

Abstract
SecA, the motor subunit of bacterial polypeptide translocase, is an RNA helicase. SecA comprises a dimerization C-terminal domain fused to an ATPase N-terminal domain containing conserved DEAD helicase motifs. We show that the N-terminal domain is organized like the motor core of DEAD proteins, encompassing two subdomains, NBD1 and IRA2. NBD1, a rigid nucleotide-binding domain, contains the minimal ATPase catalytic machinery. IRA2 binds to NBD1 and acts as an intramolecular regulator of ATP hydrolysis by controling ADP release and optimal ATP catalysis at NBD1. IRA2 is flexible and can undergo changes in its α-helical content. The C-terminal domain associates with NBD1 and IRA2 and restricts IRA2 activator function. Thus, cytoplasmic SecA is maintained in the thermally stabilized ADP-bound state and unnecessary ATP hydrolysis cycles are prevented. Two DEAD family motifs in IRA2 are essential for IRA2–NBD1 binding, optimal nucleotide turnover and polypeptide translocation. We propose that translocation ligands alleviate C-terminal domain suppression, allowing IRA2 to stimulate nucleotide turnover at NBD1. DEAD motors may employ similar mechanisms to translocate different enzymes along chemically unrelated biopolymers.
Keywords: ATPase/DEAD RNA helicase/preprotein translocation/SecA/translocase
Introduction
One third of the cellular proteome is extracytoplasmic (Gerstein et al., 2000). Most secretory and membrane proteins are synthesized as preproteins that are delivered to membranes by chaperones and/or the signal recognition particle (Danese and Silhavy, 1998; Johnson and van Waes, 1999). Bacterial translocase comprises the integral membrane proteins SecYEGDFYajC and the membrane-associated ATPase motor SecA (Economou, 1998, 2000; Manting and Driessen, 2000). SecYEA comprise the essential enzyme core. Preproteins trigger ATP-driven cycling of SecA between a ‘membrane-inserted’ and a ‘deinserted’ state (Economou and Wickner, 1994). Powered by SecA, the translocase moves processively along the polymeric substrate (Economou, 1998) in defined steps (Schiebel et al., 1991; van der Wolk et al., 1997). The precise mechanisms of SecA ATP hydrolysis and the coupling of energy to protein translocation remain elusive. Secretory preproteins cross the membrane through a SecY–SecA channel (Joly and Wickner, 1993; Matsumoto et al., 1997) that may resemble a ‘pore’ (Meyer et al., 1999; Manting et al., 2000).
SecA comprises two domains: the 68 kDa N-terminal domain and the 34 kDa dimerization C-terminal domain (Figure 1; Economou, 1998; Karamanou et al., 1999) that have been synthesized as independent polypeptides (termed N68 and C34, respectively). N68 and C34 bind to each other and reconstitute SecA (Karamanou et al., 1999; Dapic and Oliver, 2000; Nakatogawa et al., 2000). In cytoplasmic SecA, the N-terminal domain ATPase is repressed by the C-terminal domain IRA1 switch (previously IRA; Figure 1; Karamanou et al., 1999) by means of physical association.

Fig. 1. Map of SecA and truncated derivatives. SecA consensus motifs (60% identity or similarity) were determined as described (Bailey and Gribskov, 1999) and by visual inspection. The corresponding DEAD superfamily II motifs are indicated. Family motifs (dark boxes) in E.coli SecA are: motif I (aa 66–82; EAFAVVREA[S/A][R/K]RVLGXR); motif II (aa 83–139; [P/H][F/Y]DVQLIGG[M/I][V/A]LHXGXIAEM[K/R]TGEGKTL[V/T]ATL[P/A][A/V]YLNAL[S/T]GKGVHVVTVNDYLA[R/K]RD); motif III (aa 149–156; FLGL[T/S]VG[V/L]); motif IV (aa 166–191; [R/K][R/K]XAYX[A/C]DITY[A/G]TN[N/S]E[F/L]GFDYLRDN[M/L]); motif V (aa 205–227; [F/Y]AIVDEVDSILIDEARTPLIISG); motif VI (aa 333–361; V[V/L]IVDEFTGR[V/L][M/L]XGRR[Y/W]S[D/E]GLHQAIEAKE); motif VII (aa 371–397; TLA[T/S]IT[Y/F]QN[Y/F]FR[L/M]YXKL[A/S]GMTGTAXTE); motif VIII (aa 404–430; IYX[L/M]XV[V/I]XIPTNRP[M/V]XRXDXXDL[I/V]YX[T/S]); motif IX (aa 449–466; GQP[V/L]LVGT[I/T]S[V/I]EXSE[L/Y]LS); motif X (aa 494–516; IXHXVLNAKXXX[R/K]EAXI[I/V]AXAGXXGAVTIATNMAGRG TDIXLG); motif XI (aa 532–599; GGLX[I/V]IGTERHESRRID NQLRGR[A/S]GRQGD[P/A]GXSRFYLSLEDXL[M/L]R[I/L]F[A/G]); motif XII (aa 630–663; AQ[K/R]KVE[G/A]XXN[F/Y][D/E][W/L]RKQLXXYDDV[M/L]XXQRXXIYXXR); motif XIII (aa 767–818; [L/I]LXXIDXXWREHLXXMDXLRXGIXLR[G/A]YAQKDPLXE YXXE[G/A][Y/F]X[L/M]FXXM[L/M]XX[I/L]). Symbols used: X = any residue; alternative substitutions for the same residue are shown in brackets.
SecA is an ATP-dependent RNA helicase (Park et al., 1997) and contains conserved DEAD protein family motifs (Figure 1; Koonin and Gorbalenya, 1992). The collective term ‘DEAD’ represents five superfamilies of ATPases including bona fide nucleic acid helicases (de la Cruz et al., 1999; Hall and Matson, 1999). Walker box sequences A (Figure 1, motif II; Walker et al., 1982) and B (motif V) form a high-affinity nucleotide-binding site (NBD1) essential for SecA function (Matsuyama et al., 1990; Mitchell and Oliver, 1993; Economou et al., 1995). The region encompassing residues 500–650 (termed ‘NBD2’) was proposed to be responsible for a second, low-affinity nucleotide-binding activity (Mitchell and Oliver, 1993). This region is important for ATP hydrolysis (Karamanou et al., 1999) and protein translocation (Mitchell and Oliver, 1993; Economou et al., 1995). However, its function has remained unresolved since nucleotide binding to ‘NBD2’ has not been demonstrated experimentally; the proposed Walker boxes are non-canonical, with box B located outside the limits of the ATPase N-terminal domain (Karamanou et al., 1999). We now demonstrate that this region is not an independent nucleotide-binding or hydrolysis site. Instead, it is a regulatory domain that harbours two conserved DEAD family motifs. This novel intramolecular regulator of ATP hydrolysis (IRA2) is highly flexible and acts through binding to NBD1. IRA2 activates ATP hydrolysis and multiple nucleotide turnovers at NBD1 in trans and is essential for polypeptide translocation.
Results
ADP release is rate limiting for multiple ATP turnovers by SecA
To study the mechanism of ATP hydrolysis by SecA, we employed thin-layer chromatography (TLC). Full-length SecA or the isolated N68 domain completely hydrolyse stoichiometric amounts of ATP at either 37 or 4°C, within 1 min (Figure 2A, lanes 1–6). In the presence of excess ATP, both enzymes catalyse multiple rounds at 37°C (Figure 2A, lanes 7–9, and B), but SecA is less efficient due to IRA1 suppression (Figure 2B; Karamanou et al., 1999). In contrast, at 4°C, only limited hydrolysis occurs (Figure 2A, lanes 10–12, and B). Since the ATP hydrolysis chemistry per se is not affected by low temperature (Figure 2A, lanes 1–6), there must exist a rate-limiting step downstream in the reaction cycle that represses multiple rounds of ATP hydrolysis at 4°C (Figure 2B).
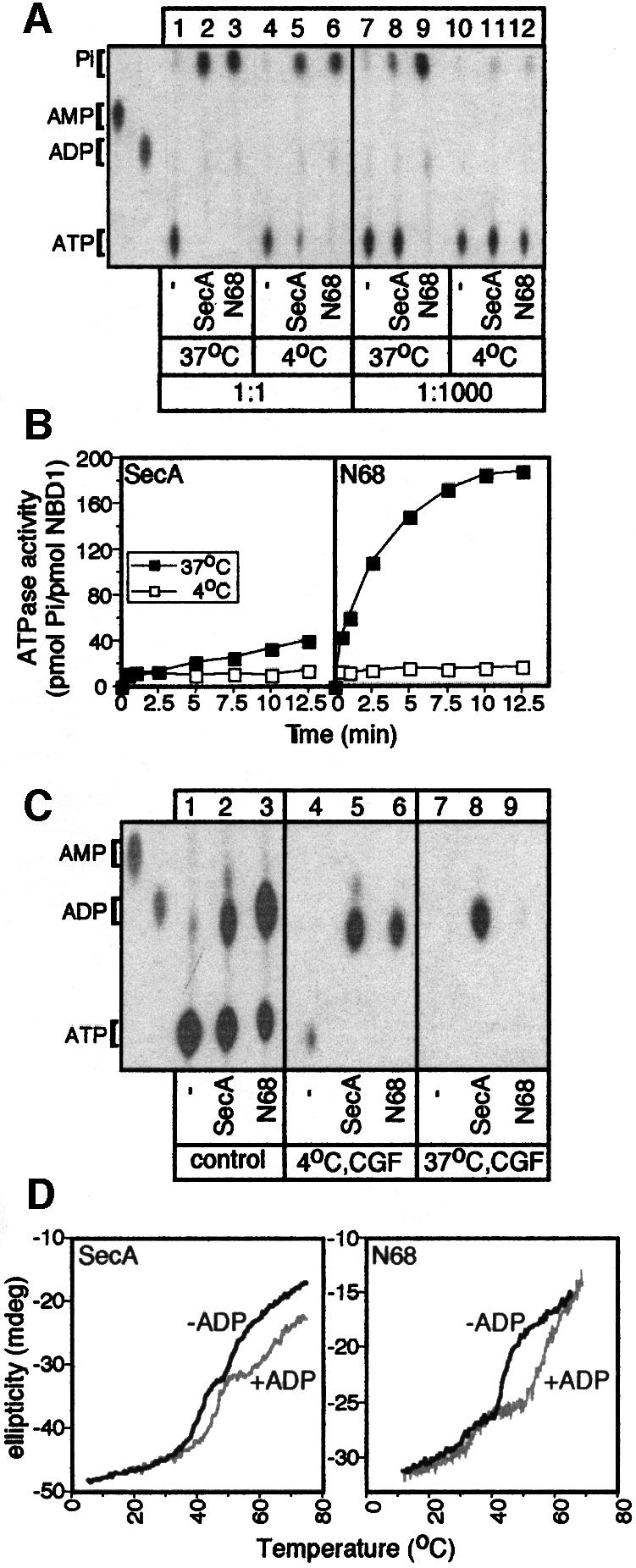
Fig. 2. ADP release from SecA is rate limiting for multiple rounds of ATP hydrolysis. (A) Single versus multiple rounds of SecA ATP hydrolysis. SecA or N68 (10 pmol) or no protein (10 µl reactions) was incubated at the indicated temperature with [γ-32P]ATP (1 µM, 1:1 molar ratio, 1 min; or 1 mM, 1:1000, 10 min). Hydrolysis was stopped by proteinase K (5 µl of 10 mg/ml stock) at 4°C and samples were analysed by TLC and autoradiography. AMP and ADP markers are indicated. (B) Effect of temperature on SecA ATP hydrolysis. SecA or N68 (50 pmol in 10 µl) was incubated with 1 mM [γ-32P]ATP (37 or 4°C, 5 min). At the indicated time point, proteinase K was added and samples were analysed by TLC (as in A) and quantitated (see Materials and methods). (C) ADP release is rate limiting for multiple ATP turnovers by SecA. SecA or N68 (100 pmol) or no protein (20 µl reactions) was incubated with 100 µM [α-32P]ATP (10 min, 4°C). One-third of the reaction was treated with proteinase K (4°C; control), one-third was subjected to centrifugal gel filtration (4°C; CGF) and one-third was incubated at 37°C (5 min) before CGF (as indicated). Samples were analysed as in (A). (D) ADP causes an increase in the melting temperatures of SecA and N68. Thermal denaturation curves, in the presence or absence of ADP (2 mM), were obtained by monitoring ellipticity at 222 nm by far-UV CD, while heating the protein samples (200 µl, 200 µg/ml in 5 mM MOPS buffer pH 7.5, 5 mM MgCl2) at 50°C/h and were analysed as described (Karamanou et al., 1999).
To determine this step, we investigated the nucleotide occupancy of SecA and N68 after ATP hydrolysis at 4°C by centrifugal gel filtration (CGF; Figure 2C). Strikingly, both enzymes are isolated in the excluded volume in an exclusively ADP-bound state (lanes 5 and 6), although 40–60% of the total nucleotide pool is ATP (lanes 2 and 3). Following CGF, most of the generated ADP remains bound to SecA and N68 (97 and 71%, respectively). A brief incubation at 37°C prior to CGF drastically reduces the amount of ADP co-isolated with N68 (only 2% remains bound; lane 9). This effect of elevated temperature is less dramatic for SecA (62% remains bound; lane 8), presumably due to the presence of the IRA1 switch (Karamanou et al., 1999). It appears that, following ATP hydrolysis at 4°C, ADP release from the N-terminal domain of SecA imposes a rate-limiting step for ATP turnover. At 37°C, the apparent ADP release rate, slow for SecA and fast for N68, could underly the observed 8-fold reduced ATP turnover of SecA compared with N68 (Table I).
Table I. Kinetic parameters of SecA and mutant derivatives.
| Protein | kcat (per min) |
|---|---|
| SecA | 4.6 |
| N68(N1–610) | 36.4 |
| N1–263 | 0.1 |
| N1–479 | 0.06 |
| N1–547 | 0.08 |
| IRA2(N462–610) | n.d. |
| N270–610 | n.d. |
| Class I | |
| SecAG510A | 42.2 |
| N68G510A | 7.8 |
| SecAR566A | 19.6 |
| N68R566A | 12.3 |
| Class II | |
| SecAR509K | 4.5 |
| N68R509K | 2.1 |
| SecAR577K | 2.6 |
| N68R577K | 0.14 |
Enzymes were incubated (5 min; 37°C; buffer B) at a range of [γ-32P]ATP concentrations (30–2000 µM). Hydrolysis was determined as in Figure 1B. n.d., not detectable, i.e. following subtraction of the ATP background, values were zero or negative.
We further investigated ADP retention by SecA or N68 using far-UV circular dichroism (CD). Thermal denaturation curves of the two enzymes reveal at least two discernible transitions centred at 39 and 48°C (Figure 2D). The more stable transition derives from a domain present in both SecA and N68. Interestingly, its apparent Tm increases dramatically by 15°C in the presence of ADP (Figure 2D; Table II). The lower transition is composite, could not be attributed to a single domain and was not analysed further. Clearly, ADP binds to the N-terminal domain and thermally stabilizes SecA.
Table II. Apparent melting temperatures (Tm) of SecA and mutant derivatives (as in Figure 1D and Karamanou et al., 1999).
| Protein |
Tm app |
|
|---|---|---|
| –ADP | +ADP | |
| SecA | 48.9 | 62.5 |
| N68(N1–610) | 48.5 | 63.0 |
| N1–263 | 49.1 | 57.0 |
| N1–479 | 47.6 | 52.5 |
| N1–547 | 48.2 | 56.1 |
| IRA2(N462–610) | 43.0 | 44.0 |
| N270–610 | 43.5 | 44.0 |
| Class I | ||
| SecAG510A | 41.9 | 48.0 |
| SecAR566A | 45.4 | 50.6 |
| Class II | ||
| SecAR509K | 48.1 | 64.0 |
| SecAR577K | 47.9 | 65.1 |
NBD1 is the minimal catalytic machinery in SecA
To delineate the N68 subdomains responsible for ADP binding and release, we aligned 68 SecA sequences. SecA contains 11 conserved motifs in the N-terminal domain (I–XI) and two in the C-terminal domain (XII and XIII; Figure 1; Karamanou et al., 1999). SecA belongs to DEAD superfamily 2 (Koonin and Gorbalenya, 1992; de la Cruz et al., 1999) and shows extensive similarity to DExH DNA repair and recombination enzymes (N.Kyrpides and A.Economou, unpublished results). We designed appropriately truncated N68 derivatives leaving conserved regions intact. The smallest fragment we generated, N1–263, fully encompasses the sequences comprising the high-affinity nucleotide-binding site NBD1 (Figure 1; Matsuyama et al., 1990; Mitchell and Oliver, 1993; den Blaauwen et al., 1999). N68 truncated derivatives were fused to oligohistidinyl tags and purified by metal affinity chromatography. All recombinant polypeptides are soluble, and their CD spectra verified that they are folded and stable (Table II and data not shown).
Thermal denaturation curves demonstrated that all NBD1-containing polypeptides (N1–263, N1–479 and N1–547; Figure 1) retain the stable 48°C transition observed for N68 (Table II). This transition can therefore be attributed to NBD1. The apparent Tm of these polypeptides is significantly increased by ADP, albeit to a lower extent than that of SecA or N68 (5–8°C; Table II). In contrast, ADP does not cause any appreciable increase in the apparent Tm of the N270–610 or N462–610 fragments (Figure 1; Table II).
We next tested the truncated N68 derivatives for the subsequent reaction step, ATP hydrolysis. N1–263, N1–479 and N1–547 can hydrolyse ATP, although very inefficiently compared with SecA or N68 (Table I). This low ATPase activity results from catalysis at NBD1 since it is eliminated by introduction of the D209N mutation (see below and data not shown) that abolishes NBD1 ATP hydrolysis in both SecA and N68 (Mitchell and Oliver, 1993; Economou et al., 1995). Fragments devoid of NBD1 (N270–610 or N462–610) display no detectable ATPase activity (Table I).
We conclude that N1–263 fully encompasses the minimal nucleotide-binding and hydrolysis machinery in SecA. Nevertheless, residues on N462–610 seem to be required for both full-scale ADP-driven stabilization of SecA and high ATP turnover. We demonstrate below that this region, hereafter termed IRA2, is an essential intramolecular regulator of ATP hydrolysis at NBD1.
IRA2 can stimulate ATP hydrolysis at NBD1
The deletion analysis presented above suggested that the N-terminal domain of SecA may comprise distinct catalytic and regulatory elements. We directly tested this hypothesis in vitro by supplying N1–479 with purified IRA2, the region missing from N1–479 but present on N68 (Figure 1). This led to a remarkable 15-fold activation of N1–479 ATPase activity, at 37°C (Figure 3A) but not at 4°C (data not shown). Analysis of these reactions by native PAGE reveals formation of N1–479–IRA2 complexes, visible as a species migrating more slowly than N1–479 or IRA2 alone (Figure 3B, lanes 1–3). In the same assay, N1–479D209N cannot become activated (Figure 3A) although it binds IRA2 as efficiently as the wild-type N1–479 (Figure 3B, compare lanes 5 and 3). This indicates that the stimulated ATP hydrolysis takes place at NBD1 and does not result from activation of a cryptic IRA2 ATPase activity.
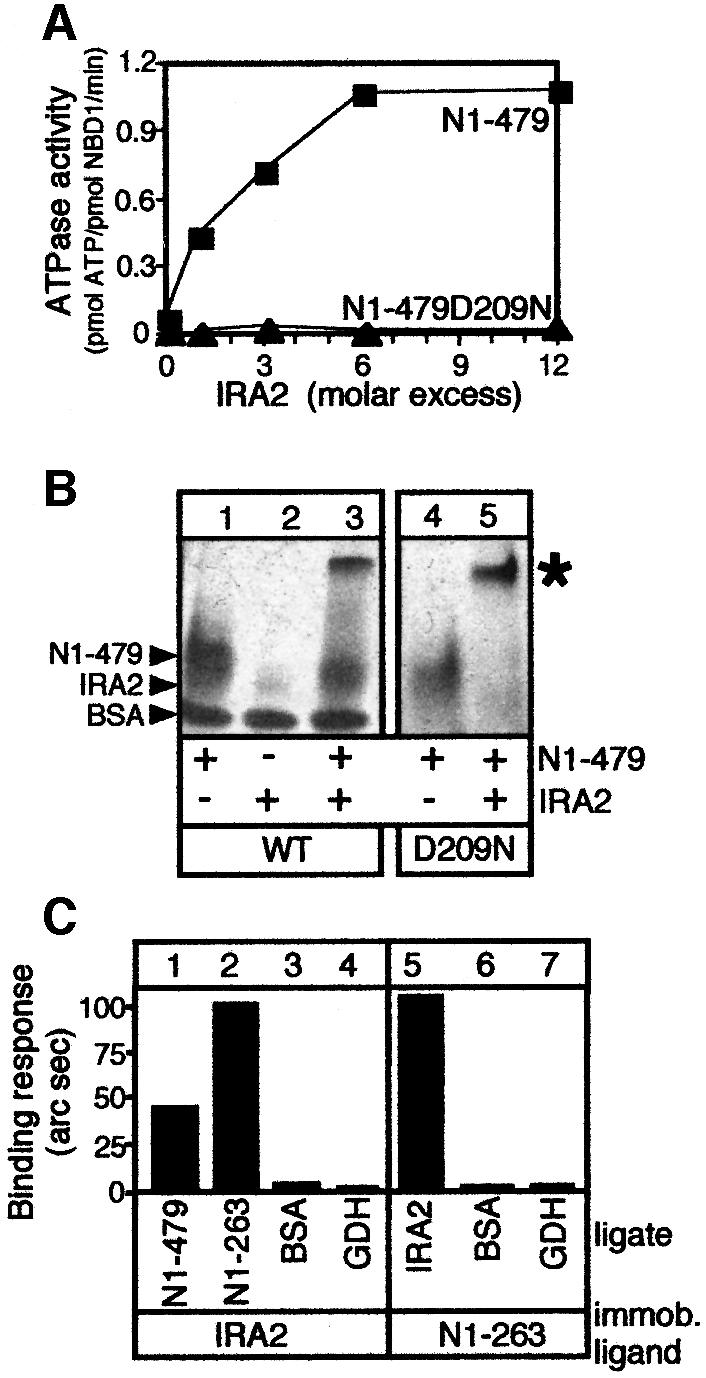
Fig. 3. NBD1 is the minimal SecA ATPase domain and is activated by IRA2. (A) IRA2 stimulates NBD1 ATP hydrolysis. N1–479 or N1–479D209N (100 pmol in 10 µl) supplemented with IRA2 (indicated molar ratios) was incubated with 0.5 mM [γ-32P]ATP (15 min; 37°C). Hydrolysis was determined as in Figure 2B. (B) IRA2 activates NBD1 ATPase through direct physical interactions. Reactions as in (A) (1:1 molar ratio shown), with BSA used as carrier, were analysed by native PAGE (4–20% gradient) and Coomassie Blue staining. An asterisk represents reconstituted complex. IRA2 migrates on native gels as a fuzzy, weakly staining band. (C) NBD1 physical association with IRA2. N1–479, N1–263 or control proteins (250 nM in 100 µl of buffer B) were added to biosensor-immobilized IRA2 on an amino surface. IRA2 (250 nM; buffer B) or control proteins were bound to immobilized N1–263. BSA, bovine serum albumin; GDH, glutamate dehydrogenase.
To quantitate IRA2 binding and to define the N1–479 region responsible for the association, we used an optical biosensor (Figure 3C). N1–263 (lane 2) and N1–479 (lane 1) bind efficiently to immobilized IRA2, while control proteins do not (lanes 3 and 4). Similarly, in the reverse experiment, IRA2 binds efficiently to immobilized N1–263 (compare lanes 5 and 2).
We conclude that IRA2 binds specifically to NBD1 and stimulates its ATPase activity.
Temperature affects the secondary structure of the IRA2 domain
Low temperature prevents ADP release from the N-terminal domain of SecA and, therefore, ATP turnover, while elevated temperature has the reverse effect (Figure 2B and C; Schmidt et al., 2000). How is temperature ‘sensed’ by N68 and SecA? To address this, we examined the secondary structure of SecA, N68, N1–263, N1–479 and IRA2 at various temperatures, by far-UV CD. All these polypeptides are predominantly α-helical at 4°C (42–46%; data not shown). Helicity values were also determined at higher temperatures and expressed as a percentage of values obtained at 4°C (taken as a 100% baseline; Table III). Significantly, the α-helicity in IRA2 decreases upon temperature increase, suggesting extensive structural flexibilty. In contrast, the helicity of N1–263 or N1–479, which are devoid of IRA2, is not affected significantly in the temperature range examined. Interestingly, the helical content of both SecA and N68 is significantly decreased within the same temperature range, suggesting that IRA2 may be the main element contributing to partial endothermal melting of SecA or N68.
Table III. Temperature-induced α-helicity changes.
| Polypeptide | Relative α-helicity (%) |
|||
|---|---|---|---|---|
| 4°C | 15°C | 24°C | 37°C | |
| SecA | 100 | 99 | 97 | 49 |
| N68(N1–610) | 100 | 96 | 95 | 52 |
| N1–263 | 100 | 95 | 92 | 75 |
| N1–479 | 100 | 100 | 99 | 92 |
| IRA2(N462–610) | 100 | 92 | 87 | 38 |
Ellipticity at 222 nm is directly related to α-helical content and was derived from far-UV CD scans as described (Karamanou et al., 1999). Helicity at 4°C is expressed as 100%. Values at higher temperatures were normalized accordingly.
Mutations in the DEAD motifs of IRA2 affect ATP turnover at NBD1
To elucidate further the molecular mechanism of IRA2 function, we sought to isolate point mutants with altered properties. SecA proteins contain only two conserved motifs (X and XI; DEAD family motifs V and VI; Figure 1) within the IRA2 domain. We generated point mutations in 100% conserved residues within SecA motifs X and XI and examined them together with the R509K mutant isolated previously (Mitchell and Oliver, 1993; Economou et al., 1995). Mutations, originally generated on His-SecA, were also transferred to N68 and IRA2 constructs. None of the mutations affects SecA or N68 secondary structure and folding (Table II and data not shown).
Of 16 mutations, three had minor phenotypic changes and were not studied further. The remaining mutants had strong phenotypes and fell sharply into two classes. Since mutations in motifs X and XI result in similar phenotypes, we treated these motifs as a functional unit and focused on one representative mutant from each motif: class I (G510A, motif X; R566A, motif XI) and class II (R509K, motif X; R577K, motif XI). Class I mutations stimulate the basal ATPase of SecA (Figure 4A, lanes 1–3), while class II mutations slightly reduce it (lanes 4 and 5). The elevated basal SecA ATPase of class I mutants takes place at NBD1 since the D209N mutation abolishes it (compare lanes 2 and 3 with 7 and 8). Interestingly, both class I and class II IRA2 mutations reduce the ATPase of N68 (Table I), indicating that the presence of C34 in SecA may suppress the primary defect of class I IRA2 mutations (see below). Since IRA2 point mutations affect ATP turnover at NBD1 in either SecA or N68 (Figure 4A; Table I), we sought to identify specific elements of the NBD1 ATPase cycle that are under IRA2 control.

Fig. 4. IRA2 is required for nucleotide binding and hydrolysis at NBD1. (A) Basal ATPase activity of SecA and IRA2 mutant derivatives determined by the malachite green method (as in Lill et al., 1990). (B) IRA2 mutations affect NBD1–IRA2 binding. IRA2 mutants (500 nM; buffer B, 100 mM NaCl, 5% glycerol) were added to biosensor-immobilized N1–479. The binding response is expressed in millidegrees. (C) ATP hydrolysis kinetics of SecA, N68 and G510A and R566A mutant derivatives (50 pmol protein in 10 µl reactions) at 4°C (1 mM [γ-32P]ATP; analysed as in Figure 2B). (D) Nucleotide occupancy of SecA IRA2 mutants. Samples (50 pmol protein in 10 µl reactions) prior to (lanes 2–7) or after CGF (lanes 8–13) were analysed as in Figure 2C. (E) ATP pre-steady-state kinetics of SecA and N68, R509K and R577K mutant derivatives (50 pmol protein, 5 µM [α-32P]ATP in 10 µl reactions analysed as in Figure 2B). Hydrolysis is expressed as a percentage of that of equimolar amounts of wild-type SecA or N68, under identical conditions.
IRA2 up-regulates ATP turnover at NBD1 by controlling ADP release
First, we examined the effect of IRA2 mutations on the ability of SecA to interact with ADP. The ADP-induced thermal stabilization of SecA is not affected by the R509K and R577K substitutions (Table II). In contrast, SecAG510A and SecAR566A undergo limited ADP-driven stabilization (5–6°C; Table II), analogous to that seen with N-terminal domain truncations devoid of IRA2 (e.g. N1–263; Table II). An increased concentration of ADP (>10 mM) fails to enhance the thermal stability of class I mutants further (data not shown), suggesting that the observed defect does not result from reduced affinity of NBD1 for ADP. In full agreement with deletion analysis, these results firmly establish that although IRA2 is not required for nucleotide binding to NBD1, it is essential for full-scale ADP-driven stabilization of N68 and SecA (Table II).
IRA2 associates with NBD1 (Figure 3B and C). To test whether class I mutations affect this property, we used an optical biosensor (Figure 4B). IRA2G510A and IRA2R566A exhibit decreased binding to immobilized N1–479 (compare lanes 2 and 3 with lane 1), while binding of IRA2 class II mutants is unaffected (compare lanes 4 and 5 with lane 1). Clearly, firm IRA2–NBD1 association is essential for full-scale ADP-mediated stabilization of SecA.
ADP binding is a property inherent to NBD1 but not to IRA2 (see above and Table II). Up-regulation of SecA ATPase by class I IRA2 mutations (Figure 4A) could result from enhanced ADP release rates, due to the altered NBD1–IRA2 interaction described above (Figure 4B). If this was the case, class I mutants would be expected to bypass the suppression of ATP turnover seen in SecA and N68 under low temperature regimes (Figure 2B). This hypothesis was tested directly. Strikingly, G510A and R566A substitutions ‘allow’ both N68 and SecA to carry out multiple rounds of ATP hydrolysis at 4°C (Figure 4C). In full agreement with these results, the amount of ADP retained by SecAG510A and SecAR566A in CGF experiments at 4°C is drastically reduced (Figure 4D, compare lanes 9 and 10 with lane 8). R509K and R577K substitutions do not alter the ability of SecA to retain ADP (compare lanes 11 and 12 with lane 8).
We conclude that IRA2 stimulates ATP turnover at NBD1 by controlling ADP release. The inability of IRA2 to release ADP from NBD1 (e.g. at 4°C or by loosened contact with NBD1) leads to repression of ATP turnover. Once ADP is released from NBD1, N68 and SecA can proceed to the next cycle of ATP hydrolysis.
IRA2 optimizes the rate of catalysis at NBD1
Study of class II mutants reveals additional properties of IRA2. The ability of SecA to retain ADP (Figure 4D) and to become thermally stabilized (Table II) is not affected by class II mutants. Despite the fact that class II mutant IRA2 domains associate with NBD1 (Figure 4B), they fail to stimulate the N1–479 ATPase (data not shown). Moreover, the R509K mutation reduces ATP turnover in N68, and the R577K substitution down-regulates ATP turnover in SecA and drastically reduces it by 250-fold in N68 (Table I). It is striking that the effect of a single point mutation (R577K) in motif XI of IRA2 is as deleterious to NBD1 turnover as deletion of the whole motif XI (i.e. N1–547) or even deletion of the whoIe IRA2 domain (i.e. N1–263 or N1–479; Figure 1; Table I).
These data indicate that in addition to controlling ADP release from NBD1, IRA2 may somehow be required for optimal catalysis. To elucidate this possibility further, we examined the effect of class II IRA2 mutations on NBD1 ATP hydrolysis by pre-steady-state kinetics (Figure 4E). This was possible since the nucleotide affinity at NBD1 is not altered by class II mutations (Km values; Mitchell and Oliver, 1993 and data not shown). Catalysis of stoichiometric amounts of ATP by SecA or N68 is complete by 1 min at either 4 or 37°C (Figure 2A). To facilitate comparison, the ATPase activity of class II mutant derivatives is expressed as a percentage of that derived from equimolar amounts of wild-type SecA or N68, under identical conditions. In agreement with steady-state kinetics (Table I), the R509K mutation does not affect catalysis in SecA but severely reduces it in N68 (Figure 4E). Catalysis of either SecA or N68 is severely affected by the R577K mutation, while low temperature exaggerates this effect (Figure 4E). These results suggest that the apparent ‘loss of function’ of class II IRA2 mutations is due to defective ATP hydrolysis at NBD1. We therefore propose that IRA2 has a dual role: it optimizes the rate of NBD1 catalysis and controls ADP release from NBD1.
C34 contribution to NBD1–IRA2 regulation
NBD1 is the minimal ATPase catalytic machinery in SecA [kcat(N1–263) = 0.1/min]. In the isolated N-terminal domain of SecA, IRA2 stimulates ATP turnover by NBD1 [kcat(N68) = 36.4/min]. However, ATP turnover by SecA is reduced 8-fold compared with N68 [kcat(SecA) = 4.6/min]. Presumably the IRA1 switch, present in SecA but absent from N68 (Figure 1; Karamanou et al., 1999), acts antagonistically to IRA2. To test whether IRA1 represses ATP turnover at NBD1 directly or by acting via IRA2, we transferred the ΔIRA1 mutation (previously Δ755–789; Karamanou et al., 1999) onto SecA IRA2 class II backgrounds. R509K or R577K substitutions abolish the observed ΔIRA1 elevated ATPase activity (Figure 5A, compare lanes 5 and 6 with 2), demonstrating that IRA1-mediated regulation in SecA requires IRA2.

Fig. 5. The C-terminal domain binds to NBD1 and IRA2. (A) Basal ATPase activity of SecA and mutant derivatives (as in Figure 4A). (B) Binding of N68 and its IRA2 mutant derivatives to biosensor-immobilized C34 (as in Figure 4B). (C) The indicated purified domains (ligate) were assayed for binding to biosensor-immobilized C34, NBD1 or IRA2 (as in Figure 4B). GDH, glutamate dehydrogenase.
IRA1 suppression is exerted via physical contact of the N- and C-terminal domains (Karamanou et al., 1999). How do SecA class I IRA2 mutants overcome IRA1 suppression and display elevated ATPase (Figure 4A)? To address this, we examined the effect of IRA2 mutations on the ability of N68 to associate with immobilized C34, using an optical biosensor (Figure 5B). Class II mutations do not affect N68 binding to C34 (lanes 1, 4 and 5). In contrast, class I mutations reduce the ability of N68 to bind C34 by 40–55% (lanes 2 and 3). We further tested whether C34 interacts physically with both NBD1 and IRA2 or only with one of them (Figure 5C). N1–263 (lane 1) or IRA2 (lane 2) binds to immobilized C34, and C34 binds to either immobilized N1–263 (lane 4) or immobilized IRA2 (lane 6). No binding is observed with a control protein (lanes 3, 5 and 7).
Class I SecA mutants are impaired in both NBD1–IRA2 association (Figure 4B) and N- to C-terminal domain binding. Loosened N- to C-terminal domain binding alleviates IRA1-mediated suppression (Karamanou et al., 1999) and could leave IRA2 unperturbed to up-regulate NBD1 ATP turnover.
IRA2 is essential for viability and protein translocation
IRA2 is a novel regulator of the NBD1 ATPase. If it is important for translocase function, IRA2 should be essential for life. To test this, secA genes with IRA2 mutations were examined for genetic complementation of the secAts strain BL21.19 (Mitchell and Oliver, 1993). Class I mutants complement BL21.19 poorly at 42°C (Figure 6A, lanes 4 and 5), while class II mutants do not (lanes 3 and 6; Mitchell and Oliver, 1993).
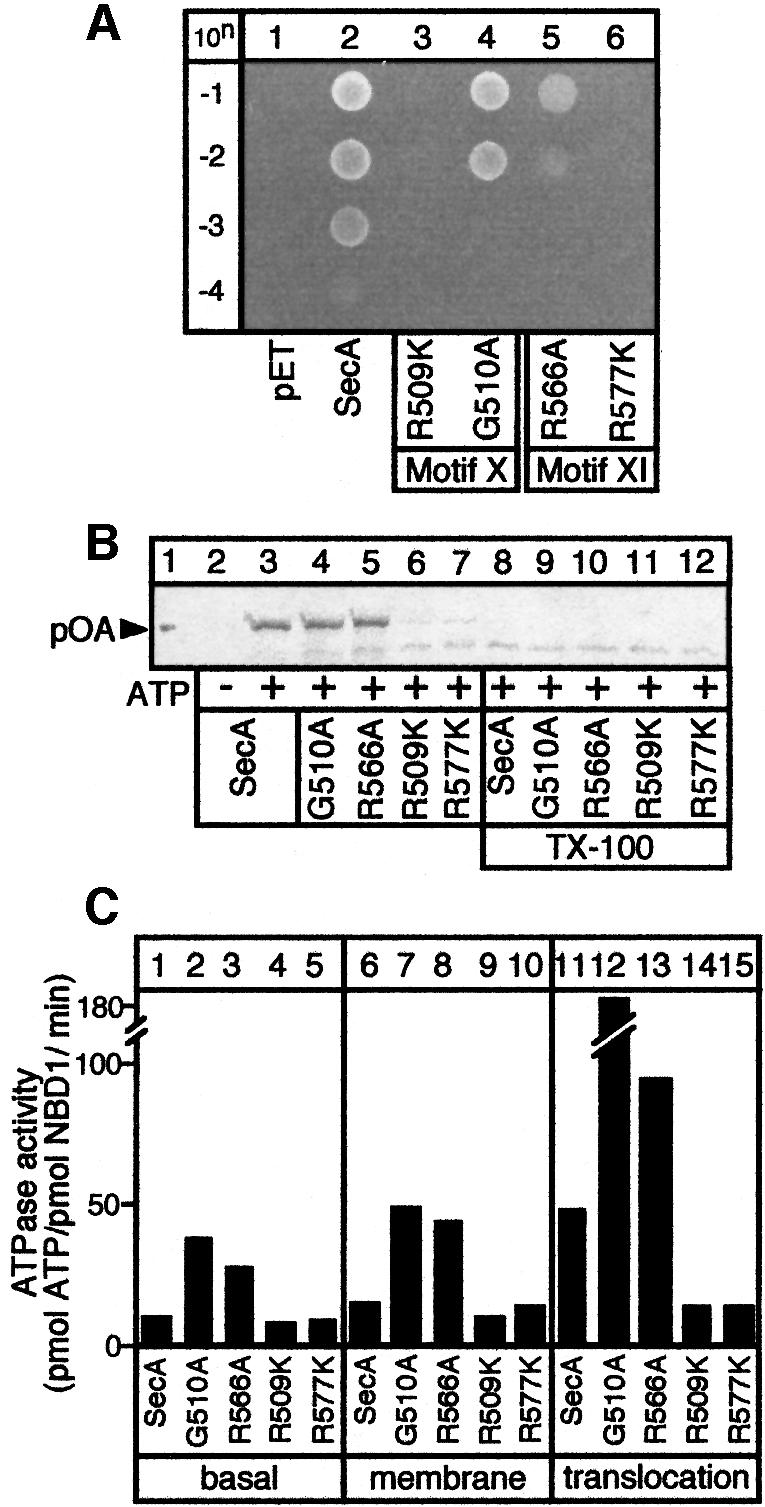
Fig. 6. The IRA2 domain is essential for protein translocation and life. (A) IRA2 mutant in vivo test. BL21.19 cultures carrying pET5 vector alone or its derivatives with cloned secA, secAR509K, secAG510A, secAR566A or secAR577K genes were adjusted to the same density. Dilutions (10n) were spotted on LB/ampicillin plates and incubated at 42°C. (B) In vitro preprotein translocation in SecYEG-proteoliposomes of SecA IRA2 mutants (as in Karamanou et al., 1999). Lane 1: 25% of input [35S]proOmpA. Triton X-100 (1% v/v) was added prior to trypsin digestion (lanes 2–12). (C) Basal, membrane and translocation ATPase activities of the IRA2 SecA mutant proteins (determined as in Lill et al., 1990).
Is IRA2 required for protein translocation per se? In a standard in vitro assay (Schiebel et al., 1991), SecAG510A and SecAR566A catalyse proOmpA translocation into SecYEG proteoliposomes (Figure 6B, compare lanes 4 and 5 with lane 3) or inner membrane vesicles (data not shown). Translocated proOmpA becomes protease accessible only upon solubilization with detergent (lanes 8–12). In contrast, the translocation ability of SecAR509K and SecAR577K is seriously compromised (compare lanes 6 and 7 with lane 3). This defect could be attributed directly to a defect of class II IRA2 mutants for a translocation-dependent stimulation of their basal NBD1 ATPase activity (Figure 6C). SecAR509K (lanes 9 and 14) and SecAR577K (lanes 10 and 15) do not show elevated ATPase activities in response to preprotein and/or membranes. In contrast, the ATPase of SecAG510A and SecAR566A becomes stimulated by translocation ligands (lanes 7, 8, 12 and 13). These results explain why class I, but not class II, mutants partially complement secAts (Figure 6A).
In summary, protein translocation by SecA is not affected by IRA2 mutations that up-regulate NBD1 ATPase (class I) but is compromised by class II mutants that down-regulate it. Clearly, the IRA2 subdomain of SecA is essential for protein translocation.
Discussion
We have determined the domain organization of the protein translocase motor SecA and unveiled the fundamental intramolecular mechanism regulating its ATPase activity. This was made possible by developing novel experimental tools that permitted study of catalytic subreactions and bimolecular interactions in solution: (i) dissection of the N-terminal domain ATPase away from IRA1 regulation and functional reconstitution (Figure 1; Karamanou et al., 1999); (ii) dissection of the N-terminal domain and functional reconstitution using subdomains (Figures 1 and 3); and (iii) isolation of kinetically poisoned mutants (Figure 4; Table I).
Four distinct modules build the SecA protomer (Figures 1 and 7): (i) NBD1, a rigid domain comprising the minimal catalytic machinery (Tables I–III); (ii) IRA2, a flexible regulatory domain (Figures 3 and 4; Table III); (iii) the region between NBD1 and IRA2, containing motifs VI–IX and the proposed preprotein-binding domain (PBD in Figure 7; Kimura et al., 1991); and (iv) the dimerization C-terminal domain (Hirano et al., 1996; Karamanou et al., 1999). The domain boundaries proposed here should be considered tentative until high-resolution structural information becomes available. Pairwise interactions between SecA modules provide a robust structural framework capable of extensive plasticity and allow formation of functional units of higher order.

Fig. 7. Domain organization model of the SecA protomer (see text for details).
In SecA, a fully functional N-terminal domain ATPase motor encompassing all seven DEAD family motifs (Figure 1) is assembled upon productive association of the nucleotide-binding and hydrolysis domain NBD1 with the regulator IRA2 (Figures 1, 3 and 4; Tables I–III). A single ATP molecule binds with high affinity to each SecA monomer (den Blaauwen et al., 1999). Intradomain communication and relative movement between the NBD1 and IRA2 modules requires motifs X and XI (Figure 4B) and could be facilitated by flexible hinges such as motif VIII, which changes its solvent accessibility in a nucleotide-dependent manner (Karamanou et al., 1999). The ATPase motor of other DEAD proteins is organized similarly from two structurally similar domains (1A and 2A). Domains 2A can exist as independent polypeptides (Koonin and Rudd, 1996; Singleton et al., 2000) or be spaced apart from the catalytic domains 1B, which are flexible (Caruthers et al., 2000) and display significant mobility relative to domain 1A (Yao et al., 1997; Velankar et al., 1999; Caruthers et al., 2000). Domain 1A (family motifs I–II; Figure 1) together with domain 2A (family motifs IV–VI; Figure 1) forms a cleft lined with all the DEAD family motifs where a single ATP molecule binds on domain 1A (Korolev et al., 1998; de la Cruz et al., 1999; Soultanas and Wigley, 2000).
ADP binding dramatically stabilizes SecA (Table II; den Blaauwen et al., 1999; Karamanou et al., 1999) without altering secondary structure (Karamanou et al., 1999) or shape (Shilton et al, 1998) through a domino-like effect: ADP binds and stabilizes NBD1 (Table II; den Blaauwen et al., 1999), which then stabilizes IRA2 (seen as proteolytic protection; Karamanou et al., 1999) leading to remarkable thermal stabilization of the whole N-terminal domain by 15°C (Table II). We anticipate that ADP binding causes localized conformational changes on NBD1 as suggested by microcalorimetry (den Blaauwen et al., 1999), CD analysis (Table II), measurements of ADP affinity and intrinsic fluorescence (Schmidt et al., 2000) and limited proteolysis studies (G.Sianidis and A.Economou, unpublished results). IRA2 ‘senses’ these changes by virtue of its direct association with NBD1 (Figure 3B and C). The inherent flexibility of IRA2 secondary structure (Table III) may facilitate the stabilization of the ADP-enhanced NBD1 conformational change by an induced fit mechanism presumably involving Gly510 and Arg566. The altered behaviour of class I mutants could result from increased IRA2 rigidity. ADP binding to SecA is stabilized by low temperature and this imposes a rate-limiting step for ATP turnover (Figure 2C). Class I point mutations in IRA2 (Table II), elevated temperature (Table III; Schmidt et al., 2000) and translocation ligands (see below) interrupt this cascade of events and trigger ADP release from NBD1 (Figure 1B and C).
An additional striking aspect of IRA2 is its participation in ATP catalysis. IRA2 motifs X and XI (V and VI in other DEAD proteins, located in domain 2A) are important for ATP hydrolysis (Figure 4C; Table I; Korolev et al., 1998; de la Cruz et al., 1999; Hall and Matson, 1999). In DNA helicases, argininyl residues in motif VI make direct contact with the ATP γ-phosphate (Velankar et al., 1999; Singleton et al., 2000). Activation of nucleotide hydrolysis in several NTPases involves ‘arginine fingers’ acting in trans (Geyer and Wittinghofer, 1997). By analogy, ATP tethered on Walker boxes A and B of NBD1 faces the apposed IRA2 motif XI residues (Figure 7). This ATP could be attacked by a catalytic ‘arginine finger’ in motif XI containing Arg577 and leading to the elevated translocation ATPase activity (Lill et al., 1990). Recently, isolation of a single hyperactivating mutation (D580V) led to the proposal that IRA2 harbours a suppressor of the NBD1 ATPase (Nakatogawa et al., 2000). However, the essential role of IRA2 in activating ATP hydrolysis at NBD1 described here is inconsistent with this proposal.
The C-terminal domain binds independently to both structural elements of the SecA DEAD motor, NBD1 and IRA2 (Figure 5C), to form SecA protomers (Karamanou et al., 1999; Dapic and Oliver, 2000; Nakatogawa et al., 2000). Through these interactions, cytoplasmic SecA is stabilized in the ADP-bound state even at 37°C (Figure 2C) and unnecessary ATP hydrolysis is prevented (Figures 2B and 6C; Karamanou et al., 1999). Three possible roles can be envisaged for the C-terminal domain: (i) it could be a molecular staple physically restricting NBD1–IRA2 movement; (ii) it could limit IRA2 plasticity through intramolecular contacts; and (iii) it could sterically prevent ADP release from NBD1. These interactions may allow the C-terminal domain to stabilize even weakly associating NBD1 and IRA2 pairs such as those of class I mutants (Figure 4B). This could explain how the C-terminal domain suppresses the reduced N68 ATPase of class I mutants (Table I).
Collectively, our data suggest a mechanism that may allow translocation conditions and ligands to switch off C-terminal domain suppression (Karamanou et al., 1999), thereby allowing IRA2 to activate the NBD1 ATPase. Interestingly, preprotein substrates (Shinkai et al., 1991), physiological temperature (Figure 2C) and the proton-motive force (Shiozuka et al., 1990) stimulate ADP release from SecA, and SecY binding to SecA stimulates its ATPase (Lill et al., 1990; Schiebel et al., 1991; Karamanou et al., 1999). Supressor analysis (Matsumoto et al., 1997; Nakatogawa et al., 2000) and binding studies (Dapic and Oliver, 2000) may even suggest that SecY acts directly on IRA2. IRA2 class I mutants may mimic SecA in its activated state during translocation. Tightly controlled activation of the NBD1 ATPase is expected to be particularly important if SecA moves processively along its polymeric substrates, as was shown for other DEAD proteins (Ali and Lohman, 1997; Bianco and Kowalczykowski, 2000; Dillingham et al., 2000; Jankowsky et al., 2000). Coupled nucleotide turnover would be essential to maintain an optimal duty ratio and cycling velocity, thus preventing accumulation of intermediates of the aminoacyl polymer in statu translocanti (Schiebel et al., 1991).
The SecA DEAD motor is essential for polypeptide translocation (Figure 6). This remarkable and exciting observation suggests that DEAD motors can operate on either nucleotidyl or aminoacyl polymers. DEAD motors are structurally conserved (Korolev et al., 1998; Caruthers et al., 2000; Singleton et al., 2000; Soultanas and Wigley, 2000) and contain non-conserved ‘add-on’ domains to provide biochemical specificities (Korolev et al., 1997, 1998; Kim et al., 1998; Velankar et al., 1999; Caruthers et al., 2000; Soultanas and Wigley, 2000). Presumably, SecA adjusted its DEAD RNA helicase motor (Park et al., 1997) to aminoacyl polymer translocation by acquiring a novel substrate recognition domain (PBD; Figure 7) and a membrane-targeting (Breukink et al., 1995; Snyders et al., 1997) C-terminal domain. Future understanding of translocase mechanics necessitates elucidation of how nucleotide-driven NBD1–IRA2 motions are communicated to the specificity domains and ‘sensed’ by preprotein substrates.
Materials and methods
Bacterial strains and recombinant DNA manipulations
Escherichia coli strains, growth and DNA manipulations were as described (Mitchell and Oliver, 1993; Ausubel et al., 1994; Karamanou et al., 1999). The identity of DNA constructs was confirmed by restriction analysis and sequencing.
Construction of His6-tagged SecA. A DNA fragment amplified by PCR using primers X26 (GCGGGATCCGCGCGCTGCTG) and X21 (AAGTGATGGTGATGGTGATGCATAATAAAATCTCAAACGCCCCG) was blunt-ended, digested with BamHI and inserted in BamHI–HpaI sites of pIMBB10, giving rise to pIMBB7. His-SecA is fully functional (not shown).
Construction of His6-tagged N-terminal N68 fragments. N1–263, N1–479 and N1–547 were constructed by PCR from pIMBB7 template, using as common forward primer X80 (5′-GGCCCGTACATATGCATCACCATCACCATCAC-3′) and reverse primers X81 (5′-CGCGACGGATCCGAAGTGGCCTTCGCCCTG-3′) for N1–263, X86 (5′-CGCGACGGA TCCCAGGACGTTGTGCTTAATAC-3′) for N1–479 and X130 (5′- CGCGACGGATCCCGGATCGTGACGTACCTGCC-3′) for N1–547. Gel-purified products digested with NdeI and BamHI were inserted into pET3a (Invitrogen), resulting in pIMBB86, pIMBB91 and pIMBB141, respectively.
Construction of His10-tagged C-terminal N68 fragments. N270–610 and N462–610 were constructed by PCR from pIMBB7 template using forward primers X88 (5′-GGCCCGTACATATGCGCCAGGTGAACCTGACC-3′) for N270–610, X93 (5′-GGCCCGTACATATGTCGGAGCTGGTGTCAAAC-3′) for N462–610 and common reverse primer X87 (5′-CCGGACCTCGAGCAGTTTACGCATCATGCC-3′). Gel-purified products digested with NdeI and XhoI were inserted into pET22b (Invitrogen), resulting in pIMBB92 and pIMBB97, respectively.
Construction of motif X/XI mutants. Mutants were constructed using Altered Sites (Promega) and pIMBB38 (secA cloned in BamHI–EcoRI sites of pALTER-EX1). For protein purification, BglII–BstXI fragments carrying secA mutations replaced the corresponding fragment of His-SecA in pIMBB7.
ATP hydrolysis determination
ATPase assays were in buffer B [50 mM Tris–HCl pH 8, 50 mM KCl, 5 mM MgCl2, 1 mM dithiothreitol (DTT)] with 1 mg/ml bovine serum albumin (BSA), supplemented with 1 mM ATP when using malachite green detection (Lill et al., 1990) or with the indicated concentrations when using TLC. [γ-32P]ATP or [α-32P]ATP was mixed with cold ATP at the indicated concentrations. Hydrolysis was stopped by proteinase K (1 mg/ml; 10 min; 4°C). CGF was performed using G-25 Ultra-Micro Spin (Amika) columns (4°C; buffer B). Protein recovery after CGF was 96–98%. Samples (1.5 µl) spotted on PEI cellulose (Merck) were developed in 0.6 M potassium phosphate pH 3.4. Hydrolysis was quantitated by scanning densitometry (Molecular Imager, Bio-Rad).
Optical biosensor assays
Surface plasmon resonance (IBIS II instrument; XanTec) and resonant mirror spectroscopy (iAsys instrument; Affinity Sensors) were at 22°C. Ligands (0.1 mg/ml in 10 mM sodium acetate pH 4.5) were immobilized on 6 kDa dextran-coated or amino surfaces following the manufacturers’ instructions. Ligate binding (0.5–1 µM; buffer B, 0.1 M NaCl, 5% glycerol) was monitored for 5 min. Binding response was calculated from 200 s.
Chemicals and biochemicals
Enzyme purification and characterization were as described (Karamanou et al., 1999). Proteases and nucleotides were from Roche. All other chemicals were from Sigma. DNA enzymes were from MINOTECH, dNTPs from Promega and Pfu polymerase from Stratagene. Sequenase, 35S-labelled dNTPs, [γ-32P]ATP (5000 Ci/mmol), [α-32P]ATP (3000 Ci/mmol), [14C]AMP (60 mCi/mmol) and [35S]methionine (1000 Ci/mmol) were from Amersham and [14C]ADP (60 mCi/mmol) was from New England Nuclear.
Acknowledgments
Acknowledgements
We are grateful to E.Gedig (Xantec), B.Pozidis, T.Roos, D.Alexandraki, G.Thireos and D.Tzamarias for suggestions and comments, G.Chalepakis for a gift of equipment, Y.Papanikolau for structural insights, and A.Kuhn, C.Stassinopoulou and M.Pelekanou for use of equipment. Our research is supported by grants (to A.E.) from the European Union (TMR-ERBFMRXCT960035, Biotech2-BIO4-CT97-2244, Biotech2-BIO4-CT98-0051, QLK3-CT-2000-00082 and RTN1-1999-00149), the Greek Secretariat of Research and Technology (GRI-088-97 and EPETII 97 EKBAN 2-17) and the University of Crete Research Fund (KA 1194).
References
- Ali J.A. and Lohman,T.M. (1997) Kinetic measurement of the step size of DNA unwinding by Escherichia coli UvrD helicase. Science, 275, 377–380. [DOI] [PubMed] [Google Scholar]
- Ausubel F.M., Brent,R., Kingston,R.E., Moore,D.D., Smith,J.A., Seidman,J.G. and Struhl,K. (1994) Current Protocols in Molecular Biology. John Wiley and Sons, NY.
- Bailey T.L. and Gribskov,M. (1999) Combining evidence using P-values: application to sequence homology searches. Bioinformatics, 14, 48–54. [DOI] [PubMed] [Google Scholar]
- Bianco P.R. and Kowalczykowski,S.C. (2000) Translocation step size and mechanism of the RecBC DNA helicase. Nature, 405, 368–372. [DOI] [PubMed] [Google Scholar]
- Breukink E., Nouwen,N., van Raalte,A., Mizushima,S., Tommassen,J. and de Kruijff,B. (1995) The C terminus of SecA is involved in both lipid binding and SecB binding. J. Biol. Chem., 270, 7902–7907. [DOI] [PubMed] [Google Scholar]
- Caruthers J.M., Johnson,E.R. and McKay,D.B. (2000) Crystal structure of yeast initiation factor 4A, a DEAD-box RNA helicase. Proc. Natl Acad. Sci. USA, 97, 13080–13085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Danese P.N. and Silhavy,T.J. (1998) Targeting and assembly of periplasmic and outer-membrane proteins in Escherichia coli. Annu. Rev. Genet., 32, 59–94. [DOI] [PubMed] [Google Scholar]
- Dapic V. and Oliver,D. (2000) Distinct membrane binding properties of N- and C-terminal domains of Escherichia coli SecA ATPase. J. Biol. Chem., 275, 25000–25007. [DOI] [PubMed] [Google Scholar]
- de la Cruz J., Kressler,D. and Linder,P. (1999) Unwinding RNA in Saccharomyces cerevisiae: DEAD-box proteins and related families. Trends Biochem. Sci., 24, 192–198. [DOI] [PubMed] [Google Scholar]
- den Blaauwen T., van der Wolk,J.P., van der Does,C., van Wely,K.H. and Driessen,A.J. (1999) Thermodynamics of nucleotide binding to NBS-I of the Bacillus subtilis preprotein translocase subunit SecA. FEBS Lett., 458, 145–150. [DOI] [PubMed] [Google Scholar]
- Dillingham M.S., Wigley,D.B. and Webb,M.R. (2000) Demonstration of unidirectional single-stranded DNA translocation by PcrA helicase: measurement of step size and translocation speed. Biochemistry, 39, 205–212. [DOI] [PubMed] [Google Scholar]
- Economou A. (1998) Bacterial preprotein translocase, mechanism and conformational dynamics of a processive enzyme. Mol. Microbiol., 27, 511–518. [DOI] [PubMed] [Google Scholar]
- Economou A. (2000) Bacterial protein translocase: a unique molecular machine with an army of substrates. FEBS Lett., 476, 18–21. [DOI] [PubMed] [Google Scholar]
- Economou A. and Wickner,W. (1994) SecA promotes preprotein translocation by undergoing ATP-driven cycles of membrane insertion and deinsertion. Cell, 78, 835–843. [DOI] [PubMed] [Google Scholar]
- Economou A., Pogliano,J.P., Beckwith,J., Oliver,D.B. and Wickner,W. (1995) SecA membrane cycling at SecYEG is driven by distinct ATP binding and hydrolysis events and is regulated by SecD and SecF. Cell, 83, 1171–1181. [DOI] [PubMed] [Google Scholar]
- Gerstein M., Lin,J. and Hegyi,H. (2000) Protein folds in the worm genome. Pac. Symp. Biocomput., 30–41. [DOI] [PubMed] [Google Scholar]
- Geyer M. and Wittinghofer,A. (1997) GEFs, GAPs, GDIs and effectors: taking a closer (3D) look at the regulation of Ras-related GTP-binding proteins. Curr. Opin. Struct. Biol., 7, 786–792. [DOI] [PubMed] [Google Scholar]
- Hall M.C. and Matson,S.W. (1999) Helicase motifs: the engine that powers DNA unwinding. Mol. Microbiol., 34, 867–877. [DOI] [PubMed] [Google Scholar]
- Hirano M., Matsuyama,S. and Tokuda,H. (1996) The carboxyl-terminal region is essential for SecA dimerization. Biochem. Biophys. Res. Commun., 229, 90–95. [DOI] [PubMed] [Google Scholar]
- Jankowsky E., Gross,C.H., Shuman,S. and Pyle,A.M. (2000) The DExH protein NPH-II is a processive and directional motor for unwinding RNA. Nature, 403, 447–451. [DOI] [PubMed] [Google Scholar]
- Johnson A.E. and van Waes,M.A. (1999) The translocon: a dynamic gateway at the ER membrane. Annu. Rev. Cell Dev. Biol., 15, 799–842. [DOI] [PubMed] [Google Scholar]
- Joly J.C. and Wickner,W. (1993) The SecA and SecY subunits of translocase are the nearest neighbors of the translocating preprotein, shielding it from phospholipids. EMBO J., 12, 255–263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karamanou S., Vrontou,E., Sianidis,G., Baud,C., Roos,T., Kuhn,A., Politou,A. and Economou,A. (1999) A molecular switch in SecA protein couples ATP hydrolysis to protein translocation. Mol. Microbiol., 34, 1133–1145. [DOI] [PubMed] [Google Scholar]
- Kim J.L., Morgenstern,K.A., Griffith,J.P., Dwyer,M.D., Thomson,J.A., Murcko,M.A., Lin,C. and Caron,P.R. (1998) Hepatitis C virus NS3 RNA helicase domain with a bound oligonucleotide: the crystal structure provides insights into the mode of unwinding. Structure, 6, 89–100. [DOI] [PubMed] [Google Scholar]
- Kimura E., Akita,M., Matsuyama,S.-i. and Mizushima,S. (1991) Determination of a region in SecA that interacts with presecretory proteins in Escherichia coli. J. Biol. Chem., 266, 6600–6606. [PubMed] [Google Scholar]
- Koonin E.V. and Gorbalenya,A.E. (1992) Autogenous translation regulation by Escherichia coli ATPase SecA may be mediated by an intrinsic RNA helicase activity of this protein. FEBS Lett., 298, 6–8. [DOI] [PubMed] [Google Scholar]
- Koonin E.V. and Rudd,K.E. (1996) Two domains of superfamily I helicases may exist as separate proteins. Protein Sci., 5, 178–180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Korolev S., Hsieh,J., Gauss,G.H., Lohman,T.M. and Waksman,G. (1997) Major domain swiveling revealed by the crystal structures of complexes of E.coli Rep helicase bound to single-stranded DNA and ADP. Cell, 90, 635–647. [DOI] [PubMed] [Google Scholar]
- Korolev S., Yao,N., Lohman,T.M., Weber,P.C. and Waksman,G. (1998) Comparisons between the structures of HCV and Rep helicases reveal structural similarities between SF1 and SF2 super-families of helicases. Protein Sci., 7, 605–610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lill R., Dowhan,W. and Wickner,W. (1990) The ATPase activity of SecA is regulated by acidic phospholipids, SecY and the leader and mature domains of precursor proteins. Cell, 60, 271–280. [DOI] [PubMed] [Google Scholar]
- Manting E.H. and Driessen,A.J. (2000) Escherichia coli translocase: the unravelling of a molecular machine. Mol. Microbiol., 37, 226–238. [DOI] [PubMed] [Google Scholar]
- Manting E.H., van Der Does,C., Remigy,H., Engel,A. and Driessen,A.J. (2000) SecYEG assembles into a tetramer to form the active protein translocation channel. EMBO J., 19, 852–861. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matsumoto G., Yoshihisa,T. and Ito,K. (1997) SecY and SecA interact to allow SecA insertion and protein translocation across the Escherichia coli plasma membrane. EMBO J., 16, 6384–6393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matsuyama S., Kimura,E. and Mizushima,S. (1990) Complementation of two overlapping fragments of SecA, a protein translocation ATPase of Escherichia coli, allows ATP binding to its amino-terminal region. J. Biol. Chem., 265, 8760–8765. [PubMed] [Google Scholar]
- Meyer T.H., Menetret,J.F., Breitling,R., Miller,K.R., Akey,C.W. and Rapoport,T.A. (1999) The bacterial SecY/E translocation complex forms channel-like structures similar to those of the eukaryotic Sec61p complex. J. Mol. Biol., 285, 1789–1800. [DOI] [PubMed] [Google Scholar]
- Mitchell C. and Oliver,D. (1993) Two distinct ATP-binding domains are needed to promote protein export by Escherichia coli SecA ATPase. Mol. Microbiol., 10, 483–497. [DOI] [PubMed] [Google Scholar]
- Nakatogawa H., Mori,H. and Ito,K. (2000) Two independent mechanisms down-regulate the intrinsic SecA ATPase activity. J. Biol. Chem., 275, 33209–33212. [DOI] [PubMed] [Google Scholar]
- Park S.K., Kim,D.W., Choe,J. and Kim,H. (1997) RNA helicase activity of Escherichia coli SecA protein. Biochem. Biophys. Res. Commun., 235, 593–597. [DOI] [PubMed] [Google Scholar]
- Schiebel E., Driessen,A.J.M., Hartl,F.-U. and Wickner,W. (1991) ΔµH+ and ATP function at different steps of the catalytic cycle of preprotein translocase. Cell, 64, 927–939. [DOI] [PubMed] [Google Scholar]
- Schmidt M., Ding,H., Ramamurthy,V., Mukerji,I. and Oliver,D. (2000) Nucleotide binding activity of SecA homodimer is conformationally regulated by temperature and altered by prlD and azi mutations. J. Biol. Chem., 275, 15440–15448. [DOI] [PubMed] [Google Scholar]
- Shilton B., Svergun,D.I., Volkov,V.V., Koch,M.H.J., Cusack,S. and Economou,A. (1998) Escherichia coli SecA shape and dimensions. FEBS Lett., 436, 277–282. [DOI] [PubMed] [Google Scholar]
- Shinkai A., Mei,L.H., Tokuda,H. and Mizushima,S. (1991) The conformation of SecA, as revealed by its protease sensitivity, is altered upon interaction with ATP, presecretory proteins, everted membrane vesicles and phospholipids. J. Biol. Chem., 266, 5827–5833. [PubMed] [Google Scholar]
- Shiozuka K., Tani,K., Mizushima,S. and Tokuda,H. (1990) The proton motive force lowers the level of ATP required for the in vitro translocation of a secretory protein in Escherichia coli. J. Biol. Chem., 265, 18843–18847. [PubMed] [Google Scholar]
- Singleton M.R., Sawaya,M.R., Ellenberger,T. and Wigley,D.B. (2000) Crystal structure of T7 gene 4 ring helicase indicates a mechanism for sequential hydrolysis of nucleotides. Cell, 101, 589–600. [DOI] [PubMed] [Google Scholar]
- Snyders S., Ramamurthy,V. and Oliver,D. (1997) Identification of a region of interaction between Escherichia coli SecA and SecY proteins. J. Biol. Chem., 272, 11302–11306. [DOI] [PubMed] [Google Scholar]
- Soultanas P. and Wigley,D.B. (2000) DNA helicases: ‘inching forward’. Curr. Opin. Struct. Biol., 10, 124–128. [DOI] [PubMed] [Google Scholar]
- van der Wolk J.P., de Wit,J.G. and Driessen,A.J. (1997) The catalytic cycle of the Escherichia coli SecA ATPase comprises two distinct preprotein translocation events. EMBO J., 16, 7297–7304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Velankar S.S., Soultanas,P., Dillingham,M.S., Subramanya,H.S. and Wigley,D.B. (1999) Crystal structures of complexes of PcrA DNA helicase with a DNA substrate indicate an inchworm mechanism. Cell, 97, 75–84. [DOI] [PubMed] [Google Scholar]
- Walker J.E., Saraste,M., Runswick,M.J. and Gay,N.J. (1982) Distantly related sequences in the α- and β-subunits of ATP synthase, myosin, kinases and other ATP-requiring enzymes and a common nucleotide binding fold. EMBO J., 1, 945–951. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yao N., Hesson,T., Cable,M., Hong,Z., Kwong,A.D., Le,H.V. and Weber,P.C. (1997) Structure of the hepatitis C virus RNA helicase domain. Nature Struct. Biol., 4, 463–467. [DOI] [PubMed] [Google Scholar]