

Abstract
The LrpA protein from the hyperthermophilic archaeon Pyrococcus furiosus belongs to the Lrp/AsnC family of transcriptional regulatory proteins, of which the Escherichia coli leucine-responsive regulatory protein is the archetype. Its crystal structure has been determined at 2.9 Å resolution and is the first for a member of the Lrp/AsnC family, as well as one of the first for a transcriptional regulator from a hyperthermophile. The structure consists of an N-terminal domain containing a helix–turn–helix (HtH) DNA-binding motif, and a C-terminal domain of mixed α/β character reminiscent of a number of RNA- and DNA-binding domains. Pyrococcus furiosus LrpA forms a homodimer mainly through interactions between the antiparallel β-sheets of the C-terminal domain, and further interactions lead to octamer formation. The LrpA structure suggests how the protein might bind and possibly distort its DNA substrate through use of its HtH motifs and control gene expression. A possible location for an effector binding site is proposed by using sequence comparisons with other members of the family coupled to mutational analysis.
Keywords: helix–turn–helix/Lrp–AsnC family/Pyrococcus furiosus/transcriptional regulator/X-ray crystallography
Introduction
Proteins from the Lrp/AsnC family, which act as global or specific regulators of transcription, have been isolated from many prokaryotes, including both bacteria and archaea (Brinkman et al., 2000 and references therein). The most extensively studied example of this family of proteins is the leucine-responsive regulatory protein (Lrp) from Escherichia coli (Calvo and Matthews, 1994; Newman and Lin, 1995). Lrp is a global regulator that acts to control gene expression. The Lrp regulon consists of ∼75 transcriptional units which are either activated or repressed by Lrp, often in response to the presence or absence of the effector leucine, which E.coli Lrp is believed to bind (Calvo and Matthews, 1994). The proteins they encode are mainly involved in transport, degradation or biosynthesis of amino acids. Lrp has been shown to exhibit negative, leucine-independent autoregulation, by binding upstream of its own promoter (–80 to –32 relative to the transcription start site as determined by DNase I footprinting; Wang et al., 1994). This large footprint region is believed to encompass a number of distinct Lrp binding sites. Lrp interacts with DNA as a homodimer, recognizing a 15 base pair imperfect inverted repeat sometimes found in multiple copies and bound in a cooperative manner (Wang and Calvo, 1993; Calvo and Matthews, 1994; Cui et al., 1995; Newman and Lin, 1995). Escherichia coli Lrp shows notable sequence similarity to the E.coli AsnC protein (25% identity) and, as a result, an evolutionary family relationship between the two proteins has been proposed (Willins et al., 1991). AsnC is responsible for the asparagine-dependent regulation of the asnA gene, the structural gene for asparagine synthetase A, and for its own autoregulation (de Wind et al., 1985; Kolling and Lother, 1985). No structures of proteins belonging to this family have been previously reported.
In Pyrococcus furiosus a putative Lrp, LrpA, the product of the lrpA gene, which exhibits 28% sequence identity to E.coli Lrp, has been isolated (Brinkman et al., 2000). Gel filtration experiments with concentrated protein samples suggest that LrpA forms a mixture of dimeric, tetrameric and octameric species at neutral pH, and an octamer below pH 6.0 (S.E.Sedelnikova, S.H.J.Smits, P.M.Leonard, A.B.Brinkman, J.van der Oost, J.B.Rafferty and D.W.Rice, in preparation). Pyrococcus furiosus LrpA has also been shown to exhibit negative autoregulation and binds to the lrpA promoter at a single site (–22 to +24 relative to the transcription start site; Brinkman et al., 2000), as determined by DNase I and hydroxyl radical footprinting. Although apparently quite a large binding site, attempts at trimming down its size from 46 to 30 bp, encompassing the most strongly protected region, result in a substantial decrease in binding by LrpA (Brinkman et al., 2000). Thus, multiple copies of LrpA may bind and possibly distort the lrpA promoter region as suggested for its E.coli homologue (Wang and Calvo, 1993). The negative autoregulation exhibited by LrpA appears to be independent of effectors, and there is no evidence for binding of leucine or any other amino acid by LrpA (Brinkman et al., 2000).
A helix–turn–helix (HtH) motif is responsible for the specific DNA interaction of many transcriptional regulators, such as the E.coli catabolite activator protein (McKay and Steitz, 1981) and the tryptophan repressor (Schevitz et al., 1985). Sequence alignments of proteins belonging to the Lrp/AsnC family as well as detailed mutagenesis studies of E.coli Lrp have suggested that these proteins also utilize an archetypal HtH motif to interact with DNA (Platko and Calvo, 1993). In P.furiosus LrpA this motif has been predicted to be located between residues 21 and 40 (Brinkman et al., 2000).
This paper reports the structure determination to 2.9 Å resolution of P.furiosus LrpA, a description of its overall fold, its structural similarity to other proteins and the possible mode of interaction between LrpA and DNA. The LrpA structure is the first example of a member of the Lrp/AsnC family, and one of the first transcriptional regulators from archaeal or hyperthermophile origin. The LrpA structure provides insights into the possible location of an effector binding site and how this widely conserved prokaryotic transcriptional regulator controls gene expression.
Results
Overall structure
The LrpA subunit has overall dimensions 60 × 30 × 45 Å and comprises two domains (Figure 1A). The N-terminal domain is formed from three α-helices (αA–αC). The C-terminal domain is formed from a four-stranded anti-parallel β-sheet (β2–β5) flanked on one face by two α-helices (αD–αE) and a short C-terminal β-strand (β6). The subunit has the connectivity β2–αD–β3–β4–αE– β5–β6 (Figure 1B). Two 310 helical turns are present, one between β1 and β2, and the other between β2 and αD. There are a limited number of contacts between the two domains, which are linked by only a single β-strand (β1).

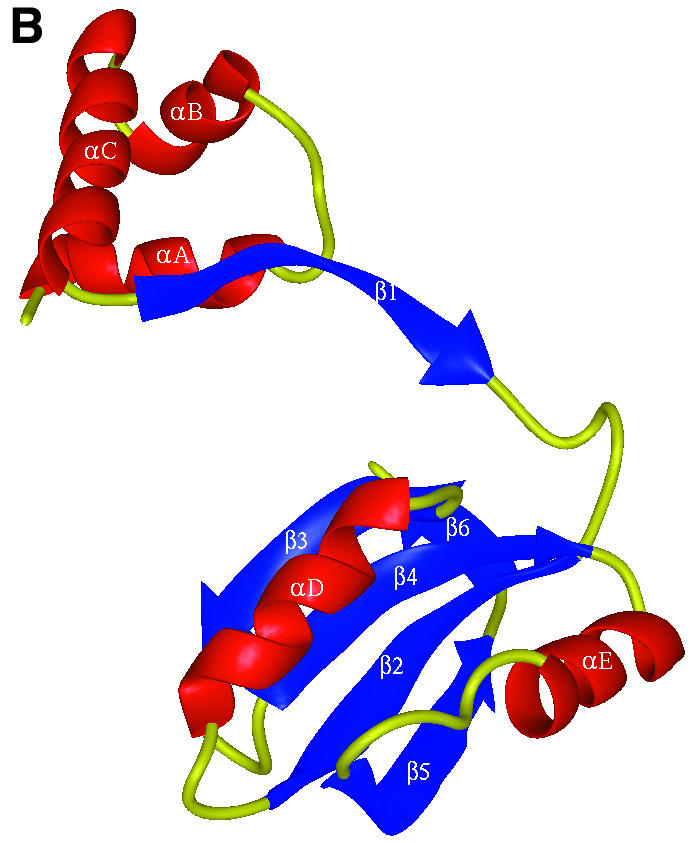

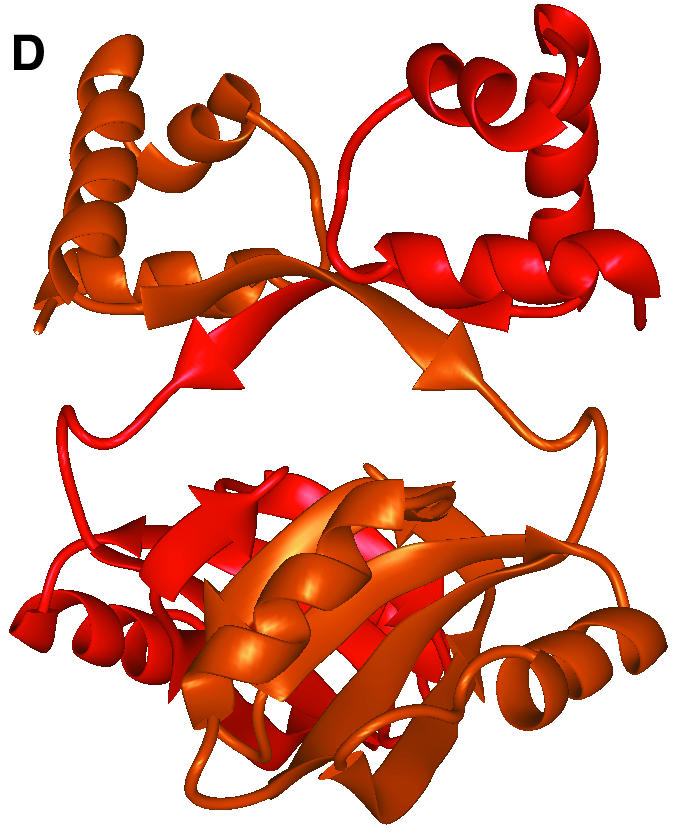
Fig. 1. The overall fold of P.furiosus LrpA. (A) Schematic stereo representation of the Cα backbone of a monomer with every 10th residue labelled. (B) Schematic representation of the fold of a monomer with α-helices and β-strands shown as labelled coils and arrows (red and blue, respectively). (C) The LrpA octamer viewed as in (B) but with subunits labelled A–H and coloured red, orange, yellow, green, blue, cyan, violet and purple, respectively. (D) Schematic representation of the fold of a dimer with the two monomers shown in red and orange. [Figures were produced using MIDAS (Ferrin et al., 1988)].
In the crystal lattice there is an obvious octamer with approximate dimensions 96 × 96 × 110 Å. The octamer has 42 symmetry and is most conveniently described as being formed from a tetramer of dimers (Figure 1C). The solvent-accessible surface area of an isolated monomer, calculated using the programme AREAIMOL with a probe radius of 1.4 Å (Lee and Richards, 1971), is 9400 Å2. On formation of the octamer, 3200 Å2 (34%) of the solvent-accessible surface is buried per monomer. Of the three distinct molecular 2-fold axes in the octamer, two are crystallographic and the other is non-crystallographic. Adjacent monomers related by the crystallographic 2-fold axes in the octamer form a dimer that buries 2100 Å2 (22%) of the solvent-accessible surface per monomer (Figure 1D). Adjacent dimers related by the non-crystallographic 2-fold axis in the octamer form a dimer–dimer interface that buries a further 1100 Å2 (12%) of the solvent-accessible surface per monomer (Figure 1C). These interfaces are described below.
Dimer interface
The interactions that form the dimer interface can be divided into three main regions. First, a hydrophobic core is held together by interactions between residues in strands β2, β3, β4 and β5 from each monomer and, in addition, the β-sheets are extended to form five-stranded antiparallel β-sheets by main chain hydrogen bonding of strand β6 to strand β3 in the other monomer. Secondly, extensive hydrogen bonding interactions can be seen in the anti-parallel β-ribbon formed by the β1 strands from both subunits. The third region of contact is hydrophobic in character and is formed between the N- and C-terminal domains of symmetry-related subunits. Specifically, residues from helix αA and the following turn in the N-terminal domain of one monomer interact with the first 310 helical turn and residues from strand β1 of the inter-domain β-ribbon in the C-terminal domain of the second monomer.
Dimer–dimer interface and octamer formation
In forming the octamer, four equivalent dimer–dimer contacts are formed that are hydrophobic in nature. For any given monomer, contacts between subunits related by a molecular 4-fold axis involve helix E and strand β5 with the 310 helical turn between strand β2 and helix D and the turn between strands β3 and β4. Helix E makes a further contact with a non-crystallographically related monomer in an adjacent dimer via interactions with the C-terminal residues.
Structure comparison
The structure of LrpA was compared with those of all the proteins in the Protein Data Bank (PDB) (Bernstein and Tasumi, 1977) using the programme PROTEP (Grindley et al., 1993). Although there was no overall match with the entire structure of LrpA, a number of hits were observed for the C-terminal domain. The three best hits were found with the N-terminal domains of the archaeal DNA polymerase B enzymes from Thermococcus gorgonarius (Hopfner et al., 1999) and Desulfurococcus strain Tok (Zhao et al., 1999), which have been proposed to bind RNA (Zhao et al., 1999), and the ribosomal protein S6 from the small ribosomal subunit of Thermus thermophilus (Lindahl et al., 1994), which, in conjunction with the S18 protein, binds the ribosomal 16s RNA (Powers and Noller, 1995). The structural motif common to these proteins consists of a four-stranded anti-parallel β-sheet with two α-helices packed on one side. This architecture is present in a number of small single-stranded RNA-binding modules including S6, which, within this structural motif, contain conserved sequences known as RNP1 and RNP2. The structure of the RNA-binding domain (RBD) of the U1A spliceosomal protein complexed with an RNA hairpin is representative of an RBD–RNA complex (Oubridge et al., 1994) and shows that the sequence motifs are positioned on the first and third β-strand of the βαββαβ fold, making strong contacts with the bound RNA molecule. Such conserved sequences are not present in LrpA and, given that the surface of the β-sheet that binds the RNA in an RBD is involved in the formation of the dimer interface in LrpA, it is unlikely that LrpA binds RNA molecules via its C-terminal domain. A βαββαβ fold similar to that in the C-terminus of LrpA has also been observed in the C-terminal DNA-binding domain of bovine papillomavirus-1 E2, whose structure has been solved at 1.7 Å bound to its smoothly bent DNA target (Hegde et al., 1992). Like the C-terminal domain of LrpA, in E2 an equivalent region of the subunit surface is also involved in the formation of a dimer, but the subunit– subunit contacts are predominantly hydrophilic. In E2 this C-terminal domain is also involved in binding DNA through interactions between the first helix of the βαββαβ fold of each monomer and the major groove of the DNA double helix. In contrast, the equivalent region in LrpA is involved in dimer–dimer interaction within the octamer, further demonstrating the versatility of this structural motif.
The structure of P.furiosus LrpA reveals the presence of an HtH motif between residues 21 and 45 in the N-terminal domain, formed by helices αB and αC. This N-terminal region appears to form a distinct ‘headpiece’ to the molecule. The HtH motif in LrpA can be superimposed on those from the E.coli catabolite activator protein (McKay and Steitz, 1981) and the tryptophan repressor (Schevitz et al., 1985) with a root mean square deviation (r.m.s.d.) of 0.51 and 1.87 Å, respectively, for the 20 Cα atoms of the motif (residues 21–40 in LrpA). The use of distinct HtH-containing ‘headpiece’ domains to bind the DNA has been observed in a number of prokaryotic transcriptional regulators including the lac repressor (Friedman et al., 1995).
Mutational analysis of Lrp/AsnC family proteins
The structure of P.furiosus LrpA is the first to be solved for a member of the Lrp/AsnC family. This structural information can be used to make a structure-based sequence alignment (Figure 2A), allowing comparisons to be made between members of the family, and facilitating the interpretation of biochemical data that exist for E.coli Lrp in the light of the current P.furiosus LrpA model. This close sequence similarity within the Lrp family combined with mutation studies carried out on E.coli Lrp (Platko and Calvo, 1993) allows us to investigate the location of both the effector and DNA binding sites. Escherichia coli Lrp has been randomly mutated and the resulting mutants tested on the basis of their effects on expression of ilvIH, one of the operons regulated positively by Lrp (Platko et al., 1990). The ilvIH operon encodes an enzyme involved in the biosynthesis of leucine, valine and isoleucine, and expression of this operon is repressed when cells are grown in the presence of leucine. Mutant strains that were resistant to the repressive effects of leucine were termed leucine response mutants. Those mutants for which binding to ilvIH DNA in vitro was markedly reduced were termed DNA binding mutants. A further class of mutants that had low ilvIH expression in vivo but apparently normal DNA binding in vitro were termed activation mutants, owing to their inability to activate transcription. The positions of these mutations have been modelled onto the LrpA structure and provide insights into the patterns of effector molecule and DNA binding. These are described in the following sections.

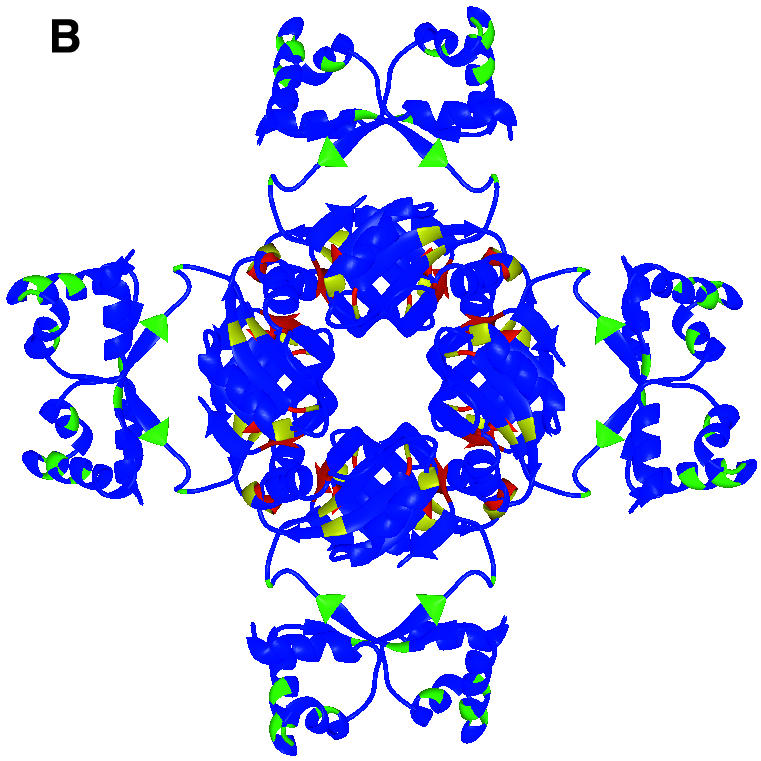

Fig. 2. Sequence alignment and location of DNA binding, activation and leucine response mutations. (A) Structure-based multiple alignment of Lrp/AsnC family sequences. Elements of secondary structure in LrpA are shown as labelled cylinders (α-helices) and arrows (β-strands). Sequences are aligned from P.furiosus LrpA, E.coli Lrp and E.coli AsnC. Residues that are conserved across all three sequences have been boxed. Those residues that are conserved between Lrp and AsnC are shaded red. Those residues that are switched from a hydrophobic side chain in Lrp to a hydrophilic side chain in AsnC are shaded blue. The positions of the E.coli Lrp DNA binding mutants, activation mutants and leucine response mutants are indicated by the symbols +, $ and #, respectively. [The figure was produced using CINEMA (Parry-Smith et al., 1998) and ALSCRIPT (Barton, 1993)]. (B) The LrpA octamer as shown in Figure 1C but with all subunits coloured blue. The positions of DNA binding mutants, activation mutants and leucine response mutants are shown in green, yellow and red, respectively. (C) An LrpA dimer viewed along its 2-fold axis (i.e. rotated 90° around the x-axis with respect to Figure 1D). The monomers are shown in blue and cyan and the equivalent residues in LrpA to those identified in E.coli Lrp as leucine response mutants are shown in red with magenta side chains. The residue sequence numbers are those of LrpA. [(B) and (C) were produced using MIDAS (Ferrin et al., 1988)].
Analysis of the pattern of leucine response mutants and location of the effector binding site in the Lrp family. A total of seven E.coli Lrp mutants were isolated that were resistant to the repressive effects of leucine (Leu107*, Asp113*, Met123*, Leu135*, Tyr146*, Val147* and Val148*; in the following discussion E.coli Lrp and AsnC sequence numbers are denoted by * and ′, respectively, and correspond to Swiss-Prot entries P19494 and P03809). When these mutations are modelled onto the structure of the LrpA octamer (the equivalent LrpA residues are Leu95, Met101, Gly111, Gly123, Ala134, Ile135 and Ile136) all seven residues are located at subunit interfaces (Figure 2B and C). Five out of the seven are found to be clustered in a single region across the dimer interface. The remaining two residues, Met123*/Gly111 and Leu 135*/Gly123, are located close to the positions of the other five mutations but in adjacent dimers of the octamer rather than within the same dimer. It is possible, therefore, that effector binding may influence formation of larger multimeric species through additional interaction with residues Met123*/Gly111 and Leu135*/Gly123, although it cannot be ruled out that mutation of these residues may have long-range effects upon leucine binding at the remote site on the dimer interface.
In addition to the above study we attempted to locate possible effector binding sites through comparison of the E.coli Lrp and AsnC sequences. We reasoned that if AsnC and Lrp bound their respective effectors at a similar site, such a site might utilize conserved interactions possibly involving charged residues on the protein and the common amino and carboxyl moieties of the effector. In addition, we might anticipate that the specificity for asparagine or leucine displayed by AsnC and Lrp, respectively, might rely on a binding pocket for the side chain of the effector which would switch in nature from hydrophilic to hydrophobic. Charged residues conserved between Lrp and AsnC (such conservation need not necessarily extend to LrpA since at present there is no evidence that P.furiosus LrpA is affected by leucine or any other molecule), and those residues that are hydrophobic in Lrp but hydrophilic in AsnC, were both plotted onto the LrpA structure. Three sites of conserved charged residues designated A, B and C, which could form possible effector molecule binding sites, were located (A, Asp 12*/Asp 7′, Asp15*/Asp10′ and Arg47*/Arg42′; B, Arg27*/Arg22′ and Glu32*/Glu27′; C, Glu104*/Glu97′ and Lys117*/Lys110′). Close to each of these sites, a residue that switched in nature between hydrophobic and hydrophilic could also be observed (A, Pro43*/Thr38′; B, Ile28*/Thr23′; C, Leu63*/Asp58′). The three sites are all situated within the N-terminal domain or immediately adjacent to it. Thus, if one of the sites represents the binding pocket for an effector molecule then it may well be that effector binding could influence the conformation of the N-terminal domain and, therefore, the relative positions of the DNA-binding helices. However, none of the three sites suggested by the sequence/structure alignment overlaps with that proposed on the basis of the leucine response mutant analysis. The sequence analysis that we have carried out does not preclude the use of main chain atoms or hydrophilic side chains in the binding of the amino and carboxyl groups on the amino acid effector molecule and, thus, these sites may be of little significance. However, it is possible that the previously observed leucine response mutants may have long-range effects and that mutation of residues in the true site is lethal and hence went undetected.
Analysis of DNA binding and activation mutants. A representation of the electrostatic surface charge potential of LrpA as computed by GRASP (Nicholls et al., 1991) reveals the residues in the recognition helix (αC) of the HtH motif to be predominantly positively charged. In contrast, helix D in the C-terminal domain, which is analogous to the DNA-binding helix in the bovine papil lomavirus-1 E2 DNA-binding domain, is predominantly negatively charged, whereas in bovine papillomavirus-1 E2 it is predominantly positively charged. This implies that P.furiosus LrpA is much more likely to bind DNA by interactions between the recognition helices of the HtH motif and the DNA. Analysis of the positions of the residues in the E.coli Lrp DNA binding mutants on the structure of LrpA reveals that, of the 10 mutants (Asp12*, Leu33*, Leu39*, Ser40*, Pro43*, Leu45*, Arg47*, Tyr60*, Leu64* and Leu69* in the E.coli Lrp sequence, equivalent to Asp3, Ile24, Ile30, Ser31, Ala34, Arg36, Arg38, Tyr51, Ile55 and Leu60 in the LrpA sequence), six are positioned in the HtH motif. Three of these are on the recognition helix αC (Pro43*, Leu45*, Arg47*) and are likely to be directly involved in DNA binding. The remaining four all lie close to the HtH motif and could disturb DNA recognition through long-range conformational effects. Analysis of the positions of the five activation mutants (Val75*, Phe89*, Phe112*, Thr118* and Ser124* in the E.coli Lrp sequence, equivalent to Thr66, Leu77, His100, Ile106 and Glu112 in the LrpA sequence) on the structure of LrpA did not reveal any obvious clustering of residues.
LrpA–DNA complex model
The two recognition helices (αC) of the HtH motifs in the dimer are separated by ∼34 Å, which corresponds well to the distance between adjacent turns of the major groove in B-form DNA. In contrast, the helices in the C-terminal domain, which are analogous to the DNA-binding helices in the bovine papillomavirus-1 E2 DNA-binding domain, are separated by ∼42 Å. Thus, we have modelled straight B-form DNA onto the surface of the LrpA dimer containing the two recognition helices, such that the 2-fold axis of the dimer is coincident with a local 2-fold in the DNA, and the two recognition helices access adjacent turns of the major groove (Figure 3). However, inspection of the interactions within this complex suggests that they may not be optimal, particularly when compared with the structures of protein–DNA complexes that utilize the HtH motif. It has been demonstrated that E.coli Lrp can induce bending of DNA upon DNA binding (Calvo and Matthews, 1994), and recent studies have suggested that P.furiosus LrpA may also be able to bend DNA (Brinkman et al., 2000). Thus, it may be that in the true complex, DNA binding would involve interaction with a bent rather than straight DNA. Alternatively, it is also possible that upon binding DNA some small rearrangement of the recognition helices occurs, as there may well be some flexibility between the N- and C-terminal domains. Consistent with this idea, there are only limited contacts between the two domains of LrpA, and this latter point is given some further support by a superposition of the two molecules in the asymmetric unit, which gives an r.m.s.d. of 0.6 Å, whereas the independent superpositions of their N- and C-terminal domains gives r.m.s.d. values of 0.5 and 0.4 Å, respectively.

Fig. 3. Modelling of DNA binding by LrpA. A straight piece of B-form DNA modelled onto an LrpA dimer such that the 2-fold axis of the dimer is coincident with a local 2-fold in the DNA and the two recognition helices access adjacent turns of the major groove. [The figure was produced using MIDAS (Ferrin et al., 1988)].
Thermostability
Differential scanning calorimetry measurements of the LrpA protein from P.furiosus have shown that it is extremely thermostable, with a melting temperature, Tm, of 111.5°C (A.B.Brinkman and J.van der Oost, unpublished data). Previous studies have suggested that an increase in the extent of ion pair networks is a frequently observed feature of proteins from hyperthermophiles (Szilagyi and Zavodsky, 2000). An analysis of the LrpA structure reveals there to be only 0.057 ion pairs per residue (using a distance limit of 4.0 Å). This is considerably lower than that observed for other hyperthermophilic proteins (Yip et al., 1998). The analysis is limited somewhat by the relatively low resolution of the LrpA structure and by the absence of a number of charged side chains on the surface of LrpA, which are disordered in the crystal but are presumably ordered upon binding DNA or in interactions with other transcriptional components such as RNA polymerase. Thus, a proper structural understanding of the thermostability of LrpA must await the determination of a comparative structure from a mesophilic member of the Lrp/AsnC family.
Discussion
The work on LrpA presented here describes the first structure of a member of the Lrp/AsnC family of proteins and one of the first structures of a transcriptional regulator from an archaeal source. It reveals a striking octameric assembly formed from a tetramer of dimers. Analysis of the structure and comparisons with sequences of other Lrp/AsnC family members has confirmed the presence of an N-terminal HtH motif and its likely role in DNA binding. In addition, this study has highlighted a potential effector binding site on LrpA via interpretation of mutational analysis of its E.coli homologue, and three further potential sites through sequence analysis. The first site appears to straddle the dimer and dimer–dimer interfaces in the octamer and suggests a possible role in effecting the multimeric state of the protein, whereas the locations of the other three possible sites suggest the potential to influence the conformation of the N-terminal domain and, therefore, the relative positions of the DNA-binding helices.
Gel retardation experiments show that LrpA binds to the lrpA promoter region as a single species (Brinkman et al., 2000). Whilst chemical cross-linking analysis has suggested the presence of an LrpA tetramer in the protein–DNA complex (Brinkman et al., 2000), solution studies (S.E.Sedelnikova, S.H.J.Smits, P.M.Leonard, A.B.Brinkman, J.van der Oost, J.B.Rafferty and D.W.Rice, in preparation) and the crystal structure have shown LrpA to exist as an octamer. Further experiments are required to resolve the discrepancy in the molecular sizes determined in these studies. One can speculate that multimerization of the LrpA dimer contributes to stabilizing the DNA–protein complex in vivo. At present it is not clear whether LrpA interacts with a single or a multiple operator (Brinkman et al., 2000), but by comparison, E.coli Lrp has been demonstrated to bind cooperatively to adjacent operators (Calvo and Matthews, 1994). The generally eukaryotic-like and hence multi-component nature of the transcriptional machinery observed in the archaea (Bell and Jackson, 1998) prompts the suggestion that LrpA may also interact with other proteins to form macromolecular complexes during transcriptional regulation. Interaction with DNA-bound TATA binding protein (TBP) (Steger et al., 1995) has been proposed for the C-terminal domain of papillomavirus-1 E2 protein, which shares the βαββαβ fold topology with the C-terminal domain of LrpA. Future biochemical and crystallographic analyses will address the intriguing possibility that LrpA is involved in the formation of larger multimeric macromolecular complexes in vivo.
Materials and methods
Crystals of LrpA were grown by the hanging-drop vapour diffusion method from buffered ammonium sulfate solutions, at both basic and acidic pH, over a range from 4 to 9 as described elsewhere (S.E.Sedelnikova, S.H.J.Smits, P.M.Leonard, A.B.Brinkman, J.van der Oost, J.B.Rafferty and D.W.Rice, in preparation). The crystals belong to space group I4122, with cell dimensions a = b = 104.5 Å and c = 245.1 Å, with one dimer in the asymmetric unit and a Vm of 5.2 Å3/Da corresponding to a high solvent content of ∼70% (Matthews, 1977). The crystals had a dmin of 2.9 Å at the CLRC Daresbury synchrotron source.
The structure was solved using MIR techniques with two isomorphous derivatives. The first of these was prepared by co-crystallizing the protein in the presence of 0.1 mM ethyl mercuri phosphate (EMP). Data were collected at room temperature on the native and derivative crystals to 4.0 Å on a Mar345 image plate mounted on a Rigaku RU200 X-ray generator. Data were processed using the DENZO suite of programs (Otwinowski and Minor, 1997) and subsequently handled using CCP4 software (CCP4, 1994). The Patterson function for this derivative was readily soluble, giving two heavy atom sites, one arising from each of the two monomers in the asymmetric unit. The two heavy atom sites were refined and a preliminary phase set calculated using MLPHARE (Otwinowski, 1991). A second derivative was produced by soaking a native crystal for 2 h in mother liquor containing 1 mM potassium tetra-cyanoplatinate [K2Pt(CN)4]. Two heavy atom sites, one arising from each of the two monomers in the asymmetric unit, were found by difference Fourier methods, and the derivative was subsequently refined and an improved phase set with an overall figure of merit of 0.53 (acentric 0.51, centric 0.61) was calculated from the two derivatives.
In order to enhance radiation stability and hence improve the resolution of the structural analysis, the crystals were cryoprotected in mineral oil by removing them from the hanging drops with a cryo loop, removing excess precipitant using absorbent dental points, dragging the loop through an oil reservoir and placing it in the cryo stream. This enabled a 2.9 Å resolution native data set and 3.8 Å resolution K2Pt(CN)4 derivative data set to be collected. Both data sets were collected on a Mar image plate detector on station 7.2 at the CLRC Daresbury Laboratory. Finally, a 3.5 Å resolution EMP derivative data set was collected on a Mar345 image plate mounted on a Rigaku RU200 X-ray generator. The cryo-cooling produced an ∼3% shrinkage of the cell in the a and b dimensions (101.3 Å) but little change in c (245.4 Å) (Table I).
Table I. Data processing and heavy atom statistics.
| Native | Native (cryo) | Hga | Hga (cryo) | Ptb | Ptb (cryo) | |
|---|---|---|---|---|---|---|
| Resolution (Å) | 4.0 | 2.9 | 4.0 | 3.5 | 4.5 | 3.8 |
| No. of observed reflections | 16 996 | 30 436 | 10 680 | 25 216 | 14 757 | 13 699 |
| No. of unique reflections | 5846 | 13 804 | 5707 | 7395 | 4200 | 6172 |
| Completeness (%) | 98.3 (99.3) | 94.7 (96.2) | 94.8 (96.2) | 87.0 (92.6) | 99.4 (100) | 91.9 (94.5) |
| Rmerge (%)c | 9.2 (34.2) | 6.5 (34.6) | 8.6 (33.3) | 6.2 (14.9) | 11.1 (28.8) | 8.5 (21.1) |
| Reflection intensities I/σI > 3 (%) | 76.8 (47.5) | 75.5 (35.4) | 69.3 (35.9) | 74.2 (30.5) | 65.3 (46.7) | 69.6 (51.1) |
| Riso (%)d | 15.1 | 18.9 | 13.3 | 29.1 | ||
| No. of heavy atom sites (monomer) | 1 | 1 | 1 | 1 | ||
| Phasing power (acentric/centric)e | 2.40/1.89 | 1.26/0.90 | 0.96/0.75 | 1.53/1.09 | ||
| Rcullis (acentric/centric)f | 0.57/0.48 | 0.79/0.77 | 0.87/0.77 | 0.74/0.70 |
The data for the highest resolution shells are given in parentheses.
aHg, ethyl mercuri phosphate (C2H5HgPO4).
bPt, potassium tetra-cyanoplatinate [K2Pt(CN)4].
cRmerge = Σ|I – <I>|/ΣI, where I is the integrated intensity of a given reflection.
dRiso = Σ|FPH – FP|/ΣFP, where FPH and FP are the derivative and native structure factor amplitudes.
ePhasing power = (r.m.s. heavy atom structure factor)/(r.m.s. lack of closure).
fRcullis = (r.m.s. lack of closure)/(r.m.s. isomorphous difference).
An electron density map was calculated to 3.5 Å resolution and improved by solvent flattening and 2-fold non-crystallographic symmetry averaging using the program DM (Cowtan, 1994). The resolution of the map was improved by phase extension to 2.9 Å and showed clearly identifiable regions of regular secondary structure. The map was skeletonized using the programme MAPMAN (Kleywegt and Jones, 1994) and a polyalanine model for a single subunit was constructed with the program O (Jones et al., 1991). This model was rotated approximately into the electron density for the second subunit in the asymmetric unit using the program PDBSET, and its position refined using rigid body refinement in O. Subsequently, the sequence was fitted into the model where it could be unambiguously assigned, and when ∼80% of the total number of side chains had been determined with confidence the structure was submitted to maximum likelihood refinement using the program REFMAC (Murshudov et al., 1997). Iterative cycles of phase combination of the partial structure phases and those from the heavy atom derivatives, model building and refinement were used to construct a complete model representing 280 out of 282 expected residues. NCS restraints were applied between the subunits and an overall average B-factor (estimated from a Wilson plot of the data) of 62 Å2 was used. The electron density for a total of 18 side chains in the A and B subunits of the asymmetric unit was not observed in the final electron density map and was all truncated at the β-carbon. The final model has an R-factor of 31.3% (Rfree, 38.2%; Brunger, 1992) for all data in the resolution range 20–2.9 Å. The model has good stereochemistry, with values for the r.m.s.d. from standard values of the bond lengths and angles of 0.012 Å and 3.0°, respectively. Model geometry was analysed using the program PROCHECK (Laskowski et al., 1993). A Ramachandran plot of the model shows all non-glycine residues inside the normally allowed regions (87.6 and 12.4% in the most favoured and additional allowed regions, respectively) and examination of χ1–χ2 plots for all residue types showed no side chains in unfavourable conformations.
The coordinates and structure factor amplitudes have been submitted to the RCSB PDB; code 111G.
Acknowledgments
Acknowledgements
We thank BBSRC, Wellcome Trust and The New Energy and Industrial Development Organization for their support. J.B.R. is the Royal Society Olga Kennard Fellow. The Krebs Institute is a BBSRC designated Biomolecular Sciences Centre, and its structural studies group is a member of the BBSRC North of England Structural Biology Centre. Part of this research was supported by grant 700-35-101 of the Council for Chemical Sciences (C.W.) of The Netherlands Organisation for Scientific Research (N.W.O.).
References
- Barton G.J. (1993) Alscript: a tool to format multiple sequence alignments. Protein Eng., 6, 37–40. [DOI] [PubMed] [Google Scholar]
- Bell S.D. and Jackson,S.P. (1998) Transcription and translation in Archaea: a mosaic of eukaryal and bacterial features. Trends Microbiol., 6, 222–228. [DOI] [PubMed] [Google Scholar]
- Bernstein F.C. and Tasumi,M. (1977) The Protein Data Bank: a computer based archival file for macromolecular structures. J. Mol. Biol., 112, 535–542. [DOI] [PubMed] [Google Scholar]
- Brinkman A.B. et al. (2000) An lrp-like transcriptional regulator from the archaeon Pyrococcus furiosus is negatively autoregulated. J. Biol. Chem., 275, 38160–38169. [DOI] [PubMed] [Google Scholar]
- Brunger A.T. (1992) Free R-value—a novel statistical quantity for assessing the accuracy of crystal structures. Nature, 355, 472–475. [DOI] [PubMed] [Google Scholar]
- Calvo J.M. and Matthews,R.G. (1994) The leucine-responsive regulatory protein, a global regulator of metabolism in Escherichia coli. Microbiol. Rev., 58, 466–449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Collaborative Computational Project No. 4 (1994) The CCP4 suite: programs for protein crystallography. Acta Crystallogr. D, 50, 760–763. [DOI] [PubMed] [Google Scholar]
- Cowtan K.D. (1994) An automated procedure for phase improvement by density modification. Joint CCP4 and ESF-EACBM Newsletter on Protein Crystallography, 31, 34–38. [Google Scholar]
- Cui Y.H., Wang,Q., Stormo,G.D. and Calvo,J.M. (1995) A consensus sequence for binding of Lrp to DNA. J. Bacteriol., 177, 4872–4880. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Wind N., de Jong,M., Meijer,M. and Stuitje,A.R. (1985) Site-directed mutagenesis of the Escherichia coli chromosome near oriC: identification and characterization of asnC, a regulatory element in E.coli asparagine metabolism. Nucleic Acids Res., 13, 8797–8811. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ferrin T.E., Huang,C.C., Jarvis,L.E. and Langridge,R. (1988) The MIDAS display system. J. Mol. Graph., 6, 13–27. [Google Scholar]
- Friedman A.M., Fischmann,T.O. and Steitz,T.A. (1995) Crystal structure of lac repressor core tetramer and its implications for DNA looping. Science, 268, 1721–1727. [DOI] [PubMed] [Google Scholar]
- Grindley H.M., Artymiuk,P.J., Rice,D.W. and Willett,P. (1993) Identification of tertiary structure resemblance in proteins using a maximal common subgraph isomorphism algorithm. J. Mol. Biol., 229, 707–721. [DOI] [PubMed] [Google Scholar]
- Hegde R.S., Grossman,S.R., Laimins,L.R. and Sigler,P.B. (1992) Crystal structure at 1.7-angstrom of the bovine papillomavirus-1 E2 DNA-binding domain bound to its DNA target. Nature, 359, 505–512. [DOI] [PubMed] [Google Scholar]
- Hopfner K.-P., Eichinger,A., Engh,R.A., Laue,F., Ankenbauer,W., Huber,R. and Angerer,B. (1999) Crystal structure of a thermostable type B DNA polymerase from Thermococcus gorgonarius. Proc. Natl Acad. Sci. USA, 96, 3600–3605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jones T.A., Zou,J.Y., Cowan,S.W. and Kjeldgaard,M. (1991) Improved methods for building protein models in electron-density maps and the location of errors in these models. Acta Crystallogr. A, 47, 110–119. [DOI] [PubMed] [Google Scholar]
- Kleywegt G.J. and Jones,T.A. (1994) Halloween … masks and bones. In Bailey,S., Hubbard,R. and Waller,D. (eds), From First Map to Final Model. SERC Daresbury Laboratory, Warrington, UK, pp. 59–66.
- Kolling R. and Lother,H. (1985) AsnC: an autogenously regulated activator of asparagine synthetase A transcription in Escherichia coli. J. Bacteriol., 164, 310–315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Laskowski R.A., MacArthur,M.W., Moss,D.S. and Thornton,J.M. (1993) PROCHECK: a program to check the stereochemical quality of protein structures. J. Appl. Crystallogr., 26, 283–291. [Google Scholar]
- Lee B. and Richards,F.M. (1971) The interpretation of protein structures: estimation of static accessibility. J. Mol. Biol., 55, 379–400. [DOI] [PubMed] [Google Scholar]
- Lindahl M. et al. (1994) Crystal-structure of the ribosomal-protein S6 from Thermus thermophilus. EMBO J., 13, 1249–1254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matthews B.W. (1977) X-ray structure of proteins. In Neurath,H. and Hill,R.L. (eds), The Proteins. Vol. 3. Academic Press, New York, NY, pp. 468–477.
- McKay D.B. and Steitz,T.A. (1981) Structure of catabolite gene activator protein at 2.9 Å resolution suggests binding to left-handed B-DNA. Nature, 290, 744–749. [DOI] [PubMed] [Google Scholar]
- Murshudov G.N., Vagin,A.A. and Dodson,E.J. (1997) Refinement of macromolecular structures by the maximum-likelihood method. Acta Crystallogr. D, 53, 240–255. [DOI] [PubMed] [Google Scholar]
- Newman E.B. and Lin,R. (1995) Leucine-responsive regulatory protein: a global regulator of gene-expression in Escherichia coli. Annu. Rev. Microbiol., 49, 747–775. [DOI] [PubMed] [Google Scholar]
- Nicholls A., Sharp,K.A. and Honig,B. (1991) Protein folding and association: insights from the interfacial and thermodynamic properties of hydrocarbons. Proteins, 11, 281–296. [DOI] [PubMed] [Google Scholar]
- Otwinowski Z. (1991) Maximum likelihood refinement of heavy atom parameters. In Wolf,W., Evans,P.R. and Leslie,A.G.W. (eds), Proceedings of the CCP4 Study Weekend: Isomorphous Replacement and Anomalous Scattering I. SERC Daresbury Laboratory, Warrington, UK, pp. 80–86.
- Otwinowski Z. and Minor,W. (1997) Processing of X-ray diffraction data collected in oscillation mode. Methods Enzymol., 276, 307–326. [DOI] [PubMed] [Google Scholar]
- Oubridge C., Ito,N., Evans,P.R., Teo,C. and Nagai,K. (1994) Crystal-structure at 1.92 Å resolution of the RNA-binding domain of the U1A spliceosomal protein. Nature, 372, 432–438. [DOI] [PubMed] [Google Scholar]
- Parry-Smith D.J., Payne,A.W.R., Mitchie,A.D. and Attwood,T.K. (1998) CINEMA—a novel colour interactive editor for multiple alignments. Gene, 221, GC57–GC63. [DOI] [PubMed] [Google Scholar]
- Platko J.V. and Calvo,J.M. (1993) Mutations affecting the ability of Escherichia coli Lrp to bind DNA, activate transcription, or respond to leucine. J. Bacteriol., 175, 1110–1117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Platko J.V., Willins,D.A. and Calvo,J.M. (1990) The IlvIH operon of Escherichia coli is positively regulated. J. Bacteriol., 172, 4563–4570. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Powers T. and Noller,H.F. (1995) Hydroxyl radical footprinting of ribosomal-proteins on 16S ribosomal RNA. RNA, 1, 194–209. [PMC free article] [PubMed] [Google Scholar]
- Schevitz R.W., Otwinowski,Z., Joachimiak,A., Lawson,C.L. and Sigler,P.B. (1985) The 3-dimensional structure of Trp repressor. Nature, 317, 782–786. [DOI] [PubMed] [Google Scholar]
- Steger G., Ham,J., Lefebvre,O. and Yaniv,M. (1995) The bovine papillomavirus-1 E2 protein contains two activation domains: one that interacts with TBP and another that functions after TBP binding. EMBO J., 14, 329–340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Szilagyi A. and Zavodszky,P. (2000) Structural differences between mesophilic, moderately thermophilic and extremely thermophilic protein subunits: results of a comprehensive survey. Struct. Fold. Des., 8, 493–504. [DOI] [PubMed] [Google Scholar]
- Wang Q. and Calvo,J.M. (1993) Lrp, a global regulatory protein of E.coli, binds cooperatively to multiple sites and activates transcription of ilvIH. J. Mol. Biol., 229, 306–318. [DOI] [PubMed] [Google Scholar]
- Wang Q., Wu,J., Friedberg,D., Platko,J.V. and Calvo,J.M. (1994) Regulation of the Escherichia coli lrp gene. J. Bacteriol., 176, 1831–1839. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Willins D.A., Ryan,C.W., Platko,J.V. and Calvo,J.M. (1991) Characterization of Lrp, an Escherichia coli regulatory protein that mediates a global response to leucine. J. Biol. Chem., 266, 10768–10774. [PubMed] [Google Scholar]
- Yip K.S.P., Britton,K.L., Stillman,T.J., Lebbink,J., De Vos,W.M., Robb,F.T., Vetriani,C., Maeder,D. and Rice,D.W. (1998) Insights into the molecular basis of thermal stability from the analysis of ion-pair networks in the glutamate dehydrogenase family. Eur. J. Biochem., 255, 336–346. [DOI] [PubMed] [Google Scholar]
- Zhao Y., Jeruzalmi,D., Moarefi,I., Leighton,L., Lasken,R. and Kuriyan,J. (1999) Crystal structure of an archaebacterial DNA polymerase. Struct. Fold. Des., 7, 1189–1199. [DOI] [PubMed] [Google Scholar]