

Abstract
While decapping plays a major role in mRNA turnover in yeast, biochemical evidence for a similar activity in mammalian cells has been elusive. We have now identified a decapping activity in HeLa cytoplasmic extracts that releases 7meGDP from capped transcripts. Decapping is activated in extracts by the addition of 7meGpppG, which specifically sequesters cap-binding proteins such as eIF4E and the deadenylase DAN/PARN. Similar to in vivo observations, the presence of a poly(A) tail represses decapping of RNAs in vitro in a poly(A)-binding protein-dependent fashion. AU-rich elements (AREs), which act as regulators of mRNA stability in vivo, are potent stimulators of decapping in vitro. The stimulation of decapping by AREs requires sequence-specific ARE-binding proteins. These data suggest that cap recognition and decapping play key roles in mediating mRNA turnover in mammalian cells.
Keywords: AU-rich elements/cap-binding proteins/decapping/mRNA turnover/poly(A)-binding proteins
Introduction
The turnover of mRNAs is a regulated process that plays a key role in determining the steady-state levels and rate of induction of transcripts (Caponigro and Parker, 1996; Jacobson and Peltz, 1996; Mitchell and Tollervey, 2000). In the yeast Saccharomyces cerevisiae, a major pathway of mRNA turnover is initiated by deadenylation (Decker and Parker, 1993). Poly(A) tail shortening leads to removal of the 5′ cap structure from the mRNA by Dcp1p and release of 7meGDP (Muhlrad et al., 1994). Auxiliary factors have been identified that appear to serve as regulators of the decapping step (Dunckley and Parker, 1999; Zhang et al., 1999a; Zuk et al., 1999; Bonnerot et al., 2000; Bouveret et al., 2000; Tharun et al., 2000). Decapped transcripts are then degraded by Xrn1p, a 5′→3′ exonuclease (Hsu and Stevens, 1993). Mutations that block this pathway of mRNA turnover revealed that a 3′→5′ exonucleolytic pathway is also functional on deadenylated transcripts in yeast (Jacobs et al., 1998; Bousquet-Antonelli et al., 2000). Additional pathways of turnover exist for certain transcripts, such as those that contain nonsense mutations (Losson and Lacroute, 1979; Czaplinski et al., 1999). The deadenylation-independent decapping pathway used in nonsense-mediated decay illustrates the pivotal role played by regulated decapping and cap recognition in mRNA turnover (Muhlrad and Parker, 1994; Hagan et al., 1995).
In mammalian cells, most mRNAs also appear to undergo poly(A) shortening as the initial step in their decay (Wilson and Treisman, 1988; Shyu et al., 1991). This deadenylation event is likely to be mediated by the poly(A)-specific exonuclease DAN/PARN (Korner et al., 1998), and recent data suggest that interaction of the 5′ cap in addition to the poly(A) tail plays an important role in determining the rate of deadenylation (Dehlin et al., 2000; Gao et al., 2000). Following deadenylation, the mechanism and factors involved in the turnover of mRNAs in mammalian cells remain largely undefined. Several sequence elements have been identified, especially AU-rich elements (AREs) in 3′-untranslated regions (UTRs), which effectively destabilize mRNAs (Shaw and Kamen, 1986; Chen and Shyu, 1995). Numerous proteins have been identified that specifically bind to AREs (Brewer, 1991; Levine et al., 1993; Ma et al., 1996), several of which may have positive (Fan and Steitz, 1998; Peng et al., 1998; Ford et al., 1999) or negative (Lai et al., 1999; Loflin et al., 1999) influences on transcript stability.
In vivo evidence suggests that decapping plays an important role in mRNA turnover in mammalian cells. PCR-based assays designed to detect cytoplasmic mRNAs with a 5′ monophosphate residue have identified such molecules that are likely to be intermediate products of the decapping reaction (Couttet et al., 1997). However, no direct biochemical evidence exists for a Dcp1p-like activity in mammalian cells. Nuss et al. (1975) have previously identified a 7-methylguanosine-specific pyrophosphatase activity in HeLa cells that cleaves 7meGMP from mRNAs, but this enzymatic activity is biochemically distinct from the yeast decapping enzyme Dcp1p, which cleaves capped mRNAs to produce 7meGDP (Beelman et al., 1996). Therefore, the enzymatic and regulatory factors that control decapping in mammalian cells have not been described.
Many general and regulatory aspects of mRNA deadenylation and decay in mammalian cells can be reproduced efficiently using HeLa cytoplasmic S100 extracts (Ford and Wilusz, 1999; Ford et al., 1999; Gao et al., 2000). In this study, we used S100 extracts to search for biochemical evidence for decapping of mammalian mRNAs. A decapping activity was identified that was found to be tightly regulated by three elements: the cap itself, the poly(A) tail and AU-rich instability elements. In all three cases, sequence-specific protein–RNA interactions were required to mediate the positive or negative effects of each element on decapping. Based on these data, we propose a model for the key role of the 5′ cap structure in the regulation of mRNA turnover.
Results
Identification of a decapping activity in HeLa cytoplasmic extracts
Following deadenylation, most yeast mRNAs are decapped prior to degradation by the 5′→3′ exonuclease Xrn1p (Muhlrad et al., 1994). Whether decapping plays a major role in the turnover of mammalian mRNAs following poly(A) tail shortening is unclear. No direct biochemical evidence exists for a bona fide Dcp1p-like decapping enzyme in mammalian cells. In order to search for a decapping activity in mammalian extracts, we used the thin-layer chromatography (TLC) assay previously developed to study the yeast enzyme (Zhang et al., 1999b). Briefly, RNA substrates were labeled exclusively at the α-phosphate of the cap structure using recombinant vaccinia capping enzyme and [α-32P]GTP, and were incubated in cytoplasmic extracts in the presence of Mg2+. Reaction products were spotted directly onto PEI cellulose sheets and small molecules were resolved in 450 mM ammonium sulfate. The positions of 7meGMP, 7meGDP and 7meGTP were identified by UV shadowing of markers run in each lane. As seen in Figure 1A, yeast whole-cell extracts contain a potent decapping activity that produces a significant amount of 7meGDP from cap-labeled RNA substrates. However, the 7meGDP product of decapping was not detected when cap-labeled RNAs were incubated in HeLa S100 extracts. In addition, no significant decrease in radioactivity at the origin was observed in these experiments. We concluded that HeLa cells are either different from yeast and lack a decapping activity, or that the decapping activity is somehow masked in the in vitro assay. Since most basic aspects of yeast gene expression are conserved in metazoans, we favored the latter suggestion.

Fig. 1. Identification of a decapping activity in HeLa cytoplasmic extracts. (A) SVARE-A0 RNA, radiolabeled exclusively at the α-phosphate of the 5′ cap structure, was incubated in standard decapping conditions using no extract, S.cerevisiae whole-cell extract (yeast lane) or HeLa S100 cytoplasmic extract. The 7meGDP product of the decapping reaction was resolved by TLC on PEI cellulose sheets developed using 0.45 M ammonium sulfate. The identification of radioactive spots was determined using markers that were visualized by UV shadowing. (B) GemARE-A60 RNA, radiolabeled exclusively at the α-phosphate of the 5′ cap structure, was incubated in the absence of extract (no extract lane) or in the presence of three independently prepared S100 cytoplasmic extracts from HeLa cells (A, B or C) in either the absence (– lanes) or presence of 20 µM cap analog (+ lanes). The 7meGDP product of the decapping reaction was resolved by chromatography on PEI cellulose sheets developed using 0.45 M ammonium sulfate. (C) GemARE-A0 RNA, radiolabeled exclusively at the α-phosphate of either a methylated 5′ cap (lane 7meGppp Cap) or an unmethylated 5′ cap (lane Gppp Cap), was incubated in a standard decapping assay in the presence of 20 µM cap analog as described above.
We hypothesized that the cap structure of mRNA substrates becomes inaccessible when the transcript is incubated in HeLa extracts due to competing cap-binding activities such as eIF4E (Sonenberg et al., 1978) or the deadenylase DAN/PARN (Dehlin et al., 2000; Gao et al., 2000). Parker and colleagues have shown previously that the yeast decapping enzyme Dcp1p is not inhibited by small cap analogs (LaGrandeur and Parker, 1998). The addition of 7meGpppG cap analog to extracts might, therefore, be able selectively to sequester these competing cap-binding activities but not inhibit the mammalian decapping enzyme. This might allow us to uncover a decapping activity in HeLa extracts. We tested this possibility by adding 20 µM 7meGpppG to reaction mixtures and repeating the decapping assay using three independently prepared HeLa cytoplasmic extracts. As seen in Figure 1B, the addition of cap analog activated decapping of RNA substrates in all of the extracts tested, as seen by the accumulation of 7meGDP. The activation of decapping on all RNA substrates we have tested required the addition of cap analog. Furthermore, decapping occurred with linear kinetics, was dependent on the presence of Mg2+ and did not require ATP (Figures 3 and 5, data not shown). Finally, the decapping activity was highly specific for 7meGppp-capped RNAs, as unmethylated cap structures were not cleaved detectably in our assays (Figure 1C). Taken together, these data suggest that HeLa cytoplasmic extracts contain a decapping activity that is repressed by cap-binding proteins that prevent its access to the cap of RNA substrates.

Fig. 3. The presence of a poly(A) tail represses decapping of three independent RNA substrates. Equimolar amounts of three independent, cap-labeled RNA substrates that either lacked a poly(A) tail (GemARE-A0, SVARE-A0 and GM-CSFT-A0) or contained 60 adenylate residues at their 3′ end (GemARE-A60, SVARE-A60 and GM-CSFT-A60) were incubated in the in vitro decapping assay in the presence of cap analog. Aliquots were removed at the indicated time points and reaction products were analyzed by TLC on PEI cellulose sheets.
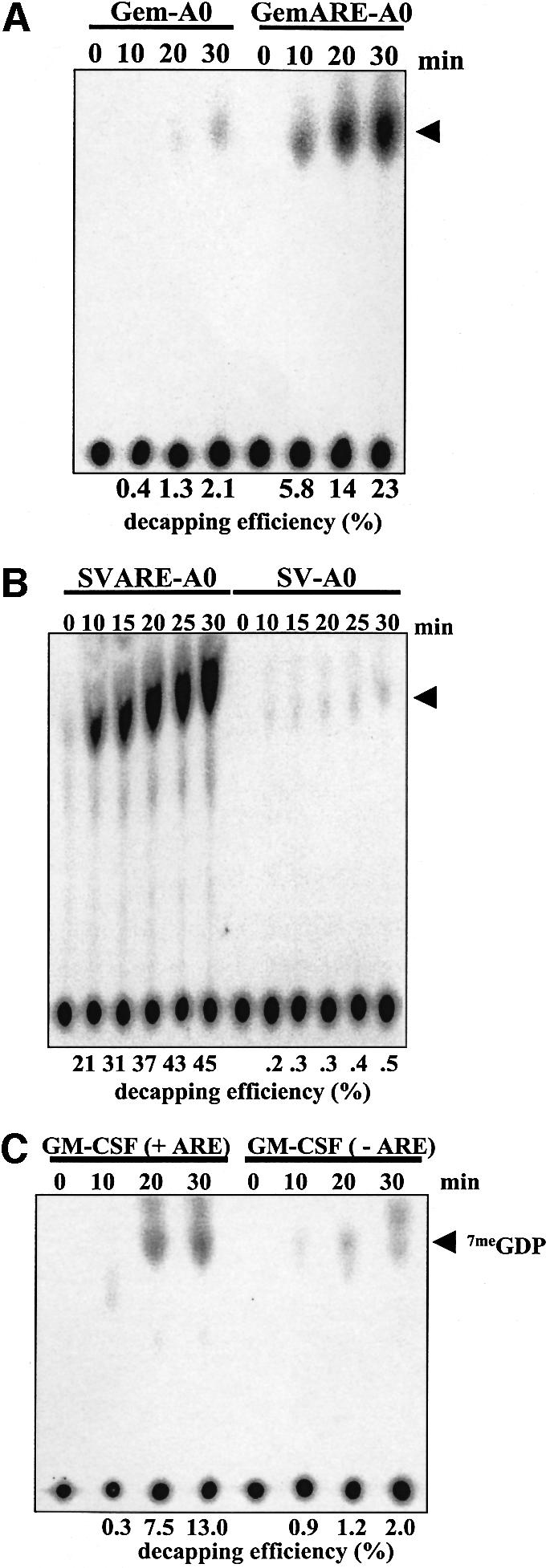
Fig. 5. The presence of an AU-rich element significantly stimulates the efficiency of decapping. (A and B) A matched pair of cap-labeled RNA substrates that either lacked (Gem-A0 or SV-A0) or contained the TNF-α AU-rich element (GemARE-A0 or SVARE-A0) were incubated in the in vitro decapping system in the presence of cap analog for the indicated amount of time. Reaction products were analyzed by TLC on PEI cellulose sheets. (C) A matched pair of cap-labeled RNA substrates that either lacked [GM-CSF(–ARE)] or contained the GM-CSF AU-rich element [GM-CSF (+ARE)] were incubated in the in vitro decapping system in the presence of cap analog for the indicated amount of time. Reaction products were analyzed by TLC on PEI cellulose sheets. For all three panels, the position of 7meGDP is indicated by an arrowhead.
In order to test the specificity of the activation of decapping in HeLa extracts by cap analog, the ability of methylated versus non-methylated cap analogs to activate decapping was compared. As seen in Figure 2A, the presence of the methyl group on the cap analog was absolutely required to activate decapping. This suggests that decapping was activated by cap analog through the specific sequestration of cap-binding proteins. In order to confirm this, UV cross-linking assays were performed using cap-labeled RNA substrates. As seen in Figure 2B, the addition of increasing amounts of methylated cap analog to HeLa S100 extracts specifically competed for a 24 kDa protein. The 24 kDa protein is likely to be eIF4E (Altmann et al., 1985) since it has a similar apparent molecular weight, specifically cross-links to 7-methylated cap structures and is only detected in cytoplasmic extracts. Interestingly, Schwartz and Parker (2000) have demonstrated recently that purified yeast eIF4E can inhibit purified yeast decapping enzyme in vitro. Similar, but not identical amounts of 7meGpppG also specifically competed for cap binding by the deadenylase DAN/PARN (Figure 2C). In order to determine whether the activation of decapping was correlated with the competition of the 24 kDa cap-binding protein and/or DAN/PARN, careful titration experiments with cap analog were performed. As seen in Figure 2D, decapping was only partially activated by the addition of 5 µM cap analog. This amount of cap analog fully sequestered the 24 kDa cap-binding protein, but not DAN/PARN, in the extracts. Full activation of decapping required 10 µM cap analog, the amount required to sequester DAN/PARN fully. These data suggest that DAN/PARN may be a key factor in preventing decapping through interaction with the 5′ cap. Specific removal of DAN/PARN from extracts by immunodepletion, however, failed to activate decapping (data not shown). We conclude that a decapping activity with properties similar to those of the yeast Dcp1p enzyme exists in mammalian cytoplasmic extracts and can be activated specifically by sequestering cap-binding activities. These data suggest that access to the cap structure is an important feature in the regulation of decapping in mammalian cells.

Fig. 2. Methylated cap analog specifically activates decapping in HeLa cytoplasmic extracts by sequestering cap-binding proteins. (A) The indicated amounts of 7meGpppG or GpppG were incubated with HeLa S100 extracts and decapping assays were performed using cap-labeled GemARE-A60 RNA. The products of decapping were analyzed on PEI cellulose sheets. The arrowhead indicates 7meGDP. (B) The indicated amounts of 7meGpppG or GpppG were incubated in HeLa S100 extracts using cap-labeled GemARE-A60 RNA under decapping conditions. After 5 min, UV cross-linking was performed, mixtures were treated with RNase, and proteins radiolabeled through cross-linking to cap-labeled RNA oligomers were analyzed by electrophoresis on a 15% acrylamide gel containing SDS. The position of eIF4E is indicated by the arrowhead. (C) The indicated amounts of 7meGpppG or GpppG were incubated in HeLa S100 extracts using cap-labeled GemARE-A60 RNA under decapping conditions. After 5 min, UV cross-linking was performed, mixtures were treated with RNase, and DAN/PARN proteins radiolabeled through cross-linking to cap-labeled RNA oligomers were immunoprecipitated and analyzed by electrophoresis on a 10% acrylamide gel containing SDS. The position of DAN/PARN is indicated by the arrowhead. (D) The indicated amounts of 7meGpppG or GpppG were incubated in HeLa S100 extracts using cap-labeled GemARE-A0 RNA under decapping conditions. The top panel shows UV cross-linking analysis of total protein as described in (B) to identify eIF4E. The middle panel shows UV cross-linking/immunoprecipitation analysis as described in (C) to identify DAN/PARN. The bottom panel shows the products of a decapping assay that were analyzed by TLC as described in (A).
A poly(A) tail represses decapping of mammalian mRNAs in a poly(A)-binding protein-dependent fashion
Poly(A) tail shortening is a prerequisite for the decapping and turnover of most yeast mRNAs (Decker and Parker, 1993). This suggests that the presence of a poly(A) tail on RNA substrates represses decapping in vivo. In order to determine whether our in vitro decapping assay faithfully reproduced this regulatory aspect of in vivo decapping, matched RNA substrates were added to HeLa S100 extracts that either lacked a poly(A) tail (GemARE-A0, SVARE-A0 or GM-CSFT-A0) or contained 60 adenylate residues at their 3′ ends (GemARE-A60, SVARE-A60 or GM-CSFT-A60). The sizes of these transcripts varied from 95 to >700 bases to ensure that results obtained were independent of the size of the transcript and could be generalized. As seen in Figure 3, all three transcripts that lacked a poly(A) tail at their 3′ end were decapped efficiently in S100 extract in the presence of cap analog. Their adenylated counterparts, however, were decapped at a dramatically reduced efficiency (10- to 20-fold). Note that the position of the 7meGDP spot varies depending on the time of incubation in the extract. In all cases, however, the radioactive spot co-migrates with 7meGDP, as determined by UV shadowing of unlabeled markers that were loaded in each lane (data not shown). The reason for this altered migration is unclear, but may be due to the generation of small molecules during the incubation of the extract, which caused alteration in the chromatographic mobility of nucleotides on PEI cellulose. We conclude that the presence of a poly(A) tail inhibits decapping in HeLa cytoplasmic extracts in a manner similar to that observed in yeast (Muhlrad et al., 1994), suggesting that the in vitro decapping assay reproduces this important regulatory aspect of mRNA turnover.
We next addressed the mechanism of poly(A) tail-mediated repression of decapping. In order to assess whether poly(A)-binding proteins (PABPs) were involved, increasing amounts of poly(A) competitor RNA were added to in vitro decapping assays using an RNA substrate that possessed a 60 base poly(A) tail. As seen in Figure 4A, the addition of cold poly(A) competitor RNA that inhibited the cross-linking of a 70 kDa PABP to the poly(A) tail (Ford and Wilusz, 1999; data not shown) effectively stimulated decapping to levels observed with deadenylated substrates. Similar data were obtained for all RNA substrates tested (data not shown). It is important to note that since our decapping assays are performed in the presence of cap analog, which sequesters the deadenylase DAN/PARN, deadenylation of the RNA substrate is not occurring under these conditions (Gao et al., 2000) and, therefore, can not account for the results obtained. Furthermore, poly(A) competitor RNA failed to stimulate decapping in the absence of cap analog (data not shown), suggesting that the presence of free cap-binding proteins in extracts is a dominant inhibitor of in vitro decapping. The stimulation of decapping was specific for poly(A), as other homopolymers such as poly(C) did not appreciably affect decapping of all RNA substrates tested (Figure 4B). Finally, we tested whether the stimulation of decapping by poly(A) competitor RNA was a general activator of decapping or was specific for adenylated transcripts. As seen in Figure 4C, the decapping of an RNA substrate that lacked a poly(A) tail (GemARE-A0) was only mildly stimulated by the addition of poly(A) competitor RNA. The small (1.5-fold) effect of poly(A) on decapping of the GemARE-A0 RNA substrate is probably due to the low affinity of PABP for the ARE contained in the 3′ portion of the transcript (D.Fritz and J.Wilusz, data not shown). Similar data were also obtained for all RNA substrates tested that lacked a 3′ poly(A) tail (data not shown). We conclude that PABPs are repressors of decapping when bound to poly(A)+ RNA substrates. Furthermore, since our decapping assays are performed in the presence of 7meGpppG, which sequesters eIF4E (Figure 2), the repression of decapping by PABPs must be occurring in a novel, eIF4E-independent fashion.
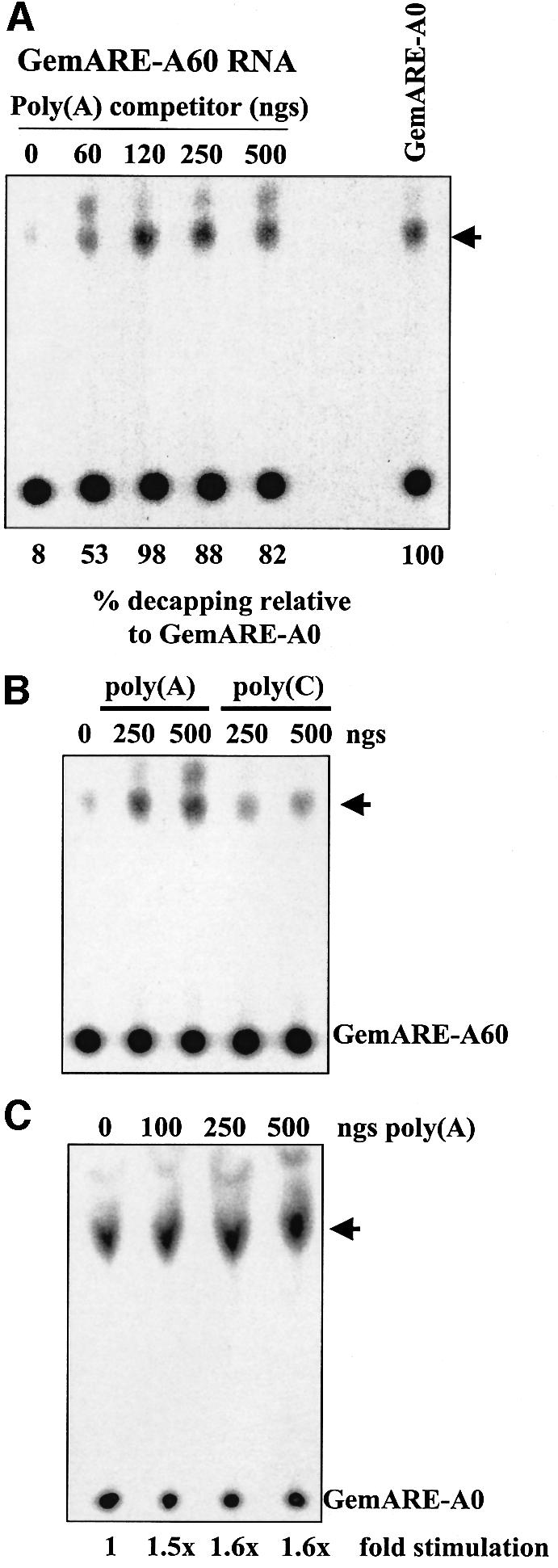
Fig. 4. The addition of poly(A) competitor RNA specifically activates decapping of polyadenylated RNA substrates. (A) Cap-labeled GemARE-A60 RNA, which contained 60 adenylate residues at its 3′ end, was incubated in the in vitro decapping system for 30 min in the presence of cap analog and the indicated amount of cold poly(A) competitor RNA. Reaction products were analyzed by TLC on PEI cellulose sheets. In the lane marked GemARE-A0, GemARE-A0 RNA [which lacks a poly(A) tail] was incubated in the in vitro decapping assay in the absence of poly(A) RNA competitor. (B) Cap-labeled GemARE-A60 RNA was incubated in the in vitro decapping system for 30 min in the presence of cap analog and the indicated amount of cold poly(A) or poly (C) competitor RNAs. Reaction products were analyzed by TLC on PEI cellulose sheets. (C) Cap-labeled GemARE-A0 RNA, which lacked a poly(A) tail, was incubated in the in vitro decapping system for 30 min in the presence of cap analog and the indicated amount of cold poly(A) competitor RNA. Reaction products were analyzed by TLC on PEI cellulose sheets. For all three panels, the position of 7meGDP is indicated by an arrowhead.
AU-rich instability elements dramatically stimulate decapping
Many short-lived mammalian mRNAs contain AREs in their 3′-UTRs that have been shown to be directly responsible for the high rate of turnover of these transcripts (Chen and Shyu, 1995). While AU-rich instability elements have been shown to increase the rate of deadenylation both in vivo (Shaw and Kamen, 1986; Shyu et al., 1989) and in vitro (Ford et al., 1999), we have recently demonstrated that efficient deadenylation requires an interaction between DAN/PARN and the 5′ cap structure (Gao et al., 2000). We hypothesized, therefore, that AREs may be capable of stimulating deadenylation by making the cap structure more accessible to the deadenylase. If this is correct, then AREs may also be able to stimulate decapping by a similar mechanism. In order to test this model, we prepared two independent RNA substrates that either lacked (Gem-A0 and SV-A0) or contained (GemARE-A0 and SVARE-A0) the ARE from the tumor necrosis factor-α (TNF-α) mRNA. As seen in Figure 5A and B, the presence of the TNF-α ARE in an RNA substrate stimulated decapping >10-fold. In order to generalize this observation to other AREs, we prepared an RNA that contained the granulocyte–macrophage colony-stimulating factor (GM-CSF) ARE or a matched control transcript that lacked the element. As seen in Figure 5C, the ARE from the GM-CSF mRNA also strongly stimulated decapping in vitro. We conclude that AU-rich instability elements dramatically stimulate decapping.
Finally, we determined whether AU-rich element binding proteins (ARE-BPs) play a role in the stimulation of decapping by the instability element. In order to sequester ARE-BPs in S100 extracts, increasing amounts of a 34 base synthetic RNA oligomer containing the TNF-α ARE were added to in vitro decapping assays. In control reactions, similar amounts of an unrelated 33 base RNA were added. As seen in Figure 6, the addition of the ARE competitor RNA significantly reduced decapping of an ARE-containing RNA substrate, while similar levels of the control competitor RNA had no effect on decapping efficiency. The reduction in decapping efficiency by the ARE synthetic RNA competitor was only observed with RNA substrates that contained an ARE (Figure 6B). Finally, we used UV cross-linking assays in an attempt to correlate the competition of a specific ARE-BP by the ARE competitor RNA with repression of decapping. Both ARE-specific RNA-binding proteins that we observed in our UV cross-linking assays, HuR (a known mRNA stabilizer) and an unidentified 40 kDa species (Ford et al., 1999), were competed by the ARE RNA oligomer with exactly the same concentration dependence (data not shown). The identity of the ARE-BP that is responsible for stimulating decapping, therefore, remains to be determined. We conclude that the stimulation of decapping by AREs occurs through trans-acting factors, which are likely to play a key regulatory role in cap-mediated events during mRNA turnover.

Fig. 6. The stimulation of decapping by AU-rich elements requires sequence-specific AU-rich element binding factors. Cap-labeled GemARE-A0 RNA (A) or Gem-A0 RNA (B) was incubated in the in vitro decapping system in the presence of cap analog and the indicated amount of a 34 base synthetic RNA competitor derived from the TNF-α AU-rich element (ARE oligo lanes) or a 33mer derived from randomly selected sequences (non-specific oligo lanes). Reaction products were analyzed by TLC on PEI cellulose sheets. The position of 7meGDP is indicated by an arrowhead. The numbers at the bottom refer to decapping efficiency relative to lane 0.
Discussion
We have identified and characterized a novel regulated decapping activity in mammalian cytoplasmic extracts, which probably plays a key role in mRNA turnover. Decapping was found to be repressed by two activities: cap-binding proteins and poly(A)-binding proteins. These data suggest that accessibility to terminal mRNA structures, as well as communication between the 3′ and 5′ ends, are important factors in mRNA stability as well as translation (Sachs and Varani, 2000). Therefore, the functional link between translation and mRNA stability is likely to be intertwined in this competition for the same cis elements of the mRNA.
The human decapping activity we have identified is similar to the well-characterized yeast Dcp1p enzyme in several aspects. Both enzymes cleave 5′ cap structures to yield 7meGDP in an Mg2+-dependent, ATP-independent fashion. Both enzymes also appear to be regulated in a similar fashion by cap-binding proteins and the poly(A) tail (Caponigro and Parker, 1995; Schwartz and Parker, 1999; Vilela et al., 2000, C.J.Wilusz, M.Gao, J.Wilusz and S.W.Peltz, unpublished results). The HeLa enzyme will, however, effectively decap short RNA substrates that have previously been shown to be inefficient substrates for the yeast enzyme (LaGrandeur and Parker, 1998; data not shown). Several auxiliary factors have been shown to influence decapping in yeast, including the putative pyrophosphatase Dcp2p (Dunckley and Parker, 1999), the complex of Lsm1–7 proteins (Bouveret et al., 2000; Tharun et al., 2000), Vps16p (Zhang et al., 1999a) and Mrt1p/Pat1p (Bonnerot et al., 2000). Unfortunately, database screens using yeast Dcp1p sequences have not uncovered any candidate mammalian enzymes that have demonstrable decapping activity. Purification and reconstitution studies are under way to characterize the protein(s) involved in mammalian decapping.
PABPs bound to the poly(A) tail effectively repress decapping in mammalian extracts (Figures 3 and 4). This observation is consistent with the premature decapping observed in vivo in yeast containing knockouts of the PAB1 gene (Caponigro and Parker, 1995; Morrissey et al., 1999). PABP has been shown previously to mediate communication between the 5′ and 3′ ends of an mRNA through a heterotrimeric complex involving eIF4G and the cap-binding protein eIF4E (Imataka et al., 1998; Wells et al., 1998). In our in vitro assays, however, PABPs repress decapping in an eIF4E-independent fashion, since the cap-binding protein is sequestered by cap analog (Figure 2). This suggests that PABP must be communicating with the 5′ cap and repressing decapping through a novel complex. The existence of such a complex was also suggested recently by genetic studies in which mutations in yeast eIF4E or eIF4G failed to stimulate deadenylation-independent decapping (Schwartz and Parker, 1999). Since this novel complex, which communicates with the 5′ cap, is not inhibited by the presence of 7meGpppG in our assays, it is likely to involve a protein that recognizes the 5′ cap in the context of a linear stretch of RNA sequence. In addition, since nonsense-mediated decay is initiated through a deadenylation-independent decapping event (Muhlrad and Parker, 1994), the innate instability of nonsense-containing mRNAs may be reflected in their inability to form this novel complex between the poly(A) tail and the 5′ cap. Finally, Gray et al. (2000) have shown recently that different regions of a tethered PABP are required for mRNA stability and translation initiation. We are currently using a biochemical approach to characterize this novel complex involved in the communication between PABP and the 5′ cap structure.
Decapping was found to be stimulated directly by AREs, sequences that were shown previously to shorten the half-lives of mRNAs dramatically and increase the rate of deadenylation (Chen and Shyu, 1995). This observation suggests that ARE-BPs involved in regulating mRNA stability may function primarily by determining the accessibility of the cap structure to the turnover machinery. Increasing access to the cap, for example, would increase deadenylation by allowing DAN/PARN to interact with the cap and increase the rate of poly(A) shortening (Dehlin et al., 2000; Gao et al., 2000). AREs and their associated binding proteins may make the cap accessible through several different mechanisms. First, they may destabilize secondary structures in the 5′-UTR (or other regions of the RNA) that effectively mask the cap. Secondly, AREs may disrupt protein–RNA interactions with the cap directly, or with the 5′-UTR that serves to restrict cap access. Thirdly, ARE-BPs may disrupt PABP interactions with the transcript and remove the repressor from one (or both) ends of the transcript. Finally, ARE-BPs may interact directly with cap-binding factors such as DAN/PARN or the decapping enzyme and stabilize (or destabilize in the case of eIF4E) their interaction with the mRNA substrate. A network of ARE-BPs exists that may positively (i.e. TTP and AUF1) or negatively (HuR) influence mRNA turnover, providing an effective repertoire to regulate mRNA stability in response to changes in cell growth and differentiation. Competition of several of these proteins by the synthetic ARE competitor RNA correlates with abrogation of ARE-stimulated decapping (data not shown). We are currently performing biochemical fractionation and reconstitution experiments to identify those ARE-BPs that specifically stimulate decapping.
Previous mutational analysis of the ARE present in the 3′-UTR of c-fos mRNA demonstrated that while the overall element played a role in increasing the rates of mRNA deadenylation and subsequent degradation, increases in the rate of poly(A) tail shortening did not require intact AUUUA portions of the element (Shyu et al., 1991). Accelerated turnover of the body of the mRNA, however, was sensitive to mutations of these AUUUA pentanucleotides. These data suggest that the ARE and associated ARE-BPs may have several independent and separable functions to stimulate first the deadenylation and then the decay of a mRNA.
In summary, our data suggest the following model for regulated decapping in mammalian cells (Figure 7). First, the highly regulated translation initiation factor eIF4E (Sonenberg and Gingras, 1998) probably serves as a general regulator that determines whether an mRNA will be translated or subject to deadenylation and decay by monitoring overall access to the 5′ cap structure (Tucker and Parker, 2000). Likewise, the presence of a poly(A) tail and associated PABPs also provides inherent stability to the RNA in both deadenylation (Ford et al., 1999; Wang et al., 1999) and decapping by mediating communication between the 5′ and 3′ ends. The process of translation probably plays an important role in overcoming these two factors, which limit the access of degradative enzymes to the 5′ cap structure. Alternatively, ARE-BPs such as TTP or AUF-1 can also regulate access of turnover factors to the cap by either competing for cap-binding proteins or directing recruiting deadenylation and/or decapping factors. When the cap of an mRNA is made accessible by the loading/movement of ribosomes or via ARE-BPs, DAN/PARN interacts with it and initiates effective deadenylation of the transcript. The affinity of DAN/PARN for the cap may represent the last hurdle for decapping of most mRNAs. Following poly(A) shortening, binding of DAN/PARN to the 5′ cap (as well as the 3′ end) of the transcript is reduced (Gao et al., 2000), allowing the deadenylation machinery to hand the transcript over to the decapping (and/or 3′→5′ exonuclease) enzymes (Allmang et al., 1999). Additional points of regulation may still occur at the level of the deadenylated mRNA if decapping requires assembly of a multicomponent complex involving Lsm proteins, Dcp2p and Mrt1p/Pat1p homologs. With all inhibitory factors now removed, decapping can proceed efficiently and the mRNA is degraded.

Fig. 7. A model for regulated mRNA decapping. The mRNA cap structure is normally stabilized by interactions with eIF4E and/or a novel PABP complex. The process of translation or the action of AU-rich element-binding proteins can disrupt these complexes involving the 5′ cap, exposing the ends of the transcript to the deadenylation machinery. Following poly(A) tail shortening, the affinity of DAN/PARN for the mRNA is dramatically reduced, allowing the decapping enzyme (as well as other degradative enzymes such as 3′→5′ exonucleases) access to the ends of the mRNA.
Materials and methods
RNAs
SVARE-A0 RNA, which contains the 34 base ARE from TNF-α inserted into the BamHI–BclI fragment representing the 3′ portion of SV40 late mRNAs, was transcribed from HindIII-linearized templates as previously described (Ford et al., 1999). SVARE-A60 RNA, a variant that contains a 60 base poly(A) tract at its 3′ end, was prepared as previously described (Ford et al., 1999). Gem-A60 RNA, which contains sequences from the pGem4 polylinker region followed by 60 A residues, was prepared as previously described (Ford et al., 1999). GemARE-A60, a variant that contains the 34 base ARE from TNF-α, was prepared as described previously (Gao et al., 2000). Transcription of HindIII-linearized templates yields GemARE-A0 RNA. Sequences encoding a 60 base poly(A) tail were added to DNA using a ligation–PCR approach described previously (Ford et al., 1997). The template for GM-CSF and GM-CSF(–ARE) was pGM-CSF (Shaw and Kamen, 1986) cut with EcoRI (to yield a 750 base transcript) or NcoI (to yield a 515 base transcript), respectively. GM-CSFT-A0 RNA was prepared by inserting the ARE from TNF-α into pGM-CSF by inserting the oligonucleotide 5′-CATGATTATTTATTATTTATTTATTATTTATTTATTTAAAC and its appropriate complement at its NcoI site. This replaced the endogenous destabilizing element of GM-CSF with the TNF-α ARE. Transcription of NcoI-linearized templates yields a 557 base GM-CSFT-A0 RNA. The addition of a 60 base poly(A) tail to GM-CSFT-A0 RNA was performed by ligation–PCR as described above.
RNAs were transcribed in vitro using SP6 polymerase as described previously (Wilusz and Shenk, 1988) in the absence of cap analog and radioactive rNTPs. To label RNAs exclusively at their cap structures, transcription products were then capped using recombinant vaccinia guanyltransferase and [α-32P]GTP (Zhang et al., 1999b). All RNAs were purified on 5% acrylamide gels prior to use. To make RNAs that were radioactively labeled at the α-phosphate of an unmethylated cap, S-adenosylhomocysteine was substituted for S-adenosylmethionine in the capping reaction.
Synthetic RNAs used in competition studies were made by the NJMS Molecular Core Facility and contained the following sequences: ARE, 5′-AUUAUUUAUUAUUUAUUUAUUAUUUAUUUAUUUA; and non- specific competitor, 5′-GGAUUAACUAAUUGAUACCGCGUAUACACGCGG. Poly(A) and poly(C) competitor RNAs, together with 7meGpppG and GpppG, were purchased from Amersham Pharmacia Biotech.
Extracts
Whole-cell yeast extracts were prepared as described (Lin et al., 1985). S100 cytoplasmic extracts were prepared from HeLa spinner cells grown in 10% horse serum as described previously (Ford and Wilusz, 1999; Ford et al., 1999). Aliquots were stored at –80°C.
In vitro decapping assay
Decapping assays were performed using conditions adapted from Zhang et al. (1999b). Cap-labeled RNAs (10–50 fmol) were incubated with 4 µl of HeLa S100 cytoplasmic extract in a 10 µl reaction in the presence of CE buffer [50 mM Tris pH 7.9, 30 mM (NH4)2SO4, 1 mM MgCl2] and 20 µM cap analog (where indicated). Reaction mixtures were incubated at 30°C for the times indicated and stopped by the addition of 1 µl of 0.25 M EDTA. Reaction products were separated and identified by TLC on PEI cellulose sheets developed in 450 mM (NH4)2SO4. Quantitation was performed using a Molecular Dynamics PhosphorImager. 7meGMP and 7meGDP (20 µg) were spotted routinely on TLC plates along with reaction samples to serve as markers that could be visualized by UV shadowing.
UV cross-linking
UV cross-linking analysis was performed as described (Wilusz and Shenk, 1988). Briefly, 20–50 fmol of cap-radiolabeled RNA were incubated in the in vitro decapping assay for 5 min and then reaction mixtures were irradiated on ice for 10 min using a 15 W germicidal light. RNases A and T1 were added, and proteins covalently attached to short radioactive RNA oligomers were analyzed on a 10% acrylamide gel containing SDS. For analysis of UV cross-linked DAN/PARN protein by immunoprecipitation using a polyclonal antibody obtained from M.Wormington (Korner et al., 1998), 400 µl of NET2 buffer (50 mM Tris pH 7.6, 150 mM NaCl, 0.01% NP-40) were added to reaction mixtures following RNase treatment, and reaction mixtures were centrifuged for 4 min. Pre-cleared samples were incubated on ice with 2–5 µl of specified rabbit polyclonal antisera for 1 h. Antigen–antibody complexes were collected on protein A-positive Staphylococcus aureus cells, washed five times in RIPA buffer (50 mM Tris pH 7.6, 150 mM NaCl, 0.1% SDS, 1% NP-40, 0.5% deoxycholate), and immunoprecipitated cross-linked proteins were analyzed on a 10 or 15% acrylamide gel containing SDS.
Acknowledgments
Acknowledgements
We wish to thank Dr Michael Wormington for the generous gift of DAN/PARN polyclonal antibodies, Dr Robert Donnelly for synthesis of synthetic RNA oligomers, and laboratory members for helpful discussions. This work was supported by grants from the National Institutes of Health to J.W. (CA80062) and to S.W.P. (GM58276).
References
- Allmang C., Petfalski,E., Podtelejnikov,A., Mann,M., Tollervey,D. and Mitchell,P. (1999) The yeast exosome and human PM-Scl are related complexes of 3′→5′ exonucleases. Genes Dev., 13, 2148–2158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Altmann M., Edery,I., Sonenberg,N. and Trachsel,H. (1985) Purification and characterization of protein synthesis initiation factor eIF-4E from the yeast Saccharomyces cerevisiae. Biochemistry, 24, 6085–6089. [DOI] [PubMed] [Google Scholar]
- Beelman C.A., Stevens,A., Caponigro,G., LaGrandeur,T.E., Hatfield,L., Fortner,D.M. and Parker,R. (1996) An essential component of the decapping enzyme required for normal rates of mRNA turnover. Nature, 382, 642–646. [DOI] [PubMed] [Google Scholar]
- Bonnerot C., Boeck,R. and Lapeyre,B. (2000) The two proteins pat1p (Mrt1p) and spb8p interact in vivo, are required for mRNA decay and are functionally linked to pab1p. Mol. Cell. Biol., 20, 5939–5946. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bousquet-Antonelli C., Presutti,C. and Tollervey,D. (2000) Identification of a regulated pathway for nuclear pre-mRNA turnover. Cell, 102, 765–775. [DOI] [PubMed] [Google Scholar]
- Bouveret E., Rigaut,G., Shevchenko,A., Wilm,M. and Seraphin,B. (2000) A Sm-like protein complex that participates in mRNA degradation. EMBO J., 19, 1661–1671. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brewer G. (1991) An A + U-rich element RNA-binding factor regulates c-myc mRNA stability in vitro. Mol. Cell. Biol., 11, 2460–2466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Caponigro G. and Parker,R. (1995) Multiple functions for the poly(A)-binding protein in mRNA decapping and deadenylation in yeast. Genes Dev., 9, 2421–2432. [DOI] [PubMed] [Google Scholar]
- Caponigro G. and Parker,R. (1996) Mechanisms and control of mRNA turnover in Saccharomyces cerevisiae. Microbiol. Rev., 60, 233–249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen C.Y. and Shyu,A.B. (1995) AU-rich elements: characterization and importance in mRNA degradation. Trends Biochem. Sci., 20, 465–470. [DOI] [PubMed] [Google Scholar]
- Couttet P., Fromont-Racine,M., Steel,D., Pictet,R. and Grange,T. (1997) Messenger RNA deadenylylation precedes decapping in mammalian cells. Proc. Natl Acad. Sci. USA, 94, 5628–5633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Czaplinski K., Ruiz-Echevarria,M.J., Gonzalez,C.I. and Peltz,S.W. (1999) Should we kill the messenger? The role of the surveillance complex in translation termination and mRNA turnover. BioEssays, 21, 685–696. [DOI] [PubMed] [Google Scholar]
- Decker C.J. and Parker,R. (1993) A turnover pathway for both stable and unstable mRNAs in yeast: evidence for a requirement for deadenylation. Genes Dev., 7, 1632–1643. [DOI] [PubMed] [Google Scholar]
- Dehlin E., Wormington,M., Korner,C.G. and Wahle,E. (2000) Cap-dependent deadenylation of mRNA. EMBO J., 19, 1079–1086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dunckley T. and Parker,R. (1999) The DCP2 protein is required for mRNA decapping in Saccharomyces cerevisiae and contains a functional MutT motif. EMBO J., 18, 5411–5422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fan X.C. and Steitz,J.A. (1998) Overexpression of HuR, a nuclear–cytoplasmic shuttling protein, increases the in vivo stability of ARE-containing mRNAs. EMBO J., 17, 3448–3460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ford L.P. and Wilusz,J. (1999) An in vitro system using HeLa cytoplasmic extracts that reproduces regulated mRNA stability. Methods, 17, 21–27. [DOI] [PubMed] [Google Scholar]
- Ford L.P., Bagga,P.S. and Wilusz,J. (1997) The poly(A) tail inhibits the assembly of a 3′-to-5′ exonuclease in an in vitro RNA stability system. Mol. Cell. Biol., 17, 398–406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ford L.P., Watson,J., Keene,J.D. and Wilusz,J. (1999) ELAV proteins stabilize deadenylated intermediates in a novel in vitro mRNA deadenylation/degradation system. Genes Dev., 13, 188–201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gao M., Fritz,D.T., Ford,L.P. and Wilusz,J. (2000) Interaction between a poly(A)-specific ribonuclease and the 5′ cap influences mRNA deadenylation rates in vitro. Mol. Cell, 5, 479–488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gray N.K., Coller,J.M., Dickson,K.S. and Wickens,M. (2000) Multiple portions of poly(A)-binding protein stimulate translation in vivo. EMBO J., 19, 4723–4733. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hagan K.W., Ruiz-Echevarria,M.J., Quan,Y. and Peltz,S.W. (1995) Characterization of cis-acting sequences and decay intermediates involved in nonsense-mediated mRNA turnover. Mol. Cell. Biol., 15, 809–823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hsu C.L. and Stevens,A. (1993) Yeast cells lacking 5′→3′ exoribonuclease 1 contain mRNA species that are poly(A) deficient and partially lack the 5′ cap structure. Mol. Cell. Biol., 13, 4826–4835. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Imataka H., Gradi,A. and Sonenberg,N. (1998) A newly identified N-terminal amino acid sequence of human eIF4G binds poly(A)-binding protein and functions in poly(A)-dependent translation. EMBO J., 17, 7480–7489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jacobs J.S., Anderson,A.R. and Parker,R.P. (1998) The 3′ to 5′ degradation of yeast mRNAs is a general mechanism for mRNA turnover that requires the SKI2 DEVH box protein and 3′ to 5′ exonucleases of the exosome complex. EMBO J., 17, 1497–1506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jacobson A. and Peltz,S.W. (1996) Interrelationships of the pathways of mRNA decay and translation in eukaryotic cells. Annu. Rev. Biochem., 65, 693–739. [DOI] [PubMed] [Google Scholar]
- Korner C.G., Wormington,M., Muckenthaler,M., Schneider,S., Dehlin,E. and Wahle,E. (1998) The deadenylating nuclease (DAN) is involved in poly(A) tail removal during the meiotic maturation of Xenopus oocytes. EMBO J., 17, 5427–5437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- LaGrandeur T.E. and Parker,R. (1998) Isolation and characterization of Dcp1p, the yeast mRNA decapping enzyme. EMBO J., 17, 1487–1496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lai W.S., Carballo,E., Strum,J.R., Kennington,E.A., Phillips,R.S. and Blackshear,P.J. (1999) Evidence that tristetraprolin binds to AU-rich elements and promotes the deadenylation and destabilization of tumor necrosis factor α mRNA. Mol. Cell. Biol., 19, 4311–4323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Levine T.D., Gao,F., King,P.H., Andrews,L.G. and Keene,J.D. (1993) Hel-N1: an autoimmune RNA-binding protein with specificity for 3′ uridylate-rich untranslated regions of growth factor mRNAs. Mol. Cell. Biol., 13, 3494–3504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin R.J., Newman,A.J., Cheng,S.C. and Abelson,J. (1985) Yeast mRNA splicing in vitro. J. Biol. Chem., 260, 14780–14792. [PubMed] [Google Scholar]
- Loflin P., Chen,C.Y. and Shyu,A.B. (1999) Unraveling a cytoplasmic role for hnRNP D in the in vivo mRNA destabilization directed by the AU-rich element. Genes Dev., 13, 1884–1897. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Losson R. and Lacroute,F. (1979) Interference of nonsense mutations with eukaryotic messenger RNA stability. Proc. Natl Acad. Sci. USA, 76, 5134–5137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ma W.J., Cheng,S., Campbell,C., Wright,A. and Furneaux,H. (1996) Cloning and characterization of HuR, a ubiquitously expressed Elav-like protein. J. Biol. Chem., 271, 8144–8151. [DOI] [PubMed] [Google Scholar]
- Mitchell P. and Tollervey,D. (2000) mRNA stability in eukaryotes. Curr. Opin. Genet. Dev., 10, 193–198. [DOI] [PubMed] [Google Scholar]
- Morrissey J.P., Deardorff,J.A., Hebron,C. and Sachs,A.B. (1999) Decapping of stabilized, polyadenylated mRNA in yeast pab1 mutants. Yeast, 15, 687–702. [DOI] [PubMed] [Google Scholar]
- Muhlrad D. and Parker,R. (1994) Premature translational termination triggers mRNA decapping. Nature, 370, 578–581. [DOI] [PubMed] [Google Scholar]
- Muhlrad D., Decker,C.J. and Parker,R. (1994) Deadenylation of the unstable mRNA encoded by the yeast MFA2 gene leads to decapping followed by 5′→3′ digestion of the transcript. Genes Dev., 8, 855–866. [DOI] [PubMed] [Google Scholar]
- Nuss D.L., Furuichi,Y., Koch,G. and Shatkin,A.J. (1975) Detection in HeLa cell extracts of a 7-methyl guanosine specific enzyme activity that cleaves m7GpppNm. Cell, 6, 21–27. [DOI] [PubMed] [Google Scholar]
- Peng S.S., Chen,C.Y., Xu,N. and Shyu,A.B. (1998) RNA stabilization by the AU-rich element binding protein, HuR, an ELAV protein. EMBO J., 17, 3461–3470. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sachs A.B. and Varani,G. (2000) Eukaryotic translation initiation: there are (at least) two sides to every story. Nature Struct. Biol., 7, 356–361. [DOI] [PubMed] [Google Scholar]
- Schwartz D.C. and Parker,R. (1999) Mutations in translation initiation factors lead to increased rates of deadenylation and decapping of mRNAs in Saccharomyces cerevisiae. Mol. Cell. Biol., 19, 5247–5256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schwartz D.C. and Parker,R. (2000) mRNA decapping in yeast requires dissociation of the cap binding protein, eukaryotic translation initiation factor 4E. Mol. Cell. Biol., 20, 7933–7942. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shaw G. and Kamen,R. (1986) A conserved AU sequence from the 3′ untranslated region of GM-CSF mRNA mediates selective mRNA degradation. Cell, 46, 659–667. [DOI] [PubMed] [Google Scholar]
- Shyu A.B., Greenberg,M.E. and Belasco,J.G. (1989) The c-fos transcript is targeted for rapid decay by two distinct mRNA degradation pathways. Genes Dev., 3, 60–72. [DOI] [PubMed] [Google Scholar]
- Shyu A.B., Belasco,J.G. and Greenberg,M.E. (1991) Two distinct destabilizing elements in the c-fos message trigger deadenylation as a first step in rapid mRNA decay. Genes Dev., 5, 221–231. [DOI] [PubMed] [Google Scholar]
- Sonenberg N. and Gingras,A.C. (1998) The mRNA 5′ cap-binding protein eIF4E and control of cell growth. Curr. Opin. Cell Biol., 10, 268–275. [DOI] [PubMed] [Google Scholar]
- Sonenberg N., Morgan,M.A., Merrick,W.C. and Shatkin,A.J. (1978) A polypeptide in eukaryotic initiation factors that crosslinks specifically to the 5′-terminal cap in mRNA. Proc. Natl Acad. Sci. USA, 75, 4843–4847. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tharun S., He,W., Mayes,A.E., Lennertz,P., Beggs,J.D. and Parker,R. (2000) Yeast Sm-like proteins function in mRNA decapping and decay. Nature, 404, 515–518. [DOI] [PubMed] [Google Scholar]
- Tucker M. and Parker,R. (2000) Mechanisms and control of mRNA decapping in Saccharomyces cerevisiae. Annu. Rev. Biochem., 69, 571–595. [DOI] [PubMed] [Google Scholar]
- Vilela C., Velasco,C., Ptushkina,M. and McCarthy,J.E. (2000) The eukaryotic mRNA decapping protein Dcp1 interacts physically and functionally with the eIF4F translation initiation complex. EMBO J., 19, 4372–4382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Z., Day,N., Trifillis,P. and Kiledjian,M. (1999) An mRNA stability complex functions with poly(A)-binding protein to stabilize mRNA in vitro. Mol. Cell. Biol., 19, 4552–4560. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wells S.E., Hillner,P.E., Vale,R.D. and Sachs,A.B. (1998) Circularization of mRNA by eukaryotic translation initiation factors. Mol. Cell, 2, 135–140. [DOI] [PubMed] [Google Scholar]
- Wilson T. and Treisman,R. (1988) Removal of poly(A) and consequent degradation of c-fos mRNA facilitated by 3′ AU-rich sequences. Nature, 336, 396–399. [DOI] [PubMed] [Google Scholar]
- Wilusz J. and Shenk,T. (1988) A 64 kd nuclear protein binds to RNA segments that include the AAUAAA polyadenylation motif. Cell, 52, 221–228. [DOI] [PubMed] [Google Scholar]
- Zhang S., Williams,C.J., Hagan,K. and Peltz,S.W. (1999a) Mutations in VPS16 and MRT1 stabilize mRNAs by activating an inhibitor of the decapping enzyme. Mol. Cell. Biol., 19, 7568–7576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang S., Williams,C.J., Wormington,M., Stevens,A. and Peltz,S.W. (1999b) Monitoring mRNA decapping activity. Methods, 17, 46–51. [DOI] [PubMed] [Google Scholar]
- Zuk D., Belk,J.P. and Jacobson,A. (1999) Temperature sensitive mutants in the Saccharomyces cerevisiae MRT4, GRC5, SLA2 and THS1 genes result in defects in mRNA turnover. Genetics, 153, 35–47. [DOI] [PMC free article] [PubMed] [Google Scholar]