

Abstract
Exposure of yeast to increases in extracellular osmolarity activates the Hog1 mitogen-activated protein kinase (MAPK), which is essential for the induction of gene expression required for cell survival upon osmotic stress. Several genes are regulated in response to osmotic stress by Sko1, a transcriptional repressor of the ATF/CREB family. We show by in vivo coprecipitation and phosphorylation studies that Sko1 and Hog1 interact and that Sko1 is phosphorylated upon osmotic stress in a Hog1-dependent manner. Hog1 phosphorylates Sko1 in vitro at multiple sites within the N-terminal region. Phosphorylation of Sko1 disrupts the Sko1–Ssn6–Tup1 repressor complex, and consistently, a mutant allele of Sko1, unphosphorylatable by Hog1, exhibits less derepression than the wild type. Interestingly, Sko1 repressor activity is further enhanced in strains with high protein kinase A (PKA) activity. PKA phosphorylates Sko1 near the bZIP domain and mutation of these sites eliminates modulation of Sko1 responses to high PKA activity. Thus, Sko1 transcriptional repression is controlled directly by the Hog1 MAPK in response to stress, and this effect is further modulated by an independent signaling mechanism through the PKA pathway.
Keywords: Hog1/mitogen-activated protein kinase/signal transduction/Sko1/stress response
Introduction
Mitogen-activated protein kinase (MAPK) cascades are common signaling modules found in both higher and lower eukaryotic cells. Activation of a MAPK results in modification of a set of target proteins, often transcription factors, that allow the generation of appropriate cellular responses to an external stimulus (reviewed in Robinson and Cobb, 1997). Yeast cells have several different MAPK cascades that transduce distinct extracellular stimuli (e.g. mating pheromone and high osmolarity) (reviewed in Gustin et al., 1998). Budding yeast (Saccharomyces cerevisiae) responds to increases in osmolarity in the extracellular environment by activating the Hog1 MAPK cascade (Boguslawski, 1992; Brewster et al., 1993).
The yeast HOG pathway is activated by two independent mechanisms, which lead to the phosphorylation and activation of the Hog1 MAPK (reviewed in Gustin et al., 1998). Once activated, Hog1 induces diverse osmo-adaptive responses such as regulation of gene expression. Recent genome-wide transcriptional analyses carried out using DNA chips and filter hybridization showed that a great number of genes are regulated by osmotic stress in a Hog1-dependent manner (Posas et al., 2000; Rep et al., 2000). Among the genes under the control of Hog1 are genes that encode proteins involved in protection against osmotic stress as well as other types of stimuli. The mechanism of gene regulation through activated Hog1 is largely unknown because transcription factors under the control of this MAPK are not well characterized. Several candidates, however, have been proposed; for example, the general stress response transcription factors Msn2 and Msn4, which are regulated by protein kinase A (PKA) upon stress. Unfortunately, their relationship with Hog1 remains unclear (Martinez-Pastor et al., 1996; Schmitt and McEntee, 1996; Görner et al., 1998). Recently it was reported that Hot1, a putative transcriptional regulator, interacts with Hog1 in the two-hybrid system and appears to act downstream of Hog1 (Rep et al., 1999). However, the direct interaction with Hog1 and its regulation by this MAPK are not yet demonstrated.
Stimulated expression of some HOG pathway-dependent genes after osmotic stress appears to be the result of transcriptional derepression (Márquez et al., 1998). The bZIP transcriptional repressor Sko1 binds to the so-called cAMP responsive element (CRE) promoter motif (Nehlin et al., 1992; Vincent and Struhl, 1992). Its implication in the transcriptional regulation of osmotic stress-inducible genes has been shown recently for the stress defense genes ENA1 (Proft and Serrano, 1999), HAL1 (Pascual-Ahuir et al., 2001) and GRE2 (Garcia-Gimeno and Struhl, 2000). Genetic data indicate that Sko1 represses stress gene expression from CRE sites by recruiting the general co-repressor complex Ssn6–Tup1. Accordingly, mutants in either one of the three components are hyperresistant to salt stress (Márquez et al., 1998; Proft and Serrano, 1999; Garcia-Gimeno and Struhl, 2000). Induction of Sko1-dependent genes involves the release of repression, and this process is completely dependent on the HOG pathway (Proft and Serrano, 1999; Garcia-Gimeno and Struhl, 2000).
In this work we demonstrate biochemically that the Hog1 MAPK regulates the Sko1 transcriptional repressor by direct phosphorylation. We show that Hog1 and Sko1 interact in vivo, we identify the relevant Hog1 phosphorylation sites and analyze their effect on stress-regulated gene expression in vivo. Our results indicate that one important function of activated Hog1 kinase is the phosphorylation and inactivation of the Sko1 repressor by disruption of its association with Ssn6 and Tup1 proteins.
Results
Sko1 represses HOG-dependent stress gene expression
The Sko1 transcriptional repressor has recently been implicated in the osmotic stress-induced expression of some stress defense genes (Proft and Serrano, 1999; Garcia-Gimeno and Struhl, 2000; Pascual-Ahuir et al., 2001). Among these, GRE2, which encodes a plant isoflavonoid reductase homolog (Garay-Arroyo and Covarrubias, 1999), shows the most pronounced expression pattern upon hyperosmotic shock. We made use of this Sko1-dependent reporter gene to investigate by northern analysis of GRE2 expression during osmotic stress the epistatic relationship between Sko1 and Hog1. As shown in Figure 1, GRE2 expression is strongly repressed when cells are grown in normal medium, while the loss of SKO1 leads to high GRE2 transcript levels (∼13-fold as compared with wild type). GRE2 transcription is induced ∼45-fold upon brief osmotic shock in wild-type cells. This induction is virtually absent (only <2-fold) in sko1Δ cells, due mainly to higher expression under non-stress conditions. In the absence of HOG1, a dramatically reduced induction of GRE2 is observed that is completely suppressed by additional deletion of SKO1 (Figure 1). It is worth noting that Sko1-mediated gene transcription is completely dependent on the kinase activity of Hog1, since expression of a catalytically impaired Hog1 allele [Hog1(KN)] was unable to restore GRE2 expression in a hog1Δ strain (data not shown). Since sko1Δ and hog1Δ sko1Δ mutants showed nearly identically high GRE2 expression, one possible explanation was that Hog1 MAPK would act exclusively through the Sko1 repressor on the transcriptional control of a subset of stress defense genes.

Fig. 1. Roles of Sko1 and Hog1 in osmotic stress-induced transcription of GRE2. Transcript levels of GRE2 were monitored by northern analysis before and during osmotic stress caused by 0.4 M NaCl. GRE2 mRNA levels were quantified, normalized for the TBP1 internal loading control and depicted in the lower panel. The different small blots presented as well as the quantification data come from the same original blot for each mutant. The isogenic yeast strains W303-1A (wt), MAP19 (sko1Δ), MAP32 (hog1Δ) and MAP33 (hog1Δ sko1Δ) were used.
Sko1 is phosphorylated in vivo upon osmotic stress in a Hog1-dependent manner
As a first approach to determine the relationship between Sko1 and Hog1, we investigated whether Sko1 is phosphorylated in vivo during hyperosmotic stress. Therefore, we fused the SKO1 gene at its chromosomal locus with a triple haemagglutinin (HA) epitope at the N-terminus. The fully functional HA–Sko1 fusion protein (HA–SKO1) was detected from wild-type and hog1Δ cells using the specific 12CA5 monoclonal antibody against HA. This immunological detection of HA–SKO1 revealed that upon brief shock with 0.4 M NaCl, Sko1 rapidly changes its mobility in SDS–polyacrylamide gels (Figure 2). To test whether this shift in mobility of Sko1 was due to phosphorylation, protein extracts were treated with alkaline phosphatase before western analysis and as expected the mobility shift was eliminated (Figure 2). This effect was reversed by the simultaneous application of phosphatase inhibitors. Moreover, phosphorylation of Sko1 in response to osmotic stress depended completely on Hog1, since HA–SKO1 expressed from hog1Δ cells did not undergo the mobility shift observed in wild-type cells. Taken together, these results show that Sko1 is rapidly phosphorylated upon hyperosmotic shock in vivo and that this modification depends on the Hog1 MAPK.

Fig. 2. In vivo phosphorylation of Sko1 upon osmotic stress depends on Hog1. HA–SKO1 was expressed from its chromosomal locus in wild-type (MAP37) and hog1Δ (MAP36) cells. Approximately 50 µg of yeast total protein extracts were separated by SDS–PAGE, and the fusion protein was detected by monoclonal HA-specific antibodies. Cells were subjected or not to brief osmotic shock (0.4 M NaCl, 10 min) and the extracts were treated or not with 10 U of alkaline phosphatase in the absence or presence of phosphatase inhibitors as indicated. Control strain (without HA–SKO1) was wild-type strain W303-1A.
Sko1 co-immunoprecipitates with Hog1
To obtain biochemical evidence for the interaction of Sko1 and Hog1 in vivo, we tested HA–Sko1 for immunoprecipitation with glutathione S-transferase (GST)–Hog1 or green fluorescent protein (GFP)–Hog1 (Figure 3).
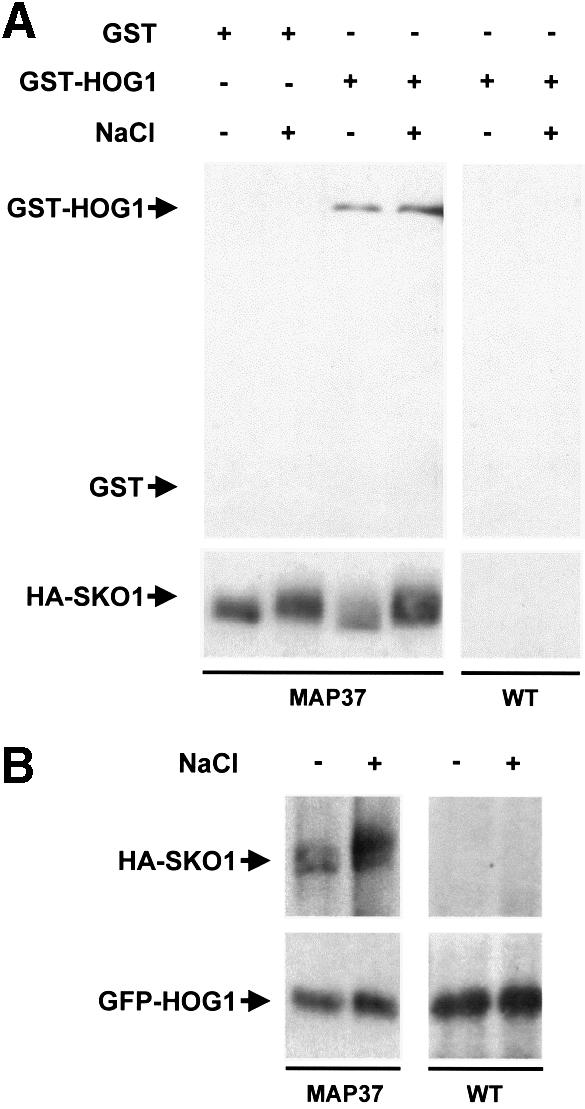
Fig. 3. In vivo binding of Hog1 to Sko1. (A) MAP37 strain (which expresses HA-tagged Sko1 from the wild-type locus) was transformed with a plasmid expressing GST or GST–HOG1 under the PGAL1 promoter. Cells were grown in the presence of galactose and samples were taken before (–) or 5 min after (+) the addition of NaCl to a final concentration of 0.4 M. HA–SKO1 was precipitated using anti-HA monoclonal antibody (12CA5) and protein A–Sepharose beads, and the presence of GST proteins in precipitates was probed by immunoblotting using anti-GST (upper panel). Antibody against HA was used to detect the HA–SKO1 fusion protein (lower panel). (B) Yeast MAP37 cells were transformed with a single copy plasmid containing GFP-tagged Hog1, expressed under its own promoter. Cells were grown in the presence of glucose and treated as in (A). HA–SKO1 and GFP–HOG1 in precipitates were probed with anti-HA and anti-GFP antibodies.
Yeast MAP37 cells (which express HA-tagged Sko1 from its own genomic locus) were transformed with a plasmid that expresses GST-tagged Hog1 from the GAL1 promoter. Cells were subjected to a brief osmotic shock and Sko1 was immunoprecipitated using specific monoclonal antibodies against HA. The presence of GST–HOG1 in the precipitates was probed by an anti-GST monoclonal antibody. As shown in Figure 3A, Sko1 was able to coprecipitate Hog1 irrespective of the environmental osmotic conditions.
The above experiments were performed using the strong GAL1 promoter. Because the amount of Hog1 in those experiments is higher than the physiological conditions, we repeated the Hog1–Sko1 coprecipitation using Hog1 expressed from a centromeric (low copy number) plasmid under the control of its own native promoter. Hog1 was fused to GFP (GFP–HOG1), and transformed into the MAP37 strain (which contains chromosomal HA-tagged Sko1). Yeast MAP37 cells transformed with GFP–HOG1 were subjected to a brief osmotic shock. GFP–HOG1 was precipitated using anti-GFP antibody, and the presence of HA–SKO1 in precipitates was detected using anti-HA monoclonal antibody. Although the levels of protein expression were lower than in the previous experiments, the Sko1–Hog1 interaction was evident. Again, the level of Sko1–Hog1 binding was not affected by osmotic shock (Figure 3B). Thus, Hog1 MAPK binds to the Sko1 transcriptional repressor.
Hog1 phosphorylates multiple sites at the N-terminus of Sko1
Sko1 is phosphorylated upon osmotic stress in a Hog1-dependent manner and interacts physically with the MAPK. We then tested whether Sko1 phosphorylation was carried out directly by the Hog1 MAPK, using purified proteins in an in vitro kinase assay. For this purpose, Hog1 and a constitutively activated version of Pbs2 [PBS2(EE)] (EE denotes Ser514→Glu and Thr518→Glu mutations in the activation loop of the Pbs2 MAPKK) were purified as GST fusion proteins from Escherichia coli. In the first step of the reaction Hog1 was activated by phosphorylation in the presence of PBS2(EE) and ATP. Then Sko1, purified from E.coli as a GST-tagged protein and [γ-32P]ATP were added to the reaction. As shown in Figure 4A, only when Sko1 was incubated with pre-activated Hog1, was phosphorylation of Sko1 observed.

Fig. 4. In vitro phosphorylation of Sko1 by Hog1. (A) In vitro activated Hog1 phosphorylates Sko1. Recombinant tagged proteins were purified from E.coli as described in Materials and methods. Hog1 and the constitutively activated Pbs2 allele [PBS2(EE)] were incubated in presence of kinase buffer and ATP. Sko1 was then added (when indicated) in the presence of radioactive ATP. Phosphorylated proteins were resolved by SDS–PAGE and detected by autoradiography. The position of tagged Sko1 is indicated on the left. (B) Hog1 phosphoryl ates the region between residues 1 and 202 of Sko1. Various Sko1 fragments were tested for their ability to be phosphorylated by an in vitro activated Hog1 (as described in Materials and methods). After in vitro kinase assay, phosphorylated proteins were resolved by SDS–PAGE and detected by autoradiography. The positions of the Sko1 fragments included in the constructs are indicated in parentheses. (C) Mutation of Sko1 Ser108, Thr113 and Ser126 to Ala abolishes Hog1 phosphorylation. Full-length Sko1 and the triple mutant SKO1(E) protein (mutant in the Ser108, Thr113 and Ser126) were tested for Hog1 phosphorylation as in (A). After phosphorylation, proteins were resolved by SDS–PAGE and transferred to a nylon membrane. Phosphorylated proteins were detected by autoradiography (upper panel). GST-tagged Sko1 proteins were detected by immunoblot using the anti-GST monoclonal antibody (lower panel).
To map the phosphorylation site(s) for Hog1 in Sko1, we created several truncated SKO1 alleles, and expressed them as GST-tagged proteins in E.coli. Truncated forms of Sko1 were purified and subjected to in vitro phosphorylation by activated Hog1 (as described above). An N-terminal fragment containing the first 214 residues was able to be phosphorylated as efficiently as the full-length protein (Figure 4B, lanes 1 and 2). Furthermore, when N-terminal deletions (fragments containing from amino acids 202, 309 and 420 to 647) were tested, phosphorylation was completely abolished (Figure 4B, lanes 3, 4 and 5), indicating that the N-terminus of Sko1 was the target for Hog1 phosphorylation.
Five sequences corresponding to the consensus phosphorylation site for MAPKs (Ser-Pro/Thr-Pro) are present in the region comprised between residues 1 and 202, namely Ser94 (KDSP), Ser108 (IISP), Thr113 (ILTP), Ser126 (LLSP) and Ser171 (SSSP). We created point mutant versions to replace Ser and Thr residues with Ala, and tested them for phosphorylation by Hog1. Single mutations of Ser94 and Ser171 did not alter Sko1 phosphorylation, and only when Ser108, Thr113 or Ser126 were mutated, was phosphorylation of Sko1 slightly reduced (data not shown). We then constructed several combinations of mutant sites and tested them for phosphorylation. Sko1 phosphorylation was only abolished in the SKO1(E) mutant, which contains mutations in all three sites, Ser108, Thr113 and Ser126 (Figure 4C).
In vivo phosphorylation assays, carried out as described previously, in wild-type cells transformed with a wild-type HA-tagged Sko1 or the mutant HA–SKO1(E) protein, demonstrated that mutation of the three sites in the SKO1(E) allele eliminated most of the mobility shift due to phosphorylation of Sko1 in response to osmotic stress (Figure 5).

Fig. 5. In vivo phosphorylation of Sko1 mutant proteins. Wild-type or hog1Δ cells were transformed with HA-tagged wild-type Sko1 (SKO1), the Sko1 mutant in the Hog1 phosphorylation sites [SKO1(E)], the Sko1 mutant in the PKA phosphorylation sites [SKO1(M)] or the Sko1 mutant in both Hog1 and PKA phosphorylation sites [SKO1(EM)]. Cells were grown in the presence of galactose for 4 h and samples were taken before (–) or 5 min after (+) the addition of NaCl to a final concentration of 0.4 M, and the extracts were treated (+) or not (–) with 10 U of alkaline phosphatase (AP). HA–SKO1 was detected by immunoblotting using anti-HA monoclonal antibody (12CA5).
Taken together, these results indicate that there is a specific cluster of three phosphorylation sites for Hog1, within 19 amino acids, in the N-terminus of Sko1 (depicted in Figure 6).

Fig. 6. Schematic overview of the phosphorylation sites in the Sko1 protein. Three Hog1 phosphorylation sites are present in the N-terminus of Sko1, whereas three PKA phosphorylation sites are located near the bZIP structural domain. (E) and (M) triple mutations, which correspond to the Hog1 and PKA phosphorylation sites, are indicated.
Sko1 is phosphorylated by PKA near the bZIP structural domain
SKO1 has been cloned as a multicopy suppressor of the growth defect caused by high PKA activity (Nehlin et al., 1992), although the connection between Sko1 and PKA remained undefined. Several putative PKA consensus motifs are present in Sko1. Thus, we tested whether Sko1 could be phosphorylated by PKA, using purified proteins in an in vitro kinase assay. Indeed, when wild-type GST-tagged Sko1 purified from E.coli was incubated with PKA in the presence of kinase buffer and [γ-32P]ATP, phosphorylation of Sko1 was observed (Figure 7A, lane 1).

Fig. 7. Sko1 is phosphorylated by PKA in vitro. (A) PKA phosphorylates the region between residues 309 and 420 of Sko1. Full-length or various Sko1 fragments were tested by their ability to be phosphorylated by PKA. Sko1 recombinant tagged proteins were purified from E.coli as described in Materials and methods, and incubated with PKA in the presence of kinase buffer and radioactive ATP. Phosphorylated proteins were resolved by SDS–PAGE and detected by autoradiography. The positions of the Sko1 fragments included in the constructs are indicated in parentheses. (B) Mutation of Sko1 Ser380, Ser393 and Ser399 to Ala abolishes PKA phosphorylation. Full-length Sko1 and the triple mutant SKO1(M) protein (mutant in Ser380, Thr393 and Ser399) were tested for PKA phosphorylation as in (A). After phosphorylation, proteins were resolved by SDS–PAGE and transferred to a nylon membrane. Phosphorylated proteins were detected by autoradiography (upper panel). GST-tagged Sko1 proteins were detected by immunoblot using the anti-GST monoclonal antibody (lower panel).
To map the phosphorylation site(s) for PKA in Sko1, we subjected to in vitro phosphorylation several truncated SKO1 alleles, expressed and purified as GST-tagged proteins from E.coli. Neither an N-terminal fragment (containing 314 amino acids) nor a C-terminal fragment (containing the last 227 residues) was able to be phosphorylated by PKA (Figure 7B, lanes 6 and 5). Moreover, a GST fusion protein containing amino acids 309–647 was phosphorylated as efficiently as the full-length protein, indicating that phosphorylation of Sko1 by PKA occurred in a segment of the protein between amino acids 309 and 420.
Three sequences corresponding to the canonical phosphorylation site for PKA (RXXS/T) are present in that region: Ser380 (RRMS), Ser393 (RKNS) and Ser399 (RKNS). We created point mutant versions to replace these Ser residues with Ala, and tested them for phosphorylation by PKA. Single mutation of any residue only slightly diminished Sko1 phosphorylation by PKA (data not shown). We then constructed several combinations of mutant sites and tested for phosphorylation. As shown in Figure 7B, phosphorylation of Sko1 by PKA was completely abolished only in the SKO1(M) mutant version, which contains mutations in all three serines (Ser380, Ser393 and Ser399). This indicates that there is a cluster of three phosphorylation sites for PKA (within 20 amino acids) near the bZIP domain of Sko1 (depicted in Figure 6).
In vivo phosphorylation assays, carried out as described previously, in wild-type cells transformed with a wild-type HA-tagged Sko1 or the mutant HA-tagged SKO1(M) allele demonstrated that mutation of the three PKA sites in the SKO1(M) allele decreased the Hog1-dependent phosphorylation of Sko1 in response to osmotic stress (Figure 5), as suggested by the absence of changes in protein migration upon stress. Consistently, a Sko1 protein containing mutations in both PKA and Hog1 phosphorylation sites [SKO1(EM)] was unable to be phosphorylated at all upon osmotic stress (Figure 5).
Hog1 phosphorylations regulate Sko1-mediated gene repression in vivo
Having found that Hog1 phosphorylates Sko1 after osmotic shock at three specific sites, we aimed at identifying the role of these phosphorylations in Sko1-mediated transcriptional repression in vivo. GST–SKO1 (wild type) and GST–SKO1(E) (unphosphorylatable by Hog1) were, therefore, expressed from yeast expression vector pYEX-4T, and the effect on transcription was measured by northern analysis of the Sko1-dependent GRE2 gene. As shown in Figure 8A, the deregulated GRE2 expression pattern of sko1Δ mutants is completely restored to wild-type levels when GST–SKO1 is expressed. We therefore concluded that the GST–SKO1 protein we used in this study functionally replaced the endogenous Sko1. Expression of GST–SKO1(E), which is no longer phosphorylatable by Hog1, resulted in a stronger repression of GRE2 and HAL1 transcript levels (Figure 8A). This enhanced repression was already observed under non-stress conditions, but was even more evident when the cells were treated with 0.4 M NaCl. This result indicated that the Hog1 phosphorylation sites identified by in vitro kinase assays indeed play a functional role in regulating Sko1 repressor activity. There is still some regulation of Sko1, independent of these sites, in agreement with the residual phosphorylation of SKO1(E) in response to osmotic stress (Figure 5).

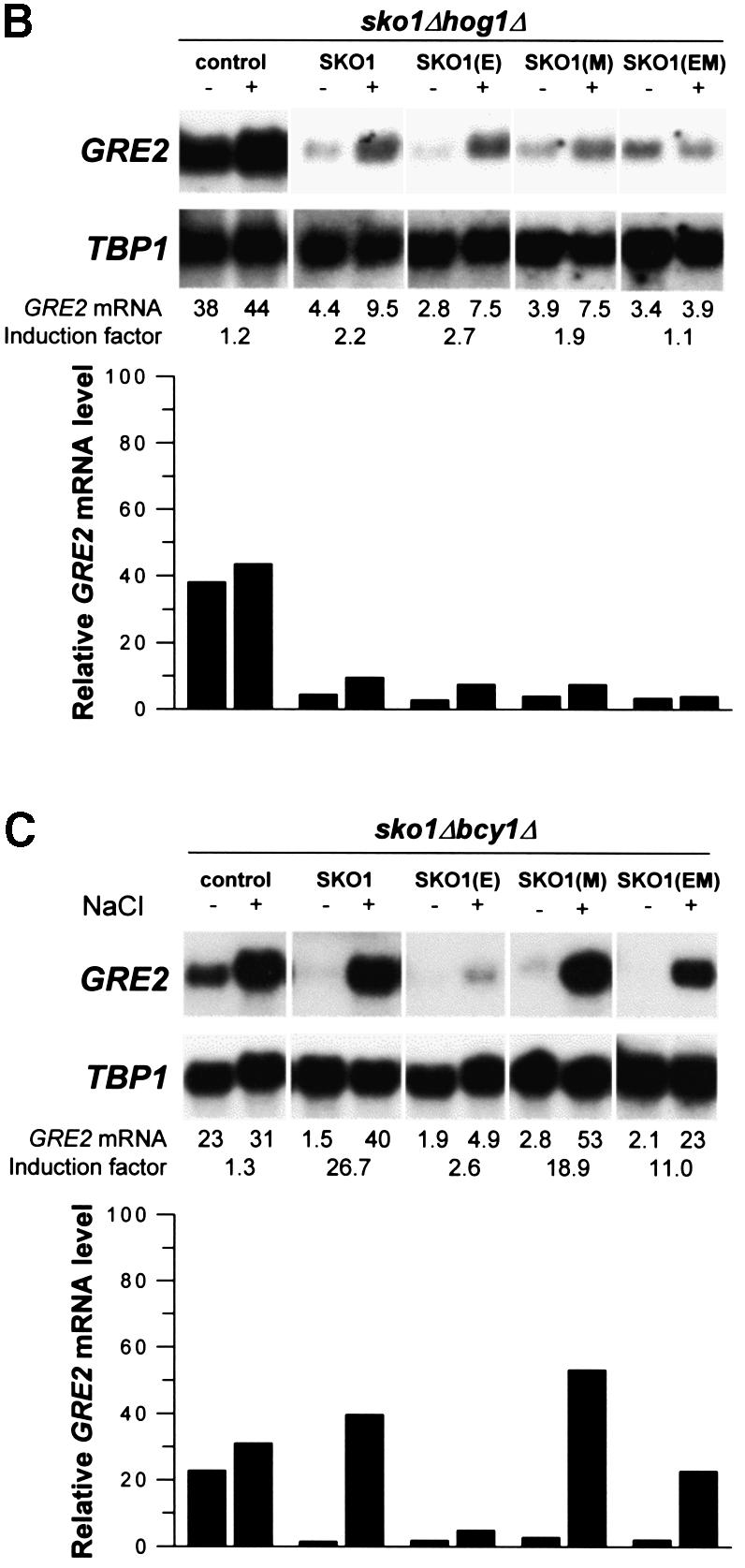
Fig. 8. Hog1 and PKA phosphorylations modulate Sko1-mediated repression in vivo. Yeast strains were transformed with the empty vector pYEX-4T (control) or the wild-type GST–Sko1 fusion (SKO1), the GST–Sko1 mutant allele in the Hog1 phosphorylation sites [SKO1(E)], the GST–Sko1 mutant allele in the PKA phosphorylation sites [SKO1(M)] or the GST–Sko1 mutant allele in both Hog1 and PKA phosphorylation sites [SKO1(EM)]. Cells were grown selectively to mid-log phase and then subjected (+) or not (–) to brief osmotic shock (0.4 M NaCl, 10 min). Total RNA was assayed by northern blot analysis for transcript levels of GRE2, HAL1 and TBP1. The induction factor represents the degree of induction plus or minus stress. The different small blots presented as well as the quantification data come from the same original blot for each mutant. (A) Hog1 phosphorylation sites are important for release from Sko1-mediated repression. GST–Sko1 fusions were expressed in wild type (W303-1A) and Δsko1 (MAP19). (B) Sko1-mediated derepression is controlled by Hog1. GST–Sko1 fusions were expressed in Δsko1Δhog1 (MAP33) cells. (C) PKA phosphorylation sites modulate Sko1 function in cells with high PKA activity. GST–Sko1 fusions were expressed in Δsko1Δbcy1 (MAP44) cells. The amount of Sko1 protein was similar for the different alleles and was not altered when cells were bcy1Δ (data not shown).
To test whether the observed difference in GRE2 expression by SKO1(E), as compared with wild-type Sko1, was dependent on Hog1 we repeated the above experiment expressing both fusions in a sko1Δ hog1Δ background. Accordingly we found that Sko1 (wild type) and Sko1(E) strongly and equally repressed the expression of GRE2 (Figure 8B). Therefore, the residual osmotic regulation of Sko1(E) is dependent on Hog1. As no Hog1 sites are present in Sko1(E), this suggests that, in addition to phosphorylating Sko1 directly, Hog1 also acts indirectly over the transcriptional regulator, perhaps through an intermediate kinase (see next section). As depicted in Figure 8A, lower panel, very similar results were obtained by northern analysis of another osmolarity-responsive gene, HAL1.
High PKA activity modulates Sko1 function
As a result of our in vitro kinase assays, a cluster of three consensus sites within Sko1 that are phosphorylated by PKA was identified. To test for the effect of these phosphorylations on Sko1-regulated stress gene expression we expressed GST–SKO1(M) (unphosphorylatable by PKA) in sko1Δ cells and compared its effect on GRE2 transcription with that of wild-type GST–SKO1. As shown in Figure 8A, GST–SKO1(M) and GST–SKO1 equally restored the salt-regulated expression pattern of GRE2. Therefore, the elimination of PKA phosphorylation sites, unlike the Hog1 phosphorylation sites, revealed no obvious phenotype concerning Sko1-regulated gene expression under these conditions (Figure 8A).
We then tested whether high cellular PKA activity, achieved by disruption of the negative regulator BCY1, had an effect on Sko1-mediated repression. We expressed GST–SKO1 (wild type and all point mutated alleles) in sko1Δbcy1Δ cells and quantified GRE2 transcription. As illustrated in Figure 8C, high PKA activity only mildly affected the stress-regulated expression of GRE2 when SKO1 (wild type) was present. However, when Hog1 phosphorylation sites were removed [SKO1(E) mutant], a complete repression of GRE2 transcription, much more pronounced than in sko1Δ cells, was observed [compare SKO1(E) in Figure 8A and C]. This led to the conclusion that high PKA activity increases Sko1-mediated repression when the Hog1 phosphorylation sites are absent. To confirm that the PKA-mediated modulation of gene expression was triggered by the PKA sites identified in Sko1, we tested the effect of the elimination of the PKA phosphorylation sites in the SKO1(E) allele by expressing the SKO1(EM) allele in yeast. Indeed, by additional deletion of the three PKA sites the negative effect of PKA on GRE2 expression was abolished (see Figure 8C, last lane). We also tested all the above-mentioned SKO1 alleles expressed in a sko1Δhog1Δbcy1Δ triple mutant strain for their effect on GRE2 transcription with a very similar result to the one obtained using a sko1Δhog1Δ mutant (data not shown). These results clearly indicate that the phosphorylation of Sko1 by PKA is important for the regulation of Sko1(E) repressing activity.
CRE-mediated expression depends on Sko1 phosphorylation by Hog1 and PKA
To confirm the effect of Sko1 mutations on gene expression we designed a second independent approach based on the CYC1–2×CRE–lacZ reporter gene to monitor Sko1 function in vivo (described in Materials and methods). As shown in Figure 9, the CRE–lacZ reporter gene is strongly repressed under normal conditions and highly expressed upon osmotic stress. Derepression upon osmotic shock is completely dependent on Hog1, and regulated expression of the CRE-mediated reporter depends on the presence of fully functional Sko1. Similar to the observations in the northern blot experiments, expression of SKO1(E) reduced gene expression significantly as compared with the wild type upon osmotic stress. A much more severe effect was observed when SKO1(E) was expressed in a bcy1Δ strain (which displays high PKA levels), and this effect was reversed by introduction of mutations of the PKA phosphorylation sites in Sko1. These results are consistent with those described previously for GRE2 gene expression.

Fig. 9. Roles of Hog1 and PKA phosphorylation sites in the regulation of a CRE-driven reporter gene. The yeast strains used in Figure 8 were co-transformed with the CYC1–(2×CRE)–lacZ reporter pMP253 and subjected (+ NaCl) or not (– NaCl) to brief osmotic shock (0.4 M NaCl, 15 min). Specific β-galactosidase activity is given in nmol/min/mg and is the result of the measurement in duplicate of three independent transformants. The amount of Sko1 protein was similar for the different alleles and was not altered when cells were bcy1Δ (data not shown).
Phosphorylation of Sko1 by the Hog1 MAPK is important for salt stress resistance
Stress genes such as ENA1 and HAL1 have been found to be negatively regulated by the Sko1 repressor and are known to play a critical role in ion homeostasis during osmotic stress. Therefore, any significant reduction in the expression levels of these stress defense genes should result in a salt-sensitive phenotype. Having found that the elimination of Hog1 phosphorylation sites renders Sko1 a more efficient repressor, we tested its influence on cellular salt resistance. As depicted in Figure 10, overexpression of SKO1(E) (unphosphorylatable by Hog1) or SKO1(EM) (unphosphorylatable by Hog1 and PKA) had a strong negative effect on the resistance of yeast cells to hyperosmotic stress caused by 0.4 M NaCl, while overexpression of SKO1(M) (deletion of PKA phosphorylation sites) did not have any effect on stress resistance. These results are in agreement with SKO1(E) and SKO1(EM) alleles having a pronounced negative effect on Sko1-mediated gene expression.
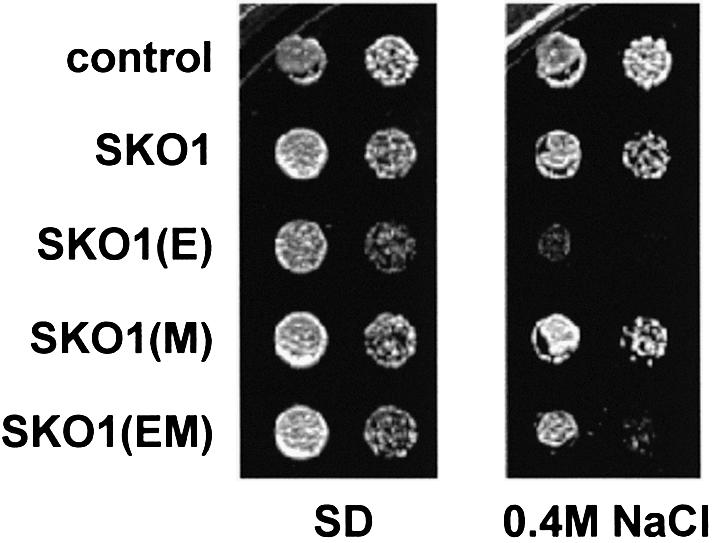
Fig. 10. Hog1 phosphorylation of Sko1 is critical for salt stress resistance. Yeast Δsko1 mutant cells (MAP19) were transformed with vector control pYEX-4T, GST–SKO1 (wild type), GST–SKO1(E) (mutant in the Hog1 phosphorylation sites), GST–SKO1(M) (mutant in the PKA phosphorylation sites) or GST–SKO1(EM) (mutant in both Hog1 and PKA phosphorylation sites) alleles. Overexpression of the GST fusions was achieved by addition of CuSO4 to a final concentration of 0.3 mM to the plates. Serial dilutions of the transformed yeast strains were spotted onto SD plates lacking uracil containing or not 0.4 M NaCl as indicated.
Interaction of Sko1 with the Ssn6 and Tup1 co-repressor is regulated by Hog1 phosphorylation upon stress
A possible molecular mechanism to regulate Sko1 transcriptional activity is to change its affinity for the co-repressor proteins Ssn6 and Tup1. To obtain biochemical evidence for a regulated interaction of Sko1 with the Ssn6 and Tup1 proteins, we tested GST–SKO1 coprecipitation with the wild-type Ssn6 and Tup1 proteins (Figure 11).

Fig. 11. In vivo binding of Sko1 to the Ssn6–Tup1 co-repressor is affected by phosphorylation upon stress. MAP19 strain (sko1Δ) was transformed with a plasmid expressing GST, GST–SKO1, GST–SKO(E) or GST–SKO1(EM). Cells were grown and subjected (+) or not (–) to a brief osmotic shock (5 min, 0.4 M NaCl). GST proteins were pulled down with glutathione–Sepharose 4B and the presence of wild-type Tup1 or Ssn6 proteins was probed by immunoblotting using specific antibodies against Tup1 (upper panel) or Ssn6 (middle panel), respectively. Total extract represents <10% of the input protein and Prec. is the total amount of Ssn6 or Tup1 coprecipitated. The amount of precipitated GST proteins was detected using anti-GST (lower panel).
Yeast sko1Δ cells were transformed with expression plasmids for GST alone, GST-tagged SKO1 (wild type) or the mutant alleles SKO1(E) and SKO1(EM) (unphosphorylatable by Hog1). Cells were subjected to a brief osmotic shock before lysis, and the presence of wild-type Ssn6 and Tup1 proteins in the GST precipitates was probed using specific antibodies. As shown in Figure 11, both Ssn6 and Tup1 proteins were coprecipitated with Sko1. Interestingly, interaction of wild-type Sko1 with the co-repressor proteins was altered in the presence of osmotic stress. A much lower amount of both Ssn6 and Tup1 was precipitated upon stress. Furthermore, when the unphosphorylatable mutants for the Hog1 MAPK, Sko1(E) and Sko1(EM) proteins, were tested for coprecipitation with Ssn6 and Tup1, the amount of either Ssn6 or Tup1 protein able to coprecipitate was similar before and after stress (Figure 11). These data clearly indicate that the interaction of Sko1 with the Ssn6–Tup1 co-repressor complex is regulated by osmotic stress dependent on the Hog1 phosphorylation sites.
Discussion
Yeast cells respond to increases in osmolarity in the extracellular environment by activating the Hog1 MAPK pathway. Activated Hog1 MAPK induces, upon translocation into the nucleus, a complex pattern of gene expression required for cell survival upon osmotic stress (Posas et al., 2000; Rep et al., 2000). To understand how the MAPK can modulate transcription of stress defense genes it is necessary to determine the molecular mechanisms by which the kinase acts. Here we report that Sko1, a transcriptional repressor of the ATF/CREB family, is controlled by the Hog1 MAPK to regulate transcription of a subset of the stress-responsive genes upon osmotic stress. Although several candidates have been proposed to be under the control of the HOG pathway (i.e. Hot1, Msn2 and Msn4), Sko1 is the first transcripitional regulator shown to be a direct substrate for the osmotic stress-activated MAPK Hog1.
Previous reports indicated that expression upon osmotic stress of some HOG pathway-dependent genes was the result of transcriptional derepression mediated by the CRE binding protein Sko1 (Proft and Serrano, 1999; Garcia-Gimeno and Struhl, 2000). Indeed, regulated derepression of a CRE–lacZ reporter gene, or CRE-containing genes GRE2, ENA1 and HAL1, is dependent on Hog1 and Sko1. Several lines of evidence suggest that Sko1 is actually a direct substrate for the MAPK. In vivo studies indicated that Hog1 was able to coprecipitate Sko1. Despite the fact that the Hog1 MAPK was able to coprecipitate Sko1 irrespective of the environmental conditions, localization studies indicate that Hog1 concentrates in the nucleus upon a brief osmotic stress (Ferrigno et al., 1998; Reiser et al., 1999) and that Sko1 is constitutively nuclear under the same conditions (see below). Thus, it seems that the interaction of Sko1 and Hog1 will occur only under stress conditions due to subcellular compartmentalization. Accordingly, it is only under those conditions that Sko1 is phosphorylated in vivo by the MAPK.
These studies of the relationship of Sko1 and Hog1 in vivo are further supported by in vitro evidence that Hog1 phosphorylates Sko1 directly. Phosphorylation by Hog1 occurs at three specific residues clustered within 19 amino acids in the N-terminus of Sko1. Remarkably, in all three cases, the residues surrounding the phosphorylation site are similar to the Hog1 site in the Rck2 kinase, the only substrate described to be phosphorylated and regulated by Hog1 to date (Bilsland-Marchesan et al., 2000). Mutation of those residues to Ala results in a Sko1 allele much less phosphorylated in vivo in response to stress. It is clear, however, that part of the Hog1 regulation of Sko1 is independent of these phosphorylation sites. It may be speculated that Hog1, in addition to phosphorylate Sko1 directly, also acts indirectly on Sko1, perhaps through an intermediate kinase. A possible candidate to mediate Hog1 phosphorylation is the kinase Rck2. In vivo phosphorylation experiments showed that Sko1 phosphorylation was not altered in rck2 mutant cells, indicating that Rck2 is not mediating the indirect effects of Hog1 over Sko1 (data not shown).
Phosphorylation by Hog1 decreased the Sko1 transcriptional repressing activity as detected by both northern blot experiments and measurements of a CRE–lacZ reporter gene. Consistently, mutation of Hog1 phosphorylation sites to Ala renders Sko1 a stronger repressor. Several mechanisms could be involved in the regulation of Sko1 repressing activity by phosphorylation in response to stress. Phosphorylation of Sko1 could regulate the recruitment of the repressor by affecting any of several steps: binding of Sko1 to the promoter, homodimerization of Sko1, localization of Sko1 to the nucleus or interaction of Sko1 with Ssn6 or Tup1. It is unlikely that DNA binding is affected, because regulated expression was achieved by Gal4DBD–Sko1 to GAL1–lacZ (Proft and Serrano, 1999). Moreover, in vitro gel retardation assays indicate that the binding capacity of Sko1 to CRE sites was independent of its phosphorylation by Hog1 (data not shown). ATF/CREB proteins bind as homodimers or heterodimers to specific DNA motifs. Indeed, Sko1 can form homodimers (Vincent and Struhl, 1992; our results), but the amount of protein able to form homodimers, quantified by coprecipitation experiments, was not affected by osmotic stress (data not shown). Localization studies based on the detection of fully functional GST–Sko1 fusion protein by indirect immunofluorescence, also did not reveal the nuclear localization of Sko1 to be regulated by osmotic stress caused by 0.4 M NaCl (A.Pascual-Ahuir, unpublished observations). Thus, the more likely possibility is that phosphorylation disrupts the interaction of Sko1 with Ssn6 or Tup1. We tested this hypothesis by coprecipitation experiments and, indeed, interaction of Sko1 with both Ssn6 and Tup1 was affected by osmotic stress. Upon stress, binding of Sko1 to the co-repressor was clearly diminished and this effect was not observed in Sko1 mutant alleles unable to be phosphorylated by Hog1. These data indicate that the integrity of the Sko1–Ssn6–Tup1 complex is regulated by osmotic stress in a Hog1-dependent fashion and indicate that Hog1 regulates gene expression by disrupting the repressor complex.
Although previous reports suggested that Sko1 function was not affected by the levels of PKA activity (Garcia-Gimeno and Struhl, 2000), CREB proteins in various species activate transcription via regulation by PKA (Montminy, 1997). Phosphorylation by PKA at three clustered sites located near the bZIP structural domain suggested that Sko1 could be regulated by this kinase. Interestingly, when the Hog1-dependent Sko1 phosphorylation sites were removed [SKO1(E)], an additional modulation by PKA was clearly revealed. Under these conditions, changes in PKA activity levels (achieved by the introduction of a bcy1Δ mutation) or the presence of osmotic stress indicated that PKA phosphorylation resulted in an increased repressor activity of Sko1. Consistently, Sko1 proteins unphosphorylatable by PKA were unable to respond to those changes in PKA activity, resulting in proteins with lower repressing activity. It seems that high PKA activity interferes with the second, indirect mode of osmotic regulation of Sko1 discussed above.
Taken together, our data allow us to speculate on a mechanism for a dual regulation of Sko1 by the Hog1 and PKA kinases. Under normal conditions, Hog1 is inactive and high levels of PKA activity are maintained within the cell, which would be responsible for the strong repressing activity of Sko1. In response to hyperosmotic stress conditions, Hog1 is activated and phosphorylates Sko1, which is, by itself, sufficient to alleviate the repressing activity of Sko1. This diminished capacity of Sko1 as a repressor is probably further enhanced by a decrease in the levels of PKA activity under stress conditions (Márquez and Serrano, 1996). Thus, the combination of opposing effects by two independent pathways under stress conditions results in changes in the pattern of Sko1 phosphorylation, which alters its capacity as a transcriptional repressor.
Materials and methods
Yeast strains
The following yeast strains were used: TM141 (MATa ura3 leu2 trp1 his3); TM233 (MATα ura3 leu2 trp1 his3 lys2 hog1::TRP1); TM261 (MATα ura3 leu2 his3 pbs2::LEU2); W303-1A (MATa ura3 leu2 trp1 his3 ade2); MAP19 (MATa ura3 leu2 trp1 his3 ade2 sko1::loxp-KAN-loxp); MAP32 (MATa ura3 leu2 trp1 his3 ade2 hog1::TRP1); MAP33 (MATa ura3 leu2 trp1 his3 ade2 sko1::loxp-KAN-loxp hog1::TRP1); MAP44 (MATa ura3 leu2 trp1 his3 ade2 sko1::loxp-KAN-loxp bcy1::LEU2); MAP37 (MATa ura3 leu2 trp1 his3 ade2 3×HA–SKO1); MAP36 (MATa ura3 leu2 trp1 his3 ade2 hog1::TRP1 3×HA–SKO1). To fuse the chromosomal SKO1 locus with a triple HA epitope (strains MAP36 and MAP37) the PCR-based strategy described by Schneider et al. (1995) was used. The correct insertion of the epitope tag at the N-terminus right after the start-ATG was confirmed by genomic PCR. The resulting HA–Sko1 fusion protein was shown to be functional by northern analysis of Sko1-dependent genes.
Plasmids
The bacterial expression vectors pGEX-KG and pGEX-4T (Pharmacia) allow the expression of GST-tagged proteins in E.coli. Full-length SKO1 was cloned into pGEX-KG. Several mutant alleles of SKO1 comprising amino acids 1–214, 202–647 and 420–647 were generated by PCR and cloned into the pGEX-4T plasmid. Construction of SKO1 site directed mutants was carried out by PCR-directed mutagenesis as described previously (Maeda et al., 1995). Each mutation was verified by DNA sequencing. The SKO1(E) mutant contains a triple amino acid substitution, Ser108, Thr113 and Ser126 to Ala. These mutations abolish phosphorylation of Sko1 by Hog1. The SKO1(M) mutant contains a triple amino acid substitution, Ser380, Ser393 and Ser399 to Ala. These mutations abolish phosphorylation of Sko1 by PKA. SKO1(E) and SKO1(M) mutant alleles were introduced into pGEX-KG for bacterial expression and into pYEX-4T (PCUP1-GST, URA3+, 2 µ; AMRAD biotech) for yeast expression. The yeast expression vector YCpIF16 (PGAL1–HA, TRP1, CEN) allowed the expression of HA fusion proteins using the GAL1 promoter (Foreman and Davis, 1994). HOG1, wild-type and mutant alleles of SKO1 were cloned into the BamHI site of YCpIF16 for expression of HA-tagged proteins. pSWM14 contains full-length HOG1 fused to GST under the GAL1 promoter (gift from H.Saito). HOG1–GFP plasmid contains HOG1 (from –419 to the termination codon) fused to GFP in a centromeric plasmid (Ferrigno et al., 1998). The CYC1–(2×CRE)–lacZ reporter pMP253 (2 µ, TRP1) was constructed by a tandem insertion of double-stranded oligonucleotide AGCTATCGATTATTTCCTACTTCTATGACGTTT (containing the CRE sequence of the ENA1 promoter) into CYC1–lacZ reporter plasmid pMP206 (Proft and Serrano, 1999) and subsequent insertion of the TRP1 marker.
Buffers and media
Buffer A is 50 mM Tris–HCl pH 7.5, 15 mM EDTA, 15 mM EGTA, 2 mM dithiothreitol (DTT), 0.1% Triton X-100, 1 mM phenylmethylsulfonyl fluoride, 1 mM benzamidine and 5 µg/ml of leupeptin. Buffer B is 50 mM Tris–HCl pH 7.5, 10 mM MgCl2, 2 mM DTT. Kinase buffer is 50 mM Tris–HCl pH 7.5, 10 mM MgCl2, 2 mM DTT. Alkaline phosphatase (AP) buffer is 50 mM Tris–HCl pH 8.0, 100 mM NaCl, 10 mM MgCl2. PS buffer is 100 mM KH2PO4/K2HPO4 pH 7.4, 1.2 M sorbitol. PBS buffer is 10 mM KH2PO4/K2HPO4 pH 7.4, 140 mM NaCl. Phosphatase inhibitor cocktail contains 10 mM NaF, 1 mM sodium pyrophosphate, 1 mM sodium vanadate and 10 mM β-glycerophosphate. SDS loading buffer is 50 mM Tris–HCl pH 6.8, 100 mM DTT, 2% SDS, 0.1% bromophenol blue and 10% glycerol.
Expression and purification of epitope-tagged proteins
GST fusion proteins encoding PBS2(EE) and HOG1 were constructed using pGEX-4T (Pharmacia). GST-tagged wild-type and mutant SKO1 proteins were constructed as described before, expressed in E.coli DH5 and purified using glutathione–Sepharose beads (Pharmacia) in buffer B as described (Posas et al., 1996). HA-tagged HOG1 and SKO1 were expressed in yeast and purification was carried out by immunoprecipitation with anti-HA monoclonal antibody 12CA5 and protein A–Sepharose beads (Roche). Beads were washed extensively with buffer A plus 150 mM NaCl, and resuspended in SDS loading buffer.
In vivo Sko1 phosphorylation assays
Wild-type (MAP37) and hog1Δ (MAP36) cells were grown in YPD and subjected or not to osmotic shock (0.4 M NaCl, 15 min). Cells were lysed in buffer A without EDTA/EGTA and treated or not with 10 U of calf intestinal phosphatase (Roche) in the presence or absence of phosphatase inhibitors.
Wild-type, pbs2Δ and hog1Δ cells containing HA–SKO1 (wild type and mutated derivatives) in yeast expression vector YCpIF16 were grown in selective medium and subjected or not to a brief osmotic shock (0.4 M NaCl, 10 min). Cell extracts were prepared in the presence of buffer A without EGTA and EDTA. Extracts were treated with or without λ phosphatase (300 U for 60 min) in the presence or absence of phosphatase inhibitors. HA–SKO1 was detected by immunoblotting using anti-HA monoclonal antibody 12CA5.
In vitro kinase assays
One microgram of recombinant GST–HOG1 from E.coli was activated by phosphorylation using 0.5 µg of GST–PBS2(EE) in the presence of kinase buffer and ATP as described in Bilsland-Marchesan et al. (2000). After 15 min at 30°C, 5 µg of GST-tagged Sko1 proteins, purified from E.coli, were added to the previous mixture together with [γ-32P]ATP (0.2 µCi/µl). The mixture was then incubated for 5 min at 30°C and the reactions were terminated by the addition of 2× SDS loading buffer. Phosphorylation of GST-tagged Sko1 proteins by PKA (Sigma) was carried out for 15 min at 30°C in the presence of kinase buffer mixture containing [γ-32P]ATP (0.2 µCi/µl). Labeled proteins were resolved by SDS–PAGE. GST-tagged proteins were probed by immunoblot with an anti-GST monoclonal antibody (Pharmacia).
In vivo coprecipitation assays
MAP37 cells carrying pSWM14 plasmid were grown in the presence of galactose and extracts were prepared. Cell extract (750 µg in buffer A plus 150 mM NaCl) was incubated with 50 µl of anti-HA monoclonal antibody 12CA5 and protein A–Sepharose beads (Roche) overnight at 4°C. Beads were washed extensively with buffer A plus 150 mM NaCl, resuspended in loading buffer and separated by SDS–PAGE. Immunoblotting was carried out using anti-HA monoclonal antibody 12CA5 (Roche) and anti-GST monoclonal antibody (Pharmacia) together with ECL reagent (Amersham). When HOG1–GFP and SKO1–HA were coprecipitated, MAP37 cells carrying the HOG–GFP plasmid were grown in the presence of glucose. Extracts were prepared as before and HOG1–GFP was precipitated by incubation overnight at 4°C with anti-GFP polyclonal antibody (gift from P.Silver), followed by the addition of 50 µl of protein A–Sepharose (Pharmacia) for 1 h at 4°C. Samples were washed with buffer A plus 150 mM NaCl. In vivo interaction of GST–SKO1 with Ssn6 or Tup1 was determined by GST pull-down experiments. Protein expression was induced for 2 h with 0.5 mM CuSO4, and cells were subjected or not to stress (0.4 M NaCl, 10 min). Cell extracts [1 mg in buffer A plus 150 mM NaCl supplemented with complete protease inhibitor cocktail (Roche)] were incubated with 20 µl of glutathione–Sepharose 4B (Pharmacia) overnight at 4°C. GST–Sko1 was detected using polyclonal anti-GST antibody (Molecular Probes, 1:5000), and Ssn6 or Tup1 was detected using anti-Ssn6 or anti-Tup1 polyclonal antibodies (kind gift from Drs S.Y.Roth and D.Edmondson, Houston, 1:4000). Signals were detected by ECL.
Northern blot analysis
Total RNA was isolated (Carlson and Botstein, 1982) from YPD- or SCD-grown yeast cells that were either untreated or subjected to the indicated osmotic stress conditions. Thirty micrograms of RNA per lane were separated in formaldehyde gels and blotted onto nylon membranes (Hybond-N, Amersham). Radioactively labelled probes were hybridized in PSE buffer (300 mM NaH2PO4/Na2HPO4 pH 7.2, 7% SDS, 1 mM EDTA). Probes used were PCR fragments spanning nucleotides 77–706 of TBP1 and 115–812 of GRE2. Signals were quantified using a Fujifilm BAS-1500 phosphoimager.
β-galactosidase assay
Transformed yeast strains were grown selectively until saturation in the appropriate SD liquid media and were then diluted in YPD. Logarithmically growing cells (optical density at 660 nm of 0.5–0.8) were treated or not with 0.4 M of NaCl for 15 min, permeabilized by ethanol/toluene treatment and β-galactosidase activity was determined as described in Gaxiola et al. (1992).
Acknowledgments
Acknowledgements
We thank G.Julià for excellent technical assistance and Dr H.Saito for plasmids, strains and advice. We thank Drs S.Y.Roth, D.Edmondson and R.J.Trumbly for generous gifts of anti-Ssn6 and anti-Tup1 antisera, and Dr E.Hidalgo and Dr L.Yenush for valuable comments on the manuscript. This work was supported by a predoctoral grant from the Spanish government to A.P.-A., the European TMR grant ERB-FMRX-CT96-0007 to M.P., and grants from the Ministerio de Ciencia y Tecnología PM99-0028 to F.P. and PB98-0565-C04-02 to J.A.
References
- Bilsland-Marchesan E., Ariño,J., Saito,H., Sunnerhagen,P. and Posas,F. (2000) Rck2 kinase is a substrate for the osmotic stress-activated mitogen-activated protein kinase Hog1. Mol. Cell. Biol., 20, 3887–3895. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boguslawski G. (1992) PBS2, a yeast gene encoding a putative protein kinase, interacts with the RAS2 pathway and affects osmotic sensitivity of Saccharomyces cerevisiae. J. Gen. Microbiol., 138, 2425–2432. [DOI] [PubMed] [Google Scholar]
- Brewster J.L., de Valoir,T., Dwyer,N.D., Winter,E. and Gustin,M.C. (1993) An osmosensing signal transduction pathway in yeast. Science, 259, 1760–1763. [DOI] [PubMed] [Google Scholar]
- Carlson M. and Botstein,D. (1982) Two differentially regulated mRNAs with different 5′-ends encode secreted and intracellular forms of yeast invertase. Cell, 28, 145–154. [DOI] [PubMed] [Google Scholar]
- DeVit M.J. and Johnston,M. (1999) The nuclear exportin Msn5 is required for nuclear export of the Mig1 glucose repressor of Saccharomyces cerevisiae. Curr. Biol., 9, 1231–1241. [DOI] [PubMed] [Google Scholar]
- Ferrigno P., Posas,F., Koepp,D., Saito,H. and Silver,P. (1998) Regulated nucleo/cytoplasmic exchange of HOG1 MAPK requires the importin β homologs NMD5 and XPO1. EMBO J., 17, 5606–5617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Foreman P.K. and Davis,R.W. (1994) Cloning vectors for the synthesis of epitope-tagged, truncated and chimeric proteins in Saccharomyces cerevisiae. Gene, 144, 63–68. [DOI] [PubMed] [Google Scholar]
- Garay-Arroyo A. and Covarrubias,A.A. (1999) Three genes whose expression is induced by stress in Saccharomyces cerevisiae. Yeast, 15, 879–892. [DOI] [PubMed] [Google Scholar]
- Garcia-Gimeno A.M. and Struhl,K. (2000) Aca1 and Aca2, ATF/CREB activators in Saccharomyces cerevisiae, are important for carbon source utilization but not the response to stress. Mol. Cell. Biol., 20, 4340–4349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gaxiola R., deLarrinoa,I.F., Villalba,J.M. and Serrano,R. (1992) A novel and conserved salt induced protein is an important determinant of salt-tolerance in yeast. EMBO J., 11, 3157–3164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Görner W., Durchschlag,E., Martinez-Pastor,M.T., Estruch,F., Ammerer,G., Hamilton,B., Ruis,H. and Schüller,C. (1998) Nuclear localization of the C2H2 zinc finger protein Msn2p is regulated by stress and protein kinase A activity. Genes Dev., 12, 586–597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gustin M.C., Albertyn,J., Alexander,M. and Davenport,K. (1998) MAP kinase pathways in the yeast Saccharomyces cerevisiae. Microbiol. Mol. Biol. Rev., 62, 1264–1300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang M., Zhou,Z. and Elledge,S.J. (1998) The DNA replication and damage checkpoint pathways induce transcription by inhibition of the Crt1 repressor. Cell, 94, 595–605. [DOI] [PubMed] [Google Scholar]
- Maeda T., Takekawa,M. and Saito,H. (1995) Activation of yeast PBS2 MAPKK by MAPKKKs or by binding of an SH3-containing osmosensor. Science, 269, 554–558. [DOI] [PubMed] [Google Scholar]
- Márquez J.A. and Serrano,R. (1996) Multiple transduction pathways regulate the sodium-extrusion gene PMR2/ENA1 during salt stress in yeast. FEBS Lett., 382, 89–92. [DOI] [PubMed] [Google Scholar]
- Márquez J.A., Pascual-Ahuir,A., Proft,M. and Serrano,R. (1998) The Ssn6–Tup1 repressor complex of Saccharomyces cerevisiae is involved in the osmotic induction of HOG-dependent and -independent genes. EMBO J., 17, 2543–2553. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martinez-Pastor M.T., Marchler,G., Schuller,C., Marchler-Bauer,A., Ruis,H. and Estruch,F. (1996) The Saccharomyces cerevisiae zinc finger proteins Msn2p and Msn4p are required for transcriptional induction through the stress response element (STRE). EMBO J., 15, 2227–2235. [PMC free article] [PubMed] [Google Scholar]
- Montminy M. (1997) Transcriptional regulation by cyclic AMP. Annu. Rev. Biochem., 66, 807–822. [DOI] [PubMed] [Google Scholar]
- Nehlin J.O., Carlberg,M. and Ronne,H. (1992) Yeast SKO1 gene encodes a bZIP protein that binds to the CRE motif and acts as a repressor of transcription. Nucleic Acids Res., 20, 5271–5278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pascual-Ahuir A., Serrano,R. and Proft,M. (2001) Sko1p-repressor and Gcn4p-activator antagonistically modulate stress-regulated transcription in S. cerevisiae. Mol. Cell. Biol., 21, 16–25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Posas F., Wurgler-Murphy,S.M., Maeda,T., Witten,E.A., Thai,T.C. and Saito,H. (1996) Yeast HOG1 MAP kinase cascade is regulated by a multistep phosphorelay mechanism in the SLN1-YPD1-SSK1 ‘two-component’ osmosensor. Cell, 86, 865–875. [DOI] [PubMed] [Google Scholar]
- Posas F., Chambers,J.R., Heyman,J.A., Hoeffler,J.P., de Nadal,E. and Ariño,J. (2000) The transcriptional response of yeast to saline stress. J. Biol. Chem., 275, 17249–17255. [DOI] [PubMed] [Google Scholar]
- Proft M. and Serrano,R. (1999) Repressors and upstream repressing sequences of the stress-regulated ENA1 gene in Saccharomyces cerevisiae: bZIP protein Sko1 confers HOG-dependent osmotic regulation. Mol. Cell. Biol., 19, 537–546. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reiser V., Ruis,H. and Ammerer,G. (1999) Kinase activity-dependent nuclear export opposes stress-induced nuclear accumulation and retention of Hog1 mitogen-activated protein kinase in the budding yeast Saccharomyces cerevisiae. Mol. Biol. Cell, 10, 1147–1161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rep M., Reiser,V., Gartner,U., Thevelein,J.M., Hohmann,S., Ammerer,G. and Ruis,H. (1999) Related osmotic stress-induced gene expression in Saccharomyces cerevisiae requires Msn1p and the novel nuclear factor Hot1p. Mol. Cell. Biol., 9, 5474–5485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rep M., Krantz,M., Thevelein,J.M. and Hohmann,S. (2000) The transcriptional response of Saccharomyces cerevisiae to osmotic shock. Hot1p and Msn2p/Msn4p are required for the induction of subsets of high osmolarity glycerol pathway-dependent genes. J. Biol. Chem., 275, 8290–8300. [DOI] [PubMed] [Google Scholar]
- Robinson M.J. and Cobb,M.H. (1997) Mitogen-activated protein kinase pathways. Curr. Opin. Cell Biol., 9, 180–186. [DOI] [PubMed] [Google Scholar]
- Schmitt A.P. and McEntee,K. (1996) Msn2p, a zinc finger DNA-binding protein, is the transcriptional activator of the multistress response in Saccharomyces cerevisiae. Proc. Natl Acad. Sci. USA, 93, 5777–5782. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schneider B.L., Seufert,W., Steiner,B., Yang,Q.H. and Futcher,A.B. (1995) Use of polymerase chain reaction epitope tagging for protein tagging in S. cerevisiae. Yeast, 11, 1265–1274. [DOI] [PubMed] [Google Scholar]
- Vincent A.C. and Struhl,K. (1992) ACR1, a yeast ATF/CREB repressor. Mol. Cell. Biol., 12, 5394–5405. [DOI] [PMC free article] [PubMed] [Google Scholar]