

Abstract
Both prokaryotes and eukaryotes respond to a decrease in temperature with the expression of a specific subset of proteins. Although a large body of information concerning cold shock-induced genes has been gathered, studies on temperature regulation have not clearly identified the key regulatory factor(s) responsible for thermosensing and signal transduction at low temperatures. Here we identified a two-component signal transduction system composed of a sensor kinase, DesK, and a response regulator, DesR, responsible for cold induction of the des gene coding for the Δ5-lipid desaturase from Bacillus subtilis. We found that DesR binds to a DNA sequence extending from position –28 to –77 relative to the start site of the temperature-regulated des gene. We show further that unsaturated fatty acids (UFAs), the products of the Δ5-desaturase, act as negative signalling molecules of des transcription. Thus, a regulatory loop composed of the DesK–DesR two-component signal transduction system and UFAs provides a novel mechanism for the control of gene expression at low temperatures.
Keywords: signal transduction/temperature regulation/thermosensing
Introduction
Variability and adaptability are crucial characteristics of organisms possessing the ability to survive and prosper in a wide variety of environmental conditions. In order for bacteria to effectively compete and survive, they have to sense environmental conditions and respond accordingly. Temperature is one of the major stresses that all living organisms face (Phadtare et al., 2000). It has been demonstrated that bacteria respond to high growth temperatures by the induction of a group of heat shock proteins, but also to low temperatures by the induction of a group of cold shock proteins (Phadtare et al., 2000). In contrast to heat shock proteins, which include chaperones required for protein folding and peptidases (Yura et al., 2000), cold-induced proteins appear to be involved in cellular functions such as general metabolism, transcription, translation and recombination (Phadtare et al., 2000). A universally conserved adaptation response observed among bacteria and most (if not all) poikilothermic organisms is the adjustment of membrane lipid composition at low temperatures (Cronan and Rock, 1996; Vigh et al., 1998). As the growth temperature is lowered, the proportion of unsaturated fatty acids (UFAs) in the membrane lipids increases. This regulatory mechanism system, called thermal control of fatty acid synthesis, is thought to be designed to ameliorate the effects of temperature changes on the physical state of the membrane phospholipids (for a review see Cronan and Rock, 1996). There are a variety of mechanisms that can alter the membrane phospholipid composition in response to a temperature change. Bacilli cells respond to a decrease in ambient growth temperature by introducing double bonds into pre-existing fatty acids of their membrane phospholipids (Grau and de Mendoza, 1993; Aguilar et al., 1998). These double bonds are inserted by specific fatty acid desaturase enzymes (de Mendoza et al., 2001). In a previous study we reported the isolation of the des gene coding for the Δ5-desaturase of Bacillus subtilis (Aguilar et al., 1998). Studies of operon fusion as well as transcriptional analysis demonstrated that des is tightly regulated during cold shock (Aguilar et al., 1998, 1999). While the des transcript is barely detected at 37°C, the production of des mRNA is transiently induced upon a temperature downshift. Derepression of des occurs exclusively at the level of transcription in a promoter-dependent fashion and does not require de novo synthesis of protein (Aguilar et al., 1999). Nevertheless, the molecular mechanism by which the transcription of the B.subtilis des gene as well as the synthesis of membrane UFAs are induced transiently by low temperature remains unsolved. Previous studies suggest that membranes can sense environmental changes and, as a consequence of changes in their phase state and microdomain organization, transmit signals that activate transcription (Vigh et al., 1998; Hoppe et al., 2000; Suzuki et al., 2000). This signalling mechanism was proposed to control the expression of cold-induced desaturases from cyanobacteria and heat shock-induced genes in Saccharomyces cerevisiae and cyanobacteria (for a review see Vigh et al., 1998). For signal transduction across the cell membrane, bacteria extensively use two-component systems, which have an input-sensing domain (histidine kinase) and an output effector domain (response regulator) (Hoch, 2000). Because sensor kinases are generally integral membrane proteins that respond to environmental signals (Dutta et al., 1999), it seems likely that temperature regulation of UFAs in bacilli could be controlled by members of the family of two-component regulatory proteins. In connection with this possibility, Suzuki et al. (2000) have recently reported that two histidine kinases and a response regulator modulate the transcription of low temperature-inducible genes from cyanobacteria.
Here we report the identification of the desK and desR genes from B.subtilis, which are directly responsible for the transcriptional regulation of the des gene. These genes encode products with similarity to an autophosphorylatable histidine kinase (DesK) and a DNA binding response regulator (DesR) of the two-component signal transduction system. In addition, we demonstrate that DesR interacts specifically with the regulatory region of the gene it controls and that UFAs act as negative regulators of des expression. Thus, a regulatory loop composed of the DesK–DesR two-component signal transduction system and UFAs provides a novel mechanism for the control of gene expression at low temperatures.
Results
Inactivation of the yocF and yocG genes prevents induction by low temperature of β-galactosidase activity in a des–lacZ fusion
A key issue in the regulation of UFAs synthesis in bacilli is to understand how a decrease in growth temperature induces the expression of the des gene required for oxygen-dependent desaturation of fatty acids. To gain insight into this regulatory pathway, the B.subtilis genome sequence (Kunst et al., 1997) was searched for potential two-component regulatory gene pairs involved in des environmental regulation. Among the 35 two-component signal transduction systems identified in B.subtilis, the two-gene operon formed by the yocF and yocG genes encodes a two-component system with no known function (Fabret et al., 1999). The predicted products of yocF and yocG exhibit structural similarity to the histidine kinases and response regulators of two-component regulatory systems, respectively (Fabret et al., 1999). The yocFG operon is located immediately downstream of the des gene (Kunst et al., 1997). This prompted us to investigate whether this signal transduction system could be involved in the regulation of the desaturase synthesis. To test the induction of desaturase expression upon a temperature shift we used the strain AKP3, which contains a fusion of the lacZ gene to the des promoter integrated ectopically at the non-essential amyE locus of B.subtilis JH642. When B.subtilis strain AKP3 is grown at 37°C the levels of β-galactosidase (β-gal) are very low (Figure 1A). However, when this strain is grown at 37°C and then shifted to 25°C, β-gal synthesis reaches induction levels ∼10-fold higher than the levels found at 37°C (Figure 1A). In addition, the induction of the des–lacZ fusion in strain AKP3 can be easily monitored in media containing X-gal, where the colonies turn blue at 25°C (Figure 1B, panel I). To determine whether the yocF and yocG genes are responsible for the control of des, we disrupted the operon with a kanamycin-resistance gene (KmR) cassette as described in Materials and methods. The yocFG gene disruption was introduced by homologous recombination into strain AKP3, giving strain AKP21. In contrast to strain AKP3, strain AKP21 did not form blue colonies at 25°C (Figure 1B, panel I) nor were its β-gal levels increased upon downshift from 37 to 25°C (Figure 1A). This result indicates that a mutation in the yocFG operon eliminates the low-temperature inducibility of the des promoter.


Fig. 1. Pattern of des–lacZ expression in wild-type and yocF–yocG– cells before and after temperature downshift. (A) Bacillus subtilis AKP3 cells (circles) and AKP21 cells (triangles) harbouring a des–lacZ transcriptional fusion were grown at 37°C to an optical density of 0.35 (at 525 nm) and then divided into two samples. One sample was transferred to 25°C (filled symbols), and the other was kept at 37°C (open symbols). Specific β-gal activities were determined at the indicated time intervals. (B) Bacterial strains [wild-type AKP3 (wt), yocF–yocG– (AKP21), yocF–yocG– pXyl:yocFG (AKP2147), yocF–yocG– pXyl:yocG (AKP2152), yocG– (AKP9) and yocG– pXyl:yocG (AKP952)] were streaked onto Luria–Bertani (LB) medium containing 30 µg/µl X-Gal, with (shaded quarter) or without (empty quarter) the addition of 0.8% l-xylose. The strains were incubated at 37°C for 12 h (left column) or for 5 h at 37°C, and then transferred to 25°C for 36 h (right column) before photography.
The yocFG mutation can be complemented in trans
To verify that the yocF and yocG genes were responsible for cold induction of des, plasmids containing the yocFG operon or the yocG gene alone under the control of the xylose-inducible Xyl promoter were integrated into the thr locus of strain AKP21 giving strains AKP2147 and AKP2152, respectively. In strain AKP2147, in which the yocF and yocG genes are provided in trans, the cold induction of the des–lacZ fusion was dependent upon the addition of xylose to the growth medium (Figure 1B, panel I). The xylose-induced expression of yocG alone, however, was unable to restablish the cold-dependent lac+ phenotype of strain AKP2152 (Figure 1B, panel II). Nevertheless, expression of this gene in strain AKP952, in which inactivation of yocG eliminated the low-temperature inducibility of the des promoter (Figure 1B, panel III), resulted in a xylose-dependent induction of the des–lacZ fusion at low growth temperatures (Figure 1B, panel III). Therefore, the yocG and yocF genes are essential for the low-temperature induction of the des gene.
Inactivation of yocG inhibits the induction of the des transcript and UFA synthesis at low temperature
We performed northern blot analysis to examine the expression of the des gene in the wild-type strain JH642 or in the yocG– strain AKP8, before and after a shift in temperature from 37 to 25°C (Figure 2A). As observed previously in the wild-type cells, the size of the des transcript is ∼1.1 kb, and this mRNA was only detected when cells were shifted to 25°C (Aguilar et al., 1999). However, the accumulation at 25°C of the des transcript was not observed in strain AKP8, directly demonstrating that inactivation of the yocG gene suppressed the low temperature-induced expression of the desaturase gene (Figure 2A).
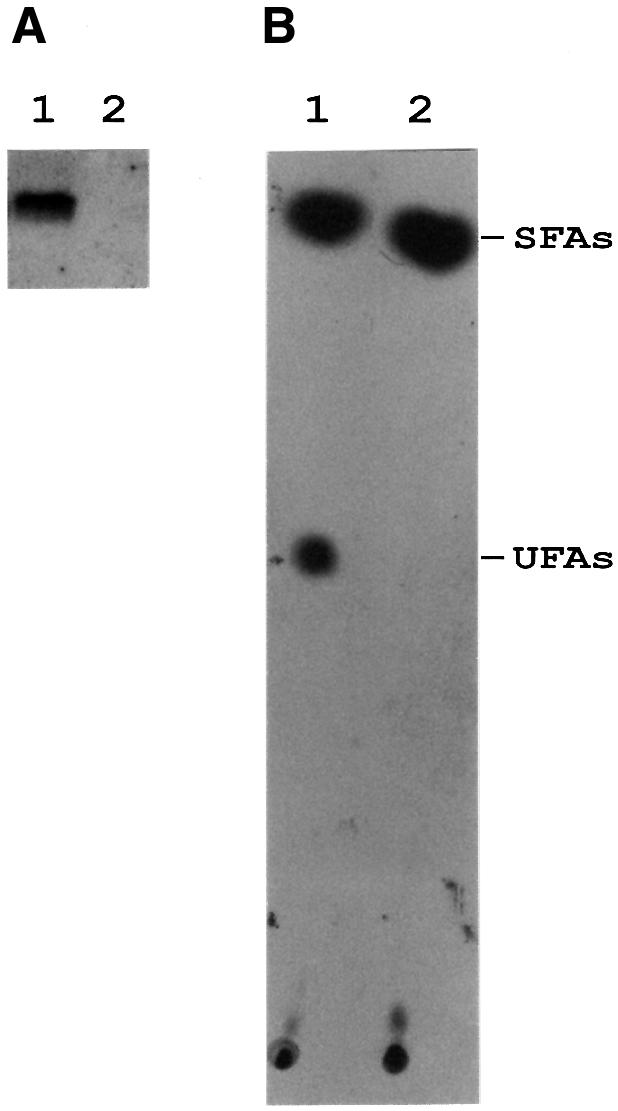
Fig. 2. des mRNA and UFA production in wild-type and yocG– strains after a downshift temperature. (A) Northern blot analysis using formaldehyde agarose gels was carried out as described in Materials and methods. Total RNA was isolated from strains JH642 (lane 1) or strain AKP8 (yocG–, lane 2) grown until mid-exponential phase at 37°C and then shifted to 25°C by 30 min. Each lane contains 10 µg of total RNA. (B) Fatty acids synthesized by strains JH642 and AKP8 at 25°C. Cultures of strains JH642 (lane 1) and AKP8 (lane 2) were grown to mid-exponential phase at 37°C, then 2 ml of these cultures were challenged with 10 µCi of [14C]acetate and further shifted to 25°C for 12 h. The lipids were then extracted and transesterified, and the resulting methyl esters were separated into saturated (SFAs) and unsaturated (UFAs) fractions by chromatography on 20% silver nitrate-impregnated silica gel thin-layers plates. The plates were developed at –17°C and autoradiographed by 7 days. The sample in lane 1 contained 15 000 c.p.m. and 2000 c.p.m. in the SFA and UFA fractions, respectively. The sample in lane 2 contained 14 000 c.p.m. in the SFA fraction, while the UFA fraction contained only background levels of radioactivity.
The fatty acid profile of the wild-type strain JH642 was compared with that of strain AKP8. The fatty acids were labelled by growth of the strains in [14C]acetate, followed by argentation chromatography of the radioactive fatty acids. While strain JH642 shifted from 37 to 20°C synthesized UFAs, strain AKP8 formed no detectable UFAs after the temperature downshift (Figure 2B). These experiments confirm that the yocG is essential for the synthesis of UFAs at low growth temperatures.
Since both the histidine kinase YocF and its cognate regulator YocG are required for des induction, we have named the genes coding for these two-component regulatory proteins as desK and desR, respectively.
h-DesR binds to the des promoter
In most cases, the response partner of the two-component transduction system is a transcriptional activator for genes whose products are specifically utilized to respond to the unique nature of a given input signal (Hoch, 2000). The genetic data presented above suggested that DesR, a response regulator for low-temperature response may bind to the des promoter region. The binding of purified His-tagged DesR (h-DesR) to the des promoter was tested using the electrophoresis mobility shift assay (EMSA). The h-DesR protein was expressed by isopropyl-β-d-thio galactopyranoside (IPTG) induction with Escherichia coli M15 (pREP4, pAR18) and purified by nickel affinity chromatography (data not shown). A PCR-amplified 367 bp DNA containing the des promoter (pdesDNA) was used as target DNA. Formation of complexes between h-DesR and labelled pdesDNA was tested by EMSA in the presence of an excess of competitor poly(dI–dC) (Figure 3A). The results show that the pdesDNA exhibited changes in mobility in the presence of 21 nM h-DesR (Figure 3A, lane 1). A more dramatic shift in the mobility of this 367 bp fragment was observed with increasing concentrations of h-DesR, and at the highest concentration of protein tested (336 nM), >90% of the input DNA was complexed with h-DesR (Figure 3A, lane 5). Two bands with shifted mobilities were usually observed, indicating that two different complexes are formed. Addition of a 3-fold excess of an unlabelled DNA fragment containing the des promoter diminishes the labelled complex formation (Figure 3B, lane 2), and a 15-fold excess of unlabelled pdesDNA abolished the labelled complex formation (Figure 3B, lane 3), suggesting specific interaction. The binding specificity was confirmed by determining that h-DesR did not shift an unrelated 290 bp fragment belonging to the repAB promoter of plasmid pLS1 (del Solar et al., 1990; data not shown).
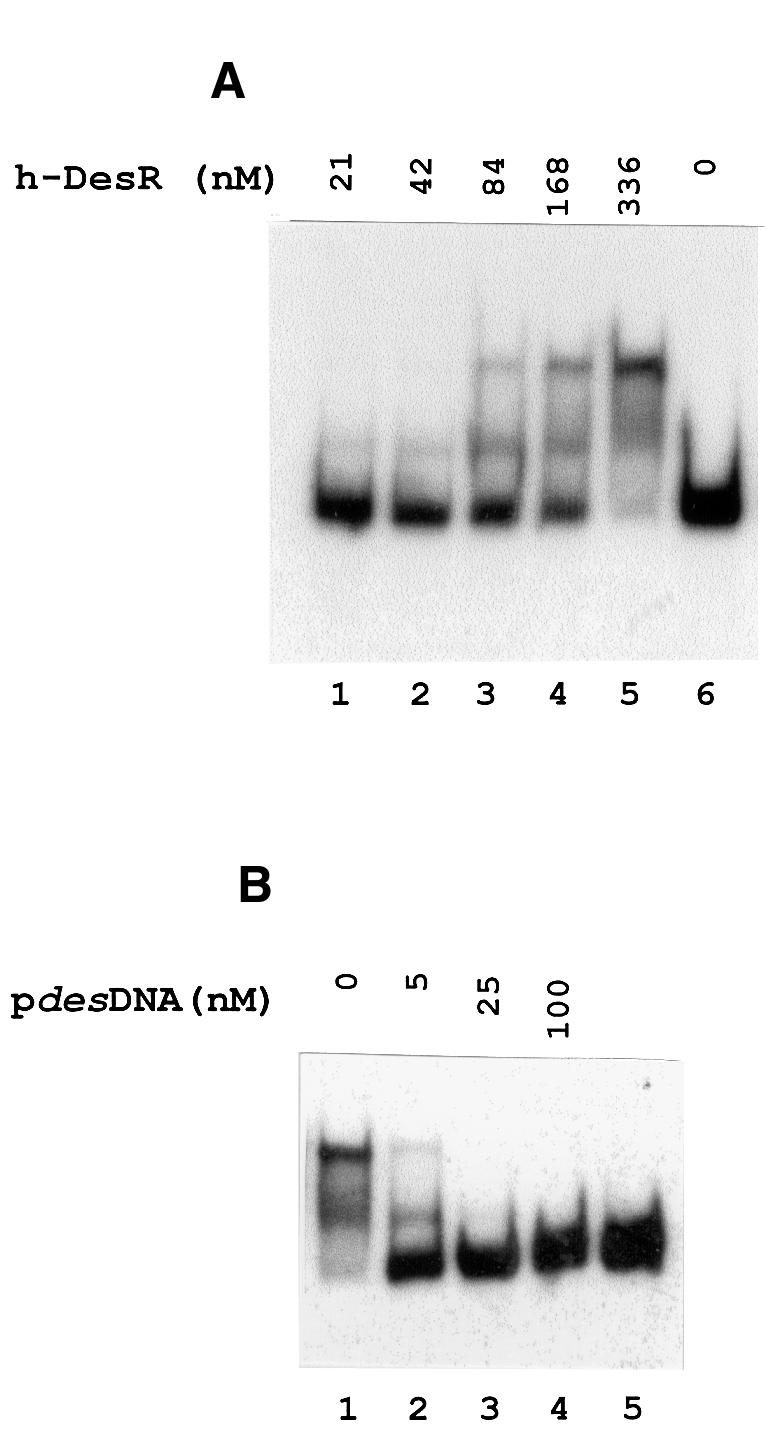
Fig. 3. Gel shift assay showing the binding of DesR to the des promoter region. (A) The 367 bp des promoter fragment (pdesDNA) was prepared by [α-32P]dATP PCR labelling as described in Materials and methods. The [32P]pdesDNA concentration in the binding mixtures was 1.7 nM in all cases. The concentration of h-DesR used in each binding reaction is indicated above the respective line. (B) Specific competition in binding reactions using 1.7 nM [32P]pdesDNA and 336 nM h-DesR. Lane 1 shows the retarded species in the absence of unlabelled homologous DNA. Lanes 2, 3 and 4 show the dissociation of the labelled complex in the presence of 3, 15 and 60-fold molar excess of unlabelled pdesDNA, respectively, added to the binding mixtures before the addition of h-DesR. Lane 5 shows [32P]pdesDNA without the addition of h-DesR.
h-DesR protects extended DNA segments upstream from its target promoter
To identify h-DesR binding sites in pdesDNA, DNase I protection analysis was performed on both strands of the des promoter. As shown in Figure 4A, binding of h-DesR resulted in the protection of DNA sequences extending from the –28 to –77 position, relative to the start of transcription. In addition to the protected regions, several hypersensitive bonds (indicative of local deformation and presumably caused by bending of the helix) were detected (Figure 4A). DNase I treatment of the h-DesR–DNA complex revealed five protected regions for both strands; these upper and lower strand protections are offset from each other towards the 3′ end of both strands. In the protected region two inverted repeats (5′-TCAT-3′) separated by nine nucleotides were found (Figure 4A). The axis of this dyad symmetry exactly matches the centre of the protected region determined by the footprinting analysis. The finding of a dyad symmetry in the centre of the protected region together with the existence of two complexes of h-DesR–DNA suggests that more than one subunit of h-DesR associates with the des promoter at one face of the DNA helix.


Fig. 4. DNase I footprinting assay of the des promoter region and in vivo characterization of the des promoter protected region. (A) DNase I footprinting of h-DesR protein on both strands of a 178 bp DNA fragment containing the des promoter (see Materials and methods). Sequencing reactions were performed on the same DNA fragment labelled at the coding (lanes 3 to 6) and non-coding (lanes 9 to 12) strands. Lanes 1, 2, 7 and 8 show the DNase I digestion products of pdesDNA in the presence (+) or absence (–) of h-DesR. Brackets mark the protected regions in each strand. The putative 17 bp symmetric region of the protected region is boxed, with the dyad axis of symmetry indicated by a dot. The inverted repeats are underlined with dots. DNase I footprints on both strands are shown. Arrows indicate hypersensitive bonds. (B) Promoter mutations. The sequence changes in the promoter variants are depicted along the protected region of the des promoter. The deleted region is indicated by dots, and the nucleotide changes to introduce mutations in the left inverted repeat are shown in bold characters. The inverted repeat sequences are underlined. The strains were grown at 37°C to an OD of 0.30 and then subjected to a downshift to 25°C. After 3 h of growth at 25°C, the cells were harvested and β-gal activities were determined. The average value of β-gal activity of strain AKP3 (bearing the wild-type promoter) was taken as 100% of promoter activity. The results shown are the average of three independent experiments.
To determine whether this dyad symmetric element is directly involved in des transcriptional regulation, promoter variants carrying a deletion or a mutation in the DesR binding site were cloned into plasmid pJM116 to create transcriptional fusions, which were then integrated at the amyE locus of the B.subtilis chromosome. Twenty-two base pairs out of the 49 bp protected region, including both inverted repeats, were deleted, yielding strain AKL59 (Figure 4B). Under cold shock conditions this deletion almost completely abolished promoter activity (Figure 4B). This result strongly suggests that the dyad symmetric element is essential for promoter activity at low temperature. Confirming this interpretation, no promoter activity was detected when the symmetry of the dyad element was disrupted by site-directed mutagenesis of the right inverted repeat (strain AKL62, Figure 4B).
DesK acts to control the activation and deactivation of DesR in response to growth temperature
Previous work has shown that the overexpression of response regulators in the absence of their cognate kinases could result in constitutive expression of the gene(s) they control (Powell and Kado, 1990). This suggests that high concentrations of unphosphorylated response regulator could bind in vivo to the target promoter and cause unregulated transcription. To determine whether an excess of DesR, without the assistance of DesK, could activate transcription of des at 37°C, we constructed the desK– strains AKP2152 and AKP20, expressing the wild-type desR from the pXyl or the pKan promoters, respectively. Antibody to DesR was generated and immunoblot assays were performed with the wild-type strain JH642 or cells overexpressing desR. As shown in Figure 5A (lanes 1–3), DesR was not detected in whole-cell lysates of strain JH642 growing at either 37 or 25°C, indicating that this protein is produced at very low levels at both temperatures. However, we found that strain AKP2152, expressing desR from the pXyl promoter, showed a significant level of DesR synthesis after induction with 0.8% xylose (Figure 5A, lane 5). Although this strain overproduced DesR in the presence of the inducer, expression of des was still repressed at either 37 or 25°C (Figure 1B, panel II). Nonetheless, we found that strain AKP20, expressing desR from the constitutive pKan promoter, showed a DesR production 5-fold greater than strain AKP2152 (Figure 5B, lane 6) and was able to express the des–lacZ fusion at 37°C (Figure 5C). This experiment demonstrates that high production of DesR promotes constitutive expression of the desaturase gene without assistance from DesK. This result, therefore, agrees with the observation that unphosphorylated response regulators can activate transcription when they are overexpressed (Powell and Kado, 1990). A somewhat surprising result was that introduction of the desKR operon, under the control of pXyl promoter, into strain AKP20 (giving strain AKP2047) restored the cold-inducible expression of des (Figure 5C). This effect could not be attributed to a reduction in the synthesis of DesR, since strain AKP2047 also overproduced the DesR protein to the same extent as AKP20 (Figure 5B, lanes 4 and 5). It should be noted that the pXyl promoter is ∼5-fold weaker than the pKan promoter (as estimated from the levels of production of DesR from desR fusions to these promoters; Figure 5A and B). It is more likely, therefore, that the cellular levels of DesK are much lower than the levels of DesR in strain AKP2047. The fact that in strain AKP2047 the xylose-induced expression of desK results in deactivation of DesR at 37°C suggests that DesK acts as a phosphatase that dephosphorylates DesR in response to an increase in growth temperature. A downshift in temperature, however, would suppress the phosphatase activity of DesK, favouring the phosphorylation of DesR by DesK, resulting in cold-induced transcriptional activation of the desaturase gene. The constitutive expression of desR in strain AKP20 could be explained by the existence of a second kinase or another phosphodonor, such as acetylphosphate (Lukat et al., 1992), capable of phosphorylating this response regulator irrespective of the growth temperature. This putative phosphate donor would have low affinity for DesR and would require a high concentration of this response regulator to phosphorylate it.

Fig. 5. Overexpression of DesR leads to constitutive expression of des. (A) JH642 cells (wild type) were cultured at 37°C until mid-exponential phase (lane 1) and then shifted to 25°C for 30 min (lane 2) or 3 h (lane 3). Samples were taken and cell extracts were analysed for the presence of DesR with DesR antiserum. AKP2152 (2152) cells were cultured at 37°C in the absence (lane 4) or presence of 0.8% l-xylose (lane 5) to mid-exponential phase and then shifted to 25°C by 2 h. Samples were taken and analysed for the presence of DesR as described above. (B) Strains JH642 (wt, lane 1), AKP2152 (2152) (lanes 2 and 3), AKP2047 (2047) (lanes 4 and 5) and AKP20 (20) (lane 6) were cultured at 37°C until mid-exponential phase and then shifted to 25°C by 2 h. AKP2152 and AKP2047 cultures were supplemented (+) or not (–) with 0.8% xylose. Samples were taken and proteins from the same quantity of cells were analysed for the presence of DesR. (C) Bacterial strains AKP3 (wt), AKP20 (desK–,pkan-desR) and AKP2047 (AKP20 pXyl-desKdesR::thr) were streaked onto LB medium containing 30 µg/µl X-Gal, with (shaded quarter) or without (empty quarter) the addition of 0.8% l-xylose. The strains were incubated at 37°C for 12 h (left) or for 5 h at 37°C, and then transferred to 25°C for 36 h (right) before photography.
UFAs regulate the expression of the des gene
Previous work has shown that a downshift of B.subtilis cells to low temperature produces a transient increase in des mRNA due to shutoff of transcription rather than to the instability of des mRNA (Aguilar et al., 1999). Since DesK and DesR are necessary for induction of des mRNA, it is possible that des expression is downregulated by shutoff of desKR transcription at low temperatures. To test this hypothesis, total cellular RNA was isolated from cultures of strain JH642 grown at 37°C and then shifted to 25°C for various times. Northern blot analysis indicated that the 1.9 kb desKR mRNA is constitutively expressed both at 37 and 25°C, although its level is slightly increased at 25°C, probably due to increased stability of the transcript at the low temperature (Figure 6). This result shows that downregulation of des transcription cannot be attributed to shutoff of desKR transcription at low temperature. Therefore, the transient induction of des should be due to inhibition of the pathway that senses or transduces low-temperature signals. Since UFAs are sophisticated signalling molecules that can mediate a myriad of cellular processes (Dowhan, 1997), including gene expression (Choi et al., 1996; Hoppe et al., 2000), we reasoned that the UFAs formed at low temperatures could act as negative regulators of the low-temperature signalling transduction system that induces the synthesis of the Δ5-desaturase. To test this hypothesis we inserted a KmR cassette into the des gene of strain AKP3, giving strain AKP4. This strain allows monitoring of the low-temperature inducibility of a des–lacZ fusion in the absence of UFAs synthesis. We assayed the β-gal activity of strains AKP3 and AKP4 upon a temperature downshift. While the β-gal levels of the des+ strain AKP3 began to decrease after 6 h at 25°C, the β-gal activity of the des–lacZ fusion contained into the des– strain AKP4 continued increasing during this incubation period, reaching levels 10-fold higher than those of strain AKP3 (Figure 7A). This experiment shows that in the absence of UFA synthesis the transcription of the desaturase promoter is increased and is not downregulated by prolonged incubation of cells at 25°C. Figure 7B shows the effects of a series of saturated fatty acids and UFAs on β-gal activity of strain AKP4. In these experiments cells were shifted from 37 to 25°C, and the fatty acids were added to the culture. Comparison of relative enzyme levels revealed that β-gal activity was repressed in all cultures containing UFAs at a concentration of 5 µM, although treatment with saturated fatty acids at this concentration did not appreciably repress the activity of the reporter (Figure 7B). Control experiments showed that UFAs do not affect the expression of lacZ fusions to promoters of other genes involved in lipid synthesis in B.subtilis (data not shown). When the regulation of the reporter was studied over a range of concentrations of several different UFAs, we found that the most effective repressor of expression of the des promoter was 16:1 Δ5 (Figure 7B). The difference in potency among 16:1 Δ5 and the other fatty acids tested strongly suggests that fatty acids with a double bond at the Δ5 position act as specific signals regulating the DesK–DesR signal transduction pathway.
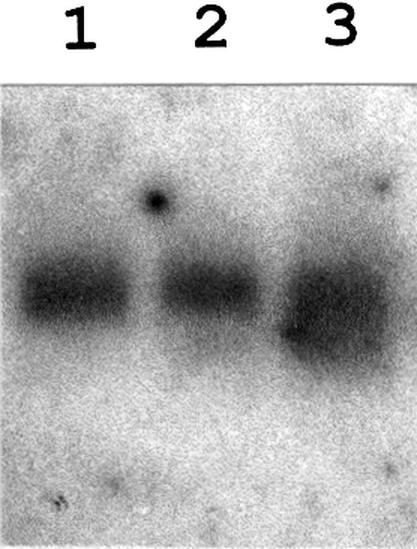
Fig. 6. desKR mRNA levels before and after a downshift temperature. Northern blot analysis using formaldehyde agarose gels was carried out as described in Materials and methods. Total RNA was isolated from strain JH642 grown until mid-exponential phase at 37°C (lane 1) and then shifted to 25°C by 30 min (lane 2) or 180 min (lane 3). Each lane contains 15 µg of total RNA.

Fig. 7. The expression of the des gene is regulated by UFAs. (A) Bacillus subtilis strains AKP3 (des+) (circles) and AKP4 (des–) (squares) harbouring a des–lacZ fusion located in the amyE locus were grown at 37°C to an OD of 0.35 and then transferred to 25°C. Aliquots were taken and specific β-gal activities were determined at the indicated time intervals. (B) Bacillus subtilis AKP4 cells were grown at 37°C to an OD of 0.27 at 525 nm and were then subjected to 0, 0.5, 2.5 or 5 µM of the indicated fatty acids before being transferred to 25°C. After 4 h of growth at 25°C, the cells were harvested and β-gal activities were determined. The average value of β-gal activity without supplement of fatty acid was taken as 100% of activity. The results shown are the average of three independent experiments.
Discussion
Although a large body of information concerning the cold shock response and cold shock proteins has been accumulated (Phadtare et al., 2000), several questions still remain unanswered. Unlike the heat shock response, a specific transcription factor governing low temperature-inducible genes has not been identified (Phadtare et al., 2000). The identification of such cellular thermosensors is an essential step in understanding the cold shock response and adaptation. However, the molecular mechanism(s) of thermosensing is far from understood, because the wide spectrum effects of temperature make it difficult to study these mechanisms directly. In contrast to other poikilothermic organisms, such as cyanobacteria (Sakamoto and Bryant, 1997) and plants (Los and Murata, 1998), B.subtilis contains a unique desaturase that is encoded by a single gene, which is tightly regulated by temperature at the transcriptional level (Aguilar et al., 1998, 1999). Due to its simplicity, the synthesis of UFAs in B.subtilis is an exceptionally well-suited system to study the sensing and the transduction of low temperature as a biological signal. We have identified a pair of two-component regulatory proteins, DesK and DesR, which recognize low-temperature signals and govern the expression of the gene coding for the desaturase of B.subtilis. Although kinase–response regulator pairs of this type were frequently reported as governors of a wide variety of pathways in response to a myriad of signals (Dutta et al., 1999; Hoch, 2000), the requirement of this system for low-temperature adaptation has only recently been suggested in cyanobacteria (Suzuki et al., 2000). In this organism it was found that inactivation of two histidine kinases moderates the low-temperature induction of the genes coding for Δ6- and ω3-desaturases. However, the transcriptional regulators of these genes were not identified (Suzuki et al., 2000). The genetic studies shown in this work indicate that the DesR protein is a transcriptional regulator controlling the low-temperature induction of the des gene. Confirming this prediction we found that the DesR protein binds specifically to the promoter region of the gene it controls, and that the dyad symmetric element found in the centre of the protected DNA region is essential for des induction. Therefore, we demonstrate here that DesR is a transcription factor directly involved in the transcriptional activation of the des gene at low temperature. Another transcription factor that has been found to stimulate transcription of two cold shock genes from E.coli, hns (La Teana et al., 1991) and gyrA (Jones et al., 1992), is CspA. Although the signal that triggers the transcriptional activity of both CspA and DesR is low temperature, the mechanism by which these transcription factors are activated is very different. While CspA mRNA is dramatically stabilized after cold shock (Phadtare et al., 2000), we suggest here that DesR is activated by a transmembrane signal transduction pathway.
Two lines of evidence lead us to propose that DesK acts to activate or deactivate DesR at 25 or 37°C, respectively: (i) the DesK histidine kinase is essential for des transcription; and (ii) DesK is required to deactivate the constitutive des transcription generated by overexpression of DesR. On the basis that no response regulator has yet been found to be active in the unphosphorylated form (Hoch, 2000), the role of DesK would then be to act as a phosphatase that dephosphorylates DesR selectively at 37°C. However, after a temperature downshift DesK would function as a specific kinase phosphorylating DesR, the cognate response regulator, which promotes des transcription. The results presented in this report strongly suggest that the sensor protein DesK is a bifunctional enzyme having both kinase and phosphatase activities. These two opposite activities of the sensor protein partner have been demonstrated in different two-component systems (for a review see Dutta et al., 1999). We have shown that the transcriptional activity of the des promoter is inhibited by either endogenously synthesized or exogenously added UFAs. Similar results have been obtained elsewhere for the OLE1 gene from S.cerevisiae coding for the Δ9-desaturase (Choi et al., 1996; Hoppe et al., 2000). The data presented here are consistent with either of two models for the control of des transcription by UFAs. One is that repression is caused by UFA-mediated dissociation of DesR. This mode of regulation has been reported for the expression of the Bacillus megaterium cytochrome P450 gene, which is controlled by the BM3R1 transcriptional repressor (Palmer et al., 1998). Binding of this negative regulator to DNA is inhibited by UFAs. A second model is that repression is mediated by inhibition of the histidine kinase activity of DesK by UFAs. Associated with the first model, we were unable to detect, in band-shift experiments, any specific displacement of DesR from its operator in the presence of UFAs (data not shown). In addition, we found that UFAs did not repress des transcription when DesR was overproduced in the absence of DesK (data not shown). Thus, UFAs do not appear to inactivate DesR directly. However, it remains possible that repression is mediated by an as yet unidentified protein that could displace DesR-P from its target DNA in the presence of UFAs.
A provisional model accounting for our results is shown in Figure 8. We envisage that DesK could assume different signalling states under varying growth temperatures. This could be accomplished by regulating the ratio of kinase to phosphatase activities, such that a phosphatase-dominant state is present at high growth temperature whereas a kinase-dominant state predominates at low growth temperature. DesK possesses four transmembrane domains, and, therefore, one or more of these domains would function to propagate a conformational change across the membrane that is sufficient to significantly alter its activity. This conformational change could be governed by the physical state of the membrane lipid bilayer. Membranes are normally in a liquid crystalline form and will undergo a transition to a gel phase state when the temperature drops (Cronan and Rock, 1996; Vigh et al., 1998). This change from a fluid (disordered) to a non-fluid state (ordered) might cause activation of the autokinase activity, resulting in autophosphorylation of a conserved histidine (His188) contained in the transmitter domain of DesK. The phosphoryl group of His188 could be directly transferred to DesR, which activates transcription of des. Derepres sion of des results in synthesis of the desaturase enzyme that specifically introduces a double bond at the Δ5-position of the acyl chains of membrane phospholipids. This metabolic pathway, therefore, generates a regulatory loop where Δ5 UFAs inhibit des transcription by favouring DesR dephosphorylation or by interacting with a UFA-responsive DNA binding protein that displaces DesR from its binding site.

Fig. 8. Model of des transcriptional control by two-component temperature signal transduction proteins. It is proposed that DesK assumes different signalling states in response to a temperature-induced change in membrane fluidity. This is accomplished by regulating the ratio of kinase to phosphatase activity such that a phosphatase-dominant state is present at 37°C, when membrane lipids are disordered (A), whereas a kinase-dominant state predominates upon an increase in the proportion of ordered membrane lipids after a temperature downshift to 25°C (B). DesK-mediated phosphorylation of DesR results in transcriptional activation of des (B). Activation of des results in synthesis of Des, which desaturates the acyl chains of membrane phospholipids (C). These newly synthesized UFAs inhibit des transcription either by favouring DesK dephosphorylation of DesR-P or by causing dissociation of DesR-P from its binding site (C) (see text for further details).
In summary, we provide evidence for the first time that a transcription factor responds directly to a temperature-regulated histidine kinase and acts as a molecular switch for the regulated transcription of a cold-induced gene.
Materials and methods
Bacterial strains and media
The bacterial strains used in this work are listed in Table I. Bacillus subtilis was propagated in Spizizen minimal salts medium (Spizizen, 1958) supplemented with glucose (0.5%), vitamin-free casein hydrolysate (0.1%), tryptophan (50 µg/ml) and phenylalanine (50 µg/ml). The parental bacterial strain was JH642 (trpC2 pheA1). All the strains derived from JH642 were obtained by transformation as described (Aguilar et al., 1998). Antibiotics were added to media at the following concentrations: 5 µg/ml chloramphenicol and 3 µg/ml kanamycin. Fatty acids were added at the concentrations indicated in Figure 7B. β-gal was assayed as described previously (Aguilar et al., 1998) and the specific activity was expressed in Miller units.
Table I. Bacillus subtilis strains used in this study.
| Strain | Relevant characteristics | Source |
|---|---|---|
| JH642 | trpC2 pheA1 | laboratory stock |
| AKP3 | JH642 amyE::[pdes(–269 to +31)a-lacZ] | this work |
| AKP4 | AKP3 des::Kmr | this work |
| AKP8 | JH642 yocG::Kmr | this work |
| AKP9 | AKP3 yocG::Kmr | this work |
| AKP952 | AKP9 thrC::(pXyl-yocG) | this work |
| AKP20 | AKP3 yocF::Kmr pKm-yocG | this work |
| AKP2047 | AKP20 thrC::(pXyl-yocFG) | this work |
| AKP21 | AKP3 yocFG::Kmr | this work |
| AKP2147 | AKP21 thrC::(pXyl-yocFG) | this work |
| AKP2152 | AKP21 thrC::(pXyl-yocG) | this work |
| AKL59 | JH642 amyE::[pdes(–269 to +31)b-lacZ] | this work |
| AKL62 | JH642 amyE::[pdes(–269 to +31)c-lacZ] | this work |
Plasmid and strain constructions
In all cases DNA fragments were obtained by PCR using the oligonucleotides (restriction sites are underlined) described in the text. Chromosomal DNA from strain JH642 was used as the template. To construct a transcriptional fusion between des and lacZ, a 301 bp DNA fragment containing the des promoter was obtained using oligo nucleotides I (AAAATGAATTCTCCGGCATCCCGATCATCGC) and II (TAGTATGGATCCTCTCATTGTGTGTCTCGGTTC). This fragment was digested with EcoRI and BamHI, and cloned into the integrational vector pJM116 (Dartois et al., 1998) generating plasmid pAR11. This plasmid was linearized with ScaI and introduced by a double cross-over event at the amyE locus of the JH642 chromosome, yielding strain AKP3. To obtain the des null mutant and the different yocFG mutants, DNA coding regions were obtained in each case and cloned in pBluescript SKII. Then, a kanamycin-resistance gene cassette obtained from pJM114 (Perego, 1993) was inserted in each of these regions, and the resulting plasmids were linearized and used to transform different B.subtilis strains by a double recombination event. To obtain the des null mutant strain a 913 bp DNA fragment was obtained using the oligonucleotides III (TTAGCGTCGACTGAACCGAGACACACAATG) and IV (ACTTCGAGCTCATAGTTGAGCACCTTTGG), and cloned in pBluescript SKII, yielding plasmid pDES913. In this plasmid a Kmr cassette was inserted between the HindIII and BclI sites, yielding pDES913KAN. This plasmid was used to transform the AKP3 strain, yielding strain AKP4. To construct the yocFG null mutant a 2378 bp DNA fragment was obtained using the oligonucleotides V (AACATGAGCTCCGGAAGAATGCCTGATG) and VI (AGTGGGTACCTTTTTCTTTATGTGCGATTC) and then cloned in pBluescript SKII, yielding plasmid pFG2378. In this plasmid the EcoRI and HindIII restriction sites were used to introduce the Kmr cassette, yielding plasmid pFG2378KAN. This plasmid was used to transform AKP3, yielding AKP21 strain. To disrupt the yocG gene, a 1466 bp DNA fragment obtained using the oligonucleotides VII (CCCGCGAGCTCCATGATACGCTTGGGCAAAAG) and VIII (TTAGTCTCGAGCAGTTGGGCATGGCAGCTTCG) was cloned in pBluescript SKII. Then, in this new construction a Kmr cassette was inserted in the ClaI–XbaI restriction sites, yielding plasmid pG1466KAN. This plasmid was used to transform the JH642 and AKP3 strains, yielding strains AKP8 and AKP9, respectively. A desK null mutant strain containing desR under the pKm promoter was constructed by first cloning a 1765 bp DNA fragment obtained with oligonucleotides V and XII (TGCTGGGTACCTGAGCGATTTCTTTTGTG) in pBluescript SKII. The resulting plasmid was digested with EcoRI and HindIII, and a Kmr cassette without its transcriptional terminator was inserted, yielding plasmid pFG1765KAN. This construction was used to transform strain AKP3, giving rise to strain AKP20. To place the yocFG operon and the yocG genes under the pXyl promoter into the thrC locus of the B.subtilis chromosome, yocFG and yocG were first amplified by PCR and then cloned under pXyl in plasmid pGS40 (G.Schujman, personal communication). Then the DNA fragments containing the fusion pXyl–yocFG or pXyl–yocG were cloned into the integrative plasmid pDG795 (Guerout-Fleury et al., 1995) and recombined into the thrC locus of the appropriate strain. A 1814 bp DNA fragment containing the yocFG genes obtained using the oligonucleotides IX (AGTAAGTCGACAAGCTGAAAATGAGGTAAGATC) and X (TTCAGGGTACCAAAAAGGATCCTGGCAGATG) was first cloned into the SalI–KpnI sites of plasmid pGS40 to obtain plasmid pGSFGXyl. This plasmid was digested with BamHI, and the DNA fragment containing yocFG under the pXyl promoter was cloned into the plasmid pDG795, yielding plasmid pAG47. This plasmid was used to transform strains AKP21 and AKP20 to give strains AKP2147 and AKP2047, respectively. A 677 bp DNA fragment containing the yocG gene was obtained using the oligonucleotides XI (TCAAAATCGATATAAAGGATGGCTTATATG) and X and then cloned into pGS40 to give plasmid pGSGXyl. This plasmid was digested with BamHI and EcoRI, and the DNA fragment containing yocG under the pXyl was cloned into plasmid pDG795, yielding plasmid pAG52. This plasmid was used to transform AKP9 and AKP21 strains, yielding strains AKP952 and AKP2152, respectively.
To delete 22 out of the 49 bp protected in the DNase I footprinting assay, leaving the –35 plus 7 bp upstream of the des gene intact (Figure 4B), oligonucleotides XXII (GTTTGGAATTCACCCCTCAAGTGAGTGGAGC), XXIII (CATTTGTCATGTCTCGTGCGGCATGCATAG), XXIV (GAGGCATGATGTGTGCTACTACAAAAGAC) and XXV (AAATCCGCGGGAGAATAAACATGATAAC) were used to generate two PCR products flanking the region to be deleted. These fragments were purified, blunt ligated, and the ligation reaction product was used as substrate for a PCR reaction using oligonucleotides XXVI (AAAATGAATTCTCCGGCATCCCGATCATCGC) and XXVII (AGTATGGATCCTCTCATTGTGTGTCTCGGTTC). This 279 nt PCR product was cloned into the integrative plasmid pJM116 to generate pAKL59.
To mutate the 3′ inverted repeat of the protected region (Figure 4B), two DNA fragments were amplified using oligonucleotides XXVIII (GTAGCACACTCTAGACCTCCTATATGACATTTGTC), XXII, XXIX (TATAGGAGGTCTAGAGTGTGCTACTACAAAAGAC) and XXV. A mix of these PCR products was used as DNA template for another PCR using oligonucleotides XXVI and XXVII. The 301 bp amplification product was cloned into plasmid pJM116 to generate pAKL62. Plasmids pAKL59 and pAKL62 were introduced into the amyE locus of strain JH642 giving strains AKL59 and AKL62, respectively.
RNA analysis
Bacillus subtilis strains were grown in supplemented Spizizen minimal salts medium and the RNA was isolated as described previously (Aguilar et al., 1999). Northern blot analysis and hybridization with a single-stranded des DNA probe were performed as previously described (Aguilar et al., 1999). The single-stranded yocFG DNA probe was synthesized with T4 DNA polymerase, [α-32P]dATP and the antisense oligonucleotide XIII (TTGGCCGGTTGTACCTTTGC) as primer of a DNA fragment obtained by PCR amplification from chromosome of strain JH642 using the oligonucleotides XIV (CAAGGAGGCCTAGCGAATGGCCCGCGATCTCCATG) and XV (AGATCTTTGGCCGGTTGTACCTTTGC). The size of the yocFG transcript was determined by comparison with Promega RNA molecular weight standards (data not shown).
Construction of expression vector and purification of His-tagged DesR
The yocG gene was amplified from genomic DNA isolated from B.subtilis. Primers XVI (GGATGGGATCCATGATTAGTATATTTATTGCAG) and XVII (AAAAAGCATGCTGGCAGATGCCAAGATC) were used. The PCR product was digested, purified and ligated into plasmid pQE30, yielding plasmid pAR18. This new construction places a His6 tag at the N-terminus of the DesR protein and was used to transform the E.coli strain M15[pREP4]. Overexpression and purification of h-DesR were performed by standard procedures (Qiagen, Inc.).
Gel shift assays
A DNA fragment including the des promoter region was prepared by PCR with plasmid pDM10 (Aguilar et al., 1998) as template. The primers used were XVIII (ATGCAGGATTCAAGCTATTTCGGGTACATC) and XIX (TCGAGGCTGAGATAAGCAAGAAACCATAGGC). The 367 bp DNA fragment was labelled by incorporation of [α-32P]dATP in the PCR amplification, and purified from 5% polyacrylamide gels at the specific activity of 2.5 × 106 c.p.m./µg. Binding of h-DesR to DNA fragments was carried out in a 25 µl reaction mix containing 25 mM Tris–HCl pH 8, 1 mM dithiothreitol (DTT), 0.25 mM EDTA, 4 mM MgCl2, 200 ng poly(dI–dC) and 5% glycerol. After 30 min at room temperature, 2.8 µl of 50% glycerol were added and the samples were applied to a 5% polyacrylamide gel, which had been pre-run for 2 h in 45 mM Tris-borate pH 8, 1 mM EDTA. Gels were dried and autoradiographed.
DNase I footprinting assays
DNA for this assay was obtained by PCR using DNA from pDM10 as template and primers XX (CGGGTACATCACGAATATGG) and XXI (TGTGTGTCTCGGTTCAGTATACGC). Both primers were previously labelled at their 5′ ends using [γ-32P]ATP and T4 polynucleotide kinase. The PCR product, a 178 bp fragment, was eluted, purified, and recovered at the specific activity of 8 × 106 c.p.m./µg. Reactions were performed in a total volume of 50 µl of a buffer containing 25 mM Tris pH 8.0, 1 mM DTT, 0.25 mM EDTA, 22 mM NaCl, 5 mM MgCl2, 2.5 mM CaCl2 and 5% glycerol. The 32P-end-labelled DNA fragments, at a final concentration of 2.6 nM, were incubated with purified h-DesR protein (700 nM) and poly(dI–dC) (4 ng/µl) at room temperature for 30 min. DNase I (0.001 U) was added, and the incubation was continued for 5 min at the same temperature. The reactions were stopped by the addition of 25 µl of a solution containing 2 M ammonium acetate, 0.15 mM EDTA, 0.8 M sodium acetate, 100 µg/ml calf thymus and 400 µg/ml tRNA. The DNA samples were ethanol precipitated and dissolved in 8 µl of sequencing loading buffer (80% deionized formamide, 10 mM sodium hydroxide, 0.1% bromophenol blue, 0.1% xylene cyanol, 1 mM EDTA). Samples (3 µl, 75 000 c.p.m.) were loaded onto 8% polyacrylamide sequencing gels and run together with sequencing reactions obtained by the dideoxy-mediated chain-termination method (T7 DNA polymerase sequencing kit, Pharmacia).
Fatty acid analysis and purification of 16:1 Δ5
Measurements of fatty acid synthesis by B.subtilis cells were performed as previously described (Aguilar et al., 1998). The 16:1 Δ5 fatty acid was prepared using the E.coli strain AK7/pDM10 (Aguilar et al., 1998). This strain was grown at 30°C in LB medium to exponential phase, and labelled with [1-14C]palmitate. Lipids were extracted and fatty acids converted to their methyl esters with sodium methoxide. The saturated and unsaturated fractions were separated into two peaks by chromatography on a 20% silver nitrate-impregnated silica gel column. The concentration of the UFA fraction was determined by gas chromatography. To release the free fatty acids, the solution containing the methyl ester was made 0.5 M in KOH and heated for 2 h at 65°C. After washing twice with water and drying under a steam of nitrogen, the residue was resuspended in dimethylsulfoxide (DMSO). The C18:1 Δ6 and C16:1 Δ9 fatty acids were purchased from Sigma. To quantify these fatty acids, both were converted to methyl esters in a 5% HCl–methanol solution followed by heating for 3 h at 80°C. The concentrations were determined by gas chromatography, and the free fatty acids were obtained as described for C16:1 Δ5.
Immunoblot analysis
Bacillus subtilis strains were grown as indicated in each case. Aliquots of 1 ml of each culture were harvested, centrifuged and frozen. The pellets were resuspended in lysis buffer (50 mM Tris–HCl pH 8.0, 5 mM EDTA, 1 mM phenylmethylsulfonyl fluoride and 0.1 mM DTT), adding 180 µl of buffer per OD525 unit. Twenty microlitres of cell resuspension were disrupted by incubating with lysozyme (500 µg/ml) for 15 min at 37°C, followed by 5 min of boiling in the presence of loading buffer. Each sample was subjected to SDS–PAGE in a 12% acrylamide gel. Proteins were electroeluted to a nitrocellulose membrane and revealed using anti-h-DesR rabbit antibody and a secondary antibody conjugated to alkaline phosphatase.
Acknowledgments
Acknowledgements
We thank M.Espinosa for generously providing facilities for part of this study, C.Nieto and G.del Solar for their valuable advice in the EMSA experiments, F.Soncini for critically reading the manuscript and S.Altabe for valuable advice in the purification of UFAs. This work was supported by the Consejo Nacional de Investigaciones Científicas y Técnicas (CONICET), Agencia de Promoción Científica y Tecnológica (FONCYT), Fundación Antorchas and the exchange program of Agencia Española de Cooperación Internacional (AECI). P.S.A. is a fellow from CONICET, L.E.C. was supported for Ministerio de Salud de la provincia del Chaco and D.d.M. is a Career Investigator from CONICET.
References
- Aguilar P.S., Cronan,J.E.,Jr and de Mendoza,D. (1998) A Bacillus subtilis gene induced by cold-shock encodes a membrane phospholipid desaturase. J. Bacteriol., 180, 2194–2200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aguilar P.S., Lopez,P. and de Mendoza,D. (1999) Transcriptional control of the low-temperature-inducible des gene, encoding the Δ5 desaturase of Bacillus subtilis. J. Bacteriol., 181, 7028–7033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Choi J.Y., Stukey,J., Hwang,S.Y. and Martin,C.E. (1996) Regulatory elements that control transcription activation and unsaturated fatty acid-mediated repression of the Saccharomyces cerevisiae OLE1 gene J. Biol. Chem., 271, 3581–3589. [DOI] [PubMed] [Google Scholar]
- Cronan J.E. Jr and Rock,C.O. (1996) Biosynthesis of membrane lipids. In Neidhart,F.C. et al. (eds), Escherichia coli and Salmonella typhimurium: Cellular and Molecular Biology. 2nd edn, Vol. 1. American Society for Microbiology, Washington, DC, pp. 612–636.
- Dartois V., Débarbouillé,F., Kunst,F. and Rapaport,G. (1998) Characterization of a novel member of the DegS–DegU regulon affected by salt stress in Bacillus subtilis. J. Bacteriol., 180, 1855–1861. [DOI] [PMC free article] [PubMed] [Google Scholar]
- del Solar G.H., Pérez-Martin,J. and Espinosa,M. (1990) Plasmid pLS1-encoded RepA protein regulates transcription from repAB promoter by binding to a DNA sequence containing a 13-base pair symmetric element. J. Biol. Chem., 265, 12569–12575. [PubMed] [Google Scholar]
- de Mendoza D., Schujman,G.S. and Aguilar,P.S. (2001) Biosynthesis and function of membrane lipids. In Sonenshein,A.L., Hoch,J.A. and Losick,R. (eds), Bacillus subtilis and its Relatives: From Genes to Cells. American Society for Microbiology, Washington DC, in press.
- Dowhan W. (1997) Molecular basis for membrane phospholipid diversity: why are there so many lipids? Annu. Rev. Biochem., 66, 199–232. [DOI] [PubMed] [Google Scholar]
- Dutta R., Qin,L. and Inouye,M. (1999) Histidine kinases: diversity of domain organisation. Mol. Microbiol., 34, 633–640. [DOI] [PubMed] [Google Scholar]
- Fabret C., Feher,V.A. and Hoch,J.A. (1999) Two component signal transduction in Bacillus subtilis: how an organism sees its world. J. Bacteriol., 181, 1975–1983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grau R. and de Mendoza,D. (1993) Regulation of the synthesis of unsaturated fatty acids in Bacillus subtilis. Mol. Microbiol., 8, 535–542. [DOI] [PubMed] [Google Scholar]
- Guerout-Fleury A.M., Shazand,K., Frandsen,N. and Stragier,P. (1995) Antibiotic-resistance cassettes for Bacillus subtilis. Gene, 167, 335–336. [DOI] [PubMed] [Google Scholar]
- Hoch J.A. (2000) Two-component and phosphorelay signal transduction. Curr. Opin. Microbiol., 3, 165–170. [DOI] [PubMed] [Google Scholar]
- Hoppe T., Matuschewsky,K., Rape,M., Schlenker,S., Ulrich,H.D. and Jentsch,S. (2000) Activation of a membrane-bound transcription factor by regulated ubiquitin/proteasome-dependent processing. Cell, 102, 577–586. [DOI] [PubMed] [Google Scholar]
- Jones P.G., Krah,R., Tafuri,S.H. and Wolffe,A.P. (1992) DNA gyrase, CS7.4, and the cold shock response in Escherichia coli. J. Bacteriol., 174, 5798–5802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kunst F. et al. (1997) The complete genome sequence of the Gram-positive bacterium Bacillus subtilis. Nature, 390, 249–256. [DOI] [PubMed] [Google Scholar]
- La Teana A., Brandi,A., Falconi,M., Spurio,R., Pon,C.L. and Gualerzi,C.O. (1991) Identification of a cold shock transcriptional enhancer of the Escherichia coli gene encoding nucleoid protein H-NS. Proc. Natl Acad. Sci. USA, 88, 10907–10911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Los D.A. and Murata,N. (1998) Structure and expression of fatty acid desaturases. Biochim. Biophys. Acta., 1394, 3–15. [DOI] [PubMed] [Google Scholar]
- Lukat G.S., Mc Clearly,W.R., Stock,A.M. and Stock,J.B. (1992) Phosphorylation of bacterial response regulator proteins by low molecular weight phospho-donors. Proc. Natl Acad. Sci. USA, 89, 718–722. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Palmer C.N.A., Axen,E. and Wolf,C.R. (1998)The repressor protein, Bm3R1, mediates an adaptive response to toxic fatty acids in Bacillus megaterium. J. Biol. Chem., 273, 18109–18116. [DOI] [PubMed] [Google Scholar]
- Perego M. (1993) Integrational vectors for genetic manipulation in Bacillus subtilis. In Sonenshein,A.L., Hoch,J.A. and Losick,R. (eds), Bacillus subtilis and Other Gram-Positive Bacteria: Biochemistry, Physiology, and Molecular Genetics. American Society for Microbiology, Washington, DC, pp. 615–625.
- Phadtare S., Yamanaka,K. and Inouye,M. (2000) The cold shock response. In Stortz,G. and Hengge-Aronis,R. (eds), Bacterial Stress Responses. American Society for Microbiology, Washington, DC, pp. 33–47.
- Powell B.S. and Kado,C.I. (1990) Specific binding of VirG to the vir box requires a C-terminal domain and exhibits a minimum concentration threshold. Mol. Microbiol., 4, 2159–2166. [DOI] [PubMed] [Google Scholar]
- Sakamoto T. and Bryant,D.A. (1997) Temperature-regulated mRNA accumulation and stabilization for fatty acid desaturase genes in the cyanobacterium Synechococcus sp. strain PCC 7002. Mol. Microbiol., 23, 1281–1292. [DOI] [PubMed] [Google Scholar]
- Spizizen J. (1958) Transformation of biochemically deficient strains of Bacillus subtilis by deoxyribonucleate. Proc. Natl Acad. Sci. USA, 44, 1072–1078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suzuki I., Los,D.A., Kanesaki,Y., Mikami,K. and Murata,N. (2000) The pathway for perception and transduction of low-temperature signals in Synechocystis. EMBO J., 19, 1327–1334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vigh L., Maresca,B. and Harwood,J.L. (1998) Does the membrane’s physical state control the expression of heat shock and other genes? Trends Biochem. Sci., 23, 369–374. [DOI] [PubMed] [Google Scholar]
- Yura T., Kanemori,M. and Morita,M.T. (2000) The heat shock response: regulation and function. In Stortz,G. and Hengge-Aronis,R. (eds), Bacterial Stress Responses. American Society for Microbiology, Washington, DC, pp. 3–18.