

Abstract
The evolutionarily conserved DnaJ proteins are essential components of Hsp70 chaperone systems. The DnaJ homologue of Escherichia coli associates with chaperone substrates and mediates their ATP hydrolysis-dependent locking into the binding cavity of its Hsp70 partner, DnaK. To determine the substrate specificity of DnaJ proteins, we screened 1633 peptides derived from 14 protein sequences for binding to E.coli DnaJ. The binding motif of DnaJ consists of a hydrophobic core of approximately eight residues enriched for aromatic and large aliphatic hydrophobic residues and arginine. The hydrophobicity of this motif explains why DnaJ itself can prevent protein aggregation. Although this motif shows differences from DnaK’s binding motif, DnaJ and DnaK share the majority of binding peptides. In contrast to DnaK, DnaJ binds peptides consisting of l- and d-amino acids, and therefore is not restricted by backbone contacts. These features allow DnaJ to scan hydrophobic protein surfaces and initiate the functional cycle of the DnaK system by associating with hydrophobic exposed patches and subsequent targeting of DnaK to these or to hydrophobic patches in spatial neighbourhood.
Keywords: heat shock proteins/Hsp40/Hsp70/protein folding/spot synthesis
Introduction
Hsp70 chaperones assist a large variety of protein folding processes in the cell, including folding of newly synthesized proteins in the cytosol, translocation of proteins into organelles, assembly and disassembly of protein complexes and control of the biological activity of folded regulatory proteins (Gething and Sambrook, 1992; Ellis and Hartl, 1999; Mayer et al., 2000). The specificity for these folding processes is provided to Hsp70 proteins through the activity of co-chaperones. The major class of co-chaperones acting as specificity factors are DnaJ proteins. They target Hsp70 proteins to their substrates by triggering ATP hydrolysis-dependent substrate association into the substrate-binding cavity of Hsp70 (Karzai and McMacken, 1996; Kelley, 1998; Misselwitz et al., 1998; Laufen et al., 1999). This activity requires the conserved J-domain (residues 2–78 in Escherichia coli DnaJ), which is essential for DnaJ’s interaction with Hsp70 partner proteins, leading to ATP hydrolysis by Hsp70 (Wall et al., 1994; Karzai and McMacken, 1996; Misselwitz et al., 1998). The adjacent G/F motif can contribute to the stimulation of ATP hydrolysis.
Escherichia coli DnaJ is capable of associating with unfolded substrates by itself. Substrate binding involves the central zinc-finger domain (residues 144–200) and the C-terminal domain (residues 201–376) (Banecki et al., 1996; Szabo et al., 1996). The individual roles for these domains in substrate recognition remain unclear despite the recent publication of information on their structure (Martinez-Yamout et al., 2000; Sha et al., 2000). Deletion of the zinc-finger domain and the C-terminal domain of DnaJ generates a fragment that contains the J domain and the G/F motif. This fragment lacks substrate-binding activity and has only low activity in stimulating DnaK’s ATPase (Wall et al., 1994), indicating that substrate recognition by DnaJ is needed for its activity in the functional cycle of DnaK.
The rate at which DnaJ associates with chaperone substrates is high enough to prevent aggregation reactions (Schröder et al., 1993; Szabo et al., 1994; Gamer et al., 1996), qualifying DnaJ as a chaperone on its own. Refolding of misfolded proteins strictly requires, however, the co-operation of DnaJ with the DnaK chaperone, which may induce conformational changes in the substrate upon interaction (Mayer et al., 2000). Based on these findings, it has been proposed that substrates first associate with DnaJ, which then allows their transfer to the Hsp70 substrate-binding cavity (Laufen et al., 1999). It is an open question whether, in this transfer reaction, the same stretch of the substrate polypeptide binds first to DnaJ and then to DnaK. Such a mechanism would require that DnaJ and DnaK had similar substrate specificities. Knowledge of the substrate-binding properties of DnaJ is therefore crucial for understanding substrate specificity and initiation of the functional cycle of the DnaK system.
We investigated the substrate-binding specificity of E.coli DnaJ by screening cellulose-bound peptides for DnaJ binding. We found that the DnaJ-binding motif shares some features with the DnaK-binding motif (Rüdiger et al., 1997b) but that it differs in some respects; we suggest that there is a mode of co-operation between DnaJ and DnaK in the selection of protein substrates.
Results
Screening of peptide scans for binding to DnaJ
To determine the binding motif within protein sequences recognized by DnaJ, we screened cellulose-bound peptide scans (Reineke et al., 1995) representing the complete sequences of 14 proteins comprising 1633 residues for DnaJ binding. This approach is justified by the finding that DnaJ binds with high affinity to peptides in solution (Feifel et al., 1998) and to the same peptides when they are bound to cellulose (not shown). The peptides constitute a subset of the sequences that were screened previously for DnaK binding (Rüdiger et al., 1997b), including sequences of in vivo protein substrates such as λP, RepE and σ32, thus allowing the substrate specificities of DnaJ and DnaK to be compared directly. We improved the detection procedure by use of a fluoroimaging system instead of chemiluminescence, which allowed better quantification and generation of statistically reliable data sets with fewer peptides than used in our previous work with DnaK. The peptide scans were composed of 13mers that overlap with adjacent peptides by 10 residues and therefore present all potential binding sites to DnaJ. They were incubated with DnaJ to equilibrium, followed by electrotransfer and immunodetection of the chaperone. Figure 1 shows selected peptide scans.
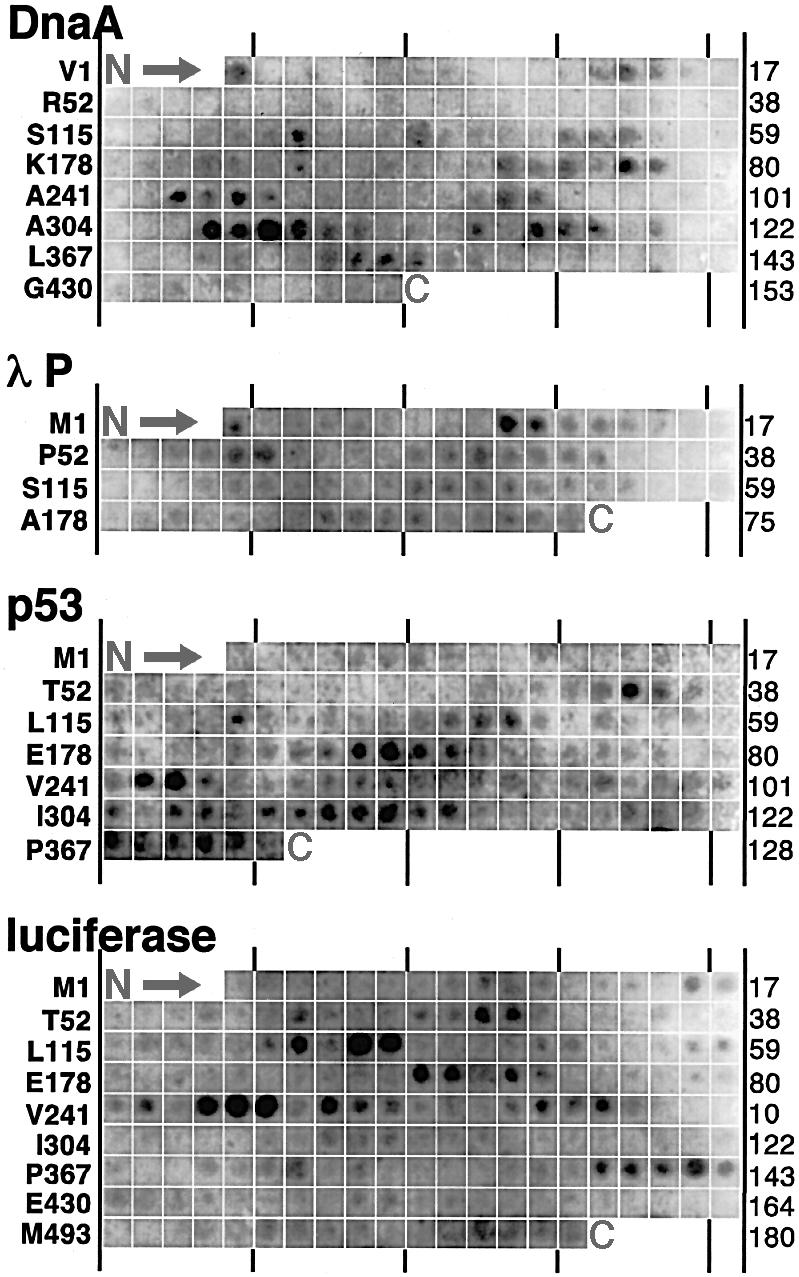
Fig. 1. DnaJ binding to cellulose-bound peptide scans. Peptide scans derived from sequences of DnaA, λP, p53 and luciferase were screened for DnaJ binding. Last spots of rows (right) and NH2-terminal residues of peptides of the first spots of rows (left) are indicated.
DnaJ bound to a subset of the peptides only, demonstrating that it discriminates between amino acid side chains. It bound frequently to neighbouring peptides with overlapping sequences in the scan, indicating that a DnaJ-binding site was shared by these peptides. DnaJ-binding peptides existed in all peptide scans tested, with no apparent clustering within scans. The frequency of DnaJ-binding sites in the library was similar to that of DnaK-binding sites [approximately one site per 36 residues (Rüdiger et al., 1997b)]. The frequency of DnaJ-binding sites was not affected by cellular or organellar origin or the size and oligomeric status of the scanned proteins.
DnaJ-binding peptides are particularly rich in aromatic residues
The size of the data set allowed reliable statistical analysis of the side-chain composition of DnaJ-binding peptides and a comparison with the known composition of DnaK-binding peptides (Rüdiger et al., 1997b). The library used was appropriate for such analysis since the relative occurrence of the 20 amino acids in the whole peptide library was similar to that found in natural proteins, except that cysteine was less frequent in the library than in nature; the library was similar to the library investigated for DnaK binding (not shown; Rüdiger et al., 1997b). The screened peptides were ordered according to their affinity for DnaJ as determined by fluoroimager quantification of the DnaJ signals (Figure 2A). Their affinity for DnaJ varied from high to low. The amino acid composition of peptides that bound DnaJ with high affinity (those with a relative DnaJ affinity of ≥40) was investigated. High-affinity DnaJ-binding peptides showed substantial differences from the total library (Figure 2B). They were strongly enriched in aromatic residues (Phe, Tyr and Trp) but also enriched in aliphatic hydrophobic residues (Leu and Ile). These enriched residues are similar to those enriched in DnaK-binding peptides (Rüdiger et al., 1997b), but differences exist with respect to the degree of enrichment. For DnaJ, aromatic residues were enriched more than aliphatic residues including Leu, while Leu is the single most prominent residue in DnaK binders. Aromatic residues were overrepresented up to nearly 2-fold in DnaJ-binding peptides while Leu was only enriched ∼1.5-fold in DnaK-binding peptides. Two-fold enrichment of a residue in a peptide library derived from protein sequences is very high. Furthermore, histidine, a polar and aromatic residue, was enriched in DnaJ-binding peptides but not in DnaK-binding peptides. Acidic (Asp, Glu) and most other residues are disfavoured by both chaperones, but the degree of disfavour of acidic residues by DnaJ is weaker and is in the same range as for alanine.

Fig. 2. Amino acid distribution in peptide-scanning libraries. For 1633 peptides representing 14 protein sequences the relative amino acid occurrence was determined. (A) Normalized affinity of DnaJ for the peptides investigated. Peptides are ordered according to their affinity for DnaJ. For the statistical analysis, two sets of peptides of high affinity for DnaJ (relative affinity >40; black bars) and low affinity for DnaJ (relative affinity <10; light bars) were selected. (B) Peptides with high affinity for DnaJ (dark bars) compared with DnaK-binding peptides identified in a previous study (Rüdiger et al., 1997b; light bars). The number for each amino acid is normalized to its occurrence in the whole peptide library (=100). (C) Sequence alignment of DnaJ-binding regions. Sixty-two DnaJ-binding regions each constituting a single strong DnaJ-binding site were aligned. Hydrophobic cores were anchored with a large hydrophobic or aromatic residue at position 10 by shifting the sequences by up to two residues. The frequency of acidic (white bars), large hydrophobic and aromatic (black bars) and basic residues (grey bars) at each position is given as a percentage. Large hydrophobic and aromatic residues are enriched between positions 10 and 17.
DnaJ recognizes a hydrophobic stretch of approximately eight residues
In the search for a consensus sequence motif of DnaJ-binding peptides, we aligned the overlapping peptides of 62 high-affinity DnaJ-binding regions in the same manner as done previously for DnaK (Rüdiger et al., 1997b), anchoring on the first N-terminal aromatic or large hydrophobic residue by shifting the binding regions within the alignment by up to two positions. Within a continuous stretch of approximately eight neighbouring residues, large hydrophobic and aromatic residues were enriched (Figure 2C). This region had an average content of 47% of such residues as compared with 28% within the whole library. The aromatic residues were particularly strongly increased (17%, compared with 8% within the whole library) while Leu was only moderately enriched (12%, compared with 9% within the whole library). Charged residues showed only small changes, with a slightly smaller proportion of acidic residues (9%) in the DnaJ-binding peptides than in the whole library (12%) and a slightly greater proportion of basic residues (13%) in the DnaJ-binding peptides than in the whole library (11%). Outside of this region we found no changes in amino acid distribution. Specific positioning of the preferred residues within the motif was not important for DnaJ binding.
This DnaJ-binding motif consisting of a hydrophobic stretch of approximately eight consecutive residues resembles the motif recognized by DnaK, although there are differences between the two motifs. The motif recognized by DnaK consists of a shorter hydrophobic core of only four or five residues, with flanking regions of lower importance for DnaK binding that are enriched in basic residues (Rüdiger et al., 1997b).
Most DnaJ-binding sites are also DnaK-binding sites
The identified differences in the consensus binding motifs of DnaJ and DnaK raise the following question: how much do the peptide-binding regions of DnaJ and DnaK overlap? Evaluation of the entire library used for screening of DnaJ and DnaK binding revealed that six out of seven DnaK-binding peptides had affinity for DnaJ, and three out of four DnaJ-binding peptides had affinity for DnaK. The degree of overlap between DnaJ- and DnaK-binding peptides is illustrated by the sequence of firefly luciferase (Figure 3A). Most DnaJ- and DnaK-binding sites in this sequence were shared, but a number of the DnaJ- or DnaK-binding peptides had good affinity for only one of these chaperones. The most prominent DnaJ-binding region that lacked affinity for DnaK was the region represented by peptides 138–141 (corresponding to residues 415–436 of the luciferase sequence).

Fig. 3. Comparison of binding of DnaK and DnaJ to peptides. (A) DnaJ- and DnaK-binding peptides in luciferase are in most cases identical. Luciferase-derived peptides that were screened for DnaJ binding (Figure 1) are represented as bars. The length of the bars corresponds to the affinity of the peptide for DnaJ. Black bars represent peptides that were classified as DnaK binders in a previous study (Rüdiger et al., 1997b). (B) Peptides that bind to DnaJ but not to DnaK contain several large hydrophobic and aromatic residues (Leu, Ile, Phe, Trp and Tyr; black boxes) with acidic residues (Glu and Asp; white circles) in between. Basic residues (Arg and Lys) are represented as grey circles. (C) DnaJ can compete with DnaK–peptide binding in solution. A complex of DnaK (0.1 µM) and the peptide σ32-Q132-Q144-C-IAANS (0.5 µM) was titrated with DnaJ (concentration as indicated). The fluorescence of the DnaK–peptide interaction was obtained by subtracting the fluorescence of the DnaJ–peptide interaction in the absence of DnaK from the total signal.
To investigate the criteria that led to differential binding of DnaJ and DnaK to peptides, we further inspected the peptides that bound to only one of the two chaperones. The peptides that bound only DnaJ showed a broad distribution of aromatic and aliphatic hydrophobic residues, usually with acidic residues in between them, and frequently contained more aromatic residues than leucine and isoleucine (Figure 3B). These characteristics, especially the existence of negatively charged residues within the binding motif, are sufficient to explain the lack of affinity of these peptides for DnaK. As an example, the above-mentioned DnaJ-binding region that lacks affinity for DnaK (represented by luciferase peptides 138–141) is shown in Figure 3B (first four peptides). We would like to emphasize that the peptides in Figure 3B differ in amino acid composition from typical DnaJ-binding peptides, especially with respect to their content of negatively charged residues (Figure 2B). Most of the peptides in Figure 3B are therefore not among the strongest DnaJ-binding peptides.
The peptides that bound only DnaK were characterized by a short hydrophobic core with a lower average content of large hydrophobic and aromatic residues (45%) than is usually found in good DnaK-binding sites (65%) within five residues of the hydrophobic core, and they had fewer such residues surrounding this core. Peptides that bound only DnaK generally had lower affinities for DnaK than peptides that bound both DnaK and DnaJ. It appears to be a general rule that peptides that bound only one of the chaperones had weaker affinities for their binding chaperone than peptides that bound both DnaK and DnaJ.
DnaJ and DnaK compete for peptide binding
Since many DnaJ-binding peptides also bind DnaK and vice versa, we investigated whether DnaJ could compete with DnaK for peptide binding in solution. We chose a fluorescently labelled peptide derived from the E.coli σ32 heat-shock transcription factor, σ32-Q132-Q144-C-IAANS, whose binding to DnaK is well characterized (McCarty et al., 1996). Such an approach is reasonable since the binding of DnaK to this peptide substrate (KD 0.08 nM) is about two orders of magnitude stronger than that to protein substrates such as σ32, and this particular peptide belongs to the DnaJ-binding class on the cellulose-bound peptide scan. The presence of DnaK strongly increased the fluorescence of σ32-Q132-Q144-C-IAANS, whereas the presence of DnaJ only weakly increased the signal. These differences in fluorescence allowed us to follow the titration of the DnaK–peptide complex with DnaJ (Figure 3C). The experiments were performed in the absence of ATP to prevent ATP-dependent substrate transfer from DnaJ to DnaK, which would generate a cooperative rather than a competitive situation. We also performed titration experiments with the DnaJ-H33Q (DnaJ259) mutant protein, which shows impaired functional interaction with the DnaK ATPase domain but has normal substrate-binding properties (Wall et al., 1994; Gässler et al., 1998; Mayer et al., 1999), yielding the same results as for wild-type DnaJ (not shown). Low concentrations of DnaJ (0.5- to 2-fold molar excess over DnaK) led to a slight increase in fluorescence for unknown reasons. Such an increase was not observed in the absence of DnaK. The titration of DnaJ at higher concentrations correlated with a decrease in fluorescence, indicating that the chaperones competed for peptide binding. When DnaJ was present in 20-fold molar excess over DnaK and in 4-fold molar excess over the peptide, the fluorescence of the DnaK–peptide complex decreased to half that of the maximum signal. Thus, although the affinity of DnaJ for this peptide is lower than that of DnaK, DnaJ can compete with DnaK for association with this peptide.
DnaJ binding does not require specific orientation of the peptide backbone
We investigated whether peptide recognition by DnaJ relies exclusively on side-chain interactions or involves backbone recognition as well. In one approach we assessed the binding of DnaJ to peptide scans composed of exclusively l- or exclusively d-amino acids [Figure 4A and B (rows a and b)]. In a second approach we assessed the binding of DnaJ to peptides synthesized with either authentic or inverse sequences [Figure 4A and B (rows a and c)]. Peptides composed of d-amino acids and synthesized with the inverse sequence have the same orientation of their side chains as peptides composed of l-amino acids, but differ with respect to their backbones. If DnaJ exclusively recognized side chains, an l-peptide synthesized with the authentic sequence should have the same DnaJ-binding properties as a d-peptide synthesized in the inverse direction.

Fig. 4. Recognition of the peptide backbone by DnaK but not DnaJ. (A) Illustration of the stereochemical differences between peptides of the same sequence but authentic or inversed sequence direction, or composed of l- or d-peptides. The hexagon and the wavy line symbolize the coupling of the C-terminus of each peptide via a linker to cellulose. The light grey forms symbolize the stereochemical connection of three different side chains of hypothetical tripeptides. The backbone atoms are indicated, except carbon. (B) Peptide scans derived from the sequences of λ CI were screened for binding to DnaJ (a–d) and DnaK (e–h). The peptides were synthesized using l- (a, c, e and g) or d-amino acids (b, d, f and h) and with authentic (e.g. H2N-STKKKPLTQEQLE-COO-cellulose; a, b, e and f) or inverse sequence (e.g. H2N-ELQEQTLPKKKTS-COO-cellulose; c, d, g and h).
We synthesized 76 peptides derived from the sequence of λCI with both authentic and inverse sequence, and using l- and d-amino acids, and investigated them for DnaJ binding. DnaJ bound to peptides with d- and l-amino acids with the same affinity and comparable binding pattern (Figure 4B, rows a–d). The patterns of DnaJ binding to authentic peptides with l- or d-amino acids were slightly more similar to each other than to those synthesized with inverse sequence with either l- or d-amino acids and vice versa. Therefore, DnaJ differentiates in the same manner between the side chains of peptides with d- or l-amino acids, indicating that the binding of DnaJ to peptides does not require a specific orientation of the backbone. The association of DnaJ with peptides therefore relies predominantly, if not exclusively, on side-chain contacts.
DnaK–peptide binding strictly requires backbone contacts and substrate directionality
We performed the same type of analysis for DnaK to compare its binding features with those of DnaJ (Figure 4B, rows e–h). In contrast to DnaJ, DnaK recognized only peptides composed of l-amino acids but not those composed of d-amino acids, indicating that specific backbone contacts are essential for substrate recognition by DnaK. However, peptides that were composed of l-amino acids and had identical amino acid composition, but differed in their authentic or inverse sequence of amino acids, were bound by DnaK to different extents. Repetitions of the experiment with independently synthesized peptide scans ruled out the possibility that synthesis problems had influenced the results. Since the differences between rows e and g of Figure 4B occurred in general for sets of adjacent peptides, we can rule out the possible artefact that the positioning of a DnaK-binding site at the N- or C-terminus or its distance from the cellulose matrix was responsible for the observed differences in affinity.
Discussion
We analysed the molecular basis for substrate recognition by E.coli DnaJ by screening cellulose-bound peptide scans. This approach avoids solubility problems of hydrophobic peptides and allows large numbers of peptides to be screened, which permits identification of the DnaJ-binding motif and all potential binding sites within the tested protein sequences.
The substrate-binding motif was determined by sequence alignment and statistical analysis of DnaJ-binding regions. It consists of a continuous stretch of approximately eight residues, particularly enriched in aromatic residues but also enriched in large hydrophobic aliphatic residues (Figure 2C). The hydrophobic nature of this motif explains why E.coli DnaJ and several prokaryotic and eukaryotic homologues exhibit chaperone activities by themselves as judged by their ability to prevent aggregation of misfolded proteins (Schröder et al., 1993; Szabo et al., 1994; Prip-Buus et al., 1996; Lu and Cyr, 1998). The hydrophobic core of the binding motif is longer than that of DnaK, and aromatic residues rather than Leu are key residues for DnaJ–substrate binding (Figure 5A) (Rüdiger et al., 1997b). Acidic residues are disfavoured in DnaJ-binding peptides, but are not excluded from the hydrophobic core as for DnaK. Despite these differences, DnaJ and DnaK have affinity for large hydrophobic and aromatic residues and therefore most peptides that bind one of these chaperones also bind the other (Figure 5B).
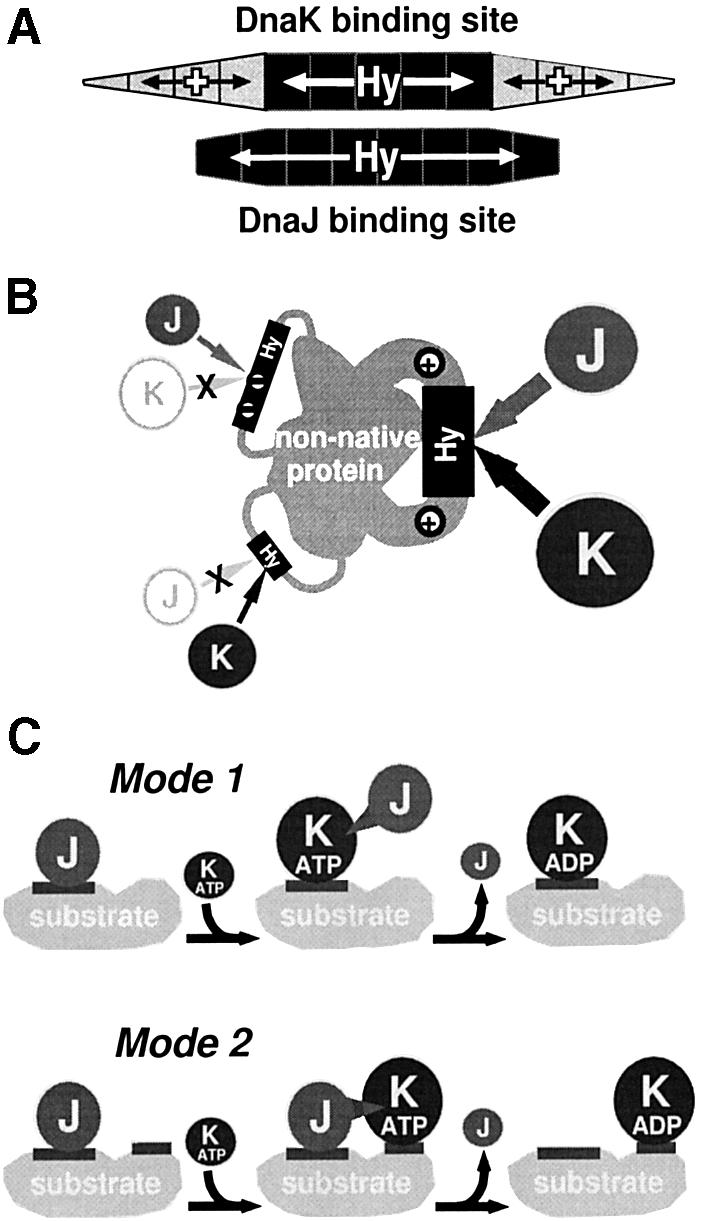
Fig. 5. Common and divergent features of substrate recognition of DnaJ and DnaK. (A) The binding motifs of DnaK and DnaJ in substrates. Positions with contributions of large hydrophobic or aromatic residues (Hy) or positively charged residues (+; the size of the area is proportional to the weight of these contributions) are indicated. While DnaK preferentially binds hydrophobic cores of four or five residues in length and positively charged residues in the flanking regions increase binding (Rüdiger et al., 1997b), DnaJ binds to longer hydrophobic segments (approximately eight residues in length). (B) Segments with denatured polypeptide sequences that bind to both DnaJ (J) and DnaK (K) are characterized by longer stretches enriched in large hydrophobic and aromatic residues (grey tubes) and basic residues (+) neighbouring such stretches, while short hydrophobic stretches bind only to DnaK. Longer stretches with acidic residues in between bind only to DnaJ. (C) Two modes of DnaJ interaction with substrates allow targeting of DnaK to unfolded proteins. Mode 1: DnaJ (J) binds to a hydrophobic segment (dark grey stretch) on the surface of a chaperone substrate that is subsequently bound by DnaK (K) (‘handover’). DnaJ dissociates after stimulation of DnaK’s ATPase; the spike indicates this functional interaction. Mode 2: DnaJ (J) binds to hydrophobic segments (dark grey stretch) on the surface of a chaperone substrate that are spatially near the stretch that is subsequently bound by DnaK (K). DnaJ dissociates after stimulation of DnaK’s ATPase; the spike indicates this functional interaction.
DnaJ and DnaK binding to side chains and the backbone
For both chaperones we determined the importance of the peptide backbone for specificity by testing the binding of DnaK and DnaJ to l- and d-peptides with sequences in authentic and inverse direction. DnaK only bound peptides with backbones of l-amino acids (Figure 4B, rows e–h), consistent with the architecture of its substrate-binding cavity (Zhu et al., 1996) and experiments with acrylodan-labelled d- and l-peptides (Feifel et al., 1998). However, until now it was not clear whether the DnaK-binding cavity allows binding of peptides in both orientations with respect to their N- and C-termini. If peptides would bind equally well in both orientations, specificity would be solely determined by side-chain contacts. However, we found that several substrate peptides changed their affinity for DnaK if they were synthesized in the inverse order (Figure 4B, rows e and g). The orientation of the backbone is thus an important determinant for DnaK’s substrate specificity. These findings have two consequences with respect to the substrate-binding cavity of DnaK. First, it has an asymmetry for binding specific side chains. Secondly, the positions of the N- and C-terminal portions of peptides in the substrate binding cavity are not variable. Thus, a peptide exhibiting lower DnaK affinity in the authentic orientation than in the inversed orientation cannot simply ‘turn around’ and bind to DnaK with the N-terminus on the other side. DnaK–peptide interactions therefore require a specific direction of the backbone.The evolutionary conservation of the binding cavity (Rüdiger et al., 1997a) suggests that this feature is conserved within the Hsp70 family. This may then have consequences for substrate recognition, e.g. by Hsp70 homologues, which interact with nascent chains that are translocating across membranes in an oriented fashion.
In contrast to DnaK, DnaJ binds peptides composed of d- as well as l-residues. This is consistent with previous results based on acrylodan-labelled peptides (Feifel et al., 1998) and demonstrates that this behaviour is independent of possible interaction of DnaJ with the acrylodan label. We showed for a large variety of peptides that the specificity of DnaJ is independent of the stereochemistry of the backbone. The difference between the authentic and inverse direction of the residues has only a minor effect on DnaJ binding. This finding is in accordance with the properties of the structures of DnaJ’s zinc-finger and C-terminal domains, which are responsible for substrate binding but lack any cavity that would allow for backbone contacts to bound substrate (see below; Figure 6) (Martinez-Yamout et al., 2000; Sha et al., 2000). The binding properties disclosed in this study indicate that DnaJ acts as a scanning factor that rapidly scans protein surfaces and recognizes side chains of hydrophobic surface patches without stable binding as found for DnaK. This interpretation is further supported by several findings. First, the substrate-binding domain of DnaJ does not appear to contain a pocket allowing for tight binding of substrates (see below). Secondly, DnaJ is known to recognize substrates in a fast, reversible and ATP-independent manner. Thus, complexes of DnaJ with the protein substrate, σ32, exhibit high association and dissociation rates (Gamer et al., 1996). Thirdly, the affinity of DnaJ for the best binding peptides published so far is ∼1 µM and thus rather low (Feifel et al., 1998) [these peptides give reasonable signals in the peptide library assay (not shown)] while the affinity of DnaK for the best binding peptides is ∼50 nM and thus rather high (McCarty et al., 1996). The preference of DnaJ for aromatic residues seems appropriate for such a scanning function since they are less flexible and more bulky than aliphatic side chains.

Fig. 6. Structures of the substrate-binding domains. (A) Domain structures of DnaJ from E.coli and Sis1 from S.cerevisiae. J, J domain; blank segments, G/F motif; grey segments, linker (DnaJ) or G/M motif (Sis1); Zn, zinc-finger domain; C, C-terminal domain. (B) Structure of the substrate-binding domain of Sis1 (C-terminal domain; Sha et al., 2000). (C and D) Structure of the substrate-binding domains of DnaJ. (C) Model of the C-terminal domain. (D) Zinc-finger domain (Martinez-Yamout et al., 2000). Hydrophobic side chains (Ile, Leu, Val, Phe, Trp, Tyr, Ala and Met) are coloured yellow in the space-filling representations (done by InsightII). Red circles indicate the sites we propose as most likely to be responsible for substrate binding according to our biochemical data. The electrostatic potential was rendered on the surface of each domain using Grasp (Nicholls et al., 1993). Red and blue indicate acidic and basic surface regions, respectively. In (C) the green quadrangles indicate the parts of the C-terminal domain of DnaJ in which residues corresponding to the N-terminal residues of the C-terminal domain of Sis1 are missing.
Structural implications of DnaJ’s binding specificity
Two recent studies (Martinez-Yamout et al., 2000; Sha et al., 2000) allowed us to correlate our data with structural features of DnaJ. Dyson and co-workers solved the NMR structure of the zinc-finger domain of E.coli DnaJ (Figure 6D) (Martinez-Yamout et al., 2000), and Sha et al. (2000) solved the crystal structure of the C-terminal domain of the yeast homologue, Sis1 (Figure 6B). On the basis of the Sis1 structure we generated a homology model of the corresponding domain of E.coli DnaJ (Figure 6C). There was one difference between the Sis1 and DnaJ C-terminal structures: in Sis1 there is a hydrophobic depression contacting a single proline side chain of another Sis1 molecule in the crystal packing, and Sha et al. (2000) proposed that this depression is responsible for substrate binding. This depression is missing in DnaJ and is replaced by residues belonging to the zinc-finger domain, which is not present in Sis1 (Figure 6).
Neither the zinc-finger domain nor the modelled C-terminal domain of DnaJ contained an obvious candidate for the substrate binding site. We therefore investigated whether the nature of the DnaJ substrate-binding motif identified in this study gives indications for such a site (Figure 6). A binding site should allow hydrophobic contacts to aliphatic and aromatic residues, preferentially in the neighbourhood of negatively charged regions, since DnaJ-binding peptides are rich in positively charged residues, although DnaJ and each of its domains has an overall negative surface charge. In the C-terminal domain the only region fulfilling these criteria is a groove at its C-terminus that is responsible for dimerization in crystallized Sis1 (Sha et al., 2000). The findings by Sha et al. that deletion of the last 15 residues at the C-terminus of Sis1 prevents both chaperone function and oligomerization of Sis1 supports this idea. From our results it would seem unlikely that, in the case of DnaJ, a single proline side chain, proposed by Sha et al. (2000) to be responsible for the interaction of substrate with Sis1, is responsible for binding of substrate to DnaJ, given that DnaJ-binding peptides are not rich in proline (Figure 2), apart from the fact that this entire depression is missing in DnaJ (see above). The finding that DnaJ did not associate with peptides having just a short hydrophobic core implies that more contacts are required. The zinc-finger domain contains no hydrophobic cavity surrounded by negatively charged residues (Martinez-Yamout et al., 2000), but it has a nearly uncharged hydrophobic tip (Figure 6D), which may contribute to DnaJ’s interactions with hydrophobic substrates. The substrate-binding properties of DnaJ’s zinc-finger and C-terminal domains seem very similar to those of the eukaryotic chaperone prefoldin, which has hydrophobic patches on the tips of ‘tentacles’ of a jellyfish-like structure (Siegert et al., 2000).
While these structural considerations are speculative at present, one important finding of our study corresponds well with the structures of both domains: they both lack any structural feature that allows specific backbone contacts with substrates. This is in accordance with our finding that DnaJ’s substrate specificity relies exclusively on side-chain interactions (Figure 4).
Targeting of substrates to DnaK
The described properties of substrate recognition by DnaJ are consistent with two, not mutually exclusive, mechanisms by which DnaJ targets substrates to DnaK (Figure 5C). In both mechanisms, DnaJ binds to surface-exposed hydrophobic stretches of a substrate, followed by, or simultaneous to, an interaction with DnaK·ATP via the J domain. In the first mechanism (mode 1), this interaction directs DnaK to a hydrophobic patch already bound by DnaJ and, in a handing-over reaction involving DnaJ- and substrate-dependent ATP hydrolysis by DnaK, DnaK associates with this patch while DnaJ dissociates from it. In the second mechanism (mode 2), DnaK associates with a different hydrophobic stretch in the spatial neighbourhood of DnaJ, followed by DnaJ- and substrate-dependent ATP hydrolysis by DnaK, which locks the substrate in a complex with DnaK. Mode 2 is supported by the fact that only the J domain, but not the substrate-binding domains, is conserved between DnaJ proteins (Laufen et al., 1998), which makes it unlikely that a conserved handing-over mechanism exists for substrate transfer from DnaJ proteins to Hsp70s (Figure 5C).
A requirement that is essential for the first mechanism and less obvious but possible for the second mechanism is that DnaJ and DnaK show substantial overlap in their binding sites in substrates. We showed here that this requirement is fulfilled. For both mechanisms, but especially for the second one, DnaJ plays crucial roles in substrate recognition by DnaK in a manner that is analogous to the ubiquitin tags that allow regulated targeting of selected proteins for proteolysis.
Materials and methods
Screening of cellulose-bound peptides
Peptide libraries were prepared by automated spot synthesis (Frank, 1992; Kramer et al., 1994; Kramer and Schneider-Mergener, 1998), in which peptides are C-terminally attached to cellulose via a (β-Ala)2 spacer. Peptides were derived from the sequences of 14 proteins: DnaA, DnaK, DnaJ and ribosomal protein L2 from E.coli, CI and P from bacteriophage λ, RepE from mini F plasmid, RepA from P1 plasmid, cytochrome b2, ATP synthase β-chain (F1-β) and ATP synthase protein 9 (Su9) from Saccharomyces cerevisiae, Photinus pyralis luciferase, bovine pancreatic trypsin inhibitor (BPTI) and human p53. The screening followed a published procedure (Rüdiger et al., 1997b) with slight modifications. Before screening the dry membranes were washed for 10 min in methanol and 3× 20 min in Tris-buffered saline (TBS; 31 mM Tris–HCl pH 7.6, 170 mM NaCl, 6.4 mM KCl). DnaJ (50 nM), purified as described (Gamer et al., 1996), was allowed to react with peptide scans in MP2 buffer [31 mM Tris–HCl pH 7.6, 100 mM KCl, 5 mM MgCl2, 0.05% (v/v) Tween 20, 5% (w/v) sucrose] for 30 min at 25°C with gentle shaking. Unbound DnaJ was removed with TBS (4°C) and peptide-bound DnaJ was electrotransferred on to polyvinylene difluoride (PVDF) membranes (Millipore) using a semi-dry blotter (Phase GmbH, Lübeck, Germany). PVDF membranes were sandwiched between blotting papers soaked with XK buffer [75 mM Tris base, 120 mM 6-aminohexane acid, 0.01% (w/v) SDS] and anode buffers XA1 (90 mM Tris base) and XA2 (300 mM Tris base) and kept at 4°C. Electrotransfer was performed at a constant power of 0.8 mA/cm2 peptide cellulose. Tranferred DnaJ was detected using DnaJ-specific polyclonal rabbit sera, an alkaline phosphatase-conjugated secondary antibody and chemifluorescence activity measurement (ECF kit, Amersham-Pharmacia) using fluoroimaging systems (FluorImager SI, Molecular Dynamics and FLA2000, Fuji). Quantification was performed using ImageQuant 1.11 (Molecular Dynamics) and the relative intensities were normalized to the signal of the reference peptide, AKTLILSHLRFVV, which was set as 100. The statistical analysis is based on evaluation of at least two independent experiments for each peptide scan.
Competition of DnaJ and DnaK in solution
Fluorescence experiments were performed as described (McCarty et al., 1996). DnaJ or DnaJ-H33Q (DnaJ259) was present during the incubation to equilibrium at 30°C.
Structural analysis
A model of the C-terminal domain of DnaJ based on the structure of Sis1 was generated using the Swiss-Model server (Peitsch, 1995, 1996; Guex and Peitsch, 1997). This model was further minimized using the Discover module of InsightII (MSI) by subsequent relaxation. Hydrogen atoms were added to the structure of Sis1. The structure of the C-terminal domain of Sis1 was minimized in the same way as for DnaJ.
Acknowledgments
Acknowledgements
We thank C.Escher and M.Seiler for excellent technical assistance, J.Gamer for providing DnaJ, T.Laufen, M.P.Mayer and A.Mogk for comments on the manuscript and S.T.Rüdiger for corrections. This work was supported by grants of the DFG and the Fonds der Chemischen Industrie to B.B.
References
- Banecki B., Liberek,K., Wall,D., Wawrzynów,A., Georgopoulos,C., Bertoli,E., Tanfani,F. and Zylicz,M. (1996) Structure–function analysis of the zinc finger region of the DnaJ molecular chaperone. J. Biol. Chem., 271, 14840–14848. [DOI] [PubMed] [Google Scholar]
- Ellis R.J. and Hartl,F.U. (1999) Principles of protein folding in the cellular environment. Curr. Opin. Struct. Biol., 9, 102–110. [DOI] [PubMed] [Google Scholar]
- Feifel B., Schönfeld,H.-J. and Christen,P. (1998) d-peptide ligands for the co-chaperone DnaJ. J. Biol. Chem., 273, 11999–12002. [DOI] [PubMed] [Google Scholar]
- Frank R. (1992) Spot synthesis: an easy technique for the positionally addressable, parallel chemical synthesis on a membrane support. Tetrahedron, 48, 9217–9232. [Google Scholar]
- Gamer J., Multhaup,G., Tomoyasu,T., McCarty,J.S., Rüdiger,S., Schönfeld,H.-J., Schirra,C., Bujard,H. and Bukau,B. (1996) A cycle of binding and release of the DnaK, DnaJ and GrpE chaperones regulates activity of the E.coli heat shock transcription factor σ32. EMBO J., 15, 607–617. [PMC free article] [PubMed] [Google Scholar]
- Gässler C.S., Buchberger,A., Laufen,T., Mayer,M.P., Schröder,H., Valencia,A. and Bukau,B. (1998) Mutations in the DnaK chaperone affecting interaction with the DnaJ co-chaperone. Proc. Natl Acad. Sci. USA, 95, 15229–15234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gething M.-J.H. and Sambrook,J.F. (1992) Protein folding in the cell. Nature, 355, 33–45. [DOI] [PubMed] [Google Scholar]
- Guex N. and Peitsch,M.C. (1997) SWISS-MODEL and the Swiss-PdbViewer: an environment for comparative protein modelling. Electrophoresis, 18, 2714–2723. [DOI] [PubMed] [Google Scholar]
- Karzai A.W. and McMacken,R. (1996) A bipartite signaling mechanism involved in DnaJ-mediated activation of the Escherichia coli DnaK protein. J. Biol. Chem., 271, 11236–11246. [DOI] [PubMed] [Google Scholar]
- Kelley W.L. (1998) The J-domain family and the recruitment of chaperone power. Trends Biochem. Sci., 23, 222–227. [DOI] [PubMed] [Google Scholar]
- Kramer A. and Schneider-Mergener,J. (1998) Synthesis and screening of peptide libraries on continuous cellulose membrane supports. Methods Mol. Biol., 87, 25–39. [DOI] [PubMed] [Google Scholar]
- Kramer A., Schuster,A., Reineke,U., Malin,R., Volkmer-Engert,R., Landgraf,C. and Schneider-Mergener,J. (1994) Combinatorial cellulose-bound peptide libraries: screening tools for the identification of peptides that bind ligands with predefined specificity. Methods, 6, 388–395. [Google Scholar]
- Laufen T., Zuber,U., Buchberger,A. and Bukau,B. (1998) DnaJ proteins. In Fink,A.L. and Goto,Y. (eds), Molecular Chaperones in Proteins: Structure, Function and Mode of Action. Marcel Dekker, New York, NY, pp. 241–274.
- Laufen T., Mayer,M.P., Beisel,C., Klostermeier,D., Reinstein,J. and Bukau,B. (1999) Mechanism of regulation of Hsp70 chaperones by DnaJ co-chaperones. Proc. Natl Acad. Sci. USA, 96, 5452–5457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu Z. and Cyr,D.M. (1998) The conserved carboxyl terminus and zinc finger-like domain of the co-chaperone Ydj1 assist Hsp70 in protein folding. J. Biol. Chem., 273, 5970–5978. [DOI] [PubMed] [Google Scholar]
- Martinez-Yamout M., Legge,G.B., Zhang,O., Wright,P.E. and Dyson,H.J. (2000) Solution structure of the cysteine-rich domain of the Escherichia coli chaperone protein DnaJ. J. Mol. Biol., 300, 805–818. [DOI] [PubMed] [Google Scholar]
- Mayer M.P., Laufen,T., Paal,K., McCarty,J.S. and Bukau,B. (1999) Investigation of the interaction between DnaK and DnaJ by surface plasmon resonance. J. Mol. Biol., 289, 1131–1144. [DOI] [PubMed] [Google Scholar]
- Mayer M.P., Rüdiger,S. and Bukau,B. (2000) Molecular basis for interactions of the DnaK chaperone with substrates. Biol. Chem., 381, 877–885. [DOI] [PubMed] [Google Scholar]
- McCarty J.S., Rüdiger,S., Schönfeld,H.-J., Schneider-Mergener,J., Nakahigashi,K., Yura,T. and Bukau,B. (1996) Regulatory region C of the E. coli heat shock transcription factor, σ32, constitutes a DnaK binding site and is conserved among eubacteria. J. Mol. Biol., 256, 829–837. [DOI] [PubMed] [Google Scholar]
- Misselwitz B., Staeck,O. and Rapoport,T.A. (1998) J proteins catalytically activate Hsp70 molecules to trap a wide range of peptide sequences. Mol. Cell, 2, 593–603. [DOI] [PubMed] [Google Scholar]
- Nicholls A., Bharadwaj,R. and Honig,B. (1993) Grasp—graphical representation and analysis of surface properties. Biophys. J., 64, A166. [Google Scholar]
- Peitsch M.C. (1995) Protein modeling by e-mail. BioTechnology, 13, 658–660. [Google Scholar]
- Peitsch M.C. (1996) ProMod and Swiss-Model: Internet-based tools for automated comparative protein modelling. Biochem. Soc. Trans, 24, 274–279. [DOI] [PubMed] [Google Scholar]
- Prip-Buus C., Westerman,B., Schmitt,M., Langer,T., Neupert,W. and Schwarz,E. (1996) Role of the mitochondrial DnaJ homologue, Mdj1p, in the prevention of heat-induced protein aggregation. FEBS Lett., 380, 142–146. [DOI] [PubMed] [Google Scholar]
- Reineke U., Sabat,R., Kramer,A., Stigler,R.-D., Seifert,M., Michel,T., Volk,H. and Schneider-Mergener,J. (1995) Mapping protein–protein contact sites using cellulose-bound peptide scans. Mol. Diversity, 1, 141–148. [DOI] [PubMed] [Google Scholar]
- Rüdiger S., Buchberger,A. and Bukau,B. (1997a) Interaction of Hsp70 chaperones with substrates. Nature Struct. Biol., 4, 342–349. [DOI] [PubMed] [Google Scholar]
- Rüdiger S., Germeroth,L., Schneider-Mergener,J. and Bukau,B. (1997b) Substrate specificity of the DnaK chaperone determined by screening cellulose-bound peptide libraries. EMBO J., 16, 1501–1507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schröder H., Langer,T., Hartl,F.-U. and Bukau,B. (1993) DnaK, DnaJ and GrpE form a cellular chaperone machinery capable of repairing heat-induced protein damage. EMBO J., 12, 4137–4144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sha B., Lee,S. and Cyr,D.M. (2000) The crystal structure of the peptide-binding fragment from the yeast Hsp40 protein Sis1. Structure, 15, 799–807. [DOI] [PubMed] [Google Scholar]
- Siegert R., Leroux,M.R., Scheufler,C., Hartl,F.-U. and Moarefi,I. (2000) Structure of the molecular chaperone prefoldin: unique interaction of multiple coiled coil tentacles with unfolded proteins. Cell, 103, 621–632. [DOI] [PubMed] [Google Scholar]
- Szabo A., Langer,T., Schröder,H., Flanagan,J., Bukau,B. and Hartl,F.U. (1994) The ATP hydrolysis-dependent reaction cycle of the Escherichia coli Hsp70 system—DnaK, DnaJ and GrpE. Proc. Natl Acad. Sci. USA, 91, 10345–10349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Szabo A., Korzun,R., Hartl,F.U. and Flanagan,J. (1996) A zinc finger-like domain of the molecular chaperone DnaJ is involved in binding to denatured protein substrates. EMBO J., 15, 408–417. [PMC free article] [PubMed] [Google Scholar]
- Wall D., Zylicz,M. and Georgopoulos,C. (1994) The NH2-terminal 108 amino acids of the Escherichia coli DnaJ protein stimulate the ATPase activity of DnaK and are sufficient for λ replication. J. Biol. Chem., 269, 5446–5451. [PubMed] [Google Scholar]
- Zhu X., Zhao,X., Burkholder,W.F., Gragerov,A., Ogata,C.M., Gottesman,M. and Hendrickson,W.A. (1996) Structural analysis of substrate binding by the molecular chaperone DnaK. Science, 272, 1606–1614. [DOI] [PMC free article] [PubMed] [Google Scholar]