

Abstract
Alternative splicing of human cystic fibrosis transmembrane conductance regulator (CFTR) exon 9 is regulated by a combination of cis-acting elements distributed through the exon and both flanking introns (IVS8 and IVS9). Several studies have identified in the IVS8 intron 3′ splice site a regulatory element that is composed of a polymorphic (TG)m(T)n repeated sequence. At present, no cellular factors have been identified that recognize this element. We have identified TDP-43, a nuclear protein not previously described to bind RNA, as the factor binding specifically to the (TG)m sequence. Transient TDP-43 overexpression in Hep3B cells results in an increase in exon 9 skipping. This effect is more pronounced with concomitant overexpression of SR proteins. Antisense inhibition of endogenous TDP-43 expression results in increased inclusion of exon 9, providing a new therapeutic target to correct aberrant splicing of exon 9 in CF patients. The clinical and biological relevance of this finding in vivo is demonstrated by our characterization of a CF patient carrying a TG10T9(ΔF508)/TG13T3(wt) genotype leading to a disease-causing high proportion of exon 9 skipping.
Keywords: alternative splicing/CFTR exon 9/cystic fibrosis/SF2/ASF/TDP-43
Introduction
RNA splicing mutations in the gene coding for the cystic fibrosis transmembrane conductance regulator (CFTR) protein have been described to lead to dysfunction of several organs such as lung, sweat glands, genital tract, intestine and pancreas, producing the complex CF symptoms (Welsh et al., 1995). CFTR mutations can also be associated with a variety of isolated clinical signs such as congenital bilateral absence of vas deferens (CBAVD) (Chillon et al., 1995; Dörk et al., 1997), nasal polyposis (Irving et al., 1997), bronchiectasis (Pignatti et al., 1995; Girodon et al., 1997), bronchopulmonary allergic aspergillosis (Cockrill and Hales, 1999) or idiopathic pancreatitis (Cohn et al., 1998; Sharer et al., 1998). In particular, the occurrence of CBAVD has been associated with production of an inactive CFTR protein following the loss of exon 9 from the coding mRNA through a process of aberrant alternative splicing (Delaney et al., 1993; Strong et al., 1993).
Several genetic studies have thus been aimed at identifying the cis-acting elements on the human CFTR gene in the vicinity of exon 9 that might explain this unusual splicing process. The elements identified so far (Figure 1A) include a (TG)m(T)n polymorphic element, the recently identified intronic splicing silencer (ISS) in IVS9, and two exon 9 enhancer and silencer elements (Pagani et al., 2000). Initially, variability in a (T)n polymorphic locus located within the 3′ splice site of IVS8 was the first element to be associated with a variable efficiency of exon 9 splicing (Chu et al., 1991). The high proportion of a T5 allele in patients affected by male infertility (caused by obstructive azoospermia, e.g. CBAVD) represents one of the better characterized correlations between the occurrence of a particular polymorphism and the associated clinical signs (Chu et al., 1993; Chillon et al., 1995; Mak et al., 1997; Rave-Harel et al., 1997; Teng et al., 1997; Cuppens et al., 1998; Larriba et al., 1998). Nonetheless, the T5 allele effect has partial penetrance, making it possible to find healthy homozygous carriers. Recently, a second polymorphic locus based on (TG)m repeats (ranging from 9 to 13 repeats in humans) localized immediately upstream of the (T)n tract was found to influence the efficiency of exon 9 splicing (Cuppens et al., 1998). In particular, T5 CFTR genes derived from CBAVD patients carried a high number of TG repeats, whilst T5 CFTR genes derived from healthy fathers carried a low number of TG repeats (Cuppens et al., 1998), suggesting that this element may play a role in the partial penetrance of the T5 allele. In previous studies we have specifically analyzed, using a minigene system, the effect of these two cis-acting elements on exon 9 splicing, and our results confirmed that the (TG)m and (T)n repeats work in concert with each other (Niksic et al., 1999). Moreover, the identification and characterization (in this work) of a CF patient carrying the TG13T3 genotype indicates that the (TG)m(T)n variability is not only associated with monosymptomatic forms of CF but that its extreme variant may also be associated with pancreatic-sufficient CF. Therefore, the identification of the cellular factors eventually binding to these elements represents a key step in understanding the complex regulation of exon 9 splicing. In this study, we identify HIV-1 TAR DNA binding protein (TDP-43) (Ou et al., 1995) as a novel factor binding to the TG element in CFTR exon 9 pre-mRNA and capable of modulating CFTR exon 9 alternative splicing. Most importantly, antisense inhibition of endogenous TDP-43 results in an upregulation of exon 9 inclusion, providing a new therapeutic target to correct aberrant splicing of exon 9 in CF patients.

Fig. 1. (A) A schematic representation of the intronic and exonic elements that affect the splicing of CFTR exon 9: the (TG)m and (T)n polymorphic regions in IVS8, the intronic splicing silencer (ISS) in IVS9, and the exonic enhancer (E) and silencer (S) sequences. (B) A schematic representation of the plasmids used: wild type (11ug/7u) and mutants selectively deleted of the (TG)m and/or (T)n repeats (Δug/Δu), (11ug/Δu) and (Δug/7u). (C) A UV cross-linking assay using HeLa nuclear extract with uniformly labeled RNA of all four constructs linearized with HindIII. The arrows indicate the position of the 50–52 kDa complex.
Results
Identification of cellular proteins binding to the (TG)m element of exon 9
To identify cellular factors binding to the (TG)m and (T)n sequences we prepared the plasmids shown in Figure 1B. The wild-type plasmid contained the CFTR exon 9 sequence, the splicing junctions and part of the flanking introns with the TG11 and T7 repeats (11ug/7u). Three mutated sequences in which we sequentially deleted both repeats (Δug/Δu), the T7 repeat alone (11ug/Δu) and the TG11 repeat alone (Δug/7u) were also prepared. These configurations were previously shown in a transient transfection system to give different proportions of exon 9(+) and 9(–) transcripts (Table I) (Niksic et al., 1999). The constructs were transcribed in vitro in the presence of [32P]UTP and equal quantities of labeled transcripts were then used in a UV cross-linking assay with HeLa nuclear extract. Figure 1C shows that among the numerous proteins that could be cross-linked to the labeled RNAs, a 50–52 kDa approximate molecular weight complex could be observed only in the RNAs that contained the (TG)m repeated sequence (11ug/7u and 11ug/Δu) but not in those without it (Δug/7u and Δug/Δu). The specificity of this interaction was tested by performing competition experiments. As expected, the 50–52 kDa complex could be readily competed away from a labeled 11ug/7u RNA by the addition of increasing amounts of cold 11ug/7u and 11ug/Δu RNA but not by equal amounts of unlabeled Δug/7u and Δug/Δu RNAs (Figure 2A). In addition, competition was not observed when we used homologous RNA transcripts from the mouse CFTR sequence (Figure 2B and C). This observation is consistent with the fact that mouse genomic sequences do not contain either the (ug)11 or the (u)7 repeated sequences (Niksic et al., 1999).
Table I. Percentage of exon 9 exclusion on mRNA derived from minigenes carrying different (TG)m(T)n configurations.
| (TG)m(T)n minigene configuration | % exon 9 exclusion |
|---|---|
| TG11T7 | 10 |
| TG11ΔT | 100 |
| ΔTGT7 | 0 |
| ΔTGΔT | 0 |
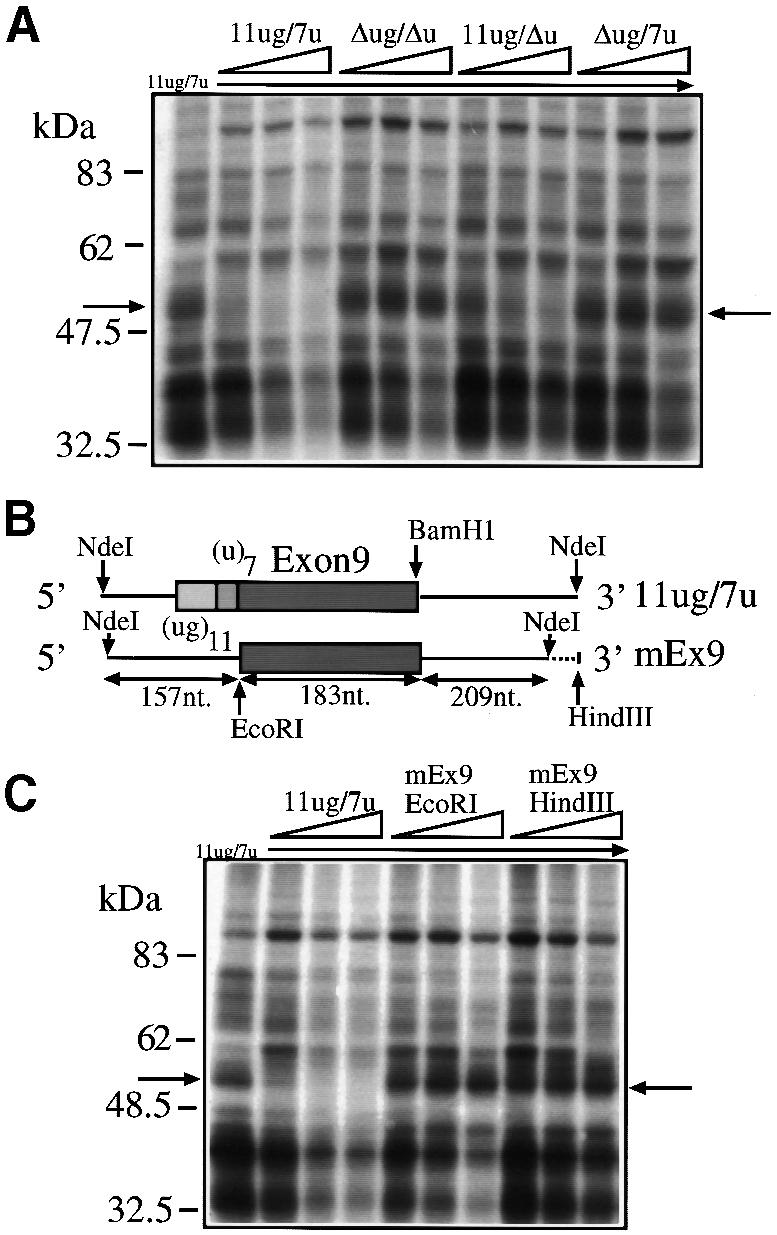
Fig. 2. (A) A competition analysis following addition of cold 11ug/7u, Δug/Δu, 11ug/Δu and Δug/7u RNAs to labeled (11ug/7u) RNA in the presence of HeLa nuclear extract. The molar ratios of cold/labeled RNA were 2, 5 and 10. (B) A schematic representation of the human (11ug/7u) and mouse (mEx9) constructs. (C) A competition analysis using labeled 11ug/7u RNA incubated with HeLa nuclear extracts following addition of cold RNAs: 11ug/7u RNA and two RNAs synthesized by cutting mEx9 with EcoRI (mEx9EcoRI) and with HindIII (mEx9HindIII). The molar ratios of cold/labeled RNA were 2, 5 and 10. The arrows indicate the 50–52 kDa complex.
(ug)m repeated sequences are both necessary and sufficient to obtain binding of the 50–52 kDa complex
The identification of (ug)m repeated sequences as the necessary and sufficient target sequence for binding of this complex was tested using a very short (ug)12 sequence. Figure 3A shows that cold (ug)12 RNA could specifically compete the 50–52 kDa complex from labeled 11ug/7u, whereas a cold (ucuu)3 RNA that contained a poly-pyrimidine sequence from the 3′ splice site of the constitutively spliced-in second exon of the Apo AI gene could not. The fact that this control RNA was not capable of binding the 50–52 kDa complex indicates that this complex is not detected by poly-pyrimidine sequences in general. This possibility had to be investigated because (ug) repeated sequences in the apolipoprotein AII gene context appear to be functionally equivalent to a continuous poly-pyrimidine tract (Shelley and Baralle, 1987) and can promote branch point selection (Coolidge et al., 1997). In addition, if we consider the apparent molecular weight of this protein, a likely candidate for its identity could be the poly-pyrimidine tract binding protein (PTB), a well-known factor involved in splicing (Mulligan et al., 1992). However, this possibility could also be ruled out by the fact that the 50–52 kDa complex was not competed by the (ucuu)3 RNA, which contains three (ucuu) motifs, the preferred SELEX binding motif for PTB binding (Singh et al., 1995).
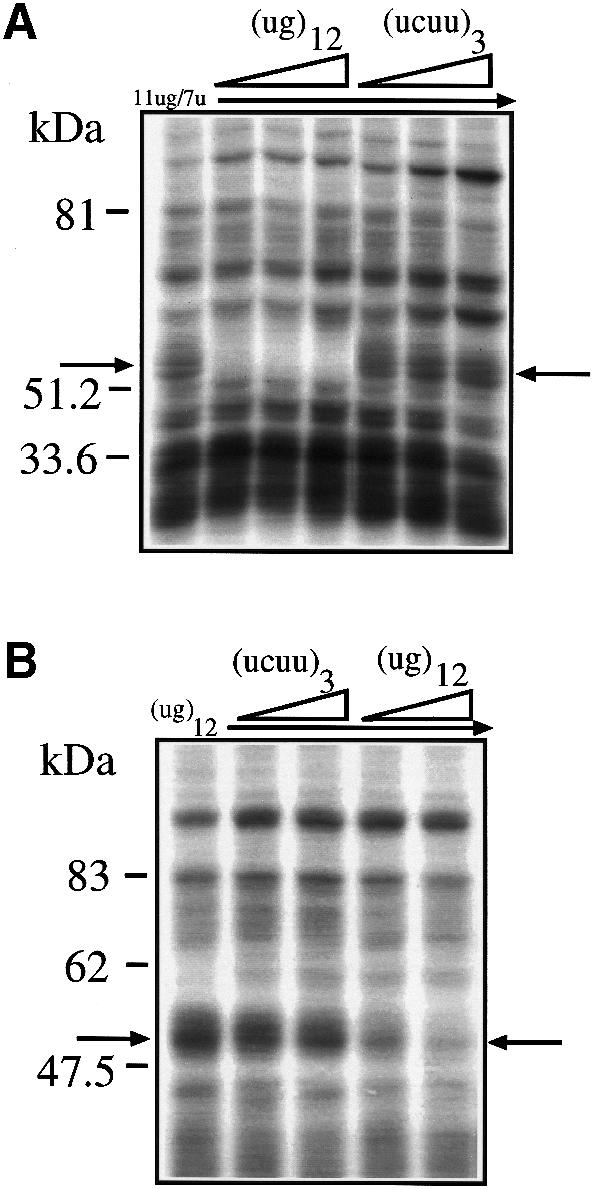
Fig. 3. (A) The effects of two short competitor RNAs, (ug)12 and (ucuu)3, in a UV cross-linking assay in the presence of HeLa nuclear extract and labeled 11ug/7u RNA. The molar ratios of cold/labeled RNA were 3, 8 and 17. (B) The results of labeling the short (ug)12 RNA and performing UV cross-linking in the presence of HeLa nuclear extract and cold (ug)12 and (ucuuu)3 RNAs. The molar ratios of cold/labeled RNA were 2 and 5. The arrows indicate the 50–52 kDa complex.
Finally, Figure 3B shows that a labeled RNA composed exclusively of (ug)12 is capable of binding a 50–52 kDa complex that possesses the same binding characteristics of the complex bound by the 11ug/7u RNA [i.e. it can be competed by cold (ug)12 RNA but not by cold (ucuu)3 RNA].
Identification of TDP-43 as a nuclear factor binding specifically to (ug) repeated sequences
In order to identify proteins that bind specifically to RNAs containing (ug) repeated motifs we set up an affinity purification procedure that involves the cross-linking of a synthetic (ug)12 RNA to adipic acid dehydrazide agarose beads. As control, a second type of beads derivatized with the poly-pyrimidine rich sequence (ucuu)3 was used. Both types of beads were separately incubated with HeLa nuclear extracts and the proteins bound were analyzed on an SDS–PAGE gel and stained with Coomassie Blue. Comparison of the binding patterns of the (ug)12- and the (ucuu)3-derivatized beads (Figure 4A) showed that the two patterns have many bands in common, together with a few (ug)12- and (ucuu)3-specific bands. Particularly, in the molecular weight range compatible with the 50–52 kDa complex a 43 kDa protein was specifically pulled down by the (ug)12 RNA but not by the (ucuu)3 RNA. On the other hand, a 57 kDa protein doublet was specifically pulled down by the (ucuu)3 RNA, which is entirely consistent with the expected PTB molecular weight (Gil et al., 1991). Internal sequence analysis of the excised 43 kDa band yielded a 17mer peptide and a search in the DDBJ/EMBL/GenBank database revealed that its sequence was 100% identical to residues 362–378 of TDP-43 (Ou et al., 1995), a cellular protein that was originally reported to bind an HIV-1 TAR DNA sequence motif and inhibit HIV-1 transcription, but had not been reported to bind RNA. Nonetheless, the presence of two RNA recognition motifs (residues 106–175 and 193–257) justifies the fact that TDP-43 may bind to RNA.

Fig. 4. (A) The results (Coomassie Blue staining) of a pull-down assay using adipic acid dehydrazide beads derivatized with (ug)12 and (ucuu)3 RNAs following incubation with HeLa nuclear extract. In the lane from the (ug)12-derivatized beads the arrow indicates the 43 kDa protein band that is absent in the lane from the (ucuu)3-derivatized beads. The arrows on the right indicate the 57 kDa doublet that is present in the (ucuu)3 lane as opposed to the (ug)12 lane. (B) The full amino acid sequence of TDP-43 with the open box corresponding to the sequenced peptide from the excised 43 kDa band. Bold and underlined sequences highlight the two RRM consensus motifs. (C) (Left panel) The purified recombinant proteins and their reactivity with labeled (ug)12 RNA in a UV cross-linking assay (right panel). (D) The reactivity of the GST–TDP-43 recombinant protein with labeled 11ug/7u, Δug/7u, 11ug/Δu and Δug/Δu RNAs.
Expression of TDP-43 as a GST fusion protein
In order to verify the specific binding of TDP-43 to (ug)12 RNA we amplified and inserted its full coding sequence as a glutathione S-transferase (GST) fusion protein in the pGEX-3X expression vector for bacterial expression. Figure 4C, left panel, shows the affinity-purified GST–TDP-43 fusion protein with the expected 70 kDa molecular weight. Figure 4C, right panel, shows that GST–TDP-43 was capable of binding labeled (ug)12 RNA and that no binding could be detected by the GST protein alone. The ability of this recombinant protein to bind the (ug)11 repeated sequences in the CF exon 9 context was then analyzed using the 11ug/7u, Δug/7u, 11ug/Δu and Δug/Δu labeled RNAs. Figure 4D shows that GST–TDP-43 displays an identical binding pattern to that of the 50–52 kDa complex obtained using the HeLa nuclear extracts (shown in Figure 1C).
The 50–52 kDa complex is formed by TDP-43 binding to RNA
The apparent discrepancy in molecular weight between the 50–52 kDa complex and TDP-43 was initially addressed by performing an immunodepletion assay using agarose A/G beads coated with the polyclonal anti-TDP-43 sera. Using these beads we immunodepleted TDP-43 from total HeLa nuclear extract before performing UV cross-linking analysis. Depletion of TDP-43 from the HeLa nuclear extract results in the disappearance of the 50–52 kDa complex (Figure 5A, upper panel), and a western blot performed after the depletion step showed that TDP-43 was effectively removed from the nuclear extract (Figure 5A, lower panel). We then performed immunoprecipitation experiments using the anti-TDP-43 antiserum and its pre-immunized serum as control. Figure 5B shows that the 50–52 kDa band can be specifically immunoprecipitated by the anti-TDP-43 antiserum following UV cross-linking with nuclear extract of a labeled 11ug/7u RNA (but not from a Δug/Δu control). It should be noted that in addition to the 50–52 kDa complex a faint additional 45 kDa band can be observed. Interestingly, the occurrence of this band is dependent on the amount of HeLa nuclear extract used in the UV cross-linking assay (Figure 5C), becoming increasingly evident with higher amounts of nuclear extract. The data shown in Figure 5C may suggest that we are detecting protein complexes consisting of TDP-43 and a still uncharacterized 50–52 kDa protein. Alternatively, this finding may indicate that the efficiency of the RNase to cleave the RNA–protein complex varies according to the amount of TDP-43 binding to the (TG)m motif. In fact, it has been previously reported that RNase treatment of a UV cross-linked RNA–protein complex leaves a variable number of residual nucleotides bound to the protein, modifying its molecular weight. For example, molecular weight shifts following UV cross-linking are a common feature for several members of the SR protein family (Ramchatesingh et al., 1995), which following UV cross-linking have been ascribed to bind RNA irreversibly and protect substantial stretches of it from RNase degradation. Indirect evidence for this is shown in Figure 5D in which the addition of increasing quantities of a recombinant GST–TDP-43 fusion protein to total nuclear extract can effectively compete for the binding of the 50–52 kDa complex to the labeled 11ug/7u RNA. It is important to note that no other protein is competed away by addition of GST–TDP-43, a result that although not formal proof is indicative that the 50–52 kDa complex contains TDP-43.

Fig. 5. (A) (Upper panel) The reactivity of labeled 11ug/7u RNA in the presence of 18 µg of HeLa nuclear extract (NE) and 18 µg of TDP-43 immunodepleted nuclear extract (NE-TDP-43). (Lower panel) A western blot assay demonstrating that immunodepletion of TDP-43 has occurred. (B) An immunoprecipitation experiment using anti-TDP-43 sera (and its pre-immune sera as control) on labeled 11ug/7u and Δug/Δu RNAs UV cross-linked with 18 µg of nuclear extract. (C) An immunoprecipitation following UV cross-linking of labeled 11ug/7u RNA with increasing quantities of nuclear extract. The arrow indicates the 50–52 kDa immunoprecipitated product whilst the asterisk indicates the second immunoprecipitated band. (D) The effect of UV cross-linking labeled 11ug/7u RNA with GST–TDP-43 alone (50 ng), with nuclear extract alone (18 µg) and nuclear extract mixed with increasing quantities of GST–TDP-43 (10, 25 and 50 ng, respectively). The 50–52 kDa complex and GST–TDP-43 protein are indicated by arrows. (E) A schematic diagram (top) of the recombinant GST–TDP-43(101–261), its expression and purification (left panel), and its reactivity with synthetic 5′ end-labeled (ug)12 RNA following UV cross-linking with (+) and without (–) RNase treatment (right panel). In the third lane (–/+) these two were mixed and loaded together in the same lane. Only 10% of the untreated sample was loaded in the (–) and (–/+) lanes. The lower amount of labeled material in the (+) lane is due to loss of the labeled 5′ end of the synthetic (ug)12 following RNase digestion.
In order to test directly the effects of residual RNA binding on TDP-43 migration we have expressed a 43 kDa TDP-43 recombinant variant, GST–TDP-43(101–261), to facilitate visualization of migration shifts (Figure 5E, top and left panel). UV cross-linking analysis was then performed with a synthetic 5′ end-labeled (ug)12 RNA oligo in the presence or absence of RNase treatment. The results demonstrate that the 43 kDa recombinant protein in the Coomassie staining (Figure 5E, left panel) migrates in the 50–52 kDa range when it is irreversibly bound to the ribonucleotide (Figure 5E, right panel). After RNase treatment there is a partial shift towards a lower molecular weight (Figure 5E, right panel) although it never reaches the original 43 kDa. The mixing of the samples (–/+) loaded in the minus (–) and plus (+) lanes shows conclusively that there is a limit product of RNase digestion, smaller than the 50–52 kDa range but higher than the original 43 kDa protein [and compatible with a sequence around (ug)8 being totally protected from RNase digestion]. It should be noted that being the synthetic 5′ end-labeled (ug)12 we can see only the molecules with the 5′ end protected. These experiments provide a direct functional evidence for significant molecular weight shift of TDP-43 in the 50–52 kDa range following UV cross-linking analysis.
Overexpression of TDP-43 and SF2/ASF in transfection assays induces CFTR exon 9 skipping
To evaluate the functional significance of the TDP-43–(ug)m interaction on CFTR exon 9 alternative splicing, Hep3B cells were transfected with different CFTR exon 9 hybrid minigene variants. Figure 6A shows a schematic representation of the hybrid minigenes prepared with different polymorphic repeats at the intron 8–exon 9 junction. Several variants, including the TG13T3 allele found in a pancreatic-sufficient CF patient (see below), were simultaneously co-transfected in Hep3B cells with plasmids coding for TDP-43 or for the splicing factor SF2/ASF, which has previously been shown to induce CFTR exon 9 skipping (Nissim-Rafinia et al., 2000; Pagani et al., 2000), or both. Figure 6B, left panel, shows that the proportion of exon 9 exclusion was strictly dependent on the composition of the polymorphic locus, being directly related to the (TG)m polymorphic number and inversely related to the (T)n polymorphic number. In fact, in the absence of overexpressed factors the highest level of exon exclusion was observed with high (TG)m repeats, ranging from 55% (TG11T3) to 86% (TG13T3). Most importantly, overexpression of TDP-43 resulted in a greater amount of CFTR exon 9(–) mRNA in all the four variants tested. To better define the CFTR exon 9 regulation we also co-expressed SF2/ASF, which binds to a different region in intron 9, the ISS (Pagani et al., 2000). Overexpression of SF2/ASF and TDP-43 with the hybrid minigene that presented the lowest rate of exon exclusion (TG11T5) resulted in an enhanced inhibitory effect, indicating that binding of the two factors on either side of the exon has an additive effect. Co-transfection with other minigenes was not performed since their high percentage of exon 9 exclusion prevented the detection of any significant additive effect. As a control, we also performed a co-transfection experiment on a hybrid minigene containing the fibronectin EDA exon (Muro et al., 1999) along with the CFTR and TDP-43 or SF2/ASF constructs. The results (Figure 6B, right panel) show that TDP-43 did not affect the splicing pattern of the EDA exon [which lacks any (TG)m structure] whereas SF2/ASF produced, as previously reported, an increase in EDA exon inclusion (Cramer et al., 1999). Finally, co-transfection of increasing amounts of TDP-43 along with the TG13T5 minigene confirms that the inhibitory effect on the inclusion of exon 9 follows a dose-dependent curve that, considering the high level of basal exclusion, was statistically significant in the 3 and 5 µg data points (Figure 6C). It is also interesting to note that a high number of TGs and a low number of Ts resulted in the appearance of an additional band between the exon 9(+) and 9(–) forms (Figure 6B, indicated by an arrow). Sequence analysis of this band showed that it was generated by the use of a cryptic AG acceptor site located in exon 9. This product has never been reported in healthy individuals, nor has it been detected in patients with T5 or in lymphoblasts from the patient with T3 alleles (see below). Its absence may be explained by the distinct sequence context of the minigene construct but its eventual presence in some of the affected tissues in CF patients can not yet be excluded.

Fig. 6. (A) A schematic representation of the hybrid minigene, hCF-(TG)m(T)n. The minimal α-globin promoter and SV40 enhancer are indicated by a small arrow at the 5′ end, the polymorphic locus (TG)m(T)n by a gray circle, and the α-globin, fibronectin EDB and human CFTR exons by black, shaded and white boxes, respectively. The primers used in the RT–PCR assay are indicated by the superimposed arrows. (B) Left panel, the expression of selected minigene variants in the presence of plasmids overexpressing either TDP-43 or SF2/ASF. Exon 9 positive (+) and negative (–) bands are indicated. The arrow indicates an aberrant splicing product originating from a cryptic 3′ splice site. The percentage of exon exclusion for each construct either alone or in the presence of SF2/ASF (500 ng), TDP-43 (3 µg) or both, is reported in the lower graph. Right panel, the effect of TDP-43 and SF2/ASF overexpression on the fibronectin EDA exon. (C) Left panel, a dose–response curve of exon 9 exclusion in the presence of increasing amounts of TDP-43 (0.5–5 µg transfected plasmid) on the TG13T5 minigene. Mean values from four independent transfection experiments performed as duplicates are shown with standard errors (right panel). The asterisks indicate statistical significance (P <0.05). M, molecular weight markers (1 kb).
Transient transfections of antisense oligos directed against TDP-43 mRNA induce CFTR exon 9 inclusion
The high level of endogenous TDP-43 found in different cell lines tested by western blotting (not shown) may be responsible for the low effect on CFTR alternative splicing of overexpressing TDP-43. An alternative approach to test its function is then to inhibit TDP-43 expression by designing several antisense phosphorothioate (PS)-oligodeoxynucleotides on different regions of the TDP-43 sequence (Figure 7A). The PS-oligos were then transfected in Hep3B along with the TG13T5 minigene. The results showed a consistent and significant increase of exon 9 inclusion in the final minigene transcript for three oligos (TIO86, TIO155, TIO1318), whilst no variation in exon 9 inclusion and/or TDP-43 endogenous levels was observed when we used a control oligo, FN56 (Figure 7B). Interestingly, the most efficient PS-oligos were TIO155 and TIO1318, which were designed to contain a sequence complementary to the GGGA motif that was recently proposed to be more accessible to antisense oligonucleotides than other sites (Tu et al., 1998). A dose–response analysis with TIO1318 shows that the inhibitory effect is dependent on oligo concentration and that there is a concomitant gradual decrease of TDP-43 endogenous levels as determined by western blot analysis (Figure 7C, upper and lower panels).

Fig. 7. Antisense inhibition of TDP-43 in Hep3B cells transfected with the TG13T5 minigene. (A) A schematic diagram of four PS-oligodeoxy nucleotides (TIO7, TIO86, TIO155 and TIO1318) used. Hep3B cells were co-transfected with 3 µg of TG13T5 minigene and each PS- or a control oligo (FN56) at a final concentration of 1 µM (B, left panel). A TG13T3 control was also included (pRc/CMV). Exon 9 inclusion levels are reported (right panel). (C) A dose–response curve with oligo TIO1318 ranging from 0.1 to 5 µM (upper panel) together with a western blot of endogenous TDP-43 levels (lower panel).
Distribution of TDP-43 and SF2/ASF mRNA in human tissues
A detailed analysis of the distribution of TDP-43 in normal human tissues was carried out by northern blotting. Previous studies had indicated that it was expressed in a variety of human tissues (Ou et al., 1995). In order to confirm and extend this observation we used polyA(+) northern blots with samples from normal human tissues. Figure 8A (upper panels) shows that the specific TDP-43 transcript (2.8 kb) could be detected in all the human tissues present. A similar result was obtained (Figure 8A, middle panels) when we used a specific probe to detect the 3.0 kb transcript of SF2/ASF (Ge et al., 1991). An attempt to normalize these values was performed by calculating the ratios of SF2/ASF and TDP-43 mRNAs to GAPDH levels (Figure 8A, lower panels). With caution due to the variability of GAPDH levels (particularly in heart and stomach muscle) the quantitation indicates that the relative expression levels of SF2/ASF and TDP-43 vary considerably among the different tissues, as reported in Figure 8B. In particular, the expression of TDP-43 and SF2/ASF was high in pancreas, placenta, lung, genital tract and spleen (all but the last are the tissues mostly affected in monosymptomatic CF and CBAVD).

Fig. 8. (A) Northern blot analysis of TDP-43 (upper panels), SF2/ASF (middle panels) and GAPDH (lower panels) performed on mRNA extracted from 16 different human tissues. The arrows indicate the major 2.8 kb mRNA species characteristic of TDP-43 and the 3.0 kb mRNA of SF2/ASF. (B) A graphical representation of the normalized TDP-43 mRNA levels (black boxes) and SF2/ASF levels (shaded boxes).
In vivo significance of the extreme TG13T3 polymorphism
Polymorphic variations in the (T)n and (TG)m elements, in particular the T5 alleles, have been associated with variable penetrance of monosymptomatic forms of CF (Chu et al., 1993; Chillon et al., 1995; Mak et al., 1997; Rave-Harel et al., 1997; Cuppens et al., 1998; Larriba et al., 1998) and can also modulate the effect of other CFTR gene mutations in cis (Kiesewetter et al., 1993). Because the TG13T3 configuration led to a particularly high proportion of exon skipping in our transfection assays, it was interesting to determine the clinical phenotype associated with this allele. In fact, the mutation screening of 100 German CF patients with at least one non-ΔF508 chromosome identified the T3 allele in one 19-year-old male who suffered from a mild CF characterized by pulmonary symptoms with recurrent infections, elevated sweat chloride concentrations and pancreatic sufficiency. His CFTR gene sequence showed no exonic mutations and the presence of a TG13T3(wt) element in trans with TG10T9(ΔF508) on the second allele (Figure 9A). The segregation analysis of the polymorphic variants at the intron 8–exon 9 junction and of the mutation ΔF508 in exon 10 proved that ΔF508 and TG13T3 are located on different alleles (Figure 9A), as the affected patient has inherited the TG10T9(ΔF508) allele from the father and the TG13T3(wt) variant from the mother whereas the healthy sister did not carry either the TG10T9(ΔF508) or the TG13T3(wt) allele. The TG13T3(wt) allele was not observed in a further 100 non-CF chromosomes from random donors, indicating that it represents a rare mutational event. The proportion of exon 9 skipping was evaluated in Epstein–Barr virus (EBV) transformed lymphocytes derived from the patient by means of RT–PCR amplification with primers located in exon 8 and exon 11, respectively (Figure 9B). Semiquantitative analysis of the RT–PCR products after their separation on a denaturing polyacrylamide gel revealed that the proportions of the four species TG10T9(ΔF508), 9(–):TG13T3 9(–):TG10T9(ΔF508), 9(+):TG13T3 9(+) were 11:61:24:4, indicating that the T3 allele in the heterozygous patient allows for some 6% normal splicing in his lymphoblasts (a result that is entirely consistent with the in vitro transfections) (Figure 9C). In another set of experiments (not shown), the patient’s non-ΔF508 cDNA was selectively amplified using a reverse primer specific for the F508 wild-type sequence within exon 10. Quantitation of the two allele-specific RT–PCR products confirmed that exon 9 skipping accounted for 96 ± 3% of the patient’s cDNA. In conclusion, this is the highest amount of exon skipping reported thus far for a naturally occurring (TG)m(T)n variant, thereby explaining the CF phenotype of this patient. A control lymphoblastoid cell line established from an individual with a TG11T7/TG10T7 genotype showed a proportion of 18 ± 1% exon 9 skipping in the same assay.

Fig. 9. (A) A family tree and a sequencing analysis of the pancreatic-sufficient CF patient carrying the TG13T3(wt) allele on one chromosome and the TG10T9(ΔF508) configuration on the other chromosome. (B) RT–PCR products spanning exons 8–11 of the CFTR cDNA, obtained from the CF patient compound heterozygous for the TG13T3 mutation and the TG10T9(ΔF508) allele (lane 1) and from a control individual compound heterozygous for the TG11T7 and TG10T7 alleles (lane 2), separated on a 2% agarose gel. The percentage of exon 9 exclusion in each CFTR transcript from the two alleles, as determined after denaturing PAGE separation, is given below. Lane 3, no template; M, 1 kb marker. (C) A semiquantitative analysis of exon 9 skipping. The RT–PCR products from the CF patient (upper profile), a control individual (middle profile), and a negative control (lower profile) were separated on a denaturing polyacrylamide gel using an ALF sequencer. Fluorescence signals were quantified using the Fragment Manager software. Peaks 1 and 3 correspond to the amplified 9(–) and 9(+) fragments from the TG10T9(ΔF508) allele whilst peaks 2 and 4 correspond to amplified 9(–) and 9(+) fragments from non-ΔF508 alleles.
Discussion
In this study we report the identification of TDP-43 (Ou et al., 1995) as a trans-acting factor binding to the (TG)m polymorphic repeat region near the 3′ splice site of human CFTR exon 9 and inducing exon skipping. This cellular protein has originally been described to bind a poly-pyrimidine-rich region of the HIV-1 TAR DNA element and function as a transcriptional inhibitor. TDP-43 has never been described as affecting the splicing process and its exact cellular function has remained elusive (Ou et al., 1995). Our analyses show that TDP-43 is involved in the formation of the 50–52 kDa complex that assembles on the (TG)m element at the 3′ splice site of CFTR exon 9. Overexpression of this protein in transfection experiments inhibits CFTR exon 9 splicing and this inhibitory role becomes more evident with a decrease in the number of (T)n repeats, a result consistent with previous studies on the (TG)m(T)n interdependence (Niksic et al., 1999; Pagani et al., 2000). Conversely, inhibition of TDP-43 expression in cultured cells by antisense PS-oligodeoxynucleotide treatment leads to an increase in exon 9 recognition. This result is particularly important because TDP-43 may represent a novel therapeutic target for some CF patients.
The importance of the (TG)m sequence and of the adjacent (T)n polymorphic region in the regulation of alternative splicing of human CFTR exon 9 has been the subject of several genetic and molecular studies, owing to the clear association of certain alleles of this characteristic polymorphism with monosymptomatic forms of CF (Chu et al., 1993; Chillon et al., 1995; Rave-Harel et al., 1997; Cuppens et al., 1998). Considering that exon 9 skipping produces a non-functional CFTR protein (Delaney et al., 1993; Strong et al., 1993) the presence of the (TG)m repeat at the 3′ end of intron 8 could be considered as a disturbing element interfering with the maturation process of the CFTR pre-mRNA. The absence of the polymorphic (TG)m repeat in mouse CFTR exon 9 (which is not subject to alternative splicing) suggests that the introduction of this region could be part of a more complex event that placed this (TG)m element adjacent to the 3′ splice site of human exon 9 early during the course of evolution (Rozmahel et al., 1997; Kazazian and Moran, 1998). In support of this hypothesis, our recent sequencing of the mouse introns flanking mouse exon 9 has shown that they lack the two intronic regulatory elements and are also of very different length when compared with the human introns (Niksic et al., 1999). The results presented in this paper suggest that the disturbing influence of the (TG)m–TDP-43 complex on the 3′ splice site can be rescued by a long poly-pyrimidine (T)n, which might distance the (TG)m repeat from the 3′ splice site. The in vivo importance of this ‘masking effect’ aggravated by lower numbers of T repeats is evident from the association of T5 alleles with certain clinical entities such as obstructive azoospermia or milder forms of CF, and is underscored by the report presented in this study of a TG13/T3(wt) allele in a pancreatic-sufficient CF patient. In fact, our in vitro and in vivo studies show that the novel TG13T3 allele leads to the largest extent of exon skipping reported thus far for a naturally occurring (TG)m(T)n variant, sufficiently high to explain the CF phenotype of this patient.
Considering that no specific cellular function has yet been ascribed to TDP-43 we lack any indication of a possible link between TDP-43 and the splicing machinery that, at some stage, must be affected by the presence of this protein. Taking into account the vicinity of the (T)n tract to the (TG)m region, one of the necessary trans-acting splicing factors that may be affected by TDP-43 is U2AF65 (Valcarcel et al., 1996), which is known to interact with the poly-pyrimidine tracts near the 3′ splice site. Further work is now aimed at clarifying this issue and whether the function of the (T)n region could be to modulate this interaction, providing us with a likely explanation for the interdependence of the (TG)m and (T)n regions on exon 9 splicing.
Finally, measurement of the levels of TDP-43 and SF2/ASF mRNAs in different human tissues shows that both proteins are abundant in pancreas, lung and genital tract, three organs that are known to be particularly affected by CF. In this respect, it should be noted that the reported distribution of the SF2/ASF protein in rat tissues has also confirmed that it is preferentially expressed in pancreas, lung and genital tract (Hanamura et al., 1998). The fact that in our transient transfection system the overexpression of SF2/ASF together with TDP-43 results in an additive inhibitory effect on exon 9 recognition, makes it tempting to speculate that in patients these two factors might act in concert to lower the 9(+) transcript. This concerted inhibitory effect would be predictably more pronounced in those organs where these two factors are most abundantly expressed. The potential variability of TDP-43 and SF2/ASF concentrations also among the same tissues of different individuals provides one possible molecular basis to explain the T5 allele partial penetration, and why genital tract, pancreas and lung involvement varies in CF patients even when they have the same mutations. However, the complexity of the regulating regions found near and inside this exon indicate that further work will be required before obtaining a complete picture of the interactions that influence this process.
Materials and methods
Plasmid construction and RNA transcription
The wild-type and mutant constructs containing the human and mouse CFTR genomic region have been described previously (Niksic et al., 1999; Pagani et al., 2000). Plasmids (ucuu)3 and (ug)12 were obtained by annealing the following sense and antisense primers: 5′-TCCTCCTCCTTCTTCTTCTTCAGG-3′, 5′-CCTGAAGAAGAAGAAGGAGGAGGA-3′, 5′-TGTGTGTGTGTGTGTGTGTGTGTGT-3′ and 5′-ACA CACACACACACACACACACAC-3′, and cloning them in pBls SK cut with ClaI and blunted with Klenow. All plasmids were linearized by digestion with an appropriate restriction enzyme. Transcription of cold and labeled RNA, nuclear extract preparation and UV cross-linking analysis have been described previously (Pagani et al., 2000). In all UV cross-linking experiments the quantity of nuclear extract used in each reaction was 18 µg unless specifically stated. Molar quantities of competitors are described in each figure legend. Samples were loaded on a 10% SDS–PAGE gel that was subsequently dried and exposed to Kodak X-Omat AR films for 12–24 h.
Affinity purification of TDP-43
One nanomole of (ucuu)3 or (ug)12 cold RNA (∼7.9 µg) was placed in a 400 µl reaction mixture containing 0.1 M NaOAc pH 5.0 and 5 mM sodium m-periodate (Sigma). Reaction mixtures were incubated for 1 h in the dark at room temperature (RT). The RNA was ethanol precipitated and resuspended in 500 µl of 0.1 M NaOAc pH 5.0. Then, 400 µl of adipic acid dehydrazide agarose bead 50% slurry (Sigma) were washed four times in 10 ml of 0.1 M NaOAc pH 5.0, and pelleted after each wash at 3000 r.p.m. for 3 min. After the final wash, 300 µl of 0.1 M NaOAc pH 5.0 were added to the beads. The slurry was then mixed with the periodate-treated RNA and incubated for 12 h at 4°C on a rotator. The beads with the bound RNA were then pelleted and washed three times in 2 ml of 2 M NaCl and three times in 3 ml of buffer D (20 mM HEPES–KOH pH 7, 6.5% v/v glycerol, 0.1 M KCl, 0.2 mM EDTA, 0.5 mM dithiothreitol). They were incubated with 0.6 mg of HeLa cell nuclear extract for 20 min at 30°C in 650 µl final volume, pelleted by centrifugation at 1000 r.p.m. for 3 min, and washed five times with 5 ml of buffer D containing 4 mM MgCl2. After the final centrifugation, 60 µl of SDS–PAGE sample buffer were added to the beads and heated for 5 min at 90°C before loading on a 10% SDS–PAGE gel. Internal sequence analysis from the Coomassie-stained band excised from the SDS–PAGE gel was performed by Eurosequence B.V. (Nijenborgh 4, NL9747 AG, Groningen, The Netherlands).
Expression of recombinant TDP-43 as GST fusion proteins
The full coding sequence of TDP-43 was amplified by PCR following RT–PCR from HeLa total RNA using the forward and reverse primers: 5′-ATGTCTGAATATATTCGGGTAACCGA-3′ and 5′-CTACATTCCCCAGCCAGAAGACTTA-3′. The central portion of TDP-43 (aa 101–261) was amplified using the following reverse and forward oligos: 5′-CAGAAAACATCCGATTTA-3′ and 5′-CTATTCGGCATTGGATATATG-3′. Both PCR products were cloned into the SmaI site of plasmid pGEX-3X (Pharmacia) and the resulting recombinant proteins were expressed and purified with glutathione S–Sepharose 4B beads (Pharmacia) according to the manufacturer’s instructions.
Immunoprecipitation and immunodepletion of TDP-43
Polyclonal antiserum against TDP-43 was obtained by immunizing a 3-month-old rabbit (New Zealand strain) according to standard protocols. Antibodies were subsequently bound to protein A/G PLUS–agarose beads (Santa Cruz Biotechnology) following 1 h incubation at RT in IP buffer (20 mM Tris pH 8.0, 300 mM NaCl, 1 mM EDTA, 0.25% NP-40). The beads were then divided in 20 µl aliquots and incubated for 2 h at RT with different UV cross-linked samples (in a final volume of 1 ml of IP buffer). Each sample was washed five times with 1 ml of IP buffer before addition of SDS-loading buffer and loaded on a 10% SDS–PAGE gel. Immunodepletion was performed by applying 72 µg of total HeLa nuclear extract in 100 µl final volume of 10 mM Tris pH 7.4, 100 mM NaCl, 2.5 mM MgCl2, 0.5% Triton X-100 buffer with anti-TDP-43 A/G beads. After 1 h incubation at RT the depleted extract was separated by centrifugation at 3000 r.p.m. for 5 min. Fractions of the supernatant (25 µl) were removed for subsequent UV cross-linking and western blotting. Western blotting was performed according to standard protocols using the rabbit anti-TDP-43 sera at a dilution of 1:2000 and developed using ECL (Amersham).
Northern blotting of TDP-43 and SF2/ASF
Northern blot analyses were performed on commercially available human poly(A)+ northern blots (Clontech). The probe for TDP-43 was a uniformly labeled EcoRI–NdeI DNA fragment corresponding to residues 59–165 of its coding region. The probe for SF2/ASF was obtained by amplifying the entire full cDNA coding sequence. Each blot was hybridized at 68°C and washed under standard conditions. A Camberra Packard Instant Imager was then used to quantify the mRNA levels and a GAPDH probe was used to normalize for different loadings of RNA in each lane.
Overexpression of TDP-43 and SF2/ASF, and antisense experiments
hCF-(TG)m(T)n minigenes, SF2/ASF-coding plasmid and fibronectin EDB minigene have been described (Pagani et al., 2000). Mammalian expression vector for TDP-43 was obtained by subcloning in pRc-CMV vector the 2743 bp TDP-43 cDNA sequence (Resource Center and Primary Database, Berlin, Germany). Minigene expression analysis was performed on Hep3B cells that were transiently transfected with DOTAP (Roche Diagnostic). The total amount of DNA transfected in each sample was taken to 6 µg (or 7.5 when the EDB minigene was included) with the control empty vector pRc/CMV. Forty-eight hours post-transfection, RT–PCR was carried out on total RNA. For quantitation of the PCRs, [α-32P]dCTP was included in the PCR mixture, the products loaded on 6% native polyacrylamide gel, dried, and exposed to a Instant Imager. The counts of each splicing band were corrected for C/G content. Experiments were performed in duplicate and repeated at least three times. Antisense PS-oligodeoxynucleotides were synthesized by MWG-Biotech: TIO7 (5′-ACCCAAGCGCAGCCCAGCCA-3′), TIO86 (5′- CCGAATATATTCAGACATCT-3′), TIO155 (5′-GAGAGCAGCACCGTCCCATC-3′), TIO1318 (5′-CTGTCTACATTCCCCAGCCA-3′) and FN56 (5′-GTCACCCGCACTCGATATCCAG-3′. Hep3B 70–80% confluent cells were co-transfected with 3 µg of minigene hCF-TG13T5 and different amounts of PS-oligodeoxynucleotide using DOTAP. Twenty-eight hours post-transfection, total RNA was extracted and protein lysates (30 µg) were used in western blot assay.
Characterization of the TG13T3 mutation
Genomic DNA was extracted from EDTA blood samples of patient and family members by a routine salting out procedure. Mutation screening of the CFTR gene was performed by SSCP analysis and direct sequencing as described (Dörk et al., 1997). Lymphoblastoid cell lines were established by EBV transformation (Neitzel, 1986). RT–PCR was carried out using 5 µg of total RNA with fluorescein-labeled primers: forward 5′- CAGAAGTAGTGATGGAGAATGTAAC-3′ (exon 8), reverse 5′-GTTGACCTCCACTCAGTGTGATTC-3′ (exon 11) or 5′-TTCATCATA GGAAACACCAAAG-3′ (exon 10, codon 508 specific). Thirty-five cycles of PCR were performed with an annealing at 62°C for 1 min, extension at 72°C for 75 s and denaturation at 94°C for 45 s. For semiquantitative analysis of exon 9 skipping, RT–PCR products were separated on either 8 or 10% denaturing polyacrylamide gels using an ALF sequencer and fluorescence signals were quantified using the Fragment Manager software (Pharmacia).
Acknowledgments
Acknowledgements
We cordially thank the CF family for taking part in this study and PD Dr K.-M.Keller for clinical information. We also wish to thank Sabine Borgmann and Regina Bendix for their help in the establishment and maintenance of lymphoblastoid cell lines, Javier Caceres for the plasmid expressing SF2/ASF, and Michela Zotti and Cristiana Stuani for skilful technical assistance. E.Z. is supported by a grant from AIRH. This work was supported by the Telethon Onlus Foundation Grant E1038.
References
- Chillon M. et al. (1995) Mutations in the cystic fibrosis gene in patients with congenital absence of the vas deferens. N. Engl. J. Med., 332, 1475–1480. [DOI] [PubMed] [Google Scholar]
- Chu C.S. et al. (1991) Variable deletion of exon 9 coding sequences in cystic fibrosis transmembrane conductance regulator gene mRNA transcripts in normal bronchial epithelium. EMBO J., 10, 1355–1363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chu C.S., Trapnell,B.C., Curristin,S., Cutting,G.R. and Crystal,R.G. (1993) Genetic basis of variable exon 9 skipping in cystic fibrosis transmembrane conductance regulator mRNA. Nature Genet., 3, 151–156. [DOI] [PubMed] [Google Scholar]
- Cockrill B.A. and Hales,C.A. (1999) Allergic bronchopulmonary aspergillosis. Annu. Rev. Med., 50, 303–316. [DOI] [PubMed] [Google Scholar]
- Cohn J.A., Friedman,K.J., Noone,P.G., Knowles,M.R., Silverman,L.M. and Jowell,P.S. (1998) Relation between mutations of the cystic fibrosis gene and idiopathic pancreatitis. N. Engl. J. Med., 339, 653–658. [DOI] [PubMed] [Google Scholar]
- Coolidge C.J., Seely,R.J. and Patton,J.G. (1997) Functional analysis of the polypyrimidine tract in pre-mRNA splicing. Nucleic Acids Res., 25, 888–896. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cramer P., Caceres,J., Cazalla,D., Kadener,S., Muro,A., Baralle,F. and Kornblihtt,A. (1999) Coupling of transcription with alternative splicing: RNA pol II promoters modulate SF2/ASF and 9G8 effects on an exonic splicing enhancer. Mol. Cell, 4, 251–258. [DOI] [PubMed] [Google Scholar]
- Cuppens H. et al. (1998) Polyvariant mutant cystic fibrosis transmembrane conductance regulator genes. The polymorphic (Tg)m locus explains the partial penetrance of the T5 polymorphism as a disease mutation. J. Clin. Invest., 101, 487–496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Delaney S.J., Rich,D.P., Thomson,S.A., Hargrave,M.R., Lovelock,P.K., Welsh,M.J. and Wainwright,B.J. (1993) Cystic fibrosis transmembrane conductance regulator splice variants are not conserved and fail to produce chloride channels. Nature Genet., 4, 426–431. [DOI] [PubMed] [Google Scholar]
- Dörk T. et al. (1997) Distinct spectrum of CFTR gene mutations in congenital absence of vas deferens. Hum. Genet., 100, 365–377. [DOI] [PubMed] [Google Scholar]
- Ge H., Zuo,P. and Manley,J.L. (1991) Primary structure of the human splicing factor ASF reveals similarities with Drosophila regulators. Cell, 66, 373–382. [DOI] [PubMed] [Google Scholar]
- Gil A., Sharp,P.A., Jamison,S.F. and Garcia-Blanco,M.A. (1991) Characterization of cDNAs encoding the polypyrimidine tract-binding protein. Genes Dev., 5, 1224–1236. [DOI] [PubMed] [Google Scholar]
- Girodon E. et al. (1997) CFTR gene mutations in adults with disseminated bronchiectasis. Eur. J. Hum. Genet., 5, 149–155. [PubMed] [Google Scholar]
- Hanamura A., Caceres,J.F., Mayeda,A., Franza,B.R.,Jr and Krainer,A.R. (1998) Regulated tissue-specific expression of antagonistic pre-mRNA splicing factors. RNA, 4, 430–444. [PMC free article] [PubMed] [Google Scholar]
- Irving R.M., McMahon,R., Clark,R. and Jones,N.S. (1997) Cystic fibrosis transmembrane conductance regulator gene mutations in severe nasal polyposis. Clin. Otolaryngol., 22, 519–521. [DOI] [PubMed] [Google Scholar]
- Kazazian H.H. Jr and Moran,J.V. (1998) The impact of L1 retrotransposons on the human genome. Nature Genet., 19, 19–24. [DOI] [PubMed] [Google Scholar]
- Kiesewetter S. et al. (1993) A mutation in CFTR produces different phenotypes depending on chromosomal background. Nature Genet., 5, 274–277. [DOI] [PubMed] [Google Scholar]
- Larriba S., Bassas,L., Gimenez,J., Ramos,M.D., Segura,A., Nunes,V., Estivill,X. and Casals,T. (1998) Testicular CFTR splice variants in patients with congenital absence of the vas deferens. Hum. Mol. Genet., 7, 1739–1743. [DOI] [PubMed] [Google Scholar]
- Mak V., Jarvi,K.A., Zielenski,J., Durie,P. and Tsui,L.C. (1997) Higher proportion of intact exon 9 CFTR mRNA in nasal epithelium compared with vas deferens. Hum. Mol. Genet., 6, 2099–2107. [DOI] [PubMed] [Google Scholar]
- Mulligan G.J., Guo,W., Wormsley,S. and Helfman,D.M. (1992) Polypyrimidine tract binding protein interacts with sequences involved in alternative splicing of β-tropomyosin pre-mRNA. J. Biol. Chem., 267, 25480–25487. [PubMed] [Google Scholar]
- Muro A.F., Caputi,M., Pariyarath,R., Pagani,F., Buratti,E. and Baralle,F.E. (1999) Regulation of fibronectin EDA exon alternative splicing: possible role of RNA secondary structure for enhancer display. Mol. Cell. Biol., 19, 2657–2671. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Neitzel H. (1986) A routine method for the establishment of permanent growing lymphoblastoid cell lines. Hum. Genet., 73, 320–326. [DOI] [PubMed] [Google Scholar]
- Niksic M., Romano,M., Buratti,E., Pagani,F. and Baralle,F.E. (1999) Functional analysis of cis-acting elements regulating the alternative splicing of human CFTR exon 9. Hum. Mol. Genet., 8, 2339–2349. [DOI] [PubMed] [Google Scholar]
- Nissim-Rafinia M., Chiba-Falek,O., Sharon,G., Boss,A. and Kerem,B. (2000) Cellular and viral splicing factors can modify the splicing pattern of CFTR transcripts carrying splicing mutations. Hum. Mol. Genet., 9, 1771–1778. [DOI] [PubMed] [Google Scholar]
- Ou S.H., Wu,F., Harrich,D., Garcia-Martinez,L.F. and Gaynor,R.B. (1995) Cloning and characterization of a novel cellular protein, TDP-43, that binds to human immunodeficiency virus type 1 TAR DNA sequence motifs. J. Virol., 69, 3584–3596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pagani F., Buratti,E., Stuani,C., Romano,M., Zuccato,E., Niksic,M., Giglio,L., Faraguna,D. and Baralle,F.E. (2000) Splicing factors induce CFTR exon 9 skipping through a non-evolutionary conserved intronic element. J. Biol. Chem., 275, 21041–21047. [DOI] [PubMed] [Google Scholar]
- Pignatti P.F., Bombieri,C., Marigo,C., Benetazzo,M. and Luisetti,M. (1995) Increased incidence of cystic fibrosis gene mutations in adults with disseminated bronchiectasis. Hum. Mol. Genet., 4, 635–639. [DOI] [PubMed] [Google Scholar]
- Ramchatesingh J., Zahler,A.M., Neugebauer,K.M., Roth,M.B. and Cooper,T.A. (1995) A subset of SR proteins activates splicing of the cardiac troponin T alternative exon by direct interactions with an exonic enhancer. Mol. Cell. Biol., 15, 4898–4907. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rave-Harel N. et al. (1997) The molecular basis of partial penetrance of splicing mutations in cystic fibrosis. Am. J. Hum. Genet., 60, 87–94. [PMC free article] [PubMed] [Google Scholar]
- Rozmahel R., Heng,H.H., Duncan,A.M., Shi,X.M., Rommens,J.M. and Tsui,L.C. (1997) Amplification of CFTR exon 9 sequences to multiple locations in the human genome. Genomics, 45, 554–561. [DOI] [PubMed] [Google Scholar]
- Sharer N., Schwarz,M., Malone,G., Howarth,A., Painter,J., Super,M. and Braganza,J. (1998) Mutations of the cystic fibrosis gene in patients with idiopathic pancreatitis. N. Engl. J. Med., 339, 645–652. [DOI] [PubMed] [Google Scholar]
- Shelley C.S. and Baralle,F.E. (1987) Deletion analysis of a unique 3′ splice site indicates that alternating guanine and thymine residues represent an efficient splicing signal. Nucleic Acids Res., 15, 3787–3799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Singh R., Valcarcel,J. and Green,M.R. (1995) Distinct binding specificities and functions of higher eukaryotic polypyrimidine tract-binding proteins. Science, 268, 1173–1176. [DOI] [PubMed] [Google Scholar]
- Strong T.V. et al. (1993) Expression of an abundant alternatively spliced form of the cystic fibrosis transmembrane conductance regulator (CFTR) gene is not associated with a cAMP-activated chloride conductance. Hum. Mol. Genet., 2, 225–230. [DOI] [PubMed] [Google Scholar]
- Teng H., Jorissen,M., Van Poppel,H., Legius,E., Cassiman,J.J. and Cuppens,H. (1997) Increased proportion of exon 9 alternatively spliced CFTR transcripts in vas deferens compared with nasal epithelial cells. Hum. Mol. Genet., 6, 85–90. [DOI] [PubMed] [Google Scholar]
- Tu G.C., Cao,Q.N., Zhou,F. and Israel,Y. (1998) Tetranucleotide GGGA motif in primary RNA transcripts. Novel target site for antisense design. J. Biol. Chem., 273, 25125–25131. [DOI] [PubMed] [Google Scholar]
- Valcarcel J., Gaur,R.K., Singh,R. and Green,M.R. (1996) Interaction of U2AF65 RS region with pre-mRNA branch point and promotion of basepairing with U2 snRNA. Science, 273, 1706–1709. [DOI] [PubMed] [Google Scholar]
- Welsh M.J., Tsui,L.-C., Boat,T.F. and Beaudet,A.L. (1995) Cystic fibrosis. In Scriver,C.R., Beaudet,A.L., Sly,W.S. and Valle,D. (eds), The Metabolic and Molecular Bases of Inherited Diseases. 7th edn. McGraw-Hill, New York, NY, pp. 3786–3799.