

Abstract
λ Integrase (Int) has the distinctive ability to bridge two different and well separated DNA sequences. This heterobivalent DNA binding is facilitated by accessory DNA bending proteins that bring flanking Int sites into proximity. The regulation of λ recombination has long been perceived as a structural phenomenon based upon the accessory protein-dependent Int bridges between high-affinity arm-type (bound by the small N-terminal domain) and low-affinity core-type DNA sites (bound by the large C-terminal domain). We show here that the N-terminal domain is not merely a guide for the proper positioning of Int protomers, but is also a context-sensitive modulator of recombinase functions. In full-length Int, it inhibits C-terminal domain binding and cleavage at the core sites. Surprisingly, its presence as a separate molecule stimulates the C-terminal domain functions. The inhibition in full-length Int is reversed or overcome in the presence of arm-type oligonucleotides, which form specific complexes with Int and core-type DNA. We consider how these results might influence models and experiments pertaining to the large family of heterobivalent recombinases.
Keywords: DNA binding domains/λ integrase/protein–DNA interactions/site-specific recombination
Introduction
The integrase protein (Int) of Escherichia coli phage λ belongs to the large tyrosine family of site-specific DNA recombinases from archaebacteria, eubacteria and yeast that catalyze rearrangements between DNA sequences with little or no sequence homology to each other. Like λ Int, many of these recombinases function in the integration and excision of viral genomes into and out of the chromosomes of their respective hosts, as first proposed by Campbell (1962). They comprise, along with other family members, a large subgroup of recombinases with the distinctive ability to simultaneously bridge two different and well-separated DNA sequences, called arm- and core-type sites, respectively. This heterobivalent DNA binding is a key architectural element in the formation of higher order complexes predicted to be ∼500 kDa in size (Better et al., 1982). Binding sites for one or more accessory DNA bending proteins (IHF, Xis and Fis in the case of λ) always separate the arm- and core-type Int binding sites. The DNA bending proteins function to bring the flanking arm- and core-type sites into close proximity so that Int, bound through its N-terminal domain to a high-affinity arm-type site, is ‘delivered’ to the lower affinity core-type sites, where the C-terminal domain binds, cleaves and re-ligates DNA strands. This architectural view of the heterobivalent recombinases has shaped the formulation of models and experiments for many years (Better et al., 1982; Richet et al., 1988; Kim and Landy 1992). Results presented here strongly indicate that the relationship between the two DNA binding domains is considerably more complex than implied by the simple architectural model. We have found that, independent of any delivery or bridging function, the small N-terminal domain greatly influences the DNA binding and cleavage functions of the large C-terminal domain, in a context-sensitive manner.
The biological functions of many tyrosine family recombinases also include decatenation or segregation of newly replicated chromosomes, conjugative transposition, regulation of plasmid copy number, and differential expression of cell surface proteins (for reviews see Sherratt, 1993; Nash, 1996; Hallet and Sherratt, 1997; Grainge and Jayaram, 1999; Azaro and Landy, 2001). Underlying this wide range of functional diversity, the unifying feature of the family members is their use of a tyrosine nucleophile to carry out site-specific recombination between two DNAs (often called att sites) in the absence of high-energy cofactors. A tyrosine hydroxyl (Tyr342 in the case of λ Int) attacks the scissile phosphate, nicking the DNA and forming a 3′ phosphotyrosine-linked DNA complex on each recombination partner. This covalent protein–DNA intermediate is resolved when the 5′-terminal hydroxyl of the invading DNA strand attacks the phosphotyrosine linkage and displaces the protein, forming a Holliday junction. The reaction is then repeated for the other strand of each DNA duplex. The locus of this chemistry on each partner DNA duplex is a pair of 9–13 bp inverted binding sites for the recombinase (called core-type sites in the case of λ) separated by a 6–8 bp ‘overlap’ region whose boundaries are defined by the staggered and precisely positioned DNA cleavage sites. For some tyrosine recombinase family members, such as the P1-encoded Cre and the yeast-encoded Flp, this 25–30 bp region is all that is required for recombination. However, for most other family members, the ‘att sites’ are considerably more complex.
In the case of the λ and all the other heterobivalent recombinases, the att sites contain additional protein binding sites in DNA sequences that comprise flanking ‘arms’, called P and P′ in the λ att sites (see Figure 1A). In λ, these two arms flanking a core region comprise the viral attP site, which can recombine with the simple bacterial attB site, consisting only of a core region. The product of this recombination is an integrated viral chromosome with prophage att sites (attL and attR) at the left and right junctions of bacterial and viral DNA. The attL and attR prophage sites are of intermediate complexity and can recombine with each other during excisive recombination to regenerate the viral and bacterial chromosomes. Both integrative and excisive recombination are executed by the virally encoded Int, and both require as an accessory protein the host-encoded IHF (integration host factor). Excisive recombination additionally requires as an accessory protein the virally encoded Xis (excision) protein, and it is stimulated by the host-encoded Fis protein (factor for inversion stimulation). All three accessory proteins introduce very sharp bends in their DNA binding sites (Thompson and Landy, 1988; Rice et al., 1996). It is these sharp bends that bring the distal ‘arm-type’ binding sites for Int into close proximity with the ‘core-type’ Int sites where DNA cleavage and ligation are executed (Figure 1B) (Moitoso de Vargas et al., 1989). The five high-affinity arm-type Int sites (P1, P2, P′1, P′2, P′3) have closely related DNA sequences comprising a consensus that is different from the consensus sequence of the four closely related low-affinity core-type Int sites (C, C′, B and B′) (Ross and Landy, 1982) (see Figure 1A).

Fig. 1. Reaction scheme, protein binding sites and basic architectural motif of λ site-specific recombination. (A) Integrative recombination between the phage and bacterial att sites, attP and attB, respectively, requires the phage-encoded integrase (Int) and the bacteria-encoded IHF. The products of this recombination, attL and attR, form the junctions between bacterial DNA (broken line) and the phage DNA (solid line). Excisive recombination between attL and attR to regenerate attP and attB requires, in addition to Int and IHF, the phage-encode Xis (excisionase) protein. Exicision is stimulated by the host-encoded FIS protein and is inhibited by excess IHF. In both directions, the reaction involves an initial top-strand cleavage (vertical downward arrow) followed by strand swapping and ligation of the new junctions. The resulting Holliday junction intermediate (not shown) is resolved to products by a second round of DNA cleavages, strand swapping and ligation on the bottom strands (vertical upward arrow). The top (vertical downward arrow) and the bottom (vertical upward arrow) strand cleavages are staggered by 7 bp and define the overlap region O (rectangle), which is flanked by inverted core-type Int binding sites (horizontal arrows), C, C′, B and B′. The enlarged diagram of attL and attR (right panel) shows the protein binding sites of the P and P′ arms: five arm-type Int binding sites P1, P2, P′1, P′2 and P′3 (ovals); three IHF sites H1, H2 and H′ (squares); two Xis sites, X1 and X2 (triangles); and one Fis F, (diamond). (B) A model for the IHF-induced bending of attL DNA. This segment of DNA binds one protomer of IHF and one protomer of λ Int. IHF binds at the H′ site and bends the DNA by >140°. Int recognizes two segments of attL DNA, namely the arm- and core-type sites (indicated by P′1 and C′) with its N- and C-domain, respectively.
The 356 amino acid λ Int protein, which was first purified by Kikuchi and Nash (1978), can be cleaved by limited proteolysis into two domains. The small N-terminal domain (residues 1–64) (hereafter referred to as the N-domain) is responsible for the high-affinity binding of Int to the arm-type sites (Moitoso de Vargas et al., 1988). The large C-terminal domain (residues 65–356) (hereafter referred to as the C-domain) is fully competent for topisomerase activity (cleaving and ligating DNA) and binding to the low-affinity core-type Int sites. It has been further dissected into two smaller domains, encompassing residues 65–169 and 170–356, respectively (Tirumalai et al., 1997, 1998). The former (central domain) is involved in binding to core-type sites. The latter comprises the minimal catalytic domain, which contains all of the conserved residues that define the tyrosine recombinase family, and is approximately the same size as the smallest family members (Nunes-Düby et al., 1998). The crystal structures of the catalytic domains of λ Int and the closely related HP1 Int highlighted the defining features of the tyrosine recombinase family (Hickman et al., 1997; Kwon et al., 1997). The crystal structures of XerD (Subramanya et al., 1997), the human type IB topoisomerases (Cheng et al., 1998; Redinbo et al., 1998), and the co-crystal structures of Cre and Flp complexed with their cognate DNA targets (Guo et al., 1997; Chen et al., 2000) suggest what the interaction between the C-domain and core-type DNA might look like. This extrapolation is especially informative because the C-domain of λ Int corresponds to the intact XerC/D, Cre and Flp recombinases.
Despite our detailed views of the structures and reactions in the region of strand exchange, relatively little is known about the extent and/or nature of interactions between the two DNA binding domains of the heterobivalent recombinases. The experiments reported here show that the N-domain is not merely a guide for the proper positioning and stabilization of Int protomers within the higher order recombinogenic complex; it is also a context-sensitive modulator of Int functions in the region of strand exchange. When the N-domain is in its normal position in intact Int, i.e. when it is ‘in cis’ with respect to the C-domain, it inhibits core-DNA binding and cleavage. However, when the N-domain is not physically linked to the C-domain via the protease-sensitive linker region, i.e. when it is present ‘in trans’, it stimulates core-DNA binding and cleavage. Interestingly, the N-domain inhibition of core binding and cleavage that is observed in cis is reversed or overcome in the presence of arm-type oligonucleotides. This oligonucleotide stimulation specifically requires an arm-type DNA sequence and the stimulating oligonucleotide participates in forming a complex with Int and core-type DNA.
Results
In cis, the N-terminal domain inhibits core-DNA binding and cleavage
The C-domain (residues 65–356) and the N-domain (residues 1–64) have both been cloned and purified (Tirumalai et al., 1997; D.Sarkar and A.Landy, in preparation). As discussed above, it has long been thought that the only function of the latter was to bind high-affinity arm-type sites (e.g. P′1,2 in Figure 1A), so that the core binding/catalytic domain could be ‘delivered’ to the sites of DNA strand exchange (core-type DNA sites) by the accessory DNA bending proteins (e.g. IHF in Figure 1B). It was, therefore, unexpected to find that, at functionally limiting protein concentrations, the C-domain protein is superior to full-length Int in topoisomerase function and DNA cleavage (Figure 2). To assay topoisomerase activity, supercoiled plasmid DNA was incubated with different amounts of either Int or C-domain and then subjected to gel electrophoresis to resolve the topoisomers. The fastest migrating band represents the supercoiled substrate DNA and the slowest migrating band represents the fully relaxed circles. Partially relaxed topoisomers of intermediate supercoiling density are distributed between these two extremes. Comparison of the left and right panels indicates that the C-domain is more efficient than Int at relaxing supercoiled DNA. Visual inspection of this gel indicates that the C-domain is 3–5 times more efficient than full-length Int in this assay.
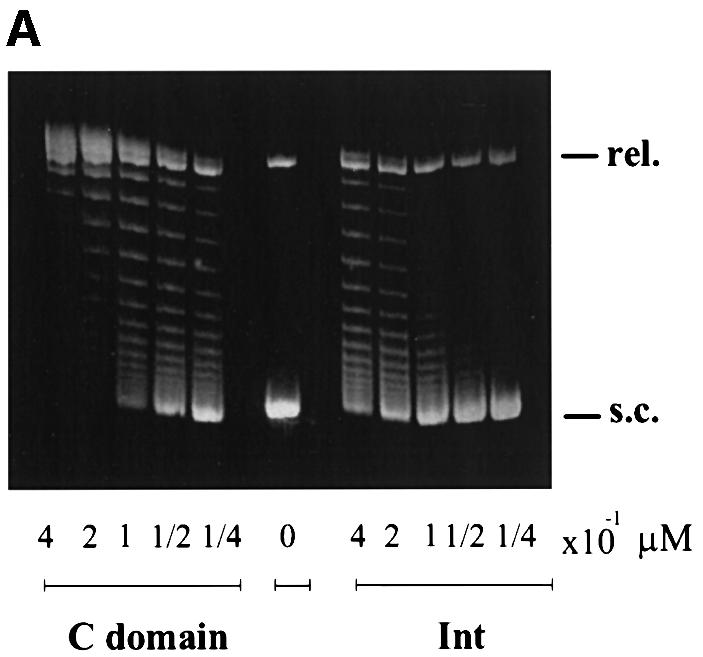

Fig. 2. Comparison of the cleavage activities by Int and its C-domain. (A) Topoisomerase activities of λ Int and the C-domain. Supercoiled pBR322 plasmid DNA (0.36 µg/assay) was incubated with the indicated amounts of protein at 25°C for 1 h (see Materials and methods). The reactions were quenched in 0.2% SDS and analyzed by agarose gel electrophoresis. The positions of supercoiled (s.c.) and relaxed (rel.) plasmid DNA comprise the two ends of the ladder of topoisomers. (B) DNA cleavage as a function of protein concentration. 32P-labeled top-strand nicked full att-site suicide substrate (0.02 µM) was incubated with the indicated amounts of Int or C-domain for 30 min at 25°C and reactions were quenched with 0.2% SDS. (C) DNA cleavage as a function of time. Int or C-domain (0.1 µM) was incubated for the indicated times and reactions were quenched in 0.2% SDS. In both (B) and (C), the reaction conditions were as described in Materials and methods and the amount of covalent complex formed was analyzed by SDS–PAGE and quantitated using a phosphorimager (Fuji).
A more direct and more precise assay of relative DNA cleavage efficiency is afforded by the use of suicide cleavage substrates (Nunes-Düby et al., 1987). The COC′ suicide substrate we used had two core-type binding sites and a nick on the top strand. When this radiolabeled substrate is cleaved by the C-domain or Int, a three-base oligomer from the top strand is lost by diffusion, thus trapping the covalent protein–DNA complex. These covalent complexes can easily be distinguished from free substrate DNA by SDS–PAGE. Figure 2B and C shows a comparison of the cleavage competence of full-length Int and C-domain as a function of either protein concentration or time. In both cases, the C-domain was found to cleave better than the full-length Int.
In order to define the basis for the difference between full-length Int and the C-domain, we compared their relative abilities to form electrophoretically stable complexes with core-type DNA. For this purpose we used mutant Ints in which the active site nucleophile Tyr342 was changed to Phe, thus eliminating the possibility of forming covalent protein–DNA intermediates (called IntF and C-domainF, respectively) (Pargellis et al., 1988). In a gel-shift assay, C-domainF is seen to form two differentially retarded bands with radiolabeled COC′ DNA (Figure 3, left panel). At lower protein concentrations, the single retarded band has the mobility expected for one C-domainF protomer bound to a 35 bp DNA fragment. At higher protein concentrations, a second slower moving band has the mobility expected for two C-domainF monomers bound to a 35 bp DNA. In contrast, the same concentrations of full-length Int fail to form any discrete complexes that are stable to gel electrophoresis (Figure 3, left panel). Under these conditions, full-length Int is at least 10 times less effective than the C-domain in forming stable complexes with core-type DNA [based on the limits of detection in this assay, and based on other gels (not shown) where a very faint Int–COC′ complex can be detected]. The same concentrations of full-length Int that fail to form complexes with COC′ are, however, very effective in forming complexes with P′1,2 arm-type DNA (Figure 3, right panel). As expected, the C-domain does not form complexes with P′1,2 DNA. The reason that the inhibition by the N-domain in cis is more pronounced for core-type DNA binding than for DNA cleavage has to do with the difference in the nature of the two assays. Whereas the binding experiments measure an equilibrium, the cleavage experiments with the suicide substrate record the irreversible accumulation (as covalent complexes) of even very transient interactions.

Fig. 3. Gel-shift assays of Int and the C-domain. The indicated amounts of protein were mixed with either 0.1 µM 32P-labeled synthetic 35 bp COC′ core-type DNA (left panel) or 36 bp P′1,2 arm-type DNA (right panel), as described in Materials and methods. ‘Zero’ lanes indicate the substrate DNA alone. The reaction was analyzed by electrophoresis in 8% polyacrylamide gel.
The topoisomerase assay is influenced by DNA binding affinity (non-specific binding and/or binding to core-like sequences), cleavage efficiency and ligation efficiency. Whereas a reduction in either of the first two would result in lower topoisomerase activity, a reduction in ligation efficiency would be scored as an increase in topoisomerase activity, since more supercoils would be relaxed per cleavage event. Therefore, the topoisomerase assays, like the suicide substrate assays, indicate that full-length Int is depressed relative to the C-domain for DNA binding and/or DNA cleavage. As might be expected, the superiority of the C-domain relative to full-length Int is not observed when the proteins are in large excess over DNA (Tirumalai et al., 1997). The very weak binding of full-length Int to core-type DNA is more than sufficient to account for its observed deficiency in DNA cleavage. However, from these particular assays, we cannot rule out the possibility that DNA cleavage by full-length Int is not also depressed relative to the C-domain. Taken together, these results suggest that the presence of residues 1–64 suppresses or inhibits the primary functions of Int, namely the binding and cleavage at core-type DNA sites. When residues 1–64 are removed, i.e. as in the C-domain protein, there is a great stimulation of these activities on core DNA sites.
In trans, the N-terminal domain stimulates core-DNA binding and cleavage
To characterize further the observed N-domain inhibition of core-DNA binding and cleavage, we investigated the effect of adding the cloned and purified N-domain protein to supplement the C-domain in trans. We were surprised to find that in a cleavage assay with the same nicked COC′ suicide substrate used in Figure 2, addition of the N-domain stimulated the activity of the C-domain. Figure 4A shows the amount of covalent complex (DNA cleavage) formed as a function of C-domain concentration in the absence of the N-domain protein or in the presence of several different concentrations of the N-domain protein. A similar stimulation by the N-domain protein is also seen in a time course carried out at low concentrations of the C-domain (Figure 4B). In both cases, the stimulation of cleavage is 3- to 5-fold. A similar level of stimulation by the N-domain is observed when the C-domain binding to COC′ DNA is assayed (Figure 4C). The fact that addition of the N-domain to full-length Int has no effect (data not shown) is a strong argument that the observed stimulation in trans is due to a specific interdomainal interface. The 3- to 5-fold stimulation of the C-domain by the N-domain in trans is especially striking when compared with the 10-fold inhibition conferred by the N-domain in cis (intact Int).

Fig. 4. Effect of N-domain in trans on cleavage activity and binding of the C-domain. (A) DNA cleavage as a function of C-domain concentration in the presence of different amounts of N-domain. Labeled suicide substrate (0.02 µM) (see legends to Figure 2B) was incubated at 20°C for 30 min with different concentrations of C-domain in either the absence (open circles) or presence of 0.5 µM (closed circles), 0.75 µM (triangles) or 1.0 µM (squares) N-domain (see Materials and methods). The reactions were quenched in 0.2% SDS. (B) DNA cleavage by a fixed concentration of C-domain as a function of time, in the presence of different amounts of N-domain. As described in (A) and Materials and methods, 0.1 µM C-domain in either the absence (open circles) or presence of 0.5 µM (closed circles), 0.75 µM (triangles) or 1.0 µM (squares) N-domain. The reactions were quenched in 0.2% SDS and analyzed by electrophoresis through 12% (w/v) SDS–PAGE (see Materials and methods). (C) Effect of N-domain in trans on binding of C-domain to core-type DNA. The indicated amounts of C-domain were incubated with 0.1 µM 32P-labeled 35 bp core-type DNA at room temperature for 20 min in the absence or presence of different concentrations of N-domain, as described in Materials and methods. The reaction was analyzed by electrophoresis in 8% polyacrylamide gel and the amount of protein–DNA complex formed was quantitated by scanning the gels on a phosphorimager (Fuji). Retarded complexes with electrophoretic mobilities corresponding to one or two bound C-domains (open or closed symbols, respectively) are plotted as a function of C-domain concentration. Reactions were either in the absence of N-domain (circles) or in the presence of 0.5 µM (triangles) or 1.0 µM (squares) N-domain.
Arm-type DNA reverses or overcomes inhibition by the N-terminal domain
The apparent dichotomy of inhibition and stimulation by the N-terminal domain prompted us to investigate whether Int binding to core-type DNA is influenced by the presence of arm-type DNA, independent of the well-established Int bridges that are dependent upon accessory DNA bending proteins (as diagrammed in Figure 1B). In other words, does arm-type DNA influence Int binding to core-type DNA even when they are not physically linked to each other (i.e. when they are in trans). To answer this question we compared Int binding to labeled COC′ DNA in the presence and absence of an unlabeled oligonucleotide containing the P′1,2 arm-type DNA sequences. Whereas Int binding to COC′ is barely detectable in the gel-shift assay, addition of a 26 bp oligonucleotide containing P′1,2 sequences greatly stimulates binding and leads to the formation of a well defined Int-dependent band (Figure 5A). A retarded band of the same mobility is observed in a reaction where COC′ DNA is unlabeled and the P′1,2 DNA is labeled. This band is formed at the expense of Int binding to P′1,2 alone, seen in lane 2 of Figure 5A, as a specific band and a general background smear. Although it is not readily visible in this particular gel photograph, Int binding to P′1,2 alone generates two retarded bands. The faster migrating band corresponds to a single bound Int and the slower migrating band corresponds to two Ints bound at P′1,2. In experiments where a 36 bp rather than a 26 bp P′1,2 oligonucleotide was used, the doubly bound oligonucleotide is clearly visible (e.g. Figure 3).

Fig. 5. The effect of arm-type oligonucleotide on Int binding to core-type DNA. (A) Int binding to core-type DNA in the presence and absence of arm-type oligonucleotide. Left panel: 32P-labeled 26 bp arm-type DNA (P′1,2*) at 0.28 µM was incubated in the absence or presence of Int (0.55 µM) and in the absence or presence of 0.1 µM unlabeled core-type DNA (COC′). Right panel: 32P-labeled core-type DNA (COC*) at 0.1 µM was incubated in the absence or presence of Int (0.55 µM) and in the absence or presence of 0.28 µM unlabeled arm-type DNA (P′1,2) for 25 min at room temperature. The reactions were analyzed by electrophoresis on native 7% polyacrylamide gels and visualized by autoradiography (see Materials and methods). One complex dependent on all three components appears to be labeled by either [32P]COC′ or [32P]P′1,2. (B) The mobility of a COC′-labeled ternary complex of Int depends on the size of the P′1,2 oligonucleo tides. Using the same conditions described in (A), Int complexes were formed with 32P-labeled COC′ in the presence of either a 26 or 36 bp P′1,2 oligonucleotide. The reactions were analyzed on a 7% polyacrylamide gel and visualized by autoradiography.
Int complexes contain, and are specific for, both arm- and core-type DNAs
The most appealing interpretation of the results shown in Figure 5 is that the Int-dependent complexes formed in the presence of COC′ and P′1,2 do in fact contain both kinds of DNA. An alternative possibility is the existence of two different Int complexes containing either P′1,2 or COC′ DNA and having very similar electrophoretic mobilities. It is, therefore, important to rule out the possibility that P′1,2 stimulates but does not participate in the formation of the observed COC′ complexes. To address this question we formed Int complexes with labeled COC′ DNA in the presence of unlabeled P′1,2 DNA either 26 or 36 bp in length. As seen in Figure 5B, the electrophoretic mobility of the Int-dependent labeled COC′ band is different for reactions containing unlabeled 26 bp versus 36 bp P′1,2 DNA. We conclude, therefore, that the Int complexes do indeed contain both COC′ and P′1,2 DNAs.
Having established that the complexes contain two types of DNA, we then asked how specific Int was in binding both types. Figure 6 shows that whereas labeled COC′ is efficiently competed out of the complexes by unlabeled COC′ DNA, it is quite resistant to competition by a non-specific competitor DNA (as detailed in Materials and methods). Labeled COC′ is 18 times more resistant to competition by non-specific competitor than by homologous competitor (Figure 6A). Similarly, labeled P′1,2 is ∼60 times more resistant to competition by non-specific competitor than by homologous competitor (Figure 6B).

Fig. 6. Arm–Int–core complex formation requires specific arm- and core-type DNA sequences. (A) 32P-labeled COC′ (0.1 µM) was incubated with 0.55 µM Int and 0.28 µM unlabeled P′1,2 in the presence of increasing amounts of unlabeled COC′ (specific competitor) or unlabeled non-specific competitor DNA for COC′ (see Materials and methods) and the percentage of labeled ternary complex was determined by gel electrophoresis as described in the legends to Figure 5A and quantitated with a phosphorimager (Fuji). The amount of ternary complex formed in the absence of any competitor was 94% and was normalized to 100% in the figure. (B) 32P-labeled P′1,2 (0.28 µM) was incubated with 0.55 µM Int and 0.1 µM unlabeled COC′ in the presence of increasing amounts of unlabeled P′1,2 (specific competitor) or unlabeled non-specific competitor DNA for P′1,2 (see Materials and methods). The percentage of labeled ternary complex was determined as in (A). The amount of ternary complex formed in the absence of any competitor was 70% and was normalized to 100% in the figure.
Arm-type DNA stimulates cleavage in the presence of heterologous DNA
The effect of P′1,2 oligonucleotide on Int binding to core-type DNA immediately raises the question of its effect on DNA cleavage. To study this we used the COC′ suicide substrate with the top-strand nick between bases three and four of the overlap region to trap covalent Int–DNA complexes, as described for Figure 2 above. In contrast to previous experiments described in this work, the assay in Figure 7 was conducted in the presence of 0.05 mg/ml herring sperm DNA (45-fold in excess over COC′ suicide substrate). Heterologous carrier DNA was used for two reasons: first to mimic more closely the in vivo conditions of core-type DNA cleavage by Int and secondly to reduce non-specific binding of the Int C-domain to the arm-type DNA, which would then reduce cleavage independent of P′1,2–Int–COC′ complex formation. Under these conditions, there is very little cleavage of COC′ by Int in the absence of P′1,2. Addition of P′1,2 greatly stimulates Int cleavage of COC′ and the extent of cleavage stimulation is dependent on the concentration of P′1,2 (Figure 7A). Moreover, the cleavage stimulation is proportional to the amount of P′1,2–Int–COC′ complexes generated in a parallel experiment (Figure 7B). This strongly suggests that, in these assay conditions, Int cleavage is dependent on P′1,2–Int–COC′ complex formation.

Fig. 7. Arm-type DNA stimulates Int cleavage and binding at COC′. (A) Time course of Int cleavage of COC′ as assayed by the formation of covalent complexes on a top-strand nicked suicide substrate. 32P-labeled COC′ covalent complexes with Int were resolved on a 7% SDS–polyacrylamide gel and quantitated on a phosphorimager (see Materials and methods). (B) Time course of ternary complexes formed with COC′, Int and P′1,2. Ternary complexes of Int, P′1,2 and 32P-labeled COC′ were resolved on 7% native polyacrylamide gels and quantitated on a phosphorimager as described in Materials and methods. In both experiments, reaction mixes contained 0.25 µM Int, 0.05 µM COC′, 0.05 mg/ml herring sperm DNA. Arm-type DNA (P′1,2) was added to a final concentration of 0.025 µM (diamonds) or 0.06 µM (triangles) or omitted (circles). Reactions were incubated at 19°C and 10 µl aliquots were taken at the indicated time points.
Discussion
The results presented here point to an additional layer of regulation on top of the already complex control of λ site-specific recombination. This web of regulation starts with expression of the Int gene, which is under both negative and positive transcriptional control and post-transcriptional ‘retro-regulation’ (Guarneros et al., 1982; Echols and Guarneros, 1983; Thompson and Landy, 1989). At the level of the reaction itself, excisive recombination is inhibited by high levels of IHF and stimulated (when Xis is limiting) by Fis, both of which are host proteins whose levels are governed by the physiology of the host cell (Thompson et al., 1987a,b; Ball and Johnson, 1991a,b; Ditto et al., 1994; Nystrom, 1995; Cassler et al., 1995). This regulation by the DNA bending accessory proteins, including the phage-encoded Xis, has, until now, been understood primarily as a structural phenomenon based upon accessory protein-dependent Int bridges between the high-affinity arm-type sites and the low-affinity core-type sites. Our present data suggest that, in addition to this structural role, a major function of the arm-type DNA sites is to reverse or overcome the N-domain inhibition of the DNA binding and cleavage activities that reside in the C-domain.
The initial suggestion that Int is a heterobivalent DNA binding protein was based upon nuclease protection experiments that not only identified two distinct and well separated classes of DNA binding sequences, but also showed that one is resistant (arm-type sites), and the other is sensitive (core-type sites), to challenge by the polyanion heparin (Ross et al., 1979; Ross and Landy, 1982; Moitoso de Vargas et al., 1988). Within an att site, Int binding to core-type sites was greatly enhanced by the presence of both arm-type sites and accessory DNA bending proteins (Richet et al., 1988; Moitoso de Vargas et al., 1989) and a model for DNA bending protein-dependent arm–Int–core bridges was proposed for the attL/attR synaptic complex of λ (Kim et al., 1990; Kim and Landy, 1992). Studies on a number of other recombination pathways have established the generality of this theme and highlighted the variations with which it is played out. Differences in the number, spacing and pattern of arm-type sites in relation to the core and accessory protein binding sites suggest a diversity of higher order recombinogenic complexes (Azaro and Landy, 2001). As seen in the recently proposed model for the bridging patterns in the recombinogenic complex of mycobacteriophage L5, the details and connectivities differ from λ, but the design elements are the same (Pena et al., 2000).
Despite the large amount of information about the C-domain, the C-domain analogs in the monovalent recombinases (Cre, Flp and XerC/D) and the nature of the higher order recombinogenic complexes, much less is known about the N-domain. The Clubb laboratory has generated NMR solution structures of the N-domain of the conjugative transposon Tn916 recombinase, alone and complexed with its cognate DNA. These have revealed an interesting double-stranded RNA binding motif, but, thus far, it has not been possible to extrapolate this structure to the N-domain of λ Int or other family members (Connolly et al., 1998; Wojciak et al., 1999). Studies from the Segall laboratory suggest that the N-domain is also involved in, or important for, intermolecular interactions between Int protomers (Jessop et al., 2000). Extensive genetic analyses of λ Int in the Gardner and Gumport laboratories have identified one group of mutants in the C-domain that affect binding to arm-type DNA and another group in the N-domain that appear to affect core-type DNA recognition (Han et al., 1994; Cheng et al., 2000). These two reciprocal classes of mutants may well point to residues involved in some of the interdomainal interactions reported here. Another particularly relevant result is the important observation by Richet et al. (1988) that attB comes naked into a preformed attP recombinogenic complex. Indeed, the failure of Int to bind attB DNA (analogous to COC′), unless the Int is part of a higher order complex, could be considered as foreshadowing the results presented here.
Inspection of the tyrosine recombinase family indicates that the large C-terminal domains of the heterobivalent recombinases, e.g. the C-domain of λ Int, correspond to the full length of their monovalent cousins such as Cre, Flp and XerC/D. Accordingly, the N-domain can be thought of as an ‘accessory domain’ added on to the basic family structure for the purposes of directionality and regulation. [The N-domain of λ Int, residues 1–64, should not be confused with the N-terminal domains of Cre, Flp and XerC/D. The latter correspond to residues 65–170 of λ Int (Tirumalai et al., 1997, 1998)]. The results reported here reveal the N-domain of λ Int as a context-sensitive modulator of DNA binding and cleavage by the catalytic C-domain. The finding that the isolated C-domain is superior to full-length Int in topoisomerase function, sequence-specific DNA cleavage and binding to core-type sites highlights the N-domain as an encumbrance, or brake, on the intrisinc functions of Int recombinase (Figures 2 and 3). How might this brake be functioning? We consider three possible mechanisms: (i) simple occlusion of surfaces required for protein–protein interactions, e.g. for dimer formation; (ii) occlusion of DNA binding surfaces; and (iii) more subtle allosteric effects on the functions of the C-domain.
The first of these potential mechanisms is our least favorite for two reasons. First, there is no evidence that Int must dimerize to bind and/or cleave at core-type sites. Secondly, when a half-att site containing only a single Int binding site is used instead of COC′, the C-domain is still superior to full-length Int in DNA cleavage, and a similar stimulation of C-domain cleavage by the N-domain is also observed (data not shown). We also do not favor as a potential mechanism the simple occlusion of the binding site for core-type DNA because such a mechanism would not easily relate to the observed stimulation of the C-domain by the N-domain in trans. The fact that the N-domain does not stimulate full-length Int in trans (data not shown) might suggest that the interdomainal interface(s) responsible for inhibition and stimulation at least partially overlap. Alternatively, this might simply reflect a dominance of inhibition over stimulation. We can not rule out the possibility that the N-domain inhibition in cis and stimulation in trans are two independent and un-related phenomena. However, we consider this an unnecessary complexity at present and therefore favor a more integrated view of the data. The third potential mechanism, involving allosteric effects of the N-domain on the functions of the C-domain, does facilitate an integrated view of our results.
The experiments in Figures 2, 3 and 4 show that the DNA binding and cleavage functions of the C-domain are either inhibited or stimulated by the N-domain, depending on whether the two are physically linked (i.e. in cis) or are unlinked (i.e. in trans). A comparison of Figures 2, 3 and 4 and data not shown indicate that the combination of separate N- and C-terminal domains (i.e. in trans) is 30- to 50-fold more active than intact native Int. The observations with individual cloned domains provided the first evidence for context-sensitive interactions between the two DNA binding domains and directed our attention to the possible role of P′1,2 DNA binding. As shown in Figures 5–7, the addition of an oligonucleotide containing the P′1,2 binding sequences to a reaction greatly stimulates the ability of Int to bind and cleave core-type (COC′) DNA. Under the conditions used here, the binding of Int to COC′ is almost undetectable in the absence of P′1,2. Based upon those experiments where faint Int–COC′ complexes have been detected (not seen in the gels shown here), we estimate that addition of the P′1,2 oligonucleotide stimulates Int binding to COC′ >10-fold. The effect of the P′1,2 oligonucleotide is not simply due to some priming or activation of a ‘dormant’ Int, but rather is the consequence of its participation in a ternary Int complex containing both kinds of DNA. This conclusion is based upon those experiments showing that the gel mobility of Int complexes with labeled COC′ DNA can be shifted by changing the length of the unlabeled P′1,2 oligonucleotide (Figure 5B). A similar result is obtained when the label is on the P′1,2 and the size of COC′ is varied (data not shown). The oligonucleotide stimulation of Int binding to core-type DNA is not the result of some ‘carrier DNA’ effects, as demonstrated by the resistance of the P′1,2–Int–COC′ complex to competition by a non-specific oligonucleotide (Figure 6) and the absence of stimulation with a control oligonucleotide lacking the P′1,2 sequence (data not shown).
The results reported here are summarized schematically in Figure 8. An extreme interpretation of these results is that the function of the N-domain is not to deliver Int protomers to the core-type sites or to position them properly within the recombinogenic and synaptic complexes. Rather, its function is to maintain recombinase in an inactive state until the N-domain has bound a cognate arm-type site. Accordingly, the requirement for accessory DNA bending proteins is not for the formation of specific recombinogenic and synaptic structures containing Int bridges between specific pairs of arm- and core-type sites. Rather, the requirement for DNA bending proteins stems from the fact that the arm- and core-type sites are too close together with respect to the persistence length of DNA. According to this view, the accessory proteins could be dispensed with if all of the arm-type DNA sites could be provided in trans. Obviously, this could not happen in nature and presumably the arm-type sites have evolved to their present locations with respect to the core-type sites precisely for the purpose of generating a dependence on accessory proteins. What had previously been viewed as a requirement for bending protein-dependent bridging of Int from arm- to core-type sites, e.g. Moitoso de Vargas et al. (1989), can now be viewed as a requirement to reverse or overcome the N-domain inhibition of recombinase functions. The formation of complexes, and low levels of recombination, between two attLs in the absence of IHF (called the straight L pathway) is potentially another example of C-domain activation by arm-type DNA sites in trans (Segall and Nash, 1996; Cassell et al., 1999).

Fig. 8. Schematic summary of the context-sensitive modulation of C-domain recombinase functions at COC′ by the N-domain and arm-type oligonucleotides.
We consider two possible mechanisms for the observed P′1,2 stimulation of C-domain functions. According to the first, P′1,2 converts or shifts the N-domain from an inhibiting to a stimulating mode or position. This is appealing because it fits well with the two observed modes of N-domain interaction with the C-domain, i.e. inhibition in cis and stimulation in trans. According to this view, the observed stimulation by the N-domain in trans is a partial mimic of what P′1,2 does for full-length Int. According to an alternative view, the role of P′1,2 is to set up Int dimers so they are favorably disposed for simultaneous binding to the two sites on COC′. In other words, P′1,2 binding overcomes, rather than reverses, the inefficiency of full-length Int (imposed by the N-domain in cis). This view requires further elaboration to accommodate the fact that the P′1,2 sites are direct repeats and the COC′ sites are inverted repeats. It also leaves unexplained the observed stimulation by the N-domain in trans. However, it should be noted that these two mechanisms are not mutually exclusive and dimer stabilization is likely to be relevant even if some version of the first model has merit. Both mechanisms are also compatible with the notion that λ Int (and the other heterobivalent recombinases) evolved from a very efficient Cre- or XerC/D-like recombinase. This efficient predecessor, corresponding to the C-domain of λ Int, would have been rendered inefficient by acquisition of the N-domain. This would have led to, or been accomplished by, a dependence on P′1,2 and the accessory DNA bending proteins.
The extreme model described above rationalizes the great diversity in the number, spacing and phasing of the DNA bending and arm-type Int sites observed in different tyrosine family site-specific recombination pathways. This apparent diversity in structures would be consistent with the notion that specific higher order structures are not essential as long as the Int protomers can simultaneously bind arm- and core-type DNA sites. We do not know the relative orientation between core- and arm-type sites, or the range of orientations that might be acceptable, but we suspect that these might be different for different integrases. It is known that imposing a 34° rotation on the phase with which an arm-type site is delivered to Int at a core-type site abolishes recombinase function and that some, but not all, 10 bp phase-preserving changes in spacing between arm- and core-type sites are well tolerated (Thompson et al., 1988; Nunes-Düby et al., 1995).
Although an extreme interpretation of the present data is useful as a heuristic device, it is an oversimplification that does not take into account all of the known features of λ recombination, such as the differential utilization of arm sites and bending-protein sites in the integrative and excisive pathways (Bushman et al., 1985; Thompson et al., 1986, 1987b; Numrych et al., 1990). We, therefore, favor a view that is a hybrid of the extreme model described above and the commonly accepted structural models. This hybrid view retains the role of intermolecular Int bridges, as proposed in a model for λ attL/attR synapsis (Kim and Landy, 1992), and it focuses attention on the importance of N-terminal domain context-sensitive modulation of recombinase functions. We also suggest that, based upon what is known about the tyrosine recombinase family, it would not be at all surprising if this critical new role for the N-terminal domain of λ Int is also a commonly shared basic feature of the other heterobivalent recombinases.
Materials and methods
Preparation of Int
Int and C-domain proteins were produced from expression plasmids under the control of a T7 promoter in E.coli BL21 (DE3, pLysS) as described previously (Tirumalai et al., 1997, 1998). To overexpress the proteins, E.coli cells carrying the appropriate expression plasmids were grown at 37°C to an A600 of 0.4–0.6, at which point protein expression was induced by the addition of isopropyl-β-d-thiogalactopyranoside to a final concentration of 1 mM and the cells were grown for a further 3 h. The cells were pelleted by centrifugation at 500 g for 10 min, washed with 50 mM Tris–HCl pH 8.0, 10% (w/v) sucrose, 100 mM sodium chloride, 10% (v/v) glycerol, 1 mM dithiothreitol (DTT), 1 mM EDTA (lysis buffer). The cell suspension was frozen as droplets directly in liquid nitrogen and the resulting ‘popcorn’ was stored at –80°C. Proteins were purified to near homogeneity by minor modifications of the protocol described by Kikuchi and Nash (1978) as detailed earlier (Tirumalai et al., 1998). The purification of the N-domain will be described elsewhere (D.Sarkar and A.Landy, in preparation). Protein concentration was estimated by the dye-binding method using bicinchoninic acid (Pierce) (Smith et al., 1985; Wiechelman et al., 1988). The purity of all the proteins (N-domain, C-domain and full-length Int) was >90% as judged by Coomassie Blue and silver staining of overloaded SDS–polyacrylamide gels.
Topoisomerase assays
Topoisomerase activities of λ Int and its C-terminal domain were assayed using supercoiled pBR322 plasmid. Supercoiled DNA (0.36 µg/assay) was incubated with the indicated amounts of Int or its C-domain in a 20 µl reaction mixture containing 10 mM Tris pH 7.5, 1 mM EDTA, 50 mM NaCl, 0.5 mg/ml bovine serum albumin (BSA), 2.5 mM DTT at 25°C for 1 h. The reactions were quenched in 0.2% SDS and electrophoresed in 1.2% agarose gel. The gel was stained with ethidium bromide, destained and then visualized on a UV-transilluminator.
Oligonucleotides
Synthetic oligos (HPLC purified) were obtained from Operon Technologies, Alameda, CA, and labeled at their 5′ OH end with [γ-32P]ATP (NEN) using T4 polynucleotide kinase (Sambrook et al., 1989). The top-strand sequences (Int binding sites are underlined) were as follows: (i) 26mer P′1,2 arm-site DNA: 5′AACAGGTCACTATCAGTCAAAATACC3′; (ii) 36mer P′1,2 arm-site DNA: 5′TAACGAACAGGTCACTATCAGTCAAATACCGATAC3′; (iii) 30mer non-specific competitor DNA for P′1,2: 5′CTAACGCCCCAGTCAGCGACAGTTCACCTA3′; (iv) 35mer COC′ core-type site: 5′GTATTGCCAGCTTTATTCAACAAAGTTGGAGCAGT3′; (v) 35mer non-specific competitor DNA for COC′: 5′GTATTGCACTAGGTATTCAACACCTGGTGAGCAGT3′; (vi) 31mer nicked attL suicide substrate: 5′TCGAGCAGCTTTTTT( )ATATTAAGTTGGAATT3′ (the empty parentheses indicate a nick). The bottom strand was unnicked and complementary to the top strand. The oligos were annealed in 10 mM Tris–HCl pH 7.5 containing 50 mM NaCl. Oligonucleotides (iii) and (v) were used as the non-specific competitors for COC′ and P′1,2 DNAs, respectively. Note that the partial palindromic structure of the core-site DNA is preserved in non-specific competitor DNA for COC′.
Gel mobility shift assays
Gel-shift assays were carried out in a volume of 10 or 20 µl. The reaction mixture consisted of 10 mM Tris–HCl pH 7.5, 75 mM NaCl, 0.5 mg/ml BSA, 5% glycerol, 2.5 mM DTT and the indicated amounts of protein and DNA. The protein was mixed with 32P-labeled arm- or core-site DNA and incubated at room temperature for 20 min unless noted otherwise. For arm–Int–core complex formation, Int was added to a binding mix (50 mM Tris–HCl pH 7.8, 80 mM NaCl, 5 mM DTT, 0.5 mg/ml BSA) already containing either cold P′1,2 and hot COC′, or hot P′1,2 and cold COC′ DNA. Binding experiments were also carried out in the presence or absence of unlabeled competitor DNA, and samples were loaded onto a native, non-denaturing 6–8% (w/v) polyacrylamide gel [acrylamide:bis acrylamide ratio 40:1.1 (w/w)]. The gels were run at 100–200 V for 3–4 h. They were then dried, autoradiographed, and bands corresponding to protein–DNA complex and substrate DNA were quantitated using a Fuji phosphorimager.
Cleavage assays by Int or C-domain
Cleavage assays were carried out using a top strand nicked attL suicide substrate for Int and the C-domain. The 5′ OH end of the top strand was labeled with [γ-32P]ATP (NEN). The cleavage reactions were carried out in reaction mixtures consisting of 10 mM Tris pH 8.0, 55–75 mM NaCl, 1 mM EDTA, 0.5 mg/ml BSA, 2.5 mM DTT, and the indicated amounts of suicide substrate and protein. The reaction mix was pre-incubated at 25°C (unless noted otherwise) and at the times indicated aliquots were quenched in 0.2% SDS and analyzed by electrophoresis through 12% (w/v) polyacrylamide [acrylamide:bisacrylamide ratio 40:1.1 (w/w)], containing 0.1% (w/v) SDS (Laemmli, 1970). Experiments to study the effect of the N-domain in trans on suicide substrate cleavage activity by the C-domain were always carried out at 20°C to obtain a comparatively lower cleavage activity by the C-domain itself. The gels were dried, autoradiographed, and bands corresponding to protein–DNA complex and substrate DNA were quantitated using a Fuji phosphorimager.
Acknowledgments
Acknowledgements
We thank Gregg Gariepy and Tina Oliveira for technical assistance, Joan Boyles for manuscript preparation, and our colleagues in the laboratory for input at many levels, especially Marco Azaro and Simone Nunes-Düby for comments on the manuscript. We also thank Jeffrey Gardner, Dick Gumport, Anca Segall and Alex Burgin for communication of results prior to publication. This work was supported by NIH grants GM62723 and GM33928.
References
- Azaro M.A. and Landy,A. (2001) Integrase and the λ Int family. In Craig,N.L., Craigie,R., Gellert,M. and Lambowitz,A. et al. (eds), Mobile DNA II. ASM Press, Washington, DC.
- Ball C.A. and Johnson,R.C. (1991a) Efficient excision of phage λ from the Escherichia coli chromosome requires the Fis protein. J. Bacteriol., 173, 4027–4031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ball C.A. and Johnson,R.C. (1991b) Multiple effects of Fis on integration and the control of lysogeny in phage λ. J. Bacteriol., 173, 4032–4038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Better M., Lu,C., Williams,R.C. and Echols,H. (1982) Site-specific DNA condensation and pairing mediated by the Int protein of bacteriophage λ. Proc. Natl Acad. Sci. USA, 79, 5837–5841. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bushman W., Thompson,J.F., Vargas,L. and Landy,A. (1985) Control of directionality in λ site-specific recombination. Science, 230, 906–911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Campbell A.M. (1962) Episomes. In Caspari,E.W. (ed.), Advances in Genetics. Academic Press, New York, NY, pp. 101–145.
- Cassell G., Moision,R., Rabani,E. and Segall,A. (1999) The geometry of a synaptic intermediate in a pathway of bacteriophage λ site-specific recombination. Nucleic Acids Res., 27, 1145–1151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cassler M.R., Grimwade,J.E. and Leonard,A.C. (1995) Cell cycle-specific changes in nucleoprotein complexes at a chromosomal replication origin. EMBO J., 14, 5833–5841. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen Y., Narendra,U., Iype,L.E., Cox,M.M. and Rice,P.A. (2000) Crystal structure of a Flp recombinase–Holliday junction complex: assembly of an active oligomer by helix swapping. Mol. Cell, 6, 885–897. [PubMed] [Google Scholar]
- Cheng C., Kussie,P., Pavletich,N. and Shuman,S. (1998) Conservation of structure and mechanism between eukaryotic topoisomerase I and site-specific recombinases. Cell, 92, 841–850. [DOI] [PubMed] [Google Scholar]
- Cheng Q., Swalla,B.M., Beck,M., Alcaraz,R.,Jr, Gumport,R.I. and Gardner,J.F. (2000) Specificity determinants for bacteriophage Hong Kong 022 integrase: analysis of mutants with relaxed core-binding specificities. Mol. Microbiol., 36, 424–436. [DOI] [PubMed] [Google Scholar]
- Connolly K.M., Wojciak,J.M. and Clubb,R.T. (1998) Site-specific DNA binding using a variation of the double stranded RNA binding motif. Nature Struct. Biol., 5, 546–550. [DOI] [PubMed] [Google Scholar]
- Ditto M.D., Roberts,D. and Weisberg,R.A. (1994) Growth phase variation of integration host factor level in Escherichia coli. J. Bacteriol., 176, 3738–3748. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Echols H. and Guarneros,G. (1983) Control of integration and excision. In Hendrix,R.W., Roberts,J.W., Stahl,F.W. and Weisberg,R.A. et al. (eds), Lambda II. Cold Spring Harbor Laboratory Press, Cold Spring Harbor, NY, pp. 75–92.
- Grainge I. and Jayaram,M. (1999) The integrase family of recombinase: organization and function of the active site. Mol. Microbiol., 33, 449–456. [DOI] [PubMed] [Google Scholar]
- Guarneros G., Montanez,C., Hernandez,T. and Court,D. (1982) Posttranscriptional control of bacteriophage λint gene expression from a site distal to the gene. Proc. Natl Acad. Sci. USA, 79, 238–242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guo F., Gopaul,D.N. and Van Duyne,G.D. (1997) Structure of Cre recombinase complexed with DNA in a site-specific recombination synapse. Nature, 389, 40–46. [DOI] [PubMed] [Google Scholar]
- Hallet B. and Sherratt,D.J. (1997) Transposition and site-specific recombination: adapting DNA cut-and-paste mechanisms to a variety of genetic rearrangements. FEMS Microbiol. Rev., 21, 157–178. [DOI] [PubMed] [Google Scholar]
- Han Y.W., Gumport,R.I. and Gardner,J.F. (1994) Mapping the functional domains of bacteriophage λ integrase protein. J. Mol. Biol., 235, 908–925. [DOI] [PubMed] [Google Scholar]
- Hickman A.B., Waninger,S., Scocca,J.J. and Dyda,F. (1997) Molecular organization in site-specific recombination: the catalytic domain of bacteriophage HP1 integrase at 2.7 Å resolution. Cell, 89, 227–237. [DOI] [PubMed] [Google Scholar]
- Jessop L., Bankhead,T., Wong,D. and Segall,A.M. (2000) The amino terminus of bacteriophage λ integrase is involved in protein–protein interactions during during recombination. J. Bacteriol., 182, 1024–1034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kikuchi Y. and Nash,H.A. (1978) The bacteriophage λint gene product. J. Biol. Chem., 253, 7149–7157. [PubMed] [Google Scholar]
- Kim S.-H. and Landy,A. (1992) Lambda Int protein bridges between higher order complexes at two distant chromosomal loci attL and attR. Science, 256, 198–203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim S., Moitoso de Vargas,L., Nunes-Düby,S.E. and Landy,A. (1990) Mapping of a higher order protein–DNA complex: two kinds of long-range interactions in λ attL. Cell, 63, 773–781. [DOI] [PubMed] [Google Scholar]
- Kwon H.J., Tirumalai,R.S., Landy,A. and Ellenberger,T. (1997) Flexibility in DNA recombination: structure of the λ integrase catalytic core. Science, 276, 126–131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Laemmli U.K. (1970) Cleavage of structural proteins during the assembly of the head of bacteriophage T4. Nature, 227, 680–685. [DOI] [PubMed] [Google Scholar]
- Moitoso de Vargas L., Pargellis,C.A., Hasan,N.M., Bushman,E.W. and Landy,A. (1988) Autonomous DNA binding domains of λ integrase recognize different sequence families. Cell, 54, 923–929. [DOI] [PubMed] [Google Scholar]
- Moitoso de Vargas L., Kim,S. and Landy,A. (1989) DNA looping generated by the DNA-bending protein IHF and the two domains of λ integrase. Science, 244, 1457–1461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nash H.A. (1996) Site-specific recombination: integration, excision, resolution and inversion of defined DNA segments. In Neidhardt,F.C. et al. (eds), Escherichia coli and Salmonella. ASM Press, Washington, DC, pp. 2363–2376.
- Numrych T.E., Gumport,R.I. and Gardner,J.F. (1990) A comparison of the effects of single-base and triple-base changes in the integrase arm-type binding sites on the site-specific recombination of bacteriophage λ. Nucleic Acids Res., 18, 3953–3959. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nunes-Düby S.E., Matsumoto,L. and Landy,A. (1987) Site-specific recombination intermediates trapped with suicide substrates. Cell, 50, 779–788. [DOI] [PubMed] [Google Scholar]
- Nunes-Düby S.E., Smith-Mungo,L.I. and Landy,A. (1995) Single base-pair precision and structural rigidity in a small IHF-induced DNA loop. J. Mol. Biol., 253, 228–242. [DOI] [PubMed] [Google Scholar]
- Nunes-Düby S., Tirumalai,R.S., Kwon,H.J., Ellenberger,T. and Landy,A. (1998) Similarities and differences among 105 members of the Int family of site-specific recombinases. Nucleic Acids Res., 26, 391–406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nystrom T. (1995) Glucose starvation stimulon of Escherichia coli: role of integration host factor in starvation survival and growth phase-dependent protein synthesis. J. Bacteriol., 177, 5707–5710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pargellis C.A., Nunes-Düby,S.E., Moitoso de Vargas,L. and Landy,A. (1988) Suicide recombination substrates yield covalent λ integrase–DNA complexes and lead to identification of the active site tyrosine. J. Biol. Chem., 263, 7678–7685. [PubMed] [Google Scholar]
- Pena C.E.A., Kahlenberg,M. and Hatfull,G.F. (2000) Assembly and activation of site-specific recombination complexes. Proc. Natl Acad. Sci. USA, 97, 7760–7765. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Redinbo M.R., Stewart,L., Kuhn,P., Champoux,J.J. and Hol,W.G.J. (1998) Crystal structures of human topoisomerase I in covalent and noncovalent complexes with DNA. Science, 279, 1504–1513. [DOI] [PubMed] [Google Scholar]
- Rice P.A., Yang,S.-W., Mizuuchi,K. and Nash,H.A. (1996) Crystal structure of an IHF–DNA complex: a protein-induced DNA u-turn. Cell, 87, 1295–1306. [DOI] [PubMed] [Google Scholar]
- Richet E., Abcarian,P. and Nash,H.A. (1988) Synapsis of attachment sites during λ integrative recombination involves capture of a naked DNA by a protein–DNA complex. Cell, 52, 9–17. [DOI] [PubMed] [Google Scholar]
- Ross W. and Landy,A. (1982) Bacteriophage λ int protein recognizes two classes of sequence in the phage att site: characterization of arm-type sites. Proc. Natl Acad. Sci. USA, 79, 7724–7728. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ross W., Landy,A., Kikuchi,Y. and Nash,H. (1979) Interaction of Int protein with specific sites on λatt DNA. Cell, 18, 297–307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sambrook J., Fritsch,E.F. and Maniatis,T. (1989) Molecular Cloning. Cold Spring Harbor Laboratory Press, Cold Spring Harbor, NY.
- Segall A.M. and Nash,H.A. (1996) Architectural flexibility in λ site-specific recombination: three alternate conformations channel the attL site into three distinct pathways. Genes Cells, 1, 453–463. [DOI] [PubMed] [Google Scholar]
- Sherratt D.J. (1993) Site-specific recombination and the segregation of circular chromosomes. In Eckstein,F. and Lilley,D.M.J. (eds), Nucleic Acids and Molecular Biology. Springer-Verlag, Berlin, pp. 202–216.
- Smith P.K. et al. (1985) Measurement of protein using bicinchoninic acid. Anal. Biochem., 150, 76–85. [DOI] [PubMed] [Google Scholar]
- Subramanya H.S., Arciszewska,L.K., Baker,R.A., Bird,L.E., Sherratt,D.J. and Wigley,D.B. (1997) Crystal structure of the site-specific recombinase, XerD. EMBO J., 16, 5178–5187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thompson J.F. and Landy,A. (1988) Empirical estimation of protein-induced DNA bending angles: applications to λ site-specific recombination complexes. Nucleic Acids Res., 16, 9687–9705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thompson J.F. and Landy,A. (1989) Regulation of λ site-specific recombination. In Berg,D.E. and Howe,M.M. (eds), Mobile DNA. American Society for Microbiology, Washington, DC, pp. 1–22.
- Thompson J.F., Waechter-Brulla,D., Gumport,R.I., Gardner,J.F., Moitoso de Vargas,L. and Landy,A. (1986) Mutations in an integration host factor-binding site: effect on λ site-specific recombination and regulatory implications. J. Bacteriol., 168, 1343–1351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thompson J.F., Moitoso de Vargas,L., Koch,C., Kahmann,R. and Landy,A. (1987a) Cellular factors couple recombination with growth phase: characterization of a new component in the λ site-specific recombination pathway. Cell, 50, 901–908. [DOI] [PubMed] [Google Scholar]
- Thompson J.F., Moitoso de Vargas,L., Skinner,S.E. and Landy,A. (1987b) Protein–protein interactions in a higher-order structure direct λ site-specific recombination. J. Mol. Biol., 195, 481–493. [DOI] [PubMed] [Google Scholar]
- Thompson J.F., Snyder,U.K. and Landy,A. (1988) Helical repeat dependence of λ integrative recombination: role of the P1 and H1 protein binding sites. Proc. Natl Acad. Sci. USA, 85, 6323–6327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tirumalai R.S., Healey,E. and Landy,A. (1997) The catalytic domain of λ site-specific recombinase. Proc. Natl Acad. Sci. USA, 94, 6104–6109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tirumalai R.S., Kwon,H., Cardente,E., Ellenberger,T. and Landy,A. (1998) The recognition of core-type DNA sites by λ Integrase. J. Mol. Biol., 279, 513–527. [DOI] [PubMed] [Google Scholar]
- Wiechelman K.J., Braun,R.D. and Fitzpatrick,J.D. (1988) Investigation of the bicinchoninic acid protein assay: identification of the groups responsible for color formation. Anal. Biochem., 175, 231–237. [DOI] [PubMed] [Google Scholar]
- Wojciak J.M., Connolly,K.M. and Clubb,R.T. (1999) NMR structure of the Tn916 integrase–DNA complex. Nature Struct. Biol., 6, 366–373. [DOI] [PubMed] [Google Scholar]