovine serum albumin (BSA), and incubated with an anti- BrdU antibody (clone 3D4, Pharmingen) followed by a FITC-conjugated anti-mouse IgG1 antibody (clone A85–1, Pharmingen). Both antibody binding reactions were carried out for 15 min at room temperature using 5 µg/ml of antibodies in PBS/0.5% BSA, and were followed by a washing step with PBS/0.5% BSA. Finally, cells were treated with 50 µg/ml PI/100 U/ml RNase A/PBS for 30 min at room temperature and analysed by flow cytometry.
Acknowledgments
Acknowledgements
We thank J.Lukas, J.Pines and S.Reed for antibodies and plasmids, S.Mittnacht for histone H1 protein and M.Truss for comments on the manuscript. C.H. would like to thank Gerhard Gaedicke for continued support. The work was supported by grants from the Deutsche Forschungsgemeinschaft to Gerhard Gaedicke and C.H. (GA167/6-2) and to C.H. (SFB421).
References
- Adams P.D., Lopez,P., Sellers,W.R. and Kaelin,W.G.,Jr (1997) Fluorescence-activated cell sorting of transfected cells. Methods Enzymol., 283, 59–72. [DOI] [PubMed] [Google Scholar]
- Agami R. and Bernards,R. (2000) Distinct initiation and maintenance mechanisms cooperate to induce G1 cell cycle arrest in response to DNA damage. Cell, 102, 55–66. [DOI] [PubMed] [Google Scholar]
- Alevizopoulos K., Vlach,J., Hennecke,S. and Amati,B. (1997) Cyclin E and c-Myc promote cell proliferation in the presence of p16INK4a and of hypophosphorylated retinoblastoma family proteins. EMBO J., 16, 5322–5333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anders D.G. and McCue,L.A. (1996) The human cytomegalovirus genes and proteins required for DNA synthesis. Intervirology, 39, 378–388. [DOI] [PubMed] [Google Scholar]
- Aparicio O.M., Stout,A.M. and Bell,S.P. (1999) Differential assembly of Cdc45p and DNA polymerases at early and late origins of DNA replication. Proc. Natl Acad. Sci. USA, 96, 9130–9135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Badie C., Itzhaki,J.E., Sullivan,M.J., Carpenter,A.J. and Porter,A.C. (2000) Repression of CDK1 and other genes with CDE and CHR promoter elements during DNA damage-induced G(2)/M arrest in human cells. Mol. Cell. Biol., 20, 2358–2366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bartek J., Bartkova,J. and Lukas,J. (1996) The retinoblastoma protein pathway and the restriction point. Curr. Opin. Cell Biol., 8, 805–814. [DOI] [PubMed] [Google Scholar]
- Benson J.D. and Huang,E.S. (1990) Human cytomegalovirus induces expression of cellular topoisomerase II. J. Virol., 64, 9–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Biron K.K., Fyfe,J.A., Stanat,S.C., Leslie,L.K., Sorrell,J.B., Lambe,C.U. and Coen,D.M. (1986) A human cytomegalovirus mutant resistant to the nucleoside analog 9-([2-hydroxy-1-(hydroxymethyl)ethoxy] methyl)guanine (BW B759U) induces reduced levels of BW B759U triphosphate. Proc. Natl Acad. Sci. USA, 83, 8769–8773. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boddy M.N. and Russell,P. (1999) DNA replication checkpoint control. Front. Biosci., 4, D841–D848. [DOI] [PubMed] [Google Scholar]
- Boldogh I., AbuBakar,S. and Albrecht,T. (1990) Activation of proto-oncogenes: an immediate early event in human cytomegalovirus infection. Science, 247, 561–564. [DOI] [PubMed] [Google Scholar]
- Bresnahan W.A., Boldogh,I., Thompson,E.A. and Albrecht,T. (1996) Human cytomegalovirus inhibits cellular DNA synthesis and arrests productively infected cells in late G1. Virology, 224, 150–160. [DOI] [PubMed] [Google Scholar]
- Bresnahan W.A., Boldogh,I., Chi,P., Thompson,E.A. and Albrecht,T. (1997) Inhibition of cellular Cdk2 activity blocks human cytomegalovirus replication. Virology, 231, 239–247. [DOI] [PubMed] [Google Scholar]
- Bresnahan W.A., Albrecht,T. and Thompson,E.A. (1998) The cyclin E promoter is activated by human cytomegalovirus 86-kDa immediate early protein. J. Biol. Chem., 273, 22075–22082. [DOI] [PubMed] [Google Scholar]
- Carnero A. and Hannon,G.J. (1998) The INK4 family of CDK inhibitors. Curr. Top. Microbiol. Immunol., 227, 43–55. [DOI] [PubMed] [Google Scholar]
- Chee M.S. et al. (1990) Analysis of the protein-coding content of the sequence of human cytomegalovirus strain AD169. Curr. Top. Microbiol. Immunol., 154, 125–169. [DOI] [PubMed] [Google Scholar]
- Chew Y.P., Ellis,M., Wilkie,S. and Mittnacht,S. (1998) pRB phosphorylation mutants reveal role of pRB in regulating S phase completion by a mechanism independent of E2F. Oncogene, 17, 2177–2186. [DOI] [PubMed] [Google Scholar]
- Dittmer D. and Mocarski,E.S. (1997) Human cytomegalovirus infection inhibits G1/S transition. J. Virol., 71, 1629–1634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dulic V., Lees,E. and Reed,S.I. (1992) Association of human cyclin E with a periodic G1–S phase protein kinase. Science, 257, 1958–1961. [DOI] [PubMed] [Google Scholar]
- Ehmann G.L., McLean,T.I. and Bachenheimer,S.L. (2000) Herpes simplex virus type 1 infection imposes a G(1)/S block in asynchronously growing cells and prevents G(1) entry in quiescent cells. Virology, 267, 335–349. [DOI] [PubMed] [Google Scholar]
- Fangman W.L. and Brewer,B.J. (1992) A question of time: replication origins of eukaryotic chromosomes. Cell, 71, 363–366. [DOI] [PubMed] [Google Scholar]
- Geng Y., Eaton,E.N., Picon,M., Roberts,J.M., Lundberg,A.S., Gifford,A., Sardet,C. and Weinberg,R.A. (1996) Regulation of cyclin E transcription by E2Fs and retinoblastoma protein. Oncogene, 12, 1173–1180. [PubMed] [Google Scholar]
- Gribaudo G. et al. (2000) Murine cytomegalovirus stimulates cellular thymidylate synthase gene expression in quiescent cells and requires the enzyme for replication. J. Virol., 74, 4979–4984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hagemeier C., Cook,A. and Kouzarides,T. (1993) The retinoblastoma protein binds E2F residues required for activation in vivo and TBP binding in vitro. Nucleic Acids Res., 21, 4998–5004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hagemeier C., Caswell,R., Hayhurst,G., Sinclair,J. and Kouzarides,T. (1994) Functional interaction between the HCMV IE2 transactivator and the retinoblastoma protein. EMBO J., 13, 2897–2903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harbour J.W. and Dean,D.C. (2000) Rb function in cell cycle regulation and apoptosis. Nature Cell Biol., 2, E65–E67. [DOI] [PubMed] [Google Scholar]
- Hartwell L.H. and Weinert,T.A. (1989) Checkpoints: controls that ensure the order of cell cycle events. Science, 246, 629–634. [DOI] [PubMed] [Google Scholar]
- Hayashi M.L., Blankenship,C. and Shenk,T. (2000) Human cytomegalovirus UL69 protein is required for efficient accumulation of infected cells in the G1 phase of the cell cycle. Proc. Natl Acad. Sci. USA, 97, 2692–2696. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hengst L. and Reed,S.I. (1998) Inhibitors of the Cip/Kip family. Curr. Top. Microbiol. Immunol., 227, 25–41. [DOI] [PubMed] [Google Scholar]
- Hurford R.K., Cobrinik,D., Lee,M.H. and Dyson,N. (1997) pRB and p107/p130 are required for the regulated expression of different sets of E2F responsive genes. Genes Dev., 11, 1447–1463. [DOI] [PubMed] [Google Scholar]
- Ikegami S., Taguchi,T., Ohashi,M., Oguro,M., Nagano,H. and Mano,Y. (1978) Aphidicolin prevents mitotic cell division by interfering with the activity of DNA polymerase-α. Nature, 275, 458–460. [DOI] [PubMed] [Google Scholar]
- Jault F.M., Spector,S.A. and Spector,D.H. (1994) The effects of cytomegalovirus on human immunodefiency virus replication in brain-derived cells correlate with permissiveness of the cells for each virus. J. Virol., 68, 959–973. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jault F.M., Jault,J.-M., Ruchti,F., Fortunato,E.A., Clark,C., Corbeil,J., Richman,D.D. and Spector,D.H. (1995) Cytomegalovirus infection induces high levels of cyclins, phosphorylated Rb and p53, leading to cell cycle arrest. J. Virol., 69, 6697–6704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiang H., Chou,H.S. and Zhu,L. (1998) Requirement of cyclin E–Cdk2 inhibition in p16(INK4a)-mediated growth suppression. Mol. Cell. Biol., 18, 5284–5290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Knudsen E.S., Buckmaster,C., Chen,T.T., Feramisco,J.R. and Wang,J.Y. (1998) Inhibition of DNA synthesis by RB: effects on G1/S transition and S-phase progression. Genes Dev., 12, 2278–2292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Knudsen K.E., Fribourg,A.F., Strobeck,M.W., Blanchard,J.M. and Knudsen,E.S. (1999) Cyclin A is a functional target of retinoblastoma tumor suppressor protein-mediated cell cycle arrest. J. Biol. Chem., 274, 27632–27641. [DOI] [PubMed] [Google Scholar]
- Knudsen K.E., Booth,D., Naderi,S., Sever-Chroneos,Z., Fribourg,A.F., Hunton,I.C., Feramisco,J.R., Wang,J.Y. and Knudsen,E.S. (2000) RB-dependent S-phase response to DNA damage. Mol. Cell. Biol., 20, 7751–7763. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koff A. et al. (1992) Formation and activation of a cyclin E–cdk2 complex during the G1 phase of the human cell cycle. Science, 257, 1689–1694. [DOI] [PubMed] [Google Scholar]
- Krakoff I.H., Brown,N.C. and Reichard,P. (1968) Inhibition of ribonucleoside diphosphate reductase by hydroxyurea. Cancer Res., 28, 1559–1565. [PubMed] [Google Scholar]
- Lembo D., Gribaudo,G., Cavallo,R., Riera,L., Angeretti,A., Hertel,L. and Landolfo,S. (1999) Human cytomegalovirus stimulates cellular dihydrofolate reductase activity in quiescent cells. Intervirology, 42, 30–36. [DOI] [PubMed] [Google Scholar]
- Lu M. and Shenk,T. (1999) Human cytomegalovirus UL69 protein induces cells to accumulate in G1 phase of the cell cycle. J. Virol., 73, 676–683. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lukac D.M. and Alwine,J.C. (1999) Effects of human cytomegalovirus major immediate-early proteins in controlling the cell cycle and inhibiting apoptosis: studies with ts13 cells. J. Virol., 73, 2825–2831. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lukac D.M., Harel,N.Y., Tanese,N. and Alwine,J.C. (1997) TAF-like functions of human cytomegalovirus immediate-early proteins. J. Virol., 71, 7227–7239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lukas C., Sorensen,C.S., Kramer,E., Santoni-Rugiu,E., Lindeneg,C., Peters,J.M., Bartek,J. and Lukas,J. (1999) Accumulation of cyclin B1 requires E2F and cyclin-A-dependent rearrangement of the anaphase-promoting complex. Nature, 401, 815–818. [DOI] [PubMed] [Google Scholar]
- Lukas J., Herzinger,T., Hansen,K., Moroni,M.C., Resnitzky,D., Helin,K., Reed,S.I. and Bartek,J. (1997) Cyclin E-induced S phase without activation of the pRb/E2F pathway. Genes Dev., 11, 1479–1492. [DOI] [PubMed] [Google Scholar]
- Ma H., Samarabandu,J., Devdhar,R.S., Acharya,R., Cheng,P.C., Meng,C. and Berezney,R. (1998) Spatial and temporal dynamics of DNA replication sites in mammalian cells. J. Cell Biol., 143, 1415–1425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mailand N., Falck,J., Lukas,C., Syljuasen,R.G., Welcker,M., Bartek,J. and Lukas,J. (2000) Rapid destruction of human Cdc25A in response to DNA damage. Science, 288, 1425–1429. [DOI] [PubMed] [Google Scholar]
- Margolis M.J., Pajovic,S., Wong,E.L., Wade,M., Jupp,R., Nelson,J.A. and Azizkhan,J.C. (1995) Interaction of the 72-kilodalton human cytomegalovirus IE1 gene product with E2F1 coincides with E2F-dependent activation of dihydrofolate reductase transcription. J. Virol., 69, 7759–7767. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McConnell B.B., Gregory,F.J., Stott,F.J., Hara,E. and Peters,G. (1999) Induced expression of p16(INK4a) inhibits both CDK4- and CDK2-associated kinase activity by reassortment of cyclin–CDK-inhibitor complexes. Mol. Cell. Biol., 19, 1981–1989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McElroy A.K., Dwarakanath,R.S. and Spector,D.H. (2000) Dysregul ation of cyclin E gene expression in human cytomegalovirus-infected cells requires viral early gene expression and is associated with changes in the Rb-related protein p130. J. Virol., 74, 4192–4206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mitra J., Dai,C.Y., Somasundaram,K., El-Deiry,W.S., Satyamoorthy,K., Herlyn,M. and Enders,G.H. (1999) Induction of p21(WAF1/CIP1) and inhibition of Cdk2 mediated by the tumor suppressor p16(INK4a). Mol. Cell. Biol., 19, 3916–3928. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murphy E.A., Streblow,D.N., Nelson,J.A. and Stinski,M.F. (2000) The human cytomegalovirus IE86 protein can block cell cycle progression after inducing transition into the S phase of permissive cells. J. Virol., 74, 7108–7118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Neu-Yilik G., Zorbas,H., Gloe,T.R., Raabe,H.M., Hopp-Christensen,T.A. and Muller,P.K. (1993) Vigilin is a cytoplasmic protein. A study on its expression in primary cells and in established cell lines of different species. Eur. J. Biochem., 213, 727–736. [DOI] [PubMed] [Google Scholar]
- Ohtani K., DeGregori,J. and Nevins,J.R. (1995) Regulation of the cyclin E gene by transcription factor E2F1. Proc. Natl Acad. Sci. USA, 92, 12146–12150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pardee A.B. (1974) A restriction point for control of normal animal cell proliferation. Proc. Natl Acad. Sci. USA, 71, 1286–1290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pari G.S. and Anders,D.G. (1993) Eleven loci encoding trans-acting factors are required for transient complementation of human cytomegalovirus oriLyt-dependent DNA replication. J. Virol., 67, 6979–6988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pines J. and Hunter,T. (1990) Human cyclin A is adenovirus E1A-associated protein p60 and behaves differently from cyclin B. Nature, 346, 760–763. [DOI] [PubMed] [Google Scholar]
- Planas-Silva M.D. and Weinberg,R.A. (1997) The restriction point and control of cell proliferation. Curr. Opin. Cell Biol., 9, 768–772. [DOI] [PubMed] [Google Scholar]
- Poma E.E., Kowalik,T.F., Zhu,L., Sinclair,J.H. and Huang,E.-S. (1996) The human cytomegalovirus IE1-72 protein interacts with the cellular p107 protein and relieves p107-mediated transcriptional repression of an E2F-responsive promoter. J. Virol., 70, 7867–7877. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Resnitzky D., Gossen,M., Bujard,H. and Reed,S.I. (1994) Acceleration of the G1/S phase transition by expression of cyclins D1 and E with an inducible system. Mol. Cell. Biol., 14, 1669–1679. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rodriguez A. and Flemington,E.K. (1999) Transfection-mediated cell cycle signaling: considerations for transient transfection-based cell cycle studies. Anal. Biochem., 272, 171–181. [DOI] [PubMed] [Google Scholar]
- Salvant B.S., Fortunato,E.A. and Spector,D.H. (1998) Cell cycle dysregulation by human cytomegalovirus: influence of the cell cycle phase at the time of infection and effects on cyclin transcription. J. Virol., 72, 3729–3741. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sarisky R.T. and Hayward,G.S. (1996) Evidence that the UL84 gene product of human cytomegalovirus is essential for promoting oriLyt-dependent DNA replication and formation of replication compartments in cotransfection assays. J. Virol., 70, 7398–7413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shadan F.F., Cowsert,L.M. and Villarreal,L.P. (1994) n-butyrate, a cell cycle blocker, inhibits the replication of polyomaviruses and papillomaviruses but not that of adenoviruses and herpesviruses. J. Virol., 68, 4785–4796. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sherr C.J. (1995) D-type cyclins. Trends Biochem. Sci., 20, 187–190. [DOI] [PubMed] [Google Scholar]
- Sherr C.J. and Roberts,J.M. (1995) Inhibitors of mammalian G1 cyclin-dependent kinases. Genes Dev., 9, 1149–1163. [DOI] [PubMed] [Google Scholar]
- Song B., Liu,J.J., Yeh,K.C. and Knipe,D.M. (2000) Herpes simplex virus infection blocks events in the G1 phase of the cell cycle. Virology, 267, 326–334. [DOI] [PubMed] [Google Scholar]
- Stenberg R.M. (1996) The human cytomegalovirus major immediate-early gene. Intervirology, 39, 343–349. [DOI] [PubMed] [Google Scholar]
- Tsurimoto T., Melendy,T. and Stillman,B. (1990) Sequential initiation of lagging and leading strand synthesis by two different polymerase complexes at the SV40 DNA replication origin. Nature, 346, 534–539. [DOI] [PubMed] [Google Scholar]
- Vousden K.H. (1995) Regulation of the cell cycle by viral oncoproteins. Semin. Cancer Biol., 6, 109–116. [DOI] [PubMed] [Google Scholar]
- Wade M., Kowalik,T.F., Mudryj,M., Huang,E.S. and Azizkhan,J.C. (1992) E2F mediates dihydrofolate reductase promoter activation and multiprotein complex formation in human cytomegalovirus infection. Mol. Cell. Biol., 12, 4364–4374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang E.H. and Tjian,R. (1994) Promoter-selective transcriptional defect in cell cycle mutant ts13 rescued by hTAFII250. Science, 263, 811–814. [DOI] [PubMed] [Google Scholar]
- Wiebusch L. and Hagemeier,C. (1999) Human cytomegalovirus 86-kilodalton IE2 protein blocks cell cycle progression in G1. J. Virol., 73, 9274–9283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Won K.A. and Reed,S.I. (1996) Site-specific autophosphorylation and ubiquitin-dependent degradation of cyclin E. EMBO J., 15, 4182–4193. [PMC free article] [PubMed] [Google Scholar]
- Zhang H.S., Gavin,M., Dahiya,A., Postigo,A.A., Ma,D., Luo,R.X., Harbour,J.W. and Dean,D.C. (2000) Exit from G1 and S phase of the cell cycle is regulated by repressor complexes containing HDAC-Rb-hSWI/SNF and Rb-hSWI/SNF. Cell, 101, 79–89. [DOI] [PubMed] [Google Scholar]
- Zhao J., Dynlacht,B., Imai,T., Hori,T. and Harlow,E. (1998) Expression of NPAT, a novel substrate of cyclin E–CDK2, promotes S-phase entry. Genes Dev., 12, 456–461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zou L. and Stillman,B. (2000) Assembly of a complex containing Cdc45p, replication protein A, and Mcm2p at replication origins controlled by S-phase cyclin-dependent kinases and Cdc7p-Dbf4p kinase. Mol. Cell. Biol., 20, 3086–3096. [DOI] [PMC free article] [PubMed] [Google Scholar]