

Abstract
The structural protein VP6 of rotavirus, an important pathogen responsible for severe gastroenteritis in children, forms the middle layer in the triple-layered viral capsid. Here we present the crystal structure of VP6 determined to 2 Å resolution and describe its interactions with other capsid proteins by fitting the atomic model into electron cryomicroscopic reconstructions of viral particles. VP6, which forms a tight trimer, has two distinct domains: a distal β-barrel domain and a proximal α-helical domain, which interact with the outer and inner layer of the virion, respectively. The overall fold is similar to that of protein VP7 from bluetongue virus, with the subunits wrapping about a central 3-fold axis. A distinguishing feature of the VP6 trimer is a central Zn2+ ion located on the 3-fold molecular axis. The crude atomic model of the middle layer derived from the fit shows that quasi-equivalence is only partially obeyed by VP6 in the T = 13 middle layer and suggests a model for the assembly of the 260 VP6 trimers onto the T = 1 viral inner layer.
Keywords: capsid protein/crystal structure/icosahedral virus/quasi-equivalence/rotavirus
Introduction
Rotaviruses are the leading cause of severe gastroenteritis in humans, resulting in the death from dehydration of >800 000 children annually throughout the world (Bern et al., 1992). No protection has been available against this pathogen since the withdrawal of the tetravalent vaccine in 1999 because of side-effects (Centers for Disease Control, 1999). Knowing the detailed architecture of the rotavirus particles and understanding the assembly process can provide a structural basis for the rational design of drugs that interfere specifically with capsid assembly. Rotaviruses are classified into several serogroups (A–G) (Estes, 1996) and form a genus in the Reoviridae family of double-stranded RNA (dsRNA) viruses (Fields, 1996). The genome is composed of 11 segments of dsRNA within an icosahedral capsid formed by three concentric protein layers (Estes, 1996). The virions lose their outer protein layer during cell entry to become transcriptionally active double-layered particles (DLPs) in which mRNAs are synthesized and actively translocated through pores in the capsid. Crystal structures of the transcriptionally competent units of two members of the Reoviridae family, the orbivirus bluetongue virus (BTV) (Grimes et al., 1998) and reovirus (Reinisch et al., 2000), have revealed their complex architecture in atomic detail. For rotaviruses, structural studies using electron cryomicroscopy (cryo-EM) and computer image processing have provided a structural description of the virion to ∼20 Å resolution, including the locations of the various proteins in the different layers of the viral capsid (Yeager et al., 1994; Prasad et al., 1996; Prasad and Estes, 1997). The organization of the two layers in the rotavirus DLPs is similar to that of the BTV cores, with a complete T = 13 layer containing 260 trimers of VP6 surrounding an inner layer of lower symmetry made up of 120 copies of VP2 (Lawton et al., 1997b).
Protein VP6 plays a key role in the organization of the virion, being in direct contact with both the inner and the outer protein shells, thus acting as a physical adaptor between two distinct biological functions of the virus, i.e. cell entry (outer layer) and genomic RNA packaging (inner layer) (Prasad and Estes, 1997). The presence of the VP6 layer is necessary for transcription (Bican et al., 1982), but the specific role of VP6 in this process is not known. Nascent transcripts have been visualized by cryo-EM emerging through the 5-fold pores formed by VP6 in the DLPs (Lawton et al., 1997a). Monoclonal antibodies against VP6 were shown to inhibit the transcription process (Ginn et al., 1992; Petitpas, 1999) and to protect mice from infection (Burns et al., 1996).
In this work, we used X-ray crystallography to determine the three-dimensional structure of VP6 from group A rotavirus to 2 Å resolution. We analyzed the VP6 interactions in the viral capsid at the amino acid level by fitting the atomic model into cryo-EM reconstructions of the viral particle. The resulting pattern of lateral interactions between VP6 trimers in the viral capsid shows that quasi-equivalence is partially obeyed in the T = 13 layer formed by VP6. Moreover, these results, combined with those of the accompanying manuscript (Lepault et al., 2001), suggest that the assembly of VP6 on top of the viral inner layer proceeds from an initial strong interaction between VP6 trimers at the 20 icosahedral 3-fold (I3) axes, where the symmetry of the two protein layers match. The VP6–VP2 interaction thus defines the correct geometry of the viral particle, while the cohesion of the second layer is ensured essentially by strong lateral inter-trimer VP6–VP6 interactions.
Results and discussion
Molecular architecture
The determination of the crystal structure is described in Materials and methods and the crystallographic statistics are listed in Table I. The VP6 trimer, depicted in Figure 1, is an elongated molecule with a shape reminiscent of a tower, as has been noted in the description of cryo-EM reconstructions of the DLPs (Prasad et al., 1996). The molecule is 95 Å long, the base has a triangular cross-section ∼60 Å per side and the head has a roughly hexagonal cross-section with a diameter of 45 Å. The three subunits that form the VP6 molecule wrap around a central 3-fold axis with a right-handed twist.
Table I. Crystallographic data and refinement statistics.
| Crystal form 1 data collection statistics |
||||||||||||||
| Data set |
Resolution limit (Å) |
|
Rsyma (%) |
Completeness (%) |
Redundancyb (%) |
<I/σ> |
||||||||
| Native | 2.5 | 6.6 (10.8) | 95.8 (89.7) | 7.6 (2.9) | 24.3 (10.7) | |||||||||
| K2PtCl4 | 2.8 | 10.0 (28.1) | 99.4 (99.8) | 7.0 (3.8) | 21.2 (6.9) | |||||||||
| CH3HgCl | 4.0 | 17.6 (27.7) | 91.2 (95.2) | 3.0 (3.0) | 6.5 (4.2) | |||||||||
| PCMBSc |
3.45 |
|
8.1 (12.5) |
98.5 (93.4) |
6.2 (5.8) |
16.8 (11.6) |
||||||||
| Phasing statistics |
||||||||||||||
| |
No. of sites |
Resolution limits (Å) |
Rcullisd cent./acent./anom. |
Phasing powere cent./acent./anom. |
Binding sites |
|||||||||
| K2PtCl4, 1 mM | 4 | 15–3 | 0.84/0.83/0.79 | 1.0/1.3/1.5 | Met295, Met300, Met342, Met394 | |||||||||
| CH3HgCl, 1 mM | 3 | 15–4 | 0.90/0.91/0.98 | 0.89/1.1/0.68 | Cys197, His316, Cys331 | |||||||||
| PCMBS, 1 mM |
1 |
15–4 |
0.95/0.96/0.98 |
0.62/0.69/0.62 |
His316 |
|||||||||
| Figure of merit |
||||||||||||||
| Resolution limit (Å) | 7.0 | 5.0 | 4.0 | 3.5 | 3.2 | 3.0 | ||||||||
| Centric | 0.730 | 0.646 | 0.489 | 0.346 | 0.310 | 0.223 | ||||||||
| Acentric |
0.511 |
0.480 |
0.327 |
0.182 |
0.181 |
0.154 |
||||||||
| Crystal form 2 data collection statistics |
||||||||||||||
| Data set |
Resolution limit (Å) |
|
Rsyma (%) |
Completeness (%) |
Redundancyb (%) |
<I/σ> |
||||||||
| High resolution | 1.95 | 7.2 (25.2) | 98.3 (97.3) | 7.1 (3.8) | 15.6 (5.1) | |||||||||
| Zinc edge |
3 |
|
7.5 (16.5) |
95.2 (96.5) |
4.1 (3.9) |
18.7 (11.6) |
||||||||
| Refinement statistics |
||||||||||||||
| Protein atoms | 3163 | |||||||||||||
| Water molecules | 266 | |||||||||||||
| Calcium ionsf | 5 | |
||||||||||||
| Zinc ionf | 1 | |
||||||||||||
| Chloride ion | 1 | |||||||||||||
| R.m.s.d. | ||||||||||||||
| bond lengths | 0.012 Å | |||||||||||||
| bond angles | 1.77° | |||||||||||||
| main chain B | 1.37 Å2 | |||||||||||||
| Resolution range (Å) | 42–1.95 (all data) | |||||||||||||
| R-factorg | 0.186 | |||||||||||||
| Free R-factorg | 0.216 | |||||||||||||
Last shell values are given in parentheses.
aRsym = Σ|Ih – <Ih>|/ΣIh, where <Ih> is the average intensity over symmetry equivalent measurements.
bRedundancy = number of measured data/number of unique data.
cPCMBS = 4-(chloromercuri)benzenesulfonic acid.
dRcullis = Σ||FPH ± FP| – FH|/Σ|FPH ± FP|, where FP, FPH and Fh are the native, derivative and heavy atom structure factor amplitudes, respectively.
ePhasing power = Σ|Fh|/Σ||FPHobs| – |FPHcalc||; anomalous phasing power = Σ|FH″|/Σ||ADobs| – |ADcalc||, where AD is the anomalous difference.
fBound metal ions: Zn2+, on the 3-fold axis at the center of the molecule (coordinated by H153 from each subunit and a Cl– ion); Ca2+ 1, on the 3-fold axis at the top (coordinated through water molecules to N250 Nδ and Y248 OH from the three subunits); Ca2+ 2, inside the big cavity at the base (coordinated by D340 Oδ, main chain carbonyl 161 and five water molecules; E134 and D337 coordinated through water molecules); Ca2+ 3, at crystal contact (coordinated by D62 and Q47 from an adjacent trimer in the crystal, and three water molecules); Ca2+ 4, at crystal contact (coordinated by D286 and N266 from two adjacent trimers in the crystal, and two water molecules); Ca2+ 5, at crystal contact (coordinated directly by E379 and through a water molecule by D380 and Q383; coordinated directly by Q268 from an adjacent trimer in the crystal; and coordinated by five water molecules).
gR-factor = Σ||Fobs| – |Fcalc||/Σ|Fobs|, where summation is over data used in the refinement; free R-factor includes only 5% of data excluded from all refinements.
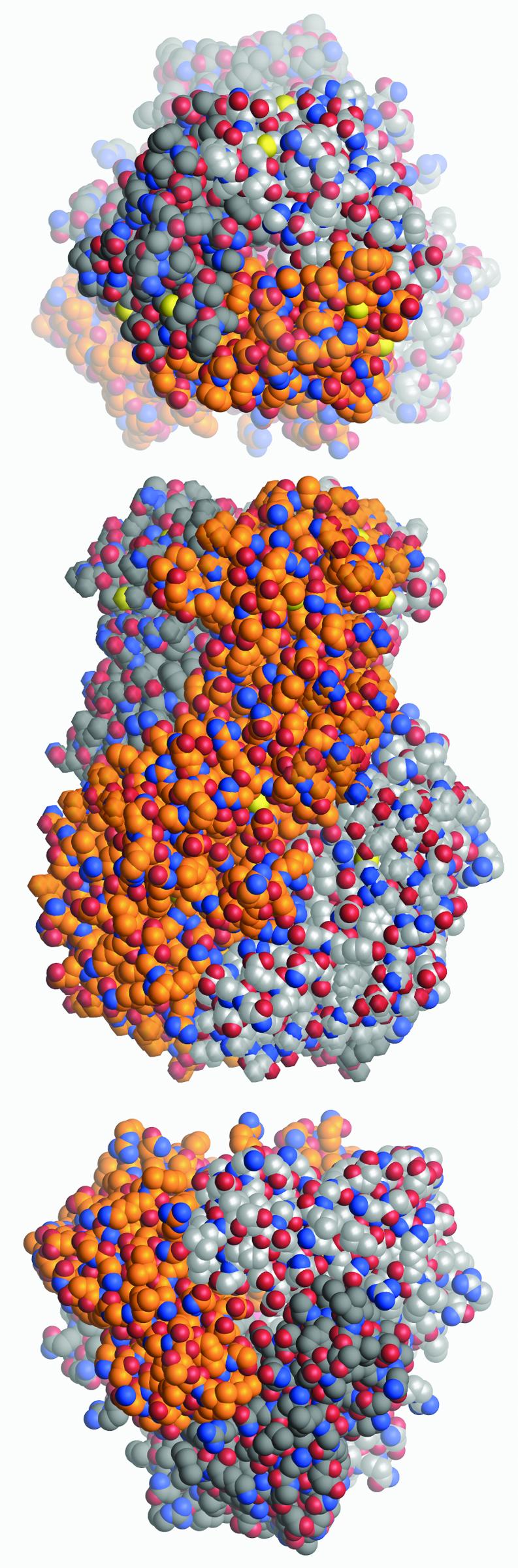
Fig. 1. Space-filling representation of the VP6 trimer in orthogonal orientations. Each atom is drawn as a sphere of the corresponding van der Waals radius, colored according to atom type (blue, nitrogen; red, oxygen; yellow, sulfur; gold, silver or gray, carbon atoms on different subunits. Top: the molecule seen from the outside of the virion (or ‘top view’). It shows a roughly hexagonal cross-section with a depression at the center. Middle: side view showing the right-handed twist of the subunits along the molecular axis. Bottom: base of the molecule, looking from underneath (from inside the virion, or ‘bottom view’). It shows a triangular cross-section, with β-hairpin βa–βb at the vertices.
The VP6 subunit
The polypeptide chain of VP6 is folded into two distinct domains, shown in the ribbon diagram of Figure 2. The overall fold exhibits two distinct domains. Domain B, at the base of the molecule, consists of a bundle of eight α-helices (αa–αh) derived from two segments of the polypeptide chain. The first segment, containing helices αa–αe, is formed by the 150 N-terminal amino acids. The second segment (residues 335–397 at the C-terminal end) contains the remaining three helices. The connection between helices αb and αc at the base of the trimer makes a β-hairpin (βa–βb) that packs vertically against the side of the molecule, forming the edges of a triangular prism at the base of the trimer (see Figure 1, bottom). Domain H (residues 151–334), at the top of the molecule, folds into a β-sandwich with the jelly-roll, or β-roll, topology frequently found in viral capsid proteins (Rossmann and Johnson, 1989). This domain is an insertion between consecutive helices αe and αf of domain B. The CHEF β-sheet lies in the outer, exposed side of the molecule, whereas the BIDG sheet lies internally. The latter β-sheet is augmented in VP6 by β-hairpin A′A″, N-terminal to strand B. The connection between strands D and E forms another β-hairpin, D′D″, running at almost 90° with respect to the strands in the A′A″BIDG sheet and participating in the packing between subunits in the trimer. This domain thus has a three-layered structure, with the six-stranded sheet A′A″BIDG in the middle, sandwiched between the CHEF sheet and the D′D″ β-hairpin. Towards the base, strand I hydrogen-bonds to the most N-terminal strand of domain H, labeled strand A, in such a way that the middle β-sheet becomes AIDG and closes the lower end of the β-roll (see Figure 2A). The top of the molecule is formed by loops A′A″, BC and HI (including the short helix αA), which form closely interacting structures at the outward facing end of the β-roll. The loops at the opposite end of the β-sandwich are very short and, in some cases (CD and GH), the hydrogen bonding pattern continues without interruption of the strands. This end of the β-sandwich contacts domains B both from the same subunit and from the clockwise subunit in the trimer.

Fig. 2. The VP6 subunit. (A) Ribbon diagram with domain H (residues 151–331) in red and the two segments that form domain B in blue (residues 1–150) and gold (residues 331–397). Secondary structural elements in domain B are labeled by lower case letters consecutively from the N-terminus, preceded by α or β for α-helices or β-strands, respectively. For domain H, the standard nomenclature for jelly-rolls is employed, using upper case letters from B to I. Additional β-strands (absent in the standard jelly-roll motif) are indicated by primed letters. (B) Stereo Cα trace of the VP6 subunit with every twentieth residue labeled.
Intra-trimer contacts
Both domains H and B participate in the contacts that stabilize the trimeric molecule. The overall area buried from solvent accessibility upon trimer formation is 4500 Å2 per subunit. The packing between the three H domains in the trimer contributes 1800 Å2 per monomer and the contacts between B domains 1700 Å2 to the total buried area, the remaining 1000 Å2 corresponding to cross-domain interactions. The amino acids participating in intra-trimer interactions are indicated in the sequence alignment of Figure 3A (which also shows the conservation of each amino acid) and the contacts are listed in Table II. VP6 has a big water-filled cavity at the center of the base, bounded at each side by the B domains of the three subunits (Figure 3B), which restricts the contact area. In contrast to the tight hydrophobic contacts between H domains, the trimer interface at the base contains mostly hydrophilic residues (see Figure 3C), suggesting that a low solvation energy would be needed for dissociation of B domains. This is in agreement with previous observations that deletion mutants of VP6 in which the β-roll is incomplete can not trimerize (Lopez et al., 1994; Affranchino and Gonzalez, 1997), whereas mutants lacking regions of VP6 that are involved in interactions between B domains can (Affranchino and Gonzalez, 1997).

Fig. 3. Intra-trimer contacts in VP6. (A) Alignment of amino acid sequences of rotavirus protein VP6 from groups A (Swissprot accession code P04509), C (P30213) and B (P26015) (top, middle and bottom lines, respectively). Human strains of rotavirus can be as close as 98.5% in sequence identity to the RF bovine group A rotavirus VP6 used in this work. However, within group A, the most distant viruses have a VP6 amino acid sequence identity of 65%, of 40% between groups A and C, and of only 16% between groups A and B (or C and B). The alignment of the last sequence is therefore only approximate. Yellow bars above the top sequence mark residues participating in intra-trimer contacts. Letters on a black background denote strictly conserved residues in group A (from analysis of 39 available sequences). For group C, a black background indicates strict identity with group A. Letters on a gray background correspond to positions displaying only conservative mutations. Secondary structure elements are labeled above the sequence in red for domain H and blue for B: rectangles denote α-helices, arrows β-strands and dotted lines connecting loops. Residues that coordinate the Zn2+ ion [His153 in group A and His339 (group A numbering) in groups C and B] are indicated. The alignment was obtained initially with the program PILEUP (Devereaux et al., 1985; Womble, 2000) and then adjusted manually based on the crystal structure. (B) Ball-and-stick representation of the trimer. Internal cavities on the 3-fold axis are shown as a blue surface. A pink ball indicates the Zn2+ ion on the 3-fold axis. There is a small cavity above the metal, and a big cavity underneath, between B domains. (C) Surface representation of the VP6 molecule with one subunit removed from the trimer. The solvent-accessible surface in the trimer is blue (including internal cavities). The inter-subunit contact surface is shown in white (main chain atoms and hydrophobic side chain atoms) and yellow (hydrophilic side chain atoms). It can be seen that contacts at the top of the molecule (above the smaller cavity) are mostly hydrophobic, whereas those at the base involve mainly hydrophilic side chains. The Zn2+ ion is shown as a red ball at the center. (D) View down the 3-fold axis from above the Zn2+ ion, shown as a solid pink sphere. A pink net shows the anomalous difference electron density corresponding to the Zn2+ ion (see Materials and methods). His153 from each of the subunits coordinates the metal as well as a Cl– ion (represented by a small silver sphere above), which closes the tetrahedral sphere of coordination. A histidine residue was modeled at position 339 instead of serine (since this position is histidine in VP6 from groups C and B rotaviruses, see A) to show that it can coordinate the metal ion equally well.
Table II. VP6 intra-trimer contactsa.
| D′D″ hairpin | H–I loop |
| D″–E loop | |
| A′ and A″ strands | F–G loop |
| D strand | |
| E strand | |
| I–αf loop | I strand |
| αf helix | AIDG sheet (bottom) |
| C-terminus | A strand |
| αf–αg loop | G strand |
| αa–αb loop | αd–αe loop |
| αb helix (N-ter) | αb helix (C-ter) |
aContacts are listed between one subunit (left column) and its clockwise neighbor in the trimer when looking from the top (right column).
Zn2+ ion
The VP6 trimer contains a metal ion located at the center of the molecule, on the 3-fold molecular axis, tetrahedrally coordinated to the Nε of His153 from each of the three subunits. The metal coordination is facilitated by a hydrogen bond between the Nδ of His153 and the main chain carbonyl of residue 339 from the adjacent subunit, an interaction that enhances the nucleophilicity of the Nε atom in the imidazole ring. We have identified this metal ion experimentally as Zn2+ by collecting a complete diffraction data set from a native crystal at the wavelength corresponding to the energy of the K absorption edge of Zn (see Materials and methods). This type of Zn2+ coordination is reminiscent of hexameric insulin, in which a histidine residue is used to coordinate Zn2+ ions on a 3-fold axis (Bentley et al., 1976). His153 is strictly conserved among group A rotaviruses but not in the other serogroups. Interestingly, all sequenced non-group A rotaviruses have a histidine residue at the position equivalent to Ser339, which can be modeled as histidine (Figure 3D) with the imidazole in position to coordinate the Zn2+ ion equally well. Interestingly, in contrast to the orbivirus cores, the capsid proteins λ1 (Reinisch et al., 2000) and σ3 (Schiff et al., 1988) of reovirus also contain Zn2+ ions.
The fourth ligand to the Zn2+ ion, which is also on the 3-fold axis above the metal, is very probably a Cl– ion. Refinement of the atomic temperature factors (B-factors) in our model converges to reasonable values for chlorine at this position, whereas the B-factor becomes negative if the site is considered as being occupied by a water molecule. This ion is coordinated by the Nζ of Lys154 from each of the three subunits in the trimer, which also donates a hydrogen bond to Glu327 from the adjacent subunit. This salt bridge helps to compensate for the accumulation of positive charges about the trimer axis.
Comparison with BTV protein VP7
A comparison between these two homologous proteins is shown in Figure 4 and a detailed description of their superposition is given in Materials and methods. The organization of the BTV VP7 trimer is similar to that of VP6, with the subunits displaying the same right-handed twist about the molecular 3-fold axis. The polypeptide chain of BTV VP7 also folds into an α-helical domain at the base and a β-roll at the top (Grimes et al., 1995). However, the β-roll contains ∼60 residues less and the strands are shorter. The connection between the two domains is also different: in rotavirus VP6, the strands of the β-roll are in direct contact with domain B, whereas in BTV VP7 there is a linker between domains that results in a molecule with a thinner region at the center, like a waist. An additional C-terminal α-helix of BTV VP7 (absent in VP6, helix 9 in Figure 4B) packs against the waist region and also gives the trimer a propeller-like contour when looking from the top (Figure 4A, top). The side view of the molecules shows differences at the base, where VP6 has a more rounded profile due to the loop joining the βa–βb hairpin to helix αc. Both proteins present cavities along the 3-fold molecular axis, but their distribution and shape are different. Whereas in BTV VP7 the bigger cavity is centered in the waist region (Basak et al., 1996), in VP6 it is at the base (as shown in Figure 3B) and is constricted in the middle by the Zn2+ ion.

Fig. 4. Comparison between rotavirus VP6 and BTV VP7. (A) Ribbon representation of rotavirus VP6 (left) and BTV VP7 (right) (Grimes et al., 1995) oriented after superposition of the trimer of B domains (see Materials and methods) in a top (upper panel) and side (bottom panel) view. In VP6, the strands in domain H extend further towards domain B. The side view shows that VP6 does not have a flat base like BTV VP7 and that the connection between the two domains is different in the two molecules. The top view shows that the propeller-like contour of BTV VP7 is absent in VP6. The BTV VP7 coordinates were obtained from the Protein Data Bank, accession code 1bvp. (B) Structural alignment of rotavirus VP6 and BTV VP7. Black letters indicate residues common to both structures, which can be superimposed as explained above. Gray letters denote residues that do not correspond to superimposable secondary structure elements. Colored dotted lines above (VP6) and below (BTV VP7) indicate regions corresponding to B (blue) and H (red) domains. The secondary structure elements of each protein are indicated (α-helices by rectangles and β-strands by arrows) above and below the rotavirus VP6 and BTV VP7 sequences, respectively. For BTV, the labels of the secondary structure elements were taken from Grimes et al. (1995).
The VP6 layer in the viral capsid
We used the cryo-EM reconstruction of the DLP extended to 17 Å resolution to fit the atomic model into density corresponding to VP6. As shown in Figure 5, the cryo-EM density clearly defines the envelope of the trimer: the towers seen in the reconstruction match the shape of the molecule and the fit is unique, as explained in Materials and methods. The chemical unit forming the T = 13 surface lattice is the VP6 polypeptide chain, but the actual building block is the trimer. For this reason, from here on, we will refer mainly to the five icosahedrally independent trimers (ITs) instead of the 13 polypeptide chains. One of the ITs sits on an icosahedral 3-fold (I3) axis and thus contributes only one subunit to the asymmetric portion, as seen in Figure 5A. Figure 5B shows a side view of VP6 fitted into the cryo-EM density corresponding to P trimers, about the icosahedral 5-fold (I5) axis. For comparison, we show the same slab of the BTV map, with VP7 trimers fitted into the 23 Å cryo-EM density, in the same orientation (Figure 5C). It is clear from this figure that the lateral contacts between trimers are different. In the case of BTV, VP7 trimers pack along a narrow band at the very base of the trimer, whereas in the rotavirus particle the contacts between trimers of VP6 are more extensive and further from the base. This is likely to be one of the reasons why VP6 can assemble into spherical or tubular particles in the absence of VP2, whereas no such particles have been reported for isolated BTV VP7. The surface electrostatic potential of the two molecules (shown in the same figure) shows that the contact regions are different in nature: while there are mainly hydrophobic contacts in BTV (Grimes et al., 1998), the corresponding regions in VP6 are negatively charged. This is in agreement with the observations that spherical particles of VP6 are formed only at low pH (Lepault et al., 2001), when the negative charges at the surface of contact are compensated by protonation.

Fig. 5. Fitting of the atomic model of VP6 into the cryo-EM reconstruction of the DLP. (A) The five icosahedrally independent trimers (ITs) are displayed in a space-filling representation as in Figure 1. The cryo-EM reconstruction is displayed as a pink net. The icosahedral symmetry axes are shown in white, with a pentagon to indicate I5, a triangle for I3 and a full ellipse for icosahedral 2-fold (I2) axes. The trimers are labeled as follows: P (for ‘pentad’) trimers, colored red, surround I5 axes; D (for ‘dyad’) trimers, colored gray, are next to an I2 axis; T (‘triad’) trimers, colored green, sit on the I3 axis and are thus shared by three consecutive icosahedral asymmetric units. The remaining two ITs are not immediately adjacent to an icosahedral axis. Trimer T′ (yellow) is the only IT to contact trimer T, and P′ (or peri-pentonal trimer, blue) is the only one to contact trimer P. All contacts between trimers involve packing of the flat faces of the triangular base (see Figure 1, bottom). The contacts are made across a local pseudo-dyad (which in the case of contacts between trimers D is the I2 axis). Q and N indicate different types of contact, as explained in the text, displaying double or single density connections in the EM reconstruction, respectively. The inset defines the labeling of each VP6 subunit in the ITs as used to describe the contacts with VP2 and VP4 in Table IV. (B) Lateral view of VP6 trimers fitted into the cryo-EM reconstructions. The inset (left) shows the surface electrostatic potential (Nichols et al., 1993) of the VP6 trimer (red and blue for negative and positive charges, respectively), in a side (top panel) and bottom (bottom panel) view. Note the strong negative charges of the regions implicated in inter-trimer contacts. (C) The same view of the BTV VP7 trimers fitted into the 23 Å resolution cryo-EM reconstruction of the core particle is shown for comparison. The lateral contacts between trimers in the two viruses are clearly different. The inset shows that the regions of contact are not charged, in contrast to rotavirus VP6.
Fitting the five ITs into cryo-EM density allowed the generation of a crude atomic model of the middle layer (Figure 6A) by applying the icosahedral symmetry operations. This model shows that trimers of VP6 in the viral particle pack by the flat faces of the base and are related by pseudo 2-fold axes, in an overall arrangement similar to that of the BTV cores (Grimes et al., 1998). The five ITs make seven different pseudo-dyad-related contacts, giving rise to the seven pairs of adjacent trimers listed in Table III. We have compared the different contacts by calculating the root mean square deviation (r.m.s.d.) between the above pairs of trimers. The resulting r.m.s.ds are listed in Table III, and show that the contacts are very similar except for pairs PP and P′D. It is generally accepted that fitting an atomic model into cryo-EM reconstructions of ∼20 Å resolution results in a precision better than 4 Å in the resulting atomic positions (Smith et al., 1993; Grimes et al., 1997). Therefore, using a cut-off of 3 Å r.m.s.d., we can classify the inter-trimer contacts into two types: quasi-equivalent (Q) contacts, which can not be differentiated at the resolution of the reconstruction; and non-equivalent (N) contacts, which are significantly different from the others. Comparison with the inter-trimer contacts found in the helical particles of VP6 (Table III), which display non-equivalent inter-trimer contacts since the 3-fold symmetry is incompatible with helical symmetry, shows that the Q contact is very similar to the common contact present in both types of tubes (Lepault et al., 2001). The Q contact thus seems to be the most thermodynamically favorable type of association between two VP6 trimers. Table III shows that the two N contacts (N1 and N2 for PP and P′D, respectively) are as different from each other and from Q contacts as are non-equivalent contacts in the tubes, indicating that the quasi-equivalence principle is partially obeyed in the rotavirus T = 13 layer. The N contacts radiate from the I5 axes (as indicated schematically in Figure 6B), suggesting that the curvature imposed by the inner VP2 layer forces this type of interaction between VP6 trimers at the vertices of the icosahedron.
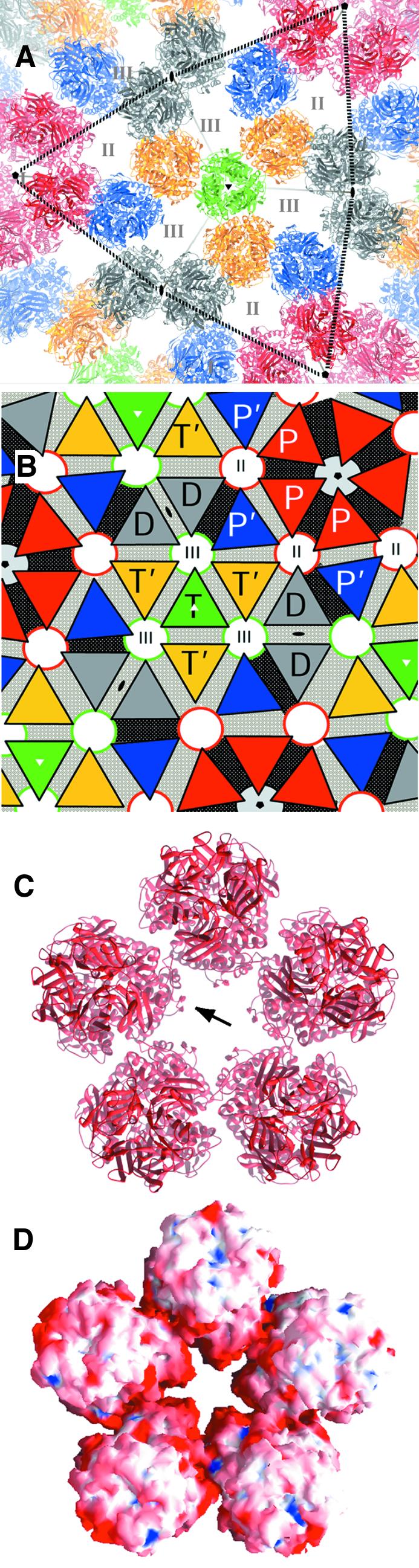
Fig. 6. Model of the VP6 layer in the viral particle based on rigid-body fitting into the cryo-EM density. (A) Each VP6 trimer is represented as a ribbon diagram colored according to the scheme described in the legend to Figure 5A. Dotted black lines joining three I5 axes delimit one of the 20 triangular faces of the icosahedron. VP6 trimers pack by the flat lateral faces of the triangular base of the molecule. Roman numerals denote the three different types of channels on the DLP (Prasad et al., 1988). (B) Schematic diagram illustrating the distribution of contacts in the viral surface. Each VP6 trimer is represented by a triangle colored according to (A). Q contacts are indicated in gray and N contacts are on a black background between adjacent triangles. Note that N contacts radiate from the I5 axes. (C) View down the 5-fold axis of the DLP (type I channel) showing that the amino acid residues lining the channel walls come exclusively from the βa–βb hairpin (arrow) at the base of the trimer. (D) Electrostatic potential (Nichols et al., 1993) calculated at the molecular surface. Negative charges are depicted in red and positive in blue. The walls lining the channel have a net negative charge. RNA transcripts have been visualized by cryo-EM to emerge through these channels (Lawton et al., 1997a). The diameter of the channel is ∼18 Å, enough to allow a single-stranded RNA molecule to pass through.
Table III. R.m.s.d. (in Å) between pseudo-dyad-related pairs of trimers after fitting the atomic model into the cryo-EM density of the DLPa and of the tubesb.
| DLP |
Large tubes |
Small tubes |
||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| PP′ | P′T′ | P′D | T′D | DD | T′T | D2 | D1 | W | d | s | w | |
| PP | 4.2 | 4.2 | 3.8 | 4.7 | 5.1 | 3.8 | 5.2 | 5.0 | 5.7 | 4.6 | 2.8 | 6.4 |
| PP′ | 1.9 | 5.8 | 2.4 | 1.5 | 1.3 | 1.4 | 1.0 | 5.8 | 0.8 | 5.4 | 3.2 | |
| P′T′ | 6.1 | 2.2 | 1.6 | 2.8 | 2.4 | 2.1 | 6.4 | 2.2 | 4.6 | 4.1 | ||
| P′D | 5.8 | 6.8 | 5.1 | 6.6 | 6.6 | 4.1 | 6.0 | 4.2 | 6.9 | |||
| T′D | 2.5 | 2.7 | 2.0 | 2.5 | 5.5 | 2.6 | 4.7 | 3.8 | ||||
| DD | 2.6 | 1.5 | 1.1 | 6.5 | 1.5 | 5.9 | 3.0 | |||||
| T′T | 2.0 | 2.2 | 4.7 | 1.9 | 5.2 | 3.0 | ||||||
aFor the DLP, values in bold correspond to Q contacts (see text). The contacts are labeled according to the adjacent trimers considered (see legend to Figure 5 for nomenclature).
bThe labels for contacts in the tubes are defined in Lepault et al. (2001).
Solvent channels in the VP6 layer
The VP6 trimers form three different solvent channels, labeled type I, II and III as described previously (Prasad et al., 1988), in the DLP. The arrangement of the VP6 trimers in the middle layer is such that hairpin βa–βb is directed towards the channels. The amino acid sequence in the βa–βb hairpin is highly conserved, especially at the tip (see Figure 3A), suggesting that these particular residues have a functional purpose. Figure 6C shows that the β-hairpin lines the walls of the channel at the base. The electrostatic potential calculated at the surface exhibits a net concentration of negative charges around the I5 axes (see Figure 6D), through which transcripts have been visualized emerging from the viral particle (Lawton et al., 1997a). The electrostatic repulsion between the negatively charged channel surface and the nucleic acid is likely to facilitate the extrusion of the mRNA transcript by increasing its fluidity. Site-directed mutagenesis of these residues should allow testing of whether they have any implication during the transcriptional process.
Contacts with the inner layer
The different triangulation of the two layers of DLPs implies that different VP6 trimers contact different regions of VP2. Our fit shows that all five ITs contact VP2, but not all the 13 subunits do, as indicated in Table IV. The EM density shows features that are different at the base of different ITs, which can be attributed to the different type of interactions made with the VP2 layer. The VP6 residues that participate in the interactions come essentially from the βb–αc loop and also from the bottom side of helix αb, which runs horizontally at the base of the trimer. The βb–αc loop at the base of the trimer (residues 60–74) is the most conserved region of the polypeptide chain, in particular the segment 65-LLGTTLL-71. The side chains of Leu65, Leu70 and Leu71 point outwards, into solvent, in the structure. This segment corresponds to the fairly loose loop seen at the base of VP6, which is absent in BTV VP7 (see Figure 4A, bottom). This loop is likely to adapt its fold to the precise type of contact made with VP2. The bottom panel of Figure 7B shows that the surface of contact with VP2 is not 3-fold symmetric. This holds true for all the ITs, except trimer T.
Table IV. Interaction of VP6 with other proteins of the viral capsid.
| Capsid protein | IT | VP6 subunit |
||
|---|---|---|---|---|
| A | B | C | ||
| VP2 | P | αb N-ter | αb N-ter | βb–αc loop |
| βb–αc loop | βb–αc loop | |||
| P′ | αb N-ter | αb N-ter | ||
| βb–αc loop | βb–αc loop | |||
| T′ | βb–αc loop | |||
| D | βb–αc loop | αb N-ter | βb–αc loop | |
| T | αb N-ter | αb N-ter | αb N-ter | |
| βb–αc loop | βb-αc loop | βb–αc loop | ||
| VP4 | P | CHEF β-sheet | ||
| E–F loop | ||||
| αc–αd loop | ||||
| βa–βb loop | ||||
| P (5)a | CHEF β-sheet | |||
| E–F loop | ||||
| αf–αg loop | ||||
| βa–βb loop | ||||
| αc | ||||
| T′ | CHEF β-sheet | |||
| βa–βb loop | ||||
| αf–αg loop | ||||
| αc–αd loop | ||||
| P′ | βa–βb loop | |||
| VP7 | A′–A″ loop | A′–A″ loop | A′–A″ loop | |
| B–C loop | B–C loop | B–C loop | ||
| H–I loop | H–I loop | H–I loop | ||
aSince two P trimers contact each VP4 spike, we have labeled P (5) the second, related to the first by an anticlockwise 5-fold rotation.
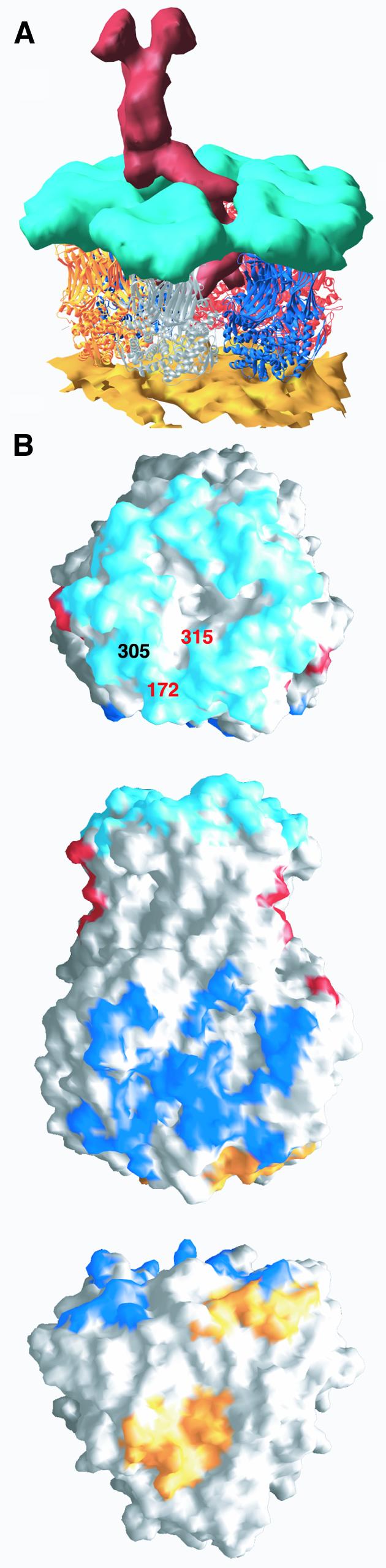
Fig. 7. Contacts with the outer layer of the viral particle. (A) View of a type II channel cut out of the cryo-EM reconstruction of the complete viral particle. The atomic model, shown as ribbons color coded as defined in Figure 5A for the different ITs, replaces the density corresponding to VP6. The EM density of the VP2 layer is shown in gold and the density of the VP7 layer in cyan. The viral hemagglutinin VP4 is shown in red. (B) Surface representation of VP6, trimer P, highlighting the regions in contact with the other layers of the viral particle. Regions contacting rotavirus VP7, VP4 and VP2 are colored cyan, red and gold, respectively. The surface in contact with neighboring trimer P′ is colored blue. Top: contacts with VP7. The amino acids involved in defining the subgroup epitopes (I and II, see text) of group A rotavirus are labeled (in different colors to indicate that they belong to different subunits). This view also shows a depression at the center, on the 3-fold axis. Residues lining the walls of this depression do not make contacts with VP7. Middle: side view of trimer P (red in Figure 5A). This is the only trimer to contact two VP4 molecules in adjacent type II channels. Regions seen in contact with trimer P′ are indicated in blue. Bottom: regions of contact between trimer P and VP2. The latter contacts do not follow the local 3-fold symmetry of the VP6 trimer.
Contacts with the outer layer
We used the cryo-EM three-dimensional reconstruction of intact virions (to ∼24 Å resolution), which contain the outer protein layer and the VP4 hemagglutinin, to examine the residues of VP6 that participate in the contacts with these proteins (illustrated in Figure 7). Rotavirus protein VP7 interacts in a one-to-one fashion with VP6: trimers of VP7 lie on top, contacting residues from loops A′A″, BC and HI at the top of domain H. Several of the amino acids involved are highly conserved, such as the stretch between Arg296 and Pro302 just before helix αA. These contacts leave a cavity around the 3-fold axis at the center of the interface between the two proteins. There is a network of main chain carbonyls pointing outwards at the top surface of VP6, towards the density corresponding to protein VP7, in an arrangement that could be matched by a precise complementary surface in VP7. At the resolution of the EM map, we have not detected any difference in the type of contacts between any of the ITs and VP7.
Contacts with the hemagglutinin spike
VP4 binds in type II channels in a unique orientation. It contacts two consecutive P, one P′ and one T′ trimers (see Table IV and Figure 7). The two consecutive P trimers are related by the I5 axis and so their surface of interaction with VP4 is presented in a different relative orientation when compared with any other pair of trimers. The globular domain of VP4 contacts the outer face of the CHEF β-sheet down to the tip of the βa–βb hairpin. The hydrophobic residues Ile270 and Leu264 in the lower half of the β-sheet have their side chains directed towards density corresponding to VP4 in the cryo-EM map. In addition, Asp286 (at the GH loop) and Asn266 (EF loop) are in the contact area. All of these residues, which point toward solvent in the structure, are strictly conserved in group A (and most of them also in group C) rotaviruses. Interestingly, in the VP6 structure, the latter two amino acids coordinate a Ca2+ ion sitting at the interface between two VP6 trimers in the crystal (the list of Ca2+ ions bound to VP6 is given in the footnotes of Table I). The same residues (Asp286 and Asn266) from the adjacent trimer in the crystal lattice participate in the coordination of the metal, so that each trimer contributes half a binding site. It would be interesting to explore the possibility that the VP6–VP4 interaction involves Ca2+, in which case VP4 would provide the second half-site needed for coordinating the metal, since chelating agents are known to lead to the loss of the outer layer (Cohen et al., 1979). Indeed, many viral particles stabilize their extracellular conformation by binding Ca2+, in both plant (Hogle et al., 1983) and animal (Liddington et al., 1991) viruses. It has been hypothesized that they make use of the cytoplasmic low Ca2+ concentration for the first steps of uncoating (Durham et al., 1977).
The antigenic structure of VP6
Group A rotaviruses are divided into four subgroups according to the presence or absence of two epitopes, named I and II, on protein VP6 (Kapikian et al., 1981; Estes and Cohen, 1989). Subgroup-specific antibodies recognize conformational epitopes that are formed only upon trimerization of VP6 (Gorziglia et al., 1988). It has been shown that a single amino acid mutation at position 172 (which is exposed at the very top of the A′A″ loop) or at position 305 (helix αA) affects binding of subgroup I antibodies (Lopez et al., 1994). A double mutation at positions 305 and 306 from helix αA affects binding of antibodies recognizing the subgroup II epitope (Lopez et al., 1994), as does a single mutation at position 315 (Tang et al., 1997). All of these residues are very close in space and form the conserved top surface of VP6, which interacts with VP7, as shown in Figure 7B, top. It thus appears that the antigenic determinants of subgroups I and II map to the same region in space and that the antibody specificity matches the chemical nature of the side chains present in this region. The amino acids that form this epitope also form the VP7 binding surface, explaining why the antigenic variability is limited and most group A rotaviruses belong to either subgroup I or II.
Concluding remarks
VP6 is a conserved protein that has the ability to interact specifically with itself and with all three other structural proteins of the virion. It presents conserved surfaces for interaction with VP2 at the bottom, VP4 at the sides of domain H, VP7 at the top and adjacent VP6 trimers at the sides of domain B. It is noteworthy that all these interacting surfaces contain the most conserved amino acids of the polypeptide chain. The lateral interactions between VP6 trimers do not contain all the information to make a closed icosahedral shell of unique size by quasi-equivalent interactions, as is the case for other viral proteins such as those forming the shell of T = 3 plant RNA viruses (Harrison et al., 1978; Abad-Zapatero et al., 1980). The inner shell thus directs the correct assembly. Three VP2 polypeptides provide a unique symmetric surface of contact for a VP6 trimer at the I3 axes. This particular trimer is likely to display the strongest interaction with the inner layer. It has been noted that other surface lattices that would result in a similar diameter for the complete particle (T = 9 or T = 12 for instance) are not observed in any of the Reoviridae (Wu et al., 2000). For particles of this size range, only T = 13 leads to a trimer sitting on the I3 axes, whereas in the other possible icosahedral surface lattices these axes are at the center of one of the hexavalent channels. In the case of phytoreovirus, for instance, the stability of the inner layer is ensured by the presence of the 20 equivalents of trimer T from the second layer, which act as a clamp (Wu et al., 2000). For rotaviruses, our data suggest a model of assembly in which the center of each of the 20 faces of the icosahedron provides nucleation sites for the middle layer, which grows by addition of trimers using the most favorable interaction (Q contacts). The result is schematized in the diagram of Figure 6B, which shows that the viral surface is a mosaic of 20 faces that present dislocations reaching the I5 axes. The curvature imposed by the Q contact between adjacent VP6 trimers is such that one T trimer can be reached from another T trimer across the I2 axes without dislocation and making exclusively quasi-equivalent contacts. This model of assembly is similar to that proposed for BTV, with the difference that the driving force for propagating the assembly is the lateral Q contacts instead of the hydrophobic interaction of each trimer with the inner layer (Grimes et al., 1998). Our results (together with those of Lepault et al., 2001) show that in contrast to BTV VP7, VP6 has the necessary information to make closed, albeit irregular, particles by itself and suggest that the strong interaction of the T trimer with a matching surface in the inner layer drives the assembly of correctly sized rotavirus particles.
Materials and methods
Structure determination and refinement
Crystals of VP6 (group A rotavirus, bovine strain RF) were obtained as described earlier (Petitpas et al., 1998). Initial diffraction data to 3.0 Å resolution from native crystals were collected at 100 K at synchrotron beamlines D41A and DW32 of the Laboratoire pour l’Utilisation du rayonnement Electromagnétique (LURE, Orsay, France) on an image plate detector (MAR Research). Diffraction data from crystal form I (see Table I) were collected to 2.5 Å resolution at beamline BM2 of the European Synchrotron Radiation Facility (ESRF, Grenoble, France) on a charged-couple device (CCD) detector. Crystals were stabilized in a cryo-protecting solution containing 30% 2-methyl-2,4-pentanediol and 100 mM maltose prior to cryo-cooling. The three-dimensional structure was determined initially by the isomorphous replacement method to 3.0 Å resolution, using the heavy atom derivatives listed in Table I, and refined to 2.5 Å. Bigger crystals of VP6 with slightly different cell parameters were obtained later, after further purification of VP6 by size exclusion chromatography in the presence of 200 mM CaCl2. These new crystals allowed data collection to 1.95 Å at beamline BW7B at synchrotron DESY (Hamburg, Germany). The refined 2.5 Å model was introduced by first carrying out a rigid body refinement (with program X-PLOR; Brünger, 1992) and then refined to 1.95 Å resolution. Additional diffraction data to 3 Å resolution at wavelength λ = 1.2815 Å, corresponding to the K absorption energy of Zn, were collected at synchrotron LURE, beamline DW21b on a MAR Research image plate detector. A single anomalous difference peak was found, which confirmed the identity of the metal ion coordinated by His153. The height of this peak corresponds to ∼10 times the r.m.s.d. of the anomalous difference map (Figure 3D).
The diffraction images were processed with programs DENZO and SCALEPACK (Otwinowski and Minor, 1996). All subsequent crystallographic analyses were carried out using the CCP4 suite of programs (CCP4, 1994) unless stated otherwise. Initial heavy atom positions were determined using RSPS (CCP4 suite). All phasing calculations and refinement were performed with SHARP (de la Fortelle and Bricogne, 1997) and solvent flattening with DM (Cowtan, 1994). The crystallographic model was built with O (Jones et al., 1991) and was refined using X-PLOR (Brünger, 1992). The final free R-factor (Brünger, 1997) is 21.4% for reflections in the 43–1.95 Å resolution range, using a bulk solvent correction to allow the introduction of the low-resolution terms. All the relevant crystallographic statistics are given in Table I.
EM image reconstructions
DLP reconstruction was carried out as described in Prasad et al. (1996) to ∼17 Å resolution from images obtained using a 400 kV electron microscope (JEOL 4000). A total of 188 particles with unique orientations were used in this reconstruction. Corrections due to contrast transfer function of the micrograph were applied as described previously (Zhou et al., 1994). The triple-layered particle (simian 4F strain) reconstruction was carried out to a nominal resolution of ∼23 Å from 128 particle images obtained from a 100 kV electron microscope.
Fitting of the crystallographic structure into the cryo-EM reconstructions
The atomic model of the VP6 trimer was initially fitted manually into the cryo-EM reconstructions. The five independent trimers were placed separately and the middle layer of the particle generated by applying the icosahedral symmetry operations. The fit was refined in reciprocal space using a fast technique first proposed by Huber and Schneider (1985). The algorithms and their implementation are essentially those described for program FITING (Castellano et al., 1992). Briefly, a normalized quadratic misfit: Q = Σ|Fem(h) – λFcal(h|R,T)|2/Σ|Fem(h)|2 is minimized with respect to the positional and translational parameters of the model (rotation R and translation T) and the scale factors λ. Fem and Fcal are the Fourier coefficients of the EM map and the model, respectively. This is the equivalent in direct space (through Parseval’s theorem) to Q = ∫ [ρem(r) – λρcal(rR,T)|2d3r, where ρem and ρcal are the electron densities of the EM map and the model, respectively. In the absence of overlap between the constituent molecules, ∫ ρcal(r)2d3r is independent of the positional variables. Consequently, given the scale factor λ, the minimum of Q corresponds to the maximum of the overlap between ρem and ρcal. The statistics obtained are R = 0.404, correlation coefficient (CC) = 0.845, where R = Σ||Fem(h)| – |Fcalc(h)||/Σ|Fem(h)| and CC = Σ Fem(h) F*cal(h)/(Σ|Fem(h)|2Σ|Fcalc(h)|1/2, where the sums are on index h.
Structural alignment and analysis of oligomeric contacts
Since the connection between the two domains in BTV VP7 and rotavirus VP6 is different (see Figure 4), the two domains have to be superposed separately. The best match is found superposing 21 Cα atoms from the five central strands of the β-roll, resulting in an r.m.s.d. of 0.92 Å. These residues are 186–191 (strand B), 225–226 (strand D), 258–264 (strand E), 288–292 (strand H) and 321–327 (strand I) superposed on BTV VP7 156–161, 186–187, 207–213, 230–235 and 242–248, respectively. When the corresponding trimers of H domains are superimposed, the r.m.s.d. becomes 1.9 Å for the 63 Cα atoms. The superposition based on B domains involves four out of the eight α-helices. The corresponding amino acids are 3–16 (helix αa), 29–40 (helix αb), 87–97(helix αc) and 385–395 (helix αh) of VP6, and 2–15, 30–41, 63–73 and 306–316 of BTV VP7, which correspond to helices 1, 2, 3 and 8, respectively (Grimes et al., 1995). Helix αc runs at an angle of 20° approximately from helix 3 in BTV VP7; the others are co-directional with the αh helix shifted so that only its C-terminal half superposes to the N-terminal half of BTV VP7 helix 8. The remaining helices do not superpose. The r.m.s.d. of this superposition is 2.3 Å for 48 Cα atoms. Superposition of the trimer of B domains results in an r.m.s.d. of 2.6 Å for the corresponding 144 Cα atoms. The molecules shown in Figure 4 are oriented according to this last superposition. The twist angle about the molecular axis in going from the center of mass of domain B to that of H (calculated from the Cα atoms that are actually superimposed for each domain in both structures) is 101° for VP6 and 115° for BTV VP7. The corresponding pitch along the molecular 3-fold axis is 39 Å for VP6 and 50 Å for BTV VP7.
The area buried from solvent at the trimer contacts was calculated using the program Quartz (C.Roberts, unpublished). The buried area per monomer in the trimer is calculated as (1/3) × (3AM – AT), where AM and AT are the accessible surface in the monomer and in the trimer, respectively. The illustrations shown in the figures were prepared using the following programs: RIBBONS (Carson, 1987) (Figures 1, 3B and D, 4A, 5A, 6A and C, and 7A); MOLSCRIPT (Kraulis, 1991) and RASTER3D (Merrit and Murphy, 1994) (Figure 2); GRASP (Nicholls et al., 1993) (Figures 3C, insets to 5B and C, 6D and 7B); and O (Jones et al., 1991) (Figure 5B and C).
The refined coordinates of VP6 have been deposited in the Protein Data Bank, accession code 1qhd.
Acknowledgments
Acknowledgements
We thank P.Alzari for orchestrating the collaboration that led to this work; J.Janin for support and encouragement; M.-C.Vaney and B.Arnoux for assistance with the computer work; M.Roth from beamline BM2 at ESRF as well as the staff from beamlines ID2 at ESRF (Grenoble, France), DW32, DW21 and D41 at LURE (Orsay, France) and BW7B at DESY (Hamburg, Germany) for help during data collection; A.Charpilienne, L.Mauny, J.Jakana, S.Mukherjee and S.Crawford for technical help; P.Vachette, C.Arias, S.Lopez, R.H.Cheng, Y.Gaudin and S.Duquerroy for helpful discussions; S.Bressanelli for critically reading the manuscript; and J.Grimes and D.Stuart for coordinates of the BTV cores before publication. We acknowledge the use of the National Center for Macromolecular Imaging funded by NIH. This work was supported by CNRS-ATIPE, SESAME, ARC and FRM grants to F.A.R., a PRFMMIP grant to F.A.R., J.C. and P.P., and an NIH grant to B.V.V.P. M.M. was the recipient of a long-term EMBO and an FRM fellowship during this work. I.P. acknowledges funding from INSERM/CNAMTS.
References
- Abad-Zapatero C., Abdel-Meguid,S.S., Johnson,J.E., Leslie,A.G.W., Rayment,I., Rossmann,M.G., Suck,D. and Tsukihara,T. (1980) Structure of southern bean mosaic virus at 2.8 Å resolution. Nature, 286, 33–39. [DOI] [PubMed] [Google Scholar]
- Affranchino J.L. and Gonzalez,S.A. (1997) Deletion mapping of functional domains in the rotavirus capsid protein VP6. J. Gen. Virol., 78, 1949–1955. [DOI] [PubMed] [Google Scholar]
- Basak A.K., Gouet,P., Grimes,J., Roy,P. and Stuart,D. (1996) Crystal structure of the top domain of African horse sickness virus VP7: comparisons with bluetongue virus VP7. J. Virol., 70, 3797–3806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bentley G., Dodson,E., Dodson,G., Hodgkin,D. and Mercola,D. (1976) Structure of insulin in 4-zinc insulin. Nature, 261, 166–168. [DOI] [PubMed] [Google Scholar]
- Bern C., Martines,J., de Zoysa,I. and Glass,R.I. (1992) The magnitude of the global problem of diarrheal disease: a ten year update. Bull. World Health Organ., 70, 705–714. [PMC free article] [PubMed] [Google Scholar]
- Bican P., Cohen,J., Charpilienne,A. and Scherrer,R. (1982) Purification and characterization of bovine rotavirus cores. J. Virol., 43, 1113–1117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brünger A.T. (1992) X-PLOR Version 3.1. A System for X-ray Crystallography and NMR. Yale University Press, New Haven, CT.
- Brünger A.T. (1997) Free R value: a novel statistical quantity for assessing the accuracy of crystal structures. Nature, 355, 472–475. [DOI] [PubMed] [Google Scholar]
- Burns J.W., Siadat-Pajouh,M., Krishnaney,A.A. and Greenberg,H.B. (1996) Protective effect of rotavirus VP6-specific IgA monoclonal antibodies that lack neutralizing activity. Science, 272, 104–107. [DOI] [PubMed] [Google Scholar]
- Carson M. (1987) Ribbon models of macromolecules. J. Mol. Graph., 5, 103–106. [Google Scholar]
- Castellano E., Oliva,G. and Navaza,J. (1992) Fast rigid-body refinement for molecular-replacement techniques. J. Appl. Crystallogr., 25, 281–284. [Google Scholar]
- CCP4 (1994) The CCP4 suite: programs for protein crystallography. Acta Crystallogr. D, 50, 760–763. [DOI] [PubMed] [Google Scholar]
- Centers for Disease Control (1999) From the Centers for Disease Control and Prevention. Withdrawal of rotavirus vaccine recommendation. J. Am. Med. Assoc., 282, 2113–2114. [PubMed] [Google Scholar]
- Cohen J., Laporte,J., Charpilienne,A. and Scherrer,R. (1979) Activation of rotavirus RNA polymerase by calcium chelation. Arch. Virol., 60, 177–186. [DOI] [PubMed] [Google Scholar]
- Cowtan K.D. (1994) DM program. Joint CCP4 ESF-EACBM Newslett., 31, 34–38. [Google Scholar]
- de la Fortelle E. and Bricogne,G. (1997) Maximum-likelihood heavy-atom parameter refinement for multiple isomorphous replacement and multiwavelength anomalous diffraction methods. Methods Enzymol., 276, 472–494. [DOI] [PubMed] [Google Scholar]
- Devereux J., Haeberli,P. and Smithies,O. (1985) A comprehensive set of sequence analysis programs for the VAX. Nucleic Acids Res, 12, 387–395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Durham A.C., Hendry,D.A. and Von Wechmar,M.B. (1977) Does calcium ion binding control plant virus disassembly? Virology, 77, 524–533. [DOI] [PubMed] [Google Scholar]
- Estes M.K. (1996) Rotaviruses and their replication. In Fields,B.N., Knipe,D.M., Howley,P.M., Chanock,R.M., Melnick,J.L., Monath,T.P., Roizman,B. and Straus,S.E. (eds), Fields Virology. Vol. 2. Lippincott-Raven, Philadelphia, PA, pp. 1625–1655.
- Estes M.K. and Cohen,J. (1989) Rotavirus gene structure and function. Microbiol. Rev., 53, 410–449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fields B.N. (1996) Reoviridae. In Fields,B.N., Knipe,D.M., Howley,P.M., Chanock,R.M., Melnick,J.L., Monath,T.P., Roizman,B. and Straus,S.E. (eds), Fields Virology. Vol. 2. Lippincott-Raven, Philadelphia, PA, pp. 1553–1555.
- Ginn D.I., Ward,R.L., Hamparian,V.V. and Hughes,J.H. (1992) Inhibition of rotavirus in vitro transcription by optimal concentrations of monoclonal antibodies specific for rotavirus VP6. J. Gen. Virol., 73, 3017–3022. [DOI] [PubMed] [Google Scholar]
- Gorziglia M., Hoshino,Y., Nishikawa,K., Maloy,W.L., Jones,R.W., Kapikian,A.Z. and Chanock,R.M. (1988) Comparative sequence analysis of the genomic segment 6 of four rotaviruses each with a different subgroup specificity. J. Gen. Virol., 69, 1659–1669. [DOI] [PubMed] [Google Scholar]
- Grimes J., Basak,A.K., Roy,P. and Stuart,D. (1995) The crystal structure of bluetongue virus VP7. Nature, 373, 167–170. [DOI] [PubMed] [Google Scholar]
- Grimes J.M., Jakana,J., Ghosh,M., Basak,A.K., Roy,P., Chiu,W., Stuart,D.I. and Prasad,B.V. (1997) An atomic model of the outer layer of the bluetongue virus core derived from X-ray crystallography and electron cryomicroscopy. Structure, 5, 885–893. [DOI] [PubMed] [Google Scholar]
- Grimes J.M., Burroughs,J.N., Gouet,P., Diprose,J.M., Malby,R., Zientara,S., Mertens,P.P. and Stuart,D.I. (1998) The atomic structure of the bluetongue virus core. Nature, 395, 470–478. [DOI] [PubMed] [Google Scholar]
- Harrison S.C., Olson,A., Schutt,C.E., Winkler,F.K. and Bricogne,G. (1978) Tomato bushy stunt virus at 2.9 Å resolution. Nature, 276, 368–373. [DOI] [PubMed] [Google Scholar]
- Hogle J., Kirchhausen,T. and Harrison,S.C. (1983) Divalent cation sites in tomato bushy stunt virus: difference maps at 2.9 Å resolution. J. Mol. Biol., 171, 95–100. [DOI] [PubMed] [Google Scholar]
- Huber R. and Schneider,M. (1985) A group refinement procedure in protein crystallography using Fourier transforms. J. Appl. Crystallogr., 18, 165–169. [Google Scholar]
- Jones T.A., Zhou,J.-Y., Cowan,S.W. and Kjeldgaard,M. (1991) Improved methods for building protein models in electron density maps and the location of errors in these models. Acta Crystallogr. A, 47, 110–119. [DOI] [PubMed] [Google Scholar]
- Kapikian A.Z., Cline,W.L., Greenberg,H.B., Wyatt,R.G., Kalica,A.R., Banks,C.E., James,H.D.,Jr, Flores,J. and Chanock,R.M. (1981) Antigenic characterization of human and animal rotaviruses by immune adherence hemagglutination assay (IAHA): evidence for distinctness of IAHA and neutralization antigens. Infect. Immun., 33, 415–425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kraulis P.E. (1991) MOLSCRIPT: a program to produce both detailed and schematic plots of protein structures. J. Appl. Crystallogr., 24, 946–950. [Google Scholar]
- Lawton J.A., Estes,M.K. and Prasad,B.V. (1997a) Three-dimensional visualization of mRNA release from actively transcribing rotavirus particles. Nature Struct. Biol., 4, 118–121. [DOI] [PubMed] [Google Scholar]
- Lawton J.A., Zeng,C.Q., Mukherjee,S.K., Cohen,J., Estes,M.K. and Prasad,B.V. (1997b) Three-dimensional structural analysis of recombinant rotavirus-like particles with intact and amino-terminal-deleted VP2: implications for the architecture of the VP2 capsid layer. J. Virol., 71, 7353–7360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lepault J., Petitpas,I., Erk,I., Navaza,J., Bigot,D., Dona,M., Vachette,P., Cohen,J. and Rey,F.A. (2001) Structural polymorphism of the major capsid protein of rotavirus. EMBO J., 20, 1498–1507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liddington R.C., Yan,Y., Moulai,J., Sahli,R., Benjamin,T.L. and Harrison,S.C. (1991) Structure of simian virus 40 at 3.8 Å resolution. Nature, 354, 278–284. [DOI] [PubMed] [Google Scholar]
- Lopez S., Espinosa,R., Greenberg,H.B. and Arias,C.F. (1994) Mapping the subgroup epitopes of rotavirus protein VP6. Virology, 204, 153–162. [DOI] [PubMed] [Google Scholar]
- Merrit E.A. and Murphy,M.E.P. (1994) Raster3D version 2.0. A program for photo-realistic molecular graphics. Acta Crystallogr. D, 50, 869–873. [DOI] [PubMed] [Google Scholar]
- Nicholls A., Bharadwaj,R. and Honig,B. (1993) GRASP—graphical representation and analysis of surface properties. Biophys. J., 64, A166. [Google Scholar]
- Otwinowski Z. and Minor,W. (1996) Processing of X-ray diffraction data collected in oscillation mode. In Carter,C.W. and Sweet,R.M. (eds), Macromolecular Crystallography—Part A. Vol. 276. Academic Press, London, UK, pp. 307–326. [DOI] [PubMed]
- Petitpas I. (1999) Etude cristallographique du rotavirus. Thesis, Faculté de Medecine et Pharmacie, Université de Bourgogne, Dijon, France.
- Petitpas I., Lepault,J., Vachette,P., Charpilienne,A., Mathieu,M., Kohli,E., Pothier,P., Cohen,J. and Rey,F.A. (1998) Crystallization and preliminary X-ray analysis of rotavirus protein VP6. J. Virol., 72, 7615–7619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prasad B.V.V. and Estes,M.K. (eds) (1997) Molecular Basis of Rotavirus Replication: Structure–Function Correlations. Oxford University Press, New York, NY.
- Prasad B.V., Wang,G.J., Clerx,J.P. and Chiu,W. (1988) Three-dimensional structure of rotavirus. J. Mol. Biol., 199, 269–275. [DOI] [PubMed] [Google Scholar]
- Prasad B.V., Rothnagel,R., Zeng,C.Q., Jakana,J., Lawton,J.A., Chiu,W. and Estes,M.K. (1996) Visualization of ordered genomic RNA and localization of transcriptional complexes in rotavirus. Nature, 382, 471–473. [DOI] [PubMed] [Google Scholar]
- Reinisch K.M., Nibert,M.L. and Harrison,S.C. (2000) Structure of the reovirus core at 3.6 Å resolution. Nature, 404, 960–967. [DOI] [PubMed] [Google Scholar]
- Rossmann M.G. and Johnson,J.E. (1989) Icosahedral RNA virus structure. Annu. Rev. Biochem., 58, 533–573. [DOI] [PubMed] [Google Scholar]
- Schiff L.A., Nibert,M.L., Co,M.S., Brown,E.G. and Fields,B.N. (1988) Distinct binding sites for zinc and double-stranded RNA in the reovirus outer capsid protein σ3. Mol. Cell. Biol., 8, 273–283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith T.J. et al. (1993) Structure of human rhinovirus complexed with Fab fragments from a neutralizing antibody. J. Virol., 67, 1148–1158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tang B., Gilbert,J.M., Matsui,S.M. and Greenberg,H.B. (1997) Comparison of the rotavirus gene 6 from different species by sequence analysis and localization of subgroup-specific epitopes using site-directed mutagenesis. Virology, 237, 89–96. [DOI] [PubMed] [Google Scholar]
- Womble D.D, (2000) GCG: The Wisconsin Package of sequence analysis programs. Methods Mol. Biol., 132, 3–22. [DOI] [PubMed] [Google Scholar]
- Wu B., Hammar,L., Xing,L., Markarian,S., Yan,J., Iwasaki,K., Fujiyoshi,Y., Omura,T. and Cheng,R.H. (2000) Phytoreovirus T = 1 core plays critical roles in organizing the outer capsid of T = 13 quasi-equivalence. Virology, 271, 18–25. [DOI] [PubMed] [Google Scholar]
- Yeager M., Berriman,J.A., Baker,T.S. and Bellamy,A.R. (1994) Three-dimensional structure of the rotavirus haemagglutinin VP4 by cryo-electron microscopy and difference map analysis. EMBO J., 13, 1011–1018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou Z.H., Prasad,B.V., Jakana,J., Rixon,F.J. and Chiu,W. (1994) Protein subunit structures in the herpes simplex virus A-capsid determined from 400 kV spot-scan electron cryomicroscopy. J. Mol. Biol., 242, 456–469. [DOI] [PubMed] [Google Scholar]