

Abstract
The Ca2+-activated pathways of Saccharomyces cerevisiae induce a delay in the onset of mitosis through the activation of Swe1, a negative regulatory kinase that inhibits the Cdc28–Clb complex. Calcineurin and Mpk1 activate Swe1 at the transcriptional and post-translational level, respectively, and both pathways are essential for the cell cycle delay. Our genetic screening identified the MCK1 gene, which encodes a glycogen synthetase kinase-3 family protein kinase, as a component of the Ca2+ signaling pathway. Genetic analyses indicated that Mck1 functions downstream of the Mpk1 pathway and down-regulates Hsl1, an inhibitory kinase of Swe1. In medium with a high concentration of Ca2+, Hsl1 was delocalized from the bud neck and destabilized in a manner dependent on both calcineurin and Mck1. Calcineurin was required for the dephosphorylation of autophosphorylated Hsl1. The E3 ubiquitin ligase complex SCFCdc4, but not the anaphase-promoting complex (APC), was essential for Hsl1 destabilization. The Ca2+-activated pathway may play a role in the rapid inactivation of Hsl1 at the cell cycle stage(s) when APC activity is low.
Keywords: calcineurin/G2 delay/GSK-3 family kinase/MAP kinase/SCF complex
Introduction
In eukaryotic cells, a change in the cytosolic Ca2+ concentration plays a key regulatory role in diverse cellular processes, such as T-cell activation, muscle contraction and neurotransmitter release (Clapham et al., 1995). In the yeast Saccharomyces cerevisiae, Ca2+ has been implicated in stress-induced expression of ion transporter genes (Haro et al., 1991; Cunningham and Fink, 1994, 1996), bud formation (Mazzoni et al., 1993; Zarzov et al., 1996; Gray et al., 1997) and viability upon pheromone-induced growth arrest (Cyert and Thorner, 1992). More recently, Ca2+ was also implicated in the regulation of cell cycle progression in G2 phase (Mizunuma et al., 1998). In S.cerevisiae, Swe1 kinase specifically inhibits a G2 form of Cdc28 by phosphorylating it at Tyr19 (Booher et al., 1993), and delays the onset of mitosis. The cell cycle regulation by Ca2+ is accomplished through the activation of calcineurin and the Mpk1 MAP kinase cascade, and these two pathways activate Swe1. Although the physiological role of Ca2+-mediated cell cycle regulation is not yet well understood, this regulation has been implicated in a mechanism that may operate under conditions where cells encounter a stress that may cause membrane stretching (Mizunuma et al., 1998). The effect of Ca2+ on cell cycle regulation is particularly obvious in a Δzds1 genetic background, in which the SWE1 expression level is higher than in wild-type cells. In this signaling process, calcineurin and the Mpk1 cascade up-regulate Swe1 at the transcriptional and post-translational level, respectively, and both of these pathways are required to mediate the cell cycle delay. The Mpk1 pathway activates Swe1 by inhibiting Hsl1, a Nim1-related kinase that negatively regulates Swe1 and leads to a delay in the entry into mitosis (Ma et al., 1996; Tanaka and Nojima, 1996; Mizunuma et al., 1998). However, the molecular mechanism underlying Hsl1 regulation by the Mpk1 pathway is as yet unknown.
Selective protein degradation is a commonly employed mechanism for the control of protein abundance. Ubiquitin-dependent proteolysis has emerged as a key mechanism for the regulation of many cellular processes, including cell cycle transitions (for reviews see Hochstrasser, 1996; Patton and Willems, 1998). The ubiquitin degradation pathway is a multistep process in which ubiquitin is covalently attached to a protein substrate by a series of enzymyatic reactions involving a ubiquitin-activating enzyme (E1), a ubiquitin-conjugating enzyme (E2) and ubiquitin ligase (E3). Ubiquitin ligases confer substrate specificity to the ubiquitylation reaction. One class of ubiquitin ligase that controls cell cycle progression in yeast and higher eukaryotes is a multisubunit complex known as the anaphase-promoting complex (APC). APC activity itself is regulated, being active from anaphase onset through G1, but then inactive in S, G2 and early M phase (Amon et al., 1994; Brandeis and Hunt, 1996; Amon, 1997; Jaspersen et al., 1998). Another major ubiquitin ligase is the SCF. The F-box domain proteins act as the specificity-determining subunit of the SCF complex (Patton et al., 1998). A central feature of the SCF-dependent ubiquitylation is its selectivity for phosphorylated substrates. Cdc6, Gcn4, Far1 and Sic1 are ubiquitylated and degraded in a manner dependent on a kinase that phosphorylates the substrate. The F-box protein Cdc4 interacts with phosphorylated substrate and recruits it to the ubiquitin ligase complex (SCFCdc4) composed of Cdc4, Skp1 and Cdc53 (Feldmann et al., 1997; Skowyra et al., 1997). Although ubiquitin-dependent proteolysis is involved in cell cycle control, the relationship of this system to the Ca2+-activated pathways is not known.
To identify a novel component(s) of the Ca2+ signaling pathways linked to cell cycle regulation, mutant strains that disrupt the signaling pathways were isolated and characterized. In this screening, MCK1, encoding a glycogen synthetase kinase-3 (GSK-3) family protein kinase, was identified as a component of this regulatory pathway. We demonstrate that Mck1 is a downstream regulator of the Mpk1 pathway. We propose a model in which Mck1 and calcineurin cooperatively regulate the delocalization of Hsl1 from the bud neck and subsequent SCFCdc4-mediated destabilization to trigger the Swe1 activation.
Results
Identification of mutations that disrupt Ca2+-induced G2 arrest and polarized bud growth
In previous work, we showed that ZDS1 null mutant (Δzds1) cells grown in medium containing a high concentration of CaCl2 (>50 mM) were delayed in the G2 phase and displayed polarized bud growth, due to the activation of cellular Ca2+ signaling pathways (Mizunuma et al., 1998). This delay is mediated by the activation of Swe1, a negative regulatory kinase of the Cdc28–Clb complex. Two pathways, calcineurin and the Mpk1 MAP kinase cascade, are implicated in this event, and both pathways are indispensable for Ca2+-triggered growth arrest. These findings indicated that a defect in either branch of the Ca2+ signaling pathways would overcome the growth arrest. To investigate the detailed mechanism of cell cycle arrest, we performed a screen for mutants that displayed a defect in this process. The Δzds1 cells are unable to grow, and show hyperpolarized bud growth on solid medium containing 200–300 mM CaCl2, whereas mutants with a defect in these pathways (e.g. Δcnb1 or Δmpk1) can grow well and show normal morphology. Our screening procedures comprised two steps. In the first screening, colonies of suppressor mutants with increased tolerance to Ca2+ were selected, which arose spontaneously from the Δzds1 strain on YPD plates containing 200 or 300 mM CaCl2. In the second screening, the mutants restored to normal morphology on Ca2+ medium were selected by microscopic observation, because the mutants with the aberrant morphology may have gained an increased tolerance to Ca2+ by mutations in other mechanisms. By genetic crosses, the recessive Ca2+-insensitive mutants were classified into 14 complementation groups designated scz1–scz14 (for suppressor of Ca2+-induced abnormalities of zds1). Details of the characterization of the mutants will be described elsewhere. In this report, we focused on characterizing the temperature-sensitive scz10 mutant.
On solid medium containing 300 mM CaCl2, the Δzds1 strain failed to grow, exhibiting an aberrant morphology, and were suppressed by the additional mutation scz10 (Figure 1A and B). To confirm that the scz10 mutation can overcome the Ca2+-induced cell cycle delay, we examined cell cycle progression by using the G1-synchronous culture treated with α-factor. The synchronized cells of wild type, Δzds1, Δzds1 scz10 and scz10 were released into YPD medium with or without 50 mM CaCl2. After the release, the DNA content, bud formation and nuclear division in synchronized cell cultures were determined (Figure 1C and D). Bud formation was slightly delayed by Ca2+, but there was no significant difference among the strains (Figure 1D). No obvious delay in S phase by Ca2+ was observed in these strains (Figure 1C). However, compared with wild type, the Δzds1 cells were delayed in the onset of mitosis by 15 min in YPD medium (without added Ca2+) and this delay was prolonged to 30–45 min by Ca2+. It was noteworthy that a significant Ca2+-induced delay (15 min) in the onset of mitosis was observed with wild-type cells using this procedure. In contrast, Ca2+ caused no significant delay in the onset of mitosis in Δzds1 Δscz10 cells (Figure 1C and D). These data indicated that the scz10 mutation suppressed the G2 delay of Δzds1 cells. They also indicated that Ca2+ can induce the G2 delay even in wild-type cells. In fact, we found by Clb2–Cdc28-dependent H1 kinase assay that the Cdc28 kinase activity was inhibited in an asynchronous cell culture of wild-type cells after 1 h of shift to high Ca2+ conditions (Figure 1E). The inhibition was transient and was restored with prolonged incubation. In addition to these suppressor phenotypes, the scz10 single mutant exhibited temperature sensitivities at >37°C and <14°C (Figure 1A).

Fig. 1. The scz10 mutation suppresses various phenotypes of the Δzds1 strain. (A) Effect of scz10 (mck1-1) on the growth of the Δzds1 mutant strain on solid medium. Wild-type (DHT22-1b), Δzds1 (YAT1), Δzds1 scz10 (YMM2144), Δzds1Δmck1 (YMM69), scz10 (YMM22) or Δmck1 (YMM68) cells were spotted on YPD plates and grown at 28°C (2 days), 37°C (2 days) or 14°C (4 days), or on YPD plates supplemented with the indicated concentration of CaCl2 and incubated at 28°C for 2 days. (B) Cell morphology (DIC), DAPI fluorescence image and flow cytometry analysis of PI-stained cells (FACS: 1C, one DNA copy; or 2C, two DNA copies) of various strains after 6 h of incubation with 100 mM CaCl2 at 28°C. (C) DNA content during cell cycle progression in various strains. Early log phase growing cells (OD600 of 0.2–0.3) of DHT22-1b, YAT1, YMM2144 or YMM22 strains were synchronized with α-factor in G1, and resuspended in YPD or YPD plus 50 mM CaCl2. Samples were taken 15 min after removing α-factor. (D) Quantification of the cumulative percentage of bud formation and nuclear division in the cell cultures prepared as in (C). For comparison, the data with Δswe1 (YMM5-8c) are shown. Open squares, DHT22-1b; open diamonds, YAT1; open triangles, YMM2144; inverted open triangles, YMM5-8c. (E) Inhibition of Clb2–Cdc28 activity by CaCl2 in vivo. CaCl2 was added to a final concentration of 100 mM to early log phase culture of wild-type cells expressing (TMY310) or not expressing (DHT22-1b) Clb2-Myc in YPD at 28°C, and incubated further. The cell extracts prepared were immunoprecipitated with anti-Myc and assayed for histone H1 kinase acitivity. In parallel, cell extracts were immunoprecipitated with anti-Myc antibody, separated by SDS–PAGE and detected with Clb2-Myc. Activity was normalized relative to the Clb2 level of each lane. Activity was expressed relative to the activity at time zero (=1.0). (F) Map of the insert ORFs in plasmid pMM2144, which fully complemented the scz10 mutation. Deletion analysis was carried out to localize the suppressor activity (indicated by the + sign).
To identify the scz10 gene, a centromeric genomic library was introduced into the scz10 strain and one plasmid that suppressed the temperature sensitivity at 37°C was recovered. The genomic DNA insert in the cloned plasmid pMM2144 was subjected further to restriction mapping and complementation analysis (Figure 1F). Partial DNA sequencing of the complementing region revealed that it contained the MCK1 gene (YNL307c ORF) encoding a protein kinase belonging to the GSK-3 family (Neigeborn and Mitchell, 1991; Shero and Hieter, 1991). Linkage of MCK1 to the SCZ10 locus was confirmed by tetrad analysis. In fact, MCK1 disruption (Δmck1) suppressed the phenotypes of Δzds1 cells in the medium containing CaCl2 (Figure 1A). The low and high temperature sensitivities of the scz10 and Δmck1 mutants were similar to those previously reported for Δmck1 mutants (Shero and Hieter, 1991; Puziss et al., 1994). Since this gene was described previously, we shall hereafter refer to SCZ10 as MCK1 and to the scz10 mutant allele as mck1-1. The mck1-1 allele, in comparison with the Δmck1 allele, exhibited qualitatively similar, but significantly more potent suppressor effects on the Ca2+-sensitive phenotypes of Δzds1, and increased sensitivities to extreme temperatures (Figure 1A). We further identified the mutation site of the mck1-1 gene amplified by PCR. A single base deletion (G) at position +706 of the coding region was found. This deletion caused a frameshift and led to the appearance of a stop codon at position +747 of the 1128 bp open reading frame. The data showing that a catalytically inactive, truncated protein is expressed from the mck1-1 gene are given below (Figure 5C). The nature of the truncated Mck1 may explain the stronger phenotypes of the mck1-1 mutation than of a Δmck1 deletion allele (see Discussion). For this reason, we used the mck1-1 mutant allele in most of the subsequent experiments.

Fig. 5. Mck1 and calcineurin cooperatively regulate Hsl1 abundance. (A) Comparison of Hsl1 abundance in various strains. Wild-type (YMM53), mck1-1 (YMM71) or Δcnb1 (YMM72), in which the genomic copy of HSL1 encodes HA-Hsl1, were grown in liquid YPD at 28°C until early log phase. CaCl2 was added to the cell cultures at the indicated times to a final concentration of 100 mM, samples were taken and western blot analysis was performed. (B) Dependence of Hsl1 destabilization on the functions of the calcineurin and Mpk1–Mck1 pathways. The strain YMM54, in which the genomic copy of HSL1 was replaced with HA-Hsl1, was transformed with the plasmids pGAL-Cmp2ΔC (pYES2::Cmp2ΔC), pGAL-Mck1 (pYES2::Mck1) or vector alone (pYES2). The transformants were grown in liquid YPR medium containing raffinose at 28°C until early log phase, and then galactose was added. Samples were taken at the indicated times and western blot analysis was performed by probing with anti-HA and anti-Cdc28 (control) antibodies. (C) Phosphorylation of Hsl1 by Mck1 in vitro. Cell extracts prepared from Δmck1 (YMM68) cells expressing HA-Mck1, HA-Mck1-1 or vector alone were immunoprecipitated with anti-HA. Kinase assay was performed by incubating the pellet with [γ-32P]ATP and recombinant GST–Hsl1(KD). Protein samples were subjected to electrophoresis and exposed for autoradiography. Western blotting was performed to detect HA-Mck1 and HA-Mck1-1, as well as Cdc28 as loading control. (D) (a) Mobility shift of HA-Hsl1 in the cells with activated Ca2+ signaling conditions in a calcineurin-dependent manner. Strain cdc4 (YMM73) or cdc4 Δcnb1 (YMM74), in which the genomic copy of HSL1 encodes HA-Hsl1, was incubated at 37°C for 2 h to inactivate cdc4. Incubation was carried out for 1 h in the medium with (+) or without (–) CaCl2 (100 mM) or FK506 (1 µg/ml) as indicated. HA-Hsl1 was detected by immunoblotting using anti-HA monoclonal antibody. (b) Phosphatase (CIP) treatment of HA-Hsl1 in the sample prepared similarly to the sample (+CaCl2, +FK506 of cdc4 cells) in (a). (E) Physical interaction of Hsl1 and calcineurin. Protein extracts prepared from the cells in which the genomic copy of HSL1 encodes HA-Hsl1 (YMM67), HA-Hsl1(KD) (YMM62) or none (DHT14) on a Δcnb1 background harboring plasmid-borne Myc-Cnb1 were immunoprecipitated with anti-HA antibody. The pellets were separated by SDS–PAGE and probed with both anti-HA and anti-Myc antibodies.
Mck1 is a downstream component of the Mpk1 branch of the Ca2+ signaling pathways and down-regulates Hsl1 kinase
We next examined whether Mck1 functions in the calcineurin branch or the Mpk1 branch of the Ca2+ signaling pathways. The activation of one of these branches by overexpression of Mpk1 or Cmp2ΔC, a hyperactive form of the calcineurin catalytic subunit (Garrett-Engele et al., 1995), on a Δzds1 background leads to polarized bud growth and G2 delay (Mizunuma et al., 1998). If a mutation is located downstream of the activated pathway, the physiological effects will not be induced by the overexpression due to the interruption of this signal. On the basis of this assumption, the plas mid containing MPK1 (pGAL::MPK1) or CMP2ΔC (pGAL::CMP2ΔC) placed under the control of the GAL promoter was introduced into the Δzds1 mck1-1 strain. The overexpression of CMP2ΔC, but not MPK1, induced physiological changes, suggesting Mck1 to be downstream of the Mpk1 pathway (Figure 2A).
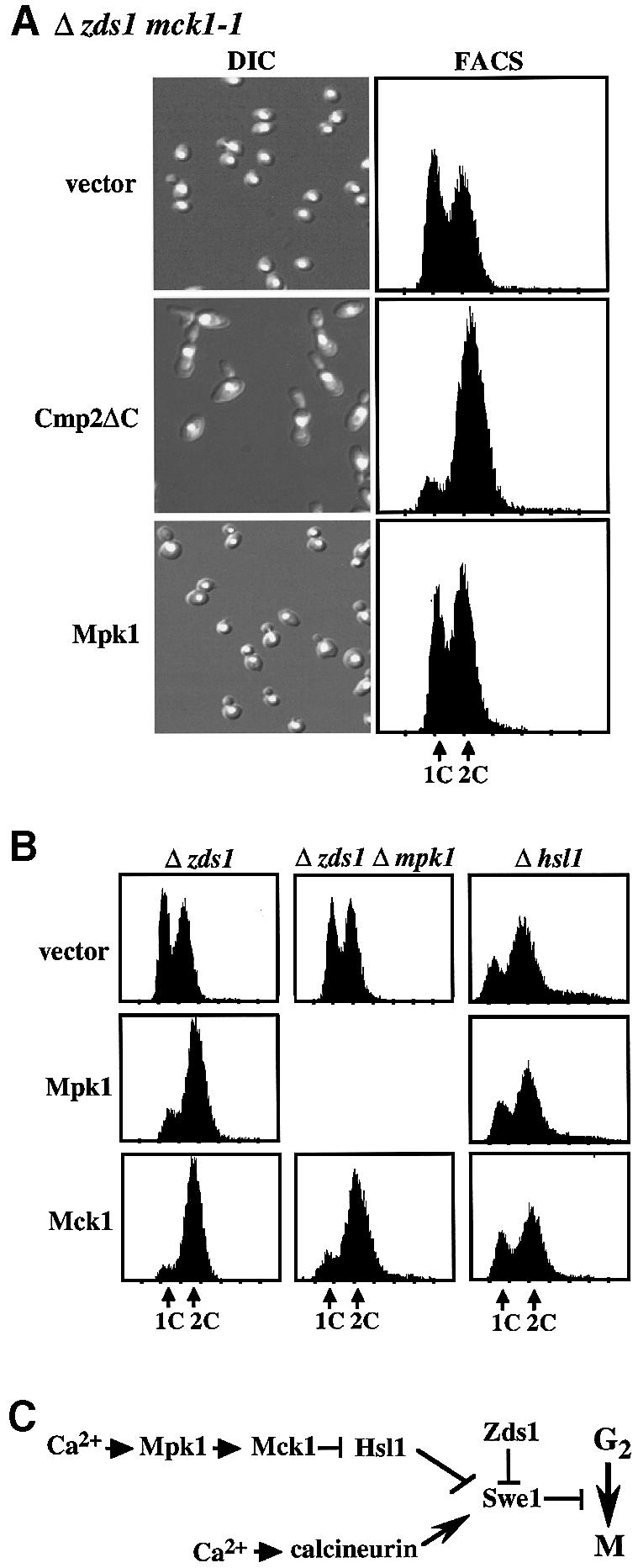
Fig. 2. Mck1 functions downstream of the Mpk1 pathway to induce G2 delay through the down-regulation of Hsl1. (A) The effect of overexpression of Cmp2ΔC (constitutively active form of the Cmp2 calcineurin catalytic subunit) or Mpk1 in the Δzds1 mck1-1 strain was examined. The Δzds1 mck1-1 (YMM2144) strain was transformed with the GAL-regulated plasmids pGAL-Cmp2ΔC (pYES2::Cmp2ΔC), pGAL-Mpk1 (pNV7::Mpk1) or vector alone (pYES2). The transformants were grown in liquid synthetic complete medium (SD minus Ura) at 28°C until early log growth phase. Cells were collected by centrifugation, washed, and suspended in SG (galactose medium, GAL promoter on) and the cells were then incubated at 28°C for 10 h. Cell morphology (DIC), FACS and the DNA content of PI-stained cells were analyzed by flow cytometry analysis. (B) The effect of overexpression of Mpk1 or Mck1 in Δzds1 (YAT1), Δzds1Δmpk1 (YMM3) or Δhsl1 (YMM52) strains was determined. These strains were transformed with pGAL-Mpk1 (pNV7::Mpk1), pGAL-Mck1 (pYES2::Mck1) or vector alone (pYES2). Experimental conditions were as described in (A). (C) Presumed pathway for the regulation of the Hsl1 kinase by Mck1 as suggested by the genetic analyses.
To examine whether Mck1 induces the G2 delay, we examined the effects of MCK1 overexpression on the Δzds1 background. Similarly to MPK1, MCK1 overexpression led to a severe G2 delay (Figure 2B). Previous genetic data indicated that the Mpk1 cascade regulates the Swe1 kinase through down-regulation of the Hsl1 kinase, a negative regulator of Swe1 (Mizunuma et al., 1998). We therefore examined whether Mck1 functions through the regulation of Hsl1. If Mck1 activates Swe1 through Hsl1 regulation, Mck1 overexpression should not enhance the G2 delay in Δhsl1 cells as found for MPK1. This was indeed the case (Figure 2B). The finding that the mck1-1 mutation could not rescue the Ca2+-induced G2 delay in Δhsl1 cells supported the above genetic relationship (data not shown). Furthermore, overexpression of MCK1 on a Δzds1 Δmpk1 (Figure 2B), but not on a Δswe1 background (data not shown) caused a strong G2 delay, confirming that MCK1 functions downstream of MPK1. Consistent with the notion that both MPK1 and MCK1 function in a common pathway, the suppressive effect of a Δmpk1 mck1-1 double mutation on the phenotype of Δzds1 was no more severe than those of the corresponding single mutations (data not shown). These genetic data established a cascade series of kinases, in which Mpk1 activates Mck1, which in turn down-regulates Hsl1 to induce Swe1 activation (Figure 2C).
Consistent with the genetic results, northern blot analysis demonstrated that Mpk1 is important for the induction of MCK1 mRNA by Ca2+, indicating that Mpk1 positively regulates MCK1 at the transcriptional level (Figure 3).

Fig. 3. Mpk1 up-regulates MCK1 transcription. CaCl2 was added to a final concentration of 100 mM to early log phase cultures of wild-type (DHT22-1b), Δzds1 (YAT1) or Δzds1 Δmpk1 (YMM3) cells in YPDat 28°C, and incubated further. RNA samples were extracted at the indicated time and northern blot analysis was performed. The intensity of MCK1 or ACT1 mRNA was measured using a BAS-1000 bio imaging analyzer and the MCK1 level was normalized to the ACT1 mRNA level.
The regulation of Hsl1 by Mck1 is not at the transcriptional level
To address whether Mck1 down-regulates HSL1 at the transcriptional level, we determined the level of HSL1 mRNA by northern blot analysis in α-factor-synchronous cell cultures in the presence or absence of 50 mM CaCl2 added at time 0 (Figure 4A). Quantification of cell morphology confirmed that >80% of the treated cells were arrested in G1. For comparison, the level of SWE1 mRNA was determined. The level of SWE1 mRNA fluctuates during the cell cycle, peaking at G1/S and declining in G2/M (Ma et al., 1996; Mizunuma et al., 1998). In medium containing CaCl2, a high level of SWE1 mRNA was sustained in G2/M by calcineurin (Mizunuma et al., 1998). As previously reported, the level of HSL1 mRNA fluctuated during the cell cycle in a manner similar to that of SWE1 mRNA (Tanaka and Nojima, 1996). However, in contrast to SWE1 mRNA, the oscillation of HSL1 mRNA in the wild-type strain was not significantly altered by Ca2+. Moreover, the fluctuation pattern was unaffected by the presence or absence of the MCK1 gene. These results indicated that HSL1 is not regulated by Ca2+ at the transcriptional level, and the regulation of Hsl1 by Mck1 is at the post-transcriptional level.

Fig. 4. Mck1 is not involved in the transcriptional regulation of HSL1. (A) The oscillation of SWE1 or HSL1 mRNA levels during cell cycle progression in various strains that were synchronized with α-factor was determined by northern blotting, using the SWE1, HSL1 and ACT1 probes. Experimental conditions were as described in the legend to Figure 1C. RNA blot hybridization with the SWE1, HSL1 and ACT1 probes. (B) Changes in the relative intensity of SWE1 or HSL1 mRNA. The amount of SWE1 or HSL1 mRNA in (A) was normalized to a constant level of ACT1 mRNA. In each case, the individual maximum value was referred to as 1. Time 0 represents the point of release from G1 phase arrest.
Mck1 and calcineurin regulate Hsl1 abundance
We next examined by immunoblot analysis whether Ca2+ down-regulates Hsl1 abundance. For this purpose, we constructed strains containing a chromosomally integrated gene coding for three copies of the influenza virus hemagglutinin (HA) epitope-tagged Hsl1. The HA-tagged Hsl1 suppressed the Ca2+-sensitive phenotype of the Δhsl1 strain, suggesting that the construct is fully functional in vivo. We compared the levels of Hsl1 in an asynchronous cell culture in medium with 100 mM CaCl2. Cell extracts were prepared, separated by SDS–PAGE and detected by immunoblotting using antibodies directed against the HA epitope (Figure 5A). As previously reported, Hsl1 protein migrated as a broad band, in which the slower migrating species were thought to correspond to the autophosphorylated, active isoform (Barral et al., 1999). Because of this property, the Hsl1 phosphorylation status provides a useful indicator of kinase activity (Barral et al., 1999). Remarkably, the HA-Hsl1 levels of wild-type and Δzds1 cells decreased upon a shift to medium with CaCl2, and the HA-Hsl1 level dropped to about <10% of the original 1 h after the shift (Figure 5A). The rate of the Ca2+-induced decrease in HA-Hsl1 abundance was not altered by the presence of 0.5 µg/ml cycloheximide, suggesting that the regulation is not at the translational level (data not shown). In contrast, HA-Hsl1 in mck1-1 cells (and Δmpk1 cells, data not shown) was sustained at higher levels, indicating that the Mpk1–Mck1 pathway was critical in inducing the decrease in the abundance of HA-Hsl1 (Figure 5A). The HA-Hsl1 protein that was present in wild-type cells during incubation with Ca2+ migrated more quickly compared with that in mck1-1 cells. The slower migrating HA-Hsl1 protein in the mck1-1 cells was verified as a phospho-isoform by treating them with calf intestinal alkaline phosphatase (CIP; data not shown). The preferential disappearance of the slower migrating species could be explained if the phosphorylated species were degraded or dephosphorylated preferentially prior to the degradation. To examine the second possibility, we asked whether calcineurin was involved in this process. The time course change in HA-Hsl1 abundance in Δcnb1 cells was slower than that in wild-type cells, and similar to that in mck1-1 cells (Figure 5A). In addition, the electrophoretic mobility of HA-Hsl1 was similar to that in mck1-1 cells. These results indicated that the calcineurin-mediated dephosphorylation of Hsl1 occurred prior to its destabilization.
When Cmp2ΔC or Mpk1 was overexpressed in Δzds1 cells by using a GAL promoter in galactose medium, the HA-Hsl1 level decreased more quickly than when the cells with control plasmid were treated similarly (Figure 5B). In contrast, when Cmp2ΔC was overexpressed in Δzds1 mck1-1 cells, as well as when Mck1 was overexpressed in Δzds1 Δcnb1 cells, the HA-Hsl1 levels decreased more slowly, confirming that the two pathways coordinately regulate the Hsl1 destabilization (Figure 5B). Basically, similar results were obtained on the wild-type (ZDS1) background, except that the rate of Hsl1 decrease was slower than on the Δzds1 background (data not shown).
Hsl1 may be a phosphorylation substrate of Mck1. To test this possibility, the genes encoding HA-tagged Mck1 or Mck1-1 mutant proteins were constructed and the fused gene was subcloned into a CEN-based plasmid. Western blot analysis following SDS–PAGE of the cell extracts detected a 60 kDa band for HA-Mck1 and a 45 kDa band for HA-Mck1-1, both considerably larger than the predicted molecular masses of the respective proteins (Figure 5C, right). The expression levels of HA-Mck1 and HA-Mck1-1 were similar. The HA-tagged proteins were immunoprecipitated and kinase activity was assayed by incubation with [γ-32P]ATP using a glutathione S-transferase (GST) fusion of Hsl1(KD) (see below for details) as substrate. A catalytically inactive Hsl1 protein [Hsl1K110A or Hsl1(KD)] was used as substrate, because Hsl1 is autophosphorylatable (Barral et al., 1999). As shown in Figure 5C, Mck1, but not Mck1-1, phosphorylated Hsl1(KD) in vitro.
The involvement of calcineurin in Hsl1 dephosphorylation was confirmed by western blot analysis of HA-Hsl1 in the cells grown in the medium containing Ca2+. In this experiment, a cdc4 mutant background was used to protect HA-Hsl1 from degradation (see later). In the cells incubated with Ca2+, the broad band was converted to a faster migrating narrow band and this mobility shift was blocked when the cells were incubated in the presence of FK506 (Figure 5D, a). Mobility shift was not induced by Ca2+ in Δcnb1 cells (Figure 5D, a). CIP treatment of HA-Hsl1 present in the wild-type cells incubated with Ca2+ in the presence of FK506 showed that the slower migrating species represent a phosphorylated form of Hsl1 (Figure 5D, b). The slow migrating species were not produced with Hsl1(KD), a catalytically inactive mutant protein (see below), indicating that Hsl1 kinase activity is essential to generate the slow migrating species (Figure 5D, a; Barral et al., 1999). These results suggested that Hsl1 is dephosphorylated prior to its degradation, and that calcineurin is responsible for the dephosphorylation in vivo.
We next examined whether calcineurin interacts with Hsl1 in vivo. Myc-tagged Cnb1 (calcineurin regulatory subunit) was co-precipitated with HA-Hsl1, but not with HA-Hsl1(KD) (see below) (Figure 5E). These results suggested that calcineurin physically interacts with the autophosphorylated, active form of Hsl1, but not with an inactive form.
Ca2+-induced Hsl1 destabilization is mediated by the SCFCdc4 complex
Since the Ca2+-induced Hsl1 destabilization appeared to occur even at the cell cycle stage(s) when APC activity is low (S, G2 and early M phases), SCF might be responsible for the regulation of Hsl1 destabilization. To address this possibility, we examined whether the rate of Hsl1 degradation is affected in the mutants cdc4 and Δgrr1, defective in different F-box proteins of SCF at 37°C, the non-permissive temperature for cdc4 (Figure 6A). In wild-type and Δzds1 cells, the Ca2+-induced Hsl1 destabilization proceeded more rapidly at 37°C than at 28°C, decreasing to a non-detectable level within 20 min of incubation. Hsl1 in the Δgrr1 cells was degraded rapidly at a rate similar to that in wild-type cells. In contrast, Hsl1 was degraded in cdc4 cells at a much slower rate, which was similar to that in mck1-1 or Δcnb1 cells. These results suggested that the Hsl1 destabilization was dependent on SCFCdc4, but not SCFGrr1. Consistent with the above result, when Cmp2ΔC or Mpk1 was overexpressed in cdc4 mutant cells, the Hsl1 level decreased more slowly than on the wild-type background (data not shown).

Fig. 6. Ca2+ induces SCFCdc4-mediated Hsl1 destabilization and Hsl1 delocalization from the bud neck in a manner dependent on both calcineurin and Mck1. (A) Comparison of Hsl1 abundance in various strains. Wild type (YMM53), mck1-1 (YMM71), Δcnb1 (YMM72), Δzds1 (YMM54), cdc4 (YMM73) or Δgrr1 (YMM76), each bearing a gene encoding HA-Hsl1, was grown in liquid YPD at 28°C until early log phase, and then shifted to 37°C for 2 h to inactivate Cdc4. CaCl2 was added to the cell cultures to a final concentration of 100 mM, samples were taken at intervals of 20 min and western blot analysis was performed. (B) Comparison of the Ca2+-induced destabilization of a kinase-negative Hsl1, HA-Hsl1(KD), in strains Δhsl1 (YMM60) or wild-type HSL1 (YMM66), each bearing a gene encoding HA-Hsl1(KD). Experimental conditions were as described in (A). (C) HA-Hsl1 localization in various strains. The cells were grown in liquid YPD at 28°C until early log phase, and then shifted to 37°C for 2 h to inactivate cdc4. CaCl2 was added to the cell cultures to a final concentration of 100 mM, and samples were then taken after 20 min of incubation. Indirect immunofluorescence microscopy was performed as described in Materials and methods. (D) Comparison of the localization of HA-Hsl1(KD) in Δhsl1 (YMM60) or HSL1 (YMM66) cells. CaCl2 was added to the cell cultures to a final concentration of 100 mM, and samples were then taken after 20 min of incubation. Experimental conditions were the same as in (C).
Ca2+ signal induces Hsl1 delocalization from the bud neck
Hsl1 is localized in the bud neck of cells and physically associates with the bud-side septin ring (Barral et al., 1999; Longtine et al., 2000). We examined whether Hsl1 localization might be affected by the Ca2+ signal. To test this possibility, we visualized Hsl1 localization microscopically using an HA-tagged version of Hsl1 expressed from a chromosomally integrated construct (Figure 6C). Strikingly, HA-Hsl1 localized to the neck was delocalized by Ca2+ in wild-type and Δzds1 strains. In contrast, in the Ca2+-treated mck1-1 or Δcnb1 cells, HA-Hsl1 still remained at the bud neck, suggesting that both Mck1 and calcineurin are required for the delocalization. In cdc4 cells, the HA signal was delocalized from the bud neck by Ca2+ and stayed in the cytoplasm (Figure 6C), suggesting that the delocalization of Hsl1 occurs prior to the degradation.
We next examined whether the Ca2+ treatment of the cells may affect septin organization, because the activity of Hsl1, which is co-localized with the septin ring, is dependent on septin function (Barral et al., 1999; Shulewitz et al., 1999; Longtine et al., 2000). In both wild-type and Δzds1 cells, Ca2+ did not cause a significant change in the localization of Cdc11, a component of the septin filament, suggesting that the organization of the septin ring is not affected significantly by Ca2+ even on the Δzds1 background (data not shown). The result suggested that the delocalization of Hsl1 by Ca2+ was not due to the perturbation of septin localization.
The localization and the Ca2+-induced delocalization of Hsl1(KD), a catalytically inactive mutant of Hsl1, depend on the presence of wild-type Hsl1
To investigate whether the delocalization and destabilization of Hsl1 are dependent on its kinase activity, we examined the stability of HA-tagged catalytically inactive Hsl1(KD). When the gene encoding HA-Hsl1(KD) was introduced into Δhsl1 cells, destabilization was not induced by Ca2+. However, when HA-Hsl1(KD) was introduced onto the wild-type HSL1 background, the destabilization of the mutant protein was induced (Figure 6B). The presence of a lower level of HA-Hsl1(KD) compared with HA-Hsl1 under the uninduced conditions could be due to the more unstable nature of unlocalized HA-Hsl1(KD). HA-Hsl1(KD) was not localized in the neck on the Δhsl1 background, suggesting that either the kinase activity or the autophosphorylation on Hsl1 is important for its proper localization (Figure 6B and D). On the wild-type HSL1 background, in contrast, HA-Hsl1(KD) was localized to the neck in ∼30% of the cells (Figure 6D). Since HA-Hsl1(KD) can be phosphorylated by wild-type Hsl1 in vivo due to a trans-autophosphorylation (Barral et al., 1999), the different behaviors of HA-Hsl1(KD) in the presence and absence of wild-type Hsl1 seem to be a reflection of the phosphorylation state of HA-Hsl1(KD). The data suggested that the Hsl1 phosphorylation, but not the kinase activity itself, is required for Hsl1 localization. It was also suggested that the Hsl1 localization to the bud neck is critically important for the Ca2+-induced Hsl1 destabilization.
Ca2+-induced Hsl1 destabilization is independent of the APC pathway
It was demonstrated recently that the Hsl1 level fluctuates during the cell cycle (McMillan et al., 1999b) via an APC-mediated degradation pathway (Burton and Solomon, 2000). We examined whether the Ca2+-induced Hsl1 destabilization requires APC function by using an APC mutant cdc23 (Irniger et al., 1995). As shown in Figure 7A, the Hsl1 destabilization was unaffected in the mutant at the restrictive temperature, indicating that the APC pathway was not significantly involved in the Ca2+-induced Hsl1 destabilization.

Fig. 7. The Ca2+-regulated Hsl1 destabilization pathway is distinct from the APC pathway. (A) Effect of Ca2+ on Hsl1 abundance in an APC mutant. Strains YMM53 or YMM65 (cdc23) with integrated HA-Hsl1 were grown in liquid YPD at 28°C until early log phase, and then shifted to 37°C for 2 h to inactivate cdc23. CaCl2 was added to the cell cultures to a final concentration of 100 mM, samples were taken at 20 min intervals and western blot analysis was performed. (B) The fluctuation of the Hsl1 abundance during the cell cycle in a calcineurin-deficient mutant. Strain YMM53 or YMM72 (Δcnb1), in which the genomic copy of HSL1 encodes an HA-tagged form of Hsl1, was grown in YPD to early log phase, and cells were synchronized as described in Figure 1C. Samples were taken at 15 min intervals after release in YPD, and whole-cell extracts were prepared. Immunoblotting was performed with antibodies against HA-Hsl1 and Cdc28 as loading control. (C) DNA content of YMM53 or YMM72 cells in a synchronous culture after release from an α-factor-induced G1 arrest.
To examine whether the Ca2+-mediated regulation is also important for the cell cycle-dependent Hsl1 fluctuation, we compared the fluctuation patterns in synchronized cell cultures of wild-type and calcineurin-deficient cells expressing HA-Hsl1. As reported previously, the abundance of Hsl1 fluctuated after release from α-factor arrest (Figure 7B and C; McMillan et al., 1999b; Burton and Solomon, 2000). Hsl1 accumulation was periodic during the cell cycle, peaking in G2 and declining prior to M. A very similar fluctuation pattern was obtained for Δcnb1 cells, indicating that the calcineurin-dependent degradation pathway did not contribute significantly to the regulation of Hsl1 stability in the normal cell cycle. These results indicated that the Ca2+-dependent, SCFCdc4-mediated pathway and the cell cycle-dependent, APC-mediated pathways are distinct mechanisms for Hsl1 degradation.
Discussion
Identification of Mck1 as the signaling component of the Ca2+-induced G2 delay
The Ca2+-activated pathways of S.cerevisiae induce a delay in the onset of mitosis through the activation of Swe1, a negative regulatory kinase that inhibits the Cdc28–Clb complex. Calcineurin and Mpk1 activate Swe1 at the transcriptional and post-translational level, respectively. In the present study, we identified the MCK1 gene, which encodes a GSK-3 family protein kinase, as a signaling component downstream of the Mpk1 pathway. The Ca2+-sensitive growth, the Ca2+-induced G2 delay and polarized bud growth of the Δzds1 strain were suppressed by mck1-1 and Δmck1 mutations. The available genetic, biochemical and imaging data are consistent with a novel signaling pathway in which Mck1 and calcineurin are activated by Ca2+, which in turn coordinately down-regulate Hsl1 by inducing the delocalization from the bud neck and subsequent destabilization via the SCFCdc4-mediated pathway (Figure 8). Therefore, calcineurin is involved in the regulation of the G2/M cell cycle progression by Swe1 activation at two distinct steps, at the transcriptional and post-transcriptional levels through the down-regulation of the Swe1 inhibitor Hsl1 by dephosphorylating it (via delocalization and destabilization). A high Ca2+ concentration was able to induce the G2 delay even in Δhsl1 cells, and the G2 delay was abolished by the deletion of either SWE1 or CNB1 (data not shown), indicating that the Ca2+-induced G2 delay in Δhsl1 cells is due mainly to the calcineurin-dependent transcriptional activation of SWE1. However, there still remains a possibility that the Ca2+ signaling pathways negatively regulate Mih1, the phosphatase that reverses the Cdc28 phosphorylation catalyzed by Swe1.

Fig. 8. Model for the role of the Ca2+ signaling pathways of budding yeast in regulating the G2 phase delay through the delocalization and destabilization of Hsl1 from the bud neck. CaN, calcineurin.
Because various phenotypes displayed by the mck1-1 allele were more severe than those of the null allele (Figure 1A), we characterized the mck1-1 mutation. A catalytically inactive, truncated Mck1-1 protein was produced at a level comparable to that of wild-type Mck1 (Figure 5C). The yeast genome encodes three additional GSK-3 family proteins, Mds1/Rim11, Mrk1 and Yol128c, which may share redundant functions with Mck1 (Andoh et al., 2000). The more potent phenotypes exhibited by the mck1-1 allele compared with the Δmck1 allele would be explained if the functional Mck1 homologs were excluded by the Mck1-1 protein from the presumed Mck1 complexes.
Some physiological roles were described previously for Mck1. Mck1 is important in mitotic chromosome segregation specific to CDEIII function (Shero and Hieter, 1991). In addition, the Mck1 function is required in diploid cells for expression of IME1, a gene encoding an early meiotic activator, and subsequent ascus maturation (Neigeborn and Mitchell, 1991). In these Mck1-mediated processes, however, the upstream and downstream regulatory mechanisms are not yet known. In addition to these, we have identified a novel event in which Mck1 plays a key regulatory role in inducing the cell cycle delay by Ca2+. Thus, Mck1 kinase seems to function in a variety of processes that operate in apparently unrelated intracellular events. Whether Mck1 has multiple target proteins in each of these events or a common target protein is responsible for all of them remains unclear. However, given that the Hsl1 kinase is a potential target of Mck1, it seems unlikely that Hsl1 is also responsible for the activation of the sporulation pathways. In fact, a Δhsl1/Δhsl1 diploid could sporulate normally, showing that Hsl1 may not be important in this event (data not shown). However, it still remains to be seen whether Hsl1 is also involved in the mitotic chromosome segregation specific to CDEIII function. The astonishingly diverse functions of Mck1 could be rationalized if the Mck1 kinase, upon activation by signals, induces destabilization of a key regulatory protein of the respective event through a ubiquitylation pathway, in a manner analogous to the mechanism described for Hsl1. The involvement of the GSK-3 family protein kinase in targeting the key regulatory protein for destabilization seems to be conserved in eukaryotes, as will be discussed for β-catenin.
An elaborate genetic screen using a Δzds1 Δzds2 strain that displays a strong mitotic defect due to a severe morphogenic defect was conducted recently by another group to isolate mutants with a defect in the signaling pathways for the morphogenesis checkpoint (McMillan et al., 1999a). In that study, the screening gave rise to SWE1 gene mutants and a dominant checkpoint defective allele of CDC28, but not to any of the mutant loci of the Ca2+ signaling pathways. This result suggested that the Ca2+ signal may not be responsible for the morphogenesis checkpoint (Lew and Reed, 1995; Sia et al., 1996; McMillan et al., 1998). It is still possible, however, that Zds2 protein may have some unknown function, in addition to its functional redundancy with Zds1.
Roles of Mck1 and calcineurin in regulating Hsl1 delocalization and destabilization
Our data demonstrated that the Hsl1 protein was destabilized via the SCFCdc4 pathway in a manner dependent on Ca2+ signaling. The physiological consequences caused by Ca2+ can be explained by the decreased abundance of Hsl1. Surprisingly, both calcineurin-deficient cells and mck1-1 cells displayed apparently similar defects in the delocalization and destabilization of Hsl1, suggesting that these events are rigorously regulated by the combined actions of calcineurin and Mck1 (Figure 8). Considering that the delocalized Hsl1 remains in the cytosol of cdc4 cells, Hsl1 delocalization may be viewed as a primary event that is regulated by calcineurin and Mck1, and it seems important for destabilization via the SCFCdc4 pathway. This view was supported further by the observation that Hsl1(KD), which was unable to localize to the bud neck, was a substrate of SCFCdc4 only when the mutant protein was localized to the neck with the aid of wild-type Hsl1 (Figure 6B and D).
The observations that phospho-Hsl1 is a calcineurin substrate and calcineurin co-immunoprecipitates with Hsl1 suggested that Hsl1 and calcineurin interact in vivo. We also demonstrated that Mck1 phosphorylates Hsl1 in vitro. In fact, Hsl1 contains as many as 54 potential phosphorylation sites (S/TXXXS/T) known as the consensus sequence for a GSK-3β phosphorylation site (Plyte et al., 1992). It still remains to be clarified whether Hsl1 phosphorylation by Mck1 requires prior phosphorylation by other protein kinases for the delocalization and destabilization of Hsl1. It is known, in some cases, that the substrate recognition by GSK-3β requires prior phosphorylation by a priming kinase. With other proteins, however, such as glycogen synthase, inhibitor-2, adenomatous polyposis coli and rAxin, prior phosphorylation is not required (Plyte et al., 1992; Ikeda et al., 1998).
Because Hsl1 is associated with the septin filament together with various other components including Hsl7 and Swe1 (Barral et al., 1999; Shulewitz et al., 1999; Longtine et al., 2000), the mechanism underlying Hsl1 delocalization that involves phosphorylation and dephosphorylation of Hsl1 seems very complex and still remains to be solved.
Conserved function of GSK-3 family kinases in inducing protein destabilization
In SCFCdc4-dependent Hsl1 destabilization, Mck1 appeared to be important for targeting Hsl1 for ubiquitylation and proteolysis by the proteasome. This scheme is reminiscent of the mechanism for β-catenin destabilization in embryos (Aberle et al., 1997). β-catenin in Wnt signaling depends on post-translational modifications and its interactions with protein partners localized in the cytoplasm and nucleus (for a review see Miller and Moon, 1996). A complex containing axin that directly associates with β-catenin, GSK-3β and adenomatous polyposis coli was implicated in negatively regulating the Wnt signaling pathway. The stability of β-catenin is controlled by phosphorylation, most probably by GSK-3β, which targets β-catenin to the unbiquitin–proteasome pathway (Aberle et al., 1997). Wnt signaling regulates interactions between axin and β-catenin and the subsequent stabilization of β-catenin. In the absence of the Wnt signal, GSK-3β targets β-catenin for a degradative pathway. Signaling by Wnt inactivates GSK-3β, and thus stabilizes β-catenin and increases availability for signaling. Thus, GSK-3 kinase regulation of protein stabilization or destabilization in response to a signal seems to be an evolutionarily conserved mechanism for regulating protein abundance through phosphorylation, ubiquitylation and proteolysis by the proteasome.
Hsl1 is a common substrate of SCF- and APC-mediated degradation depending on the signals
It was demonstrated recently that the Hsl1 level oscillates in a cell cycle-dependent manner, being absent in late M to G1, when APC activity is high, and present in S, G2 and early M phase, when APC activity is low (Burton and Solomon, 2000). The inactivation of Hsl1 by the APC-mediated degradation in late mitosis may be important to allow Swe1 to perform its role in the morphogenesis pathway in the G1 and S phases. The Ca2+ signal induces a G2 cell cycle delay through the activation of Swe1, responding to a stress that causes a membrane stretch (Mizunuma et al., 1998). Thus, the SCF-mediated Hsl1 destabilization may be viewed as a mechanism that serves to activate Swe1 rapidly through the degradation of Hsl1 in response to a stress at the cell cycle stages when the APC activity is low (S, G2 and early M phases).
Materials and methods
Strains, media and genetic methods
Yeast strains used in this study are listed in Table I. The mutant strains were all derivatives of W303-1A. Media used were as described previously (Mizunuma et al., 1998). Disruption of the HSL1 gene was performed using a hsl1 disruption plasmid Δhsl1 NS::URA3 (a gift from M.Grunstein; Ma et al., 1996). The yeast mck1::TRP1 mutant was constructed by introducing PvuII-digested pUC119-mck1::TRP1 plasmid into diploid trp1 cells.
Table I. Yeast strains used in this study.
| Strain | Genotype | Source or reference |
|---|---|---|
| W303 derivatives | ||
| DHT22-1b | MATa trp1 leu2 ade2 ura3 his3 can1-100 | W303-1A from Dr Rothstein |
| YAT1 | MATa Δzds1::TRP1 | Mizunuma et al. (1998) |
| DHT14 | MATa Δcnb1::HIS3 | Nakamura et al. (1993) |
| TNP46 | MATa Δmpk1::HIS3 | Nakamura et al. (1996) |
| YMM3 | MATa Δzds1:TRP1 Δmpk1::HIS3 | Mizunuma et al. (1998) |
| YMM5-8c | MATa Δswe1::HIS3 | Mizunuma et al. (1998) |
| YMM22 | MATa mck1-1 | this study |
| YMM36 | MATa Δzds1:TRP1 mck1 YIp-URA3-MCK1 integrated at MCK1 | this study |
| YMM51 | MATa Δhsl1::URA3 (hsl1Δ2::URA3) | Ma et al. (1996) |
| YMM52 | MATa Δhsl1::ura3 | this study |
| YMM53 | MATa Δhsl1::ura3 3HA-HSL1 integrated at HSL1 | this study |
| YMM54 | MATa Δhsl1::ura3 Δzds1::TRP1 3HA-HSL1 integrated at HSL1 | this study |
| YMM59 | MATa Δhsl1::ura3 cdc4 3HA-HSL1 integrated at HSL1 | this study |
| YMM60 | MATa Δhsl1::ura3 3HA-HSL1(KD) integrated at HSL1 | this study |
| YMM62 | MATa Δhsl1::ura3 Δcnb1::HIS3 3HA-HSL1(KD) integrated at HSL1 | this study |
| YMM65 | MATa Δhsl1::ura3 cdc23 3HA-HSL1 integrated at HSL1 | this study |
| YMM66 | MATa 3HA-HSL1(KD) integrated at HSL1 | this study |
| YMM67 | MATa Δhsl1::ura3 Δcnb1::HIS3 3HA-HSL1 integrated at HSL1 | this study |
| YMM68 | MATa Δmck1::TRP1 | this study |
| YMM69 | MATa Δmck1::TRP1 Δzds1::TRP1 | this study |
| YMM71 | MATa Δhsl1::ura3 mck1-1 3HA-HSL1 integrated at HSL1 | this study |
| YMM72 | MATa Δhsl1::ura3 Δcnb1::HIS3 3HA-HSL1 integrated at HSL1 | this study |
| YMM73 | MATa Δhsl1::ura3 cdc4 3HA-HSL1 integrated at HSL1 | this study |
| YMM74 | MATa Δhsl1::ura3 Δcnb1::HIS3 cdc4 3HA-HSL1 integrated at HSL1 | this study |
| YMM76 | MATa Δgrr1::LEU2 3HA-HSL1 integrated at HSL1 | this study |
| W1950-8c | MATα cdc4 HIS3 | A.Toh-e |
| TMY307 | MATa cdc23 | A.Toh-e |
| TMY310 | MATa Δbar1::HIS3 Clb2-Myc12-URA3 | A.Toh-e |
| TK-61 | MATa Δgrr1::LEU2 | T.Kishi |
| YMM2144 | MATa Δzds1:TRP1 mck1-1 | this study |
Plasmids
The plasmids used for creating the mck1 disruption alleles were as follows: the HindIII–SphI MCK1 fragment (pMM2144ΔSphI) was subcloned into pUC119, and the resulting plasmid was cut with NcoI, filled in with Klenow enzyme and ligated with XhoI linker (pUC119-mck1::XhoI). Finally, the SacII–XhoI TRP1 fragment was ligated to SacII–XhoI-digested pUC119-mck1::XhoI to create plasmid pUC119-mck1::TRP1 plasmid. The plasmid pYES2-Mck1 was constructed to express the MCK1 gene under the control of the GAL1 promoter. The plasmid YIp5-Mck1 was constructed to establish allelism between SCZ10 and MCK1. The plasmid pRS315-HA-Mck1 was constructed to express full-length MCK1 with three copies of the HA epitope at its N-terminus. The chromosomal HSL1 gene was replaced with a gene encoding Hsl1 with a triple HA epitope tag at its N-terminus and expressed from its own promoter, using plasmid YIP5-HA-Hsl1 (YIp5, an integrating vector containing LEU2+ as a selectable marker). Construction of the inactive kinase allele Hsl1(KD) was carried out basically as described (Barral et al., 1999). The plasmid YIp5-HA-Hsl1(KD) was constructed to integrate Hsl1(KD) with a triple HA epitope at its N-terminus. The plasmid pGEX-KG-Hsl1(KD) was constructed to express GST fused to Hsl1(KD) at its N-terminus. The plasmid YCplac22-9Myc-CNB1 was constructed to express full-length CNB1 with a Myc epitope at its C-terminus.
Flow cytometry and cell microscopy
Flow cytometoric analysis was carried out as described previously (Mizunuma et al., 1998). Yeast cells were fixed with formaldehyde to a final concentration of 3.7%, and viewed by differential interference contrast (DIC) microscopy. To visualize nuclei, cells were stained with DAPI (4′,6-diamidino-2-phenylindole).
Cell culture synchronization, RNA isolation and northern blot analysis
Cell culture synchronization and northern blot analysis were carried out essentially as described previously (Ma et al., 1996; Mizunuma et al., 1998).
Preparation of extracts, immunoprecipitations and kinase assays
Preparation of cell extracts and immunoprecipitations were carried out essentially as described previously (Barral et al., 1999). For detection of the HA-tagged proteins, Myc-tagged Cnb1 or Cdc28, monoclonal antibodies 12CA5 to the HA epitope, 9E10 to the Myc epitope or anti-PSTAIR, respectively, were used.
In Hsl1 dephosphorylation reactions, HA-Hsl1 was immunoprecipitated. CIP was added to the beads containing the HA-Hsl1 immune complex and incubated at 37°C for 30 min.
Clb2 kinase and Mck1 kinase activity were assayed as described (Barral et al., 1999) using histone H1 or GST–Hsl1(KD) as substrate, respectively.
Indirect immunofluorescence
Indirect immunofluorescence microscopy was performed as described (Pringle et al., 1991).
Acknowledgments
Acknowledgements
We thank Akio Toh-e and Tsutomu Kishi for plasmids and mutant strains, Tomoko Ohnishi for technical assistance, and Eiko Tsuchiya and Keiko Mizuta for helpful discussion. This work was supported in part by Grant-in-Aid for Scientific Research from the Ministry of Education, Science, Culture, Arts and Sports of Japan (10460044, 10217208 and 11556017), and Special Coordination Funds for Promoting Sciences and Technology from the Science and Technology Agency of the Japanese Government. M.M. is the recipient of a JSPS Fellowship (DC1).
References
- Aberle H., Bauer,A., Stappert,J., Kispert,A. and Kemler,R. (1997) β-catenin is a target for the ubiquitin–proteasome pathway. EMBO J., 16, 3797–3804. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Amon A. (1997) Regulation of B-type cyclin proteolysis by Cdc28-associated kinase in budding yeast. EMBO J., 16, 2693–2702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Amon A., Irniger,S. and Nasmyth,K. (1994) Closing the cell cycle circle in yeast: G2 cyclin proteolysis initiated at mitosis persists until the activation of G1 cyclins in the next cycle.Cell, 77, 1037–1050. [DOI] [PubMed] [Google Scholar]
- Andoh T., Hirata,Y. and Kikuchi,A. (2000) Yeast glycogen synthase kinase 3 is involved in protein degradation in cooperation with Bul1, Bul2 and Rsp5. Mol. Cell. Biol., 20, 6712–6720. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barral Y., Parra,M., Bidlingmaier,S. and Snyder,M. (1999) Nim1-related kinases coordinate cell cycle progression with the organization of the peripheral cytoskeleton in yeast. Genes Dev., 13, 176–187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Booher R.N., Deshaies,R.J. and Kirschner,M.W. (1993) Properties of Saccharomyces cerevisiae wee1 and its differential regulation of p34CDC28 in response to G1 and G2 cyclins. EMBO J., 12, 3417–3426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brandeis M. and Hunt,T. (1996) The proteolysis of mitotic cyclins in mammalian cells persists from the end of mitosis until the onset of S phase. EMBO J., 15, 5280–5289. [PMC free article] [PubMed] [Google Scholar]
- Burton J.L. and Solomon,M.J. (2000) Hsl1p, a Swe1p inhibitor, is degraded via the anaphase-promoting complex. Mol. Cell. Biol., 20, 4614–4625. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clapham D.E. (1995) Calcium signaling. Cell, 80, 259–268. [DOI] [PubMed] [Google Scholar]
- Cunningham K.W. and Fink,G.R. (1994) Calcineurin-dependent growth control in Saccharomyces cerevisiae mutants lacking PMC1, a homolog of plasma membrane Ca2+ ATPases. J. Cell Biol., 124, 351–363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cunningham K.W. and Fink,G.R. (1996) Calcineurin inhibits VCX1-dependent H+/Ca2+ exchange and induces Ca2+ ATPases in Saccharomyces cerevisiae. Mol. Cell. Biol., 16, 2226–2237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cyert M.S. and Thorner,J. (1992) Regulatory subunit (CNB1 gene product) of yeast Ca2+/calmodulin-dependent phosphoprotein phosphatases is required for adaptation to pheromone. Mol. Cell. Biol., 12, 3460–3469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feldmann R.M.R., Correll,C.C., Kaplan,K.B. and Deshaies,R.J. (1997) A complex of Cdc4p, Skp1p and Cdc53p/Cullin catalyzes ubiquitination of the phosphorylated CDK inhibitor Sic1p. Cell, 91, 221–230. [DOI] [PubMed] [Google Scholar]
- Garrett-Engele P., Moilanen,B. and Cyert,M.S. (1995) Calcineurin, the Ca2+/calmodulin-dependent protein phosphatase, is essential in yeast mutants with cell integrity defects and in mutants that lack a functional vacuolar H(+)-ATPase. Mol. Cell. Biol., 15, 4103–4114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gray J.V., Ogas,J.P., Kamada,Y., Stone,M., Levin,D.E. and Herskowitz,I. (1997) A role for the Pkc1 MAP kinase pathway of Saccharomyces cerevisiae in bud emergence and identification of a putative upstream regulator. EMBO J., 16, 4924–4937. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haro R., Graciadebles,B. and Rodriguez-Navarro,A. (1991) A novel P-type ATPase from yeast involved in sodium transport. FEBS Lett., 291, 189–191. [DOI] [PubMed] [Google Scholar]
- Hochstrasser M. (1996) Ubiqitin-dependent protein degradation. Annu. Rev. Genet., 30, 405–439. [DOI] [PubMed] [Google Scholar]
- Ikeda S., Kishida,S., Yamamoto,H., Murai,H., Koyama,S. and Kikuchi,A. (1998) Axin, a negative regulator of the Wnt signaling pathway, forms a complex with GSK-3β and β-catenin and promotes GSK-3 β-dependent phosphorylation of β-catenin. EMBO J., 17, 1371–1384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Irniger S., Piatti,S., Michaelis,C. and Nasmyth,K. (1995) Genes involved in sister chromatid separation are needed for B-type cyclin proteolysis in budding yeast. Cell, 81, 269–278. [DOI] [PubMed] [Google Scholar]
- Jaspersen S.L., Charles,J.F., Tinker-Kulberg,R.L. and Morgan,D.O. (1998) A late mitotic regulatory network controlling cyclin destruction in Saccharomyces cerevisiae. Mol. Biol. Cell, 9, 2803–2817. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lew D.J. and Reed,S.I. (1995) A cell cycle checkpoint monitors cell morphogenesis in budding yeast. J. Cell Biol., 129, 739–749. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Longtine M.S., Theesfeld,C.L., McMillan,J.N., Weaver,E., Pringle,J.R. and Lew,D.J. (2000) Septin-dependent assembly of a cell cycle-regulatory module in Saccharomyces cerevisiae. Mol. Cell. Biol., 20, 4049–4061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ma X.-J., Lu,Q. and Grunstein,M. (1996) A search for proteins that interact genetically with histone H3 and H4 amino termini uncovers novel regulators of the Swe1 kinase in Saccharomyces cerevisiae. Genes Dev., 10, 1327–1340. [DOI] [PubMed] [Google Scholar]
- Mazzoni C., Zarov,P., Rambourg,A. and Mann,C. (1993) The Slt2(Mpk1) MAP kinase homolog is involved in polarized cell growth in Saccharomyces cerevisiae. J. Cell Biol., 123, 1821–1833. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McMillan J.N., Sia,R.A.L. and Lew,D.J. (1998) A morphogenesis checkpoint monitors the actin cytoskeleton in yeast. J. Cell Biol., 142, 1487–1499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McMillan J.N., Sia,R.A., Bardes,E.S. and Lew,D.J. (1999a) Phosphorylation-independent inhibition of cdc28p by the tyrosine kinase Swe1p in the morphogenesis checkpoint. Mol. Cell. Biol., 19, 5981–5990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McMillan J.N., Longtine,M.S., Sia,R.A., Theesfeld,C.L., Bardes,E.S., Pringle,J.R. and Lew,D.J. (1999b) The morphogenesis checkpoint in Saccharomyces cerevisiae: cell cycle control of Swe1p degradation by Hsl1p and Hsl7p. Mol. Cell. Biol., 19, 6929–6939. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miller J.R. and Moon,R.T. (1996) Signal transduction through β-catenin and specification of cell fate during embryogenesis. Genes Dev., 10, 2527–2539. [DOI] [PubMed] [Google Scholar]
- Mizunuma M., Hirata,D., Miyahara,K., Tsuchiya,E. and Miyakawa,T. (1998) Role of calcineurin and Mpk1 in regulating the onset of mitosis in budding yeast. Nature, 392, 303–306. [DOI] [PubMed] [Google Scholar]
- Nakamura T., Liu,Y., Hirata,D., Namba,H., Harada,S., Hirokawa,T. and Miyakawa,T. (1993) Protein phosphatase type 2B (calcineurin)-mediated, FK506-sensitive regulation of intracellular ions in yeast is an important determinant for adaptation to high salt stress conditions. EMBO J., 12, 4063–4071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakamura T., Ohmoto,T., Hirata,D., Tsuchiya,E. and Miyakawa,T. (1996) Genetic evidence for the functional redundancy of the calcineurin- and Mpk1-mediated pathways in the regulation of cellular events important for growth in Saccharomyces cerevisiae. Mol. Gen. Genet., 251, 211–219. [DOI] [PubMed] [Google Scholar]
- Neigeborn L. and Mitchell,A.P. (1991) The yeast MCK1 gene encodes a protein kinase homolog that activates early meiotic gene expression. Genes Dev., 5, 533–548. [DOI] [PubMed] [Google Scholar]
- Patton E.E. and Willems,A.R. (1998) Combinatorial control in ubiquitin-dependent proteolysis: don’t Skp the F-box hypothesis. Trends Genet., 14, 236–243. [DOI] [PubMed] [Google Scholar]
- Patton E.E., Willems,A.R., Sa,D., Kuras,L., Thomas,D., Craig,K.L. and Tyers,M. (1998) Cdc53 is a scaffold protein for multiple Cdc34/Skp1/F-box protein complexes that regulate cell division and methionine biosynthesis in yeast. Genes Dev., 12, 692–705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Plyte S.E., Hughes,K., Nikolakaki,E., Pulverer,B.J. and Woodgett,J.R. (1992) Glycogen synthase kinase-3: functions in oncogenesis and development. Biochim. Biophys. Acta, 1114, 147–162. [DOI] [PubMed] [Google Scholar]
- Pringle J.R., Adams,A.E.M., Drubin,D.G. and Haarer,B.K. (1991) Immunofluorescence methods for yeast. Methods Enzymol., 194, 565–602. [DOI] [PubMed] [Google Scholar]
- Puziss J.W., Hardy,T.A., Johnson,R.B., Roach,P.J. and Hieter,P. (1994) MDS1, a dosage suppressor of an mck1 mutant, encodes a putative yeast homolog of glycogen synthase kinase 3. Mol. Cell. Biol., 14, 831–839. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shero J.H. and Hieter,P.A. (1991) Suppressor of a centromere DNA mutation encodes a putative protein kinase (MCK1). Genes Dev., 5, 549–560. [DOI] [PubMed] [Google Scholar]
- Shulewitz M.J., Inouye,C.J. and Thorner,J. (1999) Hsl7 localizes to a septin ring and serves as an adapter in a regulatory pathway that relieves tyrosine phosphorylation of Cdc28 protein kinase in Saccharomyces cerevisiae. Mol. Cell. Biol., 19, 7123–7137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sia R.A.L., Herald,H.A. and Lew,D.J. (1996) Cdc28 tyrosine phosphorylation and the morphogenesis checkpoint in budding yeast. Mol. Biol. Cell, 7, 1657–1666. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Skowyra D., Craig,K.L., Tyers,M., Elledge,S.J. and Harper,J.W. (1997) F-box proteins are receptors that recruit phosphorylated substrates to the SCF ubiquitin-ligase complex. Cell, 91, 209–219. [DOI] [PubMed] [Google Scholar]
- Tanaka S. and Nojima,H. (1996) Nik1: a Nim1-like protein kinase of S.cerevisiae interacts with the Cdc28 complex and regulates cell cycle progression. Genes Cells, 1, 905–921. [DOI] [PubMed] [Google Scholar]
- Zarzov P., Mazzoni,C. and Mann,C. (1996) The Slt2(Mpk1) MAP kinase is activated during periods of polarized cell growth in yeast. EMBO J., 15, 83–91. [PMC free article] [PubMed] [Google Scholar]