

Abstract
In higher eukaryotic cells, the p53 protein is degraded by the ubiquitin–26S proteasome system mediated by Mdm2 or the human papilloma virus E6 protein. Here we show that COP9 signalosome (CSN)-specific phosphorylation targets human p53 to ubiquitin–26S proteasome-dependent degradation. As visualized by electron microscopy, p53 binds with high affinity to the native CSN complex. p53 interacts via its N-terminus with CSN subunit 5/Jab1 as shown by far-western and pull-down assays. The CSN-specific phosphorylation sites were mapped to the core domain of p53 including Thr155. A phosphorylated peptide, Δp53(145–164), specifically inhibits CSN-mediated phosphorylation and p53 degradation. Curcumin, a CSN kinase inhibitor, blocks E6-dependent p53 degradation in reticulocyte lysates. Mutation of Thr155 to valine is sufficient to stabilize p53 against E6-dependent degradation in reticulocyte lysates and to reduce binding to Mdm2. The p53T155V mutant accumulates in both HeLa and HL 60 cells and exhibits a mutant (PAb 240+) conformation. It induces the cyclin-dependent inhibitor p21. In HeLa and MCF-7 cells, inhibition of CSN kinase by curcumin or Δp53(145–164) results in accumulation of endogenous p53.
Keywords: COP9 signalosome/Jab1/Mdm2/proteasome/ubiquitin
Introduction
The COP9 signalosome complex is conserved from fission yeast to human (Mundt et al., 1999). Originally it was identified in plant cells as a repressor of light-controlled development and called the COP9 complex (Wei et al., 1994; Wei and Deng, 1999). Recently the complex has been isolated from many different organisms (Seeger et al., 1998; Wei et al., 1998; Freilich et al., 1999; Mundt et al., 1999) and renamed the COP9 signalosome (CSN) (Deng et al., 2000).
The core CSN complex consists of eight subunits (CSN1–CSN8) that exhibit significant sequence homologies to the eight subunits of the 26S proteasome lid complex (Glickman et al., 1998; Henke et al., 1999). The lid and the base are subcomplexes of the 19S regulator that associates with the 20S proteasome to form the 26S particle (for a recent review see Ferrell et al., 2000). The 26S proteasome is part of the ubiquitin (Ub) system, the major proteolytic system in eukaryotic cells, and is known to be the only protease that degrades polyubiquitylated proteins (Hershko and Ciechanover, 1998).
The exact function of CSN presently is not clear. CSN is essential for development of Drosophila melanogaster (Freilich et al., 1999), and insufficiency of CSN subunit 3 might be responsible for developmental disorders in the Smith–Magenis syndrome (Elsea et al., 1999; Potocki et al., 1999). The purified complex from human red blood cells possesses kinase activity that phosphorylates transcriptional regulators such as c-Jun, IκBα and p105 (Seeger et al., 1998). Interestingly, curcumin, a major active component of the food flavoring turmeric and known to be anti-tumorigenic (Huang et al., 1995) and anti-angiogenic (Arbiser et al., 1998), has been identified as the most effective inhibitor of CSN kinase activity (Henke et al., 1999). Overexpression of CSN2, a subunit of CSN, leads to activation of the c-Jun signaling pathway independently of the Jun N-terminal kinase (Naumann et al., 1999). These data indicate that CSN has a function in signal transduction.
There is increasing evidence for a functional cooperation between CSN and the Ub–26S proteasome system in regulating the stability of important cellular proteins. The abundance of the cyclin-dependent kinase inhibitor p27Kip1 is regulated by its degradation via the Ub–26S proteasome pathway (Pagano et al., 1995). Jab1, the CSN subunit 5, specifically binds p27Kip1 and promotes its degradation by the Ub–26S proteasome system (Tomoda et al., 1999). Recently it has been shown that interaction of CSN5 with the precursor of the lutropin/choriogonadotropin receptor (rLHR) also accelerates degradation of the protein (Li et al., 2000). A clear example of CSN and Ub–26S proteasome system cooperation is the transcription factor c-Jun, which also binds to CSN5/Jab1 (Claret et al., 1996). CSN phosphorylates c-Jun at the N-terminal transactivation domain including Ser63 and Ser73 (Seeger et al., 1998), which prevents ubiquitylation and degradation of the transcription factor (Musti et al., 1997). Here we show that the stability of the tumor suppressor p53 also depends on the cooperation between CSN and the Ub–26S proteasome system.
The importance of p53 was deduced from the finding that ∼50% of all human cancers contain mutations within the p53 gene. The stability/activity of p53 is crucial for prevention of tumorigenesis and tumor growth. The tumor suppressor p53 is also a substrate of the Ub pathway (Maki et al., 1996) and its abundance is tightly regulated. Under normal growth conditions, it is maintained at very low levels through continuous degradation by the Ub–26S proteasome system. Its degradation is mediated by Mdm2 serving as both a nuclear–cytoplasmic shuttle (Freedman and Levine, 1998) and a p53-specific Ub ligase (Honda and Yasuda, 1999). Preventing the interaction between p53 and Mdm2 is sufficient to promote p53 stabilization (Honda and Yasuda, 1999). In the presence of the human papilloma virus (HPV) type 16, the viral E6 protein forms a complex with the cellular E6-AP protein that acts as a Ub ligase on p53 (Scheffner et al., 1993). The accelerated HPV-dependent degradation of p53 might be one reason for the occurrence of cervical carcinoma (for a review see Rapp and Chen, 1998).
DNA damage induces specific phosphorylation that stabilizes p53 and enables it to act as a tumor suppressor by increasing the efficiency of DNA repair and by eliminating cells with genome instability via apoptosis. Phosphorylation of p53 Ser15 and Ser20 has been detected in response to ionizing radiation. Ser15 is phosphorylated by ataxia telangiectasia mutated (ATM) kinase, which acts upstream of the checkpoint kinase Chk2 (Banin et al., 1998). Just recently, Chk2 has been identified as the enzyme that phosphorylates p53 at Ser20 (Chehab et al., 2000; Hirao et al., 2000), which prevents binding to Mdm2 (for a review see Carr, 2000). Active p53 induces p21 to cause inhibition of G1 cyclin-dependent kinases.
Here we show that CSN-specific phosphorylation of the tumor suppressor dedicates the protein to rapid degradation by the Ub–26S proteasome system. We also demonstrate that mutation of a single CSN-specific phosphorylation site stabilizes p53 against the Ub–26S proteasome-dependent degradation in vitro and in vivo. In addition, inhibition of CSN-specific p53 phosphorylation in HeLa and MCF-7 cells leads to stabilization of the endogenous tumor suppressor.
Results
CSN phosphorylates wild-type human p53 in the sequence-specific DNA-binding domain
As shown in Figure 1A, human full-length, wild-type p53 (p53wt) is phosphorylated by highly purified CSN in vitro. To test whether phosphorylation is CSN specific, we carried out the reaction in the presence of 50 µM curcumin, the most effective inhibitor of CSN kinase we had identified previously (Henke et al., 1999). Under these conditions, phosphorylation of p53wt was inhibited. In addition, we probed an N-terminal fragment of p53, Δp53(1–154), in the kinase assay. The N-terminal fragment was not phosphorylated by CSN, despite containing CSN-specific phosphorylation sites (see below). To see whether Δp53(1–154) can compete with full-length p53wt in the kinase reaction, we incubated constant amounts of p53wt with increasing concentrations of Δp53(1–154), which caused inhibition of full-length p53wt phosphorylation (Figure 1B).

Fig. 1. Full-length p53wt is phosphorylated at the sequence-specific DNA-binding domain by purified CSN. (A) Recombinant His6-tagged, full-length p53wt and Δp53(1–154) were incubated in the presence of purified human CSN and [γ-32P]ATP. Curcumin and Δp53(145–164) were added to assays with p53wt at final concentrations of 50 and 200 µM, respectively. For Δp53(145–164), a Ki of ∼40 µM was estimated. After 60 min, the reaction mixture was separated by SDS–PAGE and stained with Coomassie. The dried gels were analyzed by autoradiography. (B) Competition of CSN-dependent phosphorylation of full-length p53wt with increasing amounts of Δp53(1–154) using kinase assay conditions as in (A). SDS–poly acrylamide gels were stained with Coomassie, dried, and 32P-labeled p53 was visualized by autoradiography. (C) Functional domains of p53 and the major phospho-peptide identified by mass spectrometry and peptide sequence analysis. Phospho-peptide analysis was carried out as described in Materials and methods. The putative phosphorylation sites Ser149, Thr150 and Thr155 indicated are phosphorylated in Δp53(145–164).
To identify CSN-specific phosphorylation sites, we performed a phospho-peptide analysis of p53. Separation of chymotryptic fragments by HPLC, analysis of radioactivity and identification by peptide sequencing as well as mass spectrometry revealed one major 32P-labeled peptide (amino acids 147–160) shown in Figure 1C. The peptide fragment containing putative phosphorylation sites Ser149, Thr150 and Thr155 is localized in the sequence-specific DNA-binding region of p53 (core domain). A p53 peptide [Δp53(145–164)] was synthesized with phosphorylated amino acid residues Ser149, Thr150 and Thr155. The peptide was used as a product inhibitor in the kinase assay. As shown in Figure 1A, Δp53(145–164) reduced p53wt phosphorylation by the CSN. The Ki was estimated to be ∼40 µM (data not shown).
CSN preferentially binds p53 oligomers
Phosphorylation of p53 by CSN requires binding of the tumor suppressor to the complex. To demonstrate this, recombinant His6-tagged p53wt was incubated with purified CSN at a molar ratio of ∼1:1 before the mixture was loaded on a glycerol gradient. After centrifugation, fractions were collected, and proteins were separated by SDS–PAGE and then blotted onto nitrocellulose. Blots then were tested with anti-p53 and anti-CSN2 antibodies. As seen in Figure 2A, most recombinant p53 co-sedimented with the CSN complex in glycerol gradient fractions 9–13.

Fig. 2. p53wt binds to the purified CSN complex. (A) Recombinant p53wt tightly associates with purified human CSN complex in glycerol gradients. Purified CSN and His6-tagged p53 were mixed in a molar ratio of ∼1:1 and the mixture was analyzed by glycerol gradient centrifugation (p53+CSN). In control gradients, p53wt alone was centrifuged. The gradients were fractionated and analyzed by western blotting with an anti-p53 antibody (Anti-p53). After stripping, the same blots were re-probed with a CSN-specific antibody (Anti-CSN2). p53 alone sediments at 200 kDa, indicating that most tumor suppressor is oligomerized. (B) Electron microscopy of gold-labeled p53 alone (p53 alone) and of purified CSN complexes incubated with gold-labeled p53 (p53+CSN). The sections of electron micrographs shown were recorded at 2 µm defocus. About one-third of the CSN complexes are labeled with p53–gold particles (black arrowhead). Free CSN complexes (white arrowhead) show a wide variability in shapes, as well as diffuse boundaries. A gallery of extracted p53–gold-labeled CSN complexes is shown.
To confirm CSN–p53 binding, electron microscopy studies with p53 conjugated to electron-dense colloidal gold particles (diameter ∼3.5 nm) were performed. Free p53–gold particles preferentially appeared in monomeric form with slight aggregation (Figure 2B, p53 alone). When incubated with the CSN complex (Figure 2B, p53+CSN), the arrangement of p53–gold particles was altered. Under our conditions, approximately one-third of the CSN complexes were tagged with gold-labeled p53. Most of the bound p53–gold particles appeared in oligomeric clusters, whereby tetrameric gold clusters comprised almost 50% as estimated by statistical analysis of 330 p53–gold-labeled complexes. However, one, two, three and occasionally five p53–gold particles were also found associated with the CSN complexes. In agreement with previous data, unlabeled CSN complexes occurred in a variety of two-dimensional views (Kapelari et al., 2000). Despite the short incubation time of 5 min, unbound p53–gold particles were rarely detected, which indicates a high affinity of p53 for CSN. A separate gallery of enlarged single CSN complexes labeled with p53–gold is shown in Figure 2B, which demonstrates the organization of gold particles on the CSN complexes in more detail, and stresses that oligomeric association was the most prominent arrangement observed.
p53 binds via its N-terminus to the CSN subunit 5/Jab1
To determine which of the core CSN subunits is involved in p53 binding, far-western blots were carried out with all recombinant subunits of CSN, except CSN4. The data clearly revealed the binding of p53 to CSN5/Jab1 (Figure 3A). Similar data were obtained when p53 was immobilized on nitrocellulose and then incubated with recombinant CSN5/Jab1 (Figure 3B). In these experiments, Δp53(1–154) was also utilized as a potential target for CSN5/Jab1. The data demonstrate that CSN5/Jab1 interacts with the N-terminal 154 amino acids of p53. To make sure that p53 binds to CSN5/Jab1, we performed pull-down experiments. p53wt or Δp53(1–154) was bound to Ni-NTA magnetic agarose and incubated with reticulocyte lysate containing in vitro translated 35S-labeled CSN5/Jab1. Data shown in Figure 3C support p53wt and Δp53(1–154) interaction with CSN5/Jab1, which explains the competition of the two polypeptides demonstrated in Figure 1B.

Fig. 3. p53wt and Δp53(1–154) bind to the CSN subunit 5/Jab1. (A) Far-western blots performed with immobilized, recombinant CSN subunits. Recombinant subunits used were separated by SDS–PAGE and stained with Coomassie. The same proteins were immobilized on nitrocellulose and incubated with p53wt. After washings, the blots were tested with an anti-p53 antibody (Anti-p53). (B) Far-western blots performed with immobilized p53wt and Δp53(1–154). ΔCSN3(111–403) was used as a negative control. Immobilized proteins were incubated with recombinant CSN5/Jab1. The blots were tested with a specific anti-CSN5 antibody (Anti-CSN5). To avoid false-positive interactions, blots were stripped and re-probed with the same antibody. All specific interactions disappeared after stripping (data not shown). (C) Pull-down assays with p53wt or Δp53(1–154) and in vitro translated, 35S-labeled CSN5/Jab1. CSN5/Jab1 was translated in reticulocyte lysate using a CSN5 cDNA-pcDNA3.1 construct possessing a T7 promotor and coding for an N-terminal Flag tag (translated JAB1). The occurrence of three different bands might be due to internal starts of translation. Recombinant p53wt, Δp53(1–154) or Mdm2 (control) was bound to Ni-NTA magnetic agarose and incubated with 35S-labeled Jab1-containing lysate. After SDS–PAGE, 35S-labeled Jab1 was visualized by autoradiography. Weak bands seen in the control indicate unspecific binding of CSN5/Jab1. Coomassie of JAB1 denotes recombinant His6-tagged Jab1 separated by SDS–PAGE and stained with Coomassie. (D) Sequence alignment of the regions of c-Jun (Claret et al., 1996) and p27Kip1 (Tomoda et al., 1999) that bind to CSN5/Jab1 with Δp53(1–154). The region with the highest homology is shown.
Since interacting amino acid regions of p27Kip1 and c-Jun with CSN5/Jab1 have been characterized (Claret et al., 1996; Tomoda et al., 1999), Δp53(1–154) was aligned with the two peptides (Figure 3D). The alignment revealed a similar region in the N-terminal portion of p53 (amino acids 82–105) that might form the interacting surface with CSN5/Jab1.
Site-directed mutagenesis confirms the CSN-specific phosphorylation sites of p53
We were curious to determine whether all, or just particular, putative phosphorylation sites are real targets of the CSN kinase. For that purpose, Ser149, Thr150 and Thr155 were substituted with alanine or valine using site-directed mutagenesis, and recombinant proteins were tested in kinase assays with isolated CSN. Single mutations of Ser149 or Thr150 had little impact on CSN-specific phosphorylation of p53 (data not shown). Therefore, a double mutant (p53S149A, T150V) was produced in bacteria and tested in the kinase assay. In this case, Thr150 was changed to valine, since the exchange with alanine led to an interruption of translation at the mutation site. In addition, valine exhibits a structure that is closer to that of threonine than that of alanine. In the kinase assay, p53S149A, T150V was still phosphorylated (Figure 4), indicating that Thr155 is an additional target of CSN kinase. Since the mutant protein p53T155V is phosphorylated by CSN, we concluded that Ser149 and/or Thr150 are true CSN-specific phosphorylation sites. As shown in Figure 4, the triple mutant p53S149A, T150V, T155V showed the lowest grade of CSN-dependent phosphorylation of all the mutants tested, although there still remained a curcumin-sensitive background of phosphorylation (data not shown). In summary, these data demonstrate that Ser149, Thr150 and Thr155 are CSN-specific phosphorylation sites. Moreover, p53 possesses additional phosphorylation sites that are modified by CSN. A phospho-peptide analysis of triple mutant p53S149A, T150V, T155V revealed that the chymotryptic peptide 94–103 contains additional CSN-specific phosphorylation sites. Mutation of all serine and threonine residues of peptide 94–103 produced a p53 version that is phosphorylated on peptide 147–160 (data not shown).

Fig. 4. CSN-specific phosphorylation of p53wt and p53 mutants. The kinase assays with purified CSN and recombinant p53wt or p53 mutants were carried out as outlined in Materials and methods. The putative CSN-specific phosphorylation sites Ser149, Thr150 and Thr155 were mutated to alanine or valine as indicated by site-directed mutagenesis (see Materials and methods). The intensity of p53S149A, T150V, T155V phosphorylation was ∼25% of that with p53wt as estimated by densitometry using Aida 2.1.
Impact of CSN-specific phosphorylation on degradation of p53wt and p53 mutants by the Ub–26S proteasome system
Ub–26S proteasome-dependent degradation of p53 was studied in reticulocyte lysate containing an intact Ub–26S proteasome system and the CSN complex. In vitro translated p53 was found to be comparatively stable in reticulocyte lysate so, in order to see degradation within minutes, the HPV protein E6 was added. Under these conditions, in vitro expressed 35S-labeled p53wt was removed almost completely after 1–2 h (see Figure 5A). As expected, the addition of the proteasome inhibitor lactacystin led to stabilization of p53, indicating that the degradation is mostly proteasome dependent. A similarly effective stabilization of p53 was obtained in the presence of curcumin. The CSN inhibitor curcumin has no direct effect on the activity of the 26S proteasome, as tested in vitro with fluorogenic peptides and Ub conjugates as substrates (data not shown). To make sure that degradation depends on CSN-mediated phosphorylation, we added the specific competitor Δp53(145–164) to the degradation assay. Under these conditions, p53wt was also stabilized.

Fig. 5. Impact of CSN-specific phosphorylation on Ub–26S proteasome-dependent p53 degradation in reticulocyte lysate. (A) E6-dependent degradation of p53wt is inhibited by lactacystin (20 µM), curcumin (50 µM) and Δp53(145–164) (200 µM). p53T155V, p53S149A, T150V, T155V and Δp53(1–154) mutants are stabilized against HPV E6- and proteasome-dependent degradation. p53wt and p53 mutant cDNAs were in vitro translated in the presence of [35S]methionine and degradation assays were performed as outlined in Materials and methods. 35S-labeled p53wt or p53 mutant proteins were visualized by autoradiography. (B) Binding to Mdm2 is reduced with p53 mutants possessing a substitution of Thr155 to valine. Pull-down assays were performed with in vitro translated, 35S-labeled p53wt or p53 mutants and with recombinant, unlabeled Mdm2 as described in Materials and methods. As control, recombinant Mdm2 and p53wt were separated on an SDS–polyacrylamide gel, which was stained with Coomassie (Coomassie of Mdm2 and of p53). The negative control without Mdm2 (p53 without Mdm2) was obtained by incubating 35S-labeled, in vitro translated p53wt with magnetic beads in the absence of recombinant Mdm2. Radioactive p53wt or p53 mutants bound to Mdm2 were visualized by autoradiography. Binding of p53T155V to Mdm2 is approximately four times less than that of p53wt as estimated by densitometry using Aida 2.1.
Mutations of Ser149 and Thr150 had no impact on the Ub–26S proteasome-dependent degradation of p53, and the double mutant p53S149A, T150V also behaved like p53wt (Figure 5A). In contrast, mutation of Thr155 to valine was sufficient to stabilize p53 in that system. We assume that the stabilization of the triple mutant p53S149A, T150V, T155V is due solely to the substitution of Thr155 (Figure 5A). Interestingly, the N-terminal fragment Δp53(1–154) was also stable under the conditions used.
To assess the impact of CSN phosphorylation on the Mdm2-dependent degradation of p53, the Mdm2 binding ability of p53wt and p53 mutants was tested by pull-down assays (see Figure 5B). Equivalent to the HPV E6 protein-dependent degradation data, the mutants p53S149A and p53T150A and the double mutant p53S149A, T150V behaved like p53wt in terms of binding to Mdm2. In contrast, the mutation of Thr155 to valine caused a reduction of p53 binding to Mdm2 by ∼75% as compared with p53wt.
Mutation of Thr155 to valine stabilizes p53 in cells and induces p21, the inhibitor of G1 cyclin-dependent kinases
To test stability in vivo, p53wt and p53 mutants were transfected into HeLa as well as HL 60 cells. HeLa cells have been transformed with HPV and express the E6 protein (Beer-Romero et al., 1997), whereas HL 60 cells lack p53 and express Mdm2 after transfection with p53 (data not shown). As shown by western blotting in Figure 6A, 24 h after transfection p53T155Val and p53S149A, T150V, T155V were stabilized in both HeLa and HL 60 cells as compared with p53wt or p53S149A, T150V. To test whether p53wt and p53 mutants possess transactivational activity, western blots were performed with lysates from HL 60 cells obtained 24 h after transfection using an anti-p21 antibody (Figure 6B). Since p21 was induced by p53wt and by all p53 mutants tested, we assumed that the mutation of Thr155 to valine had no impact on the ability of p53 to cause G1 cell cycle arrest.

Fig. 6. p53T155V and p53S149A, T150V, T155V are stabilized in HeLa and HL 60 cells and induce p21. (A) HeLa cells and HL 60 cells were transiently transfected with vector alone (control), p53wt, the double mutant p53S149A, T150V, the mutant p53T155V or the triple mutant p53S149A, T150V, T155V expression constructs. Lysates of HeLa and HL 60 cells were tested 24 h after transfection by western blotting using an anti-p53 antibody (Anti-p53). The same protein amounts were loaded as estimated by Bradford assay. As expected, there is no p53 in HL 60 cell lysates transfected with the vector alone and the endogenous p53 levels in HeLa cells are not detectable. As estimated by densitometry using Aida 2.1, p53T155V and p53S149A, T150V, T155V proteins accumulate up to five times in HeLa and more than twice in HL 60 cells as compared with p53wt or p53S149A, T150V proteins. (B) p53wt and p53 mutants transfected into HL 60 cells induce the cyclin-dependent kinase inhibitor p21. Lysates of HL 60 cells obtained 24 h after transfection were tested by western blotting using an anti-p21 antibody (Anti-p21). There is no p21 seen after transfecting HL 60 cells with the vector alone (control). (C) Immunoprecipitation of in vitro translated 35S-labeled p53wt in the absence (p53wt) and presence of curcumin (+Curcumin) and of p53 mutants by the conformation-specific anti-p53 antibodies PAb 1620 and PAb 240. The immunoprecipitates were analyzed by SDS–PAGE and autoradiography.
We were interested to see whether our mutations exert any impact on the conformation of the p53 molecule. Therefore, p53wt and p53 mutants were 35S-labeled in reticulocyte lysate and subsequently immunoprecipitated using the anti-p53 conformation-specific antibodies PAb 1620 and PAb 240 (Webley et al., 2000). As seen in Figure 6C, p53wt and p53S149A, T150V were recognized by PAb 1620, indicating a wild-type conformation, whereas p53T155V and p53S149A, T150V, T155V had a rather mutant conformation, as indicated by binding of PAb 240 and a weak interaction with PAb 1620. Most interestingly, inhibition of CSN-specific phosphorylation by curcumin also induced a change of p53wt from wild-type to mutant conformation (Figure 6C).
Inhibition of CSN-specific phosphorylation in HeLa and MCF-7 cells stabilizes endogenous p53
Due to the presence of the E6- and Ub–26S proteasome-dependent proteolytic system in HeLa cells, intracellular p53 levels are very low (Beer-Romero et al., 1997) and can hardly be detected with common anti-p53 antibodies. If CSN-specific phosphorylation targets intracellular p53 to degradation by the E6- and Ub–26S proteasome-dependent pathway, inhibition of CSN should increase endogenous p53 levels. To test this possibility, HeLa cells were treated with 50 µM curcumin for 6 h. As shown in Figure 7, endogenous p53 increased dramatically after curcumin treatment. To make sure that curcumin does not affect the Ub–26S proteasome system in cells, we tested the same blots with an anti-c-Jun antibody. The transcription factor c-Jun is a substrate of the Ub–26S proteasome system (Hershko and Ciechanover, 1998) and it was degraded in the presence of curcumin.
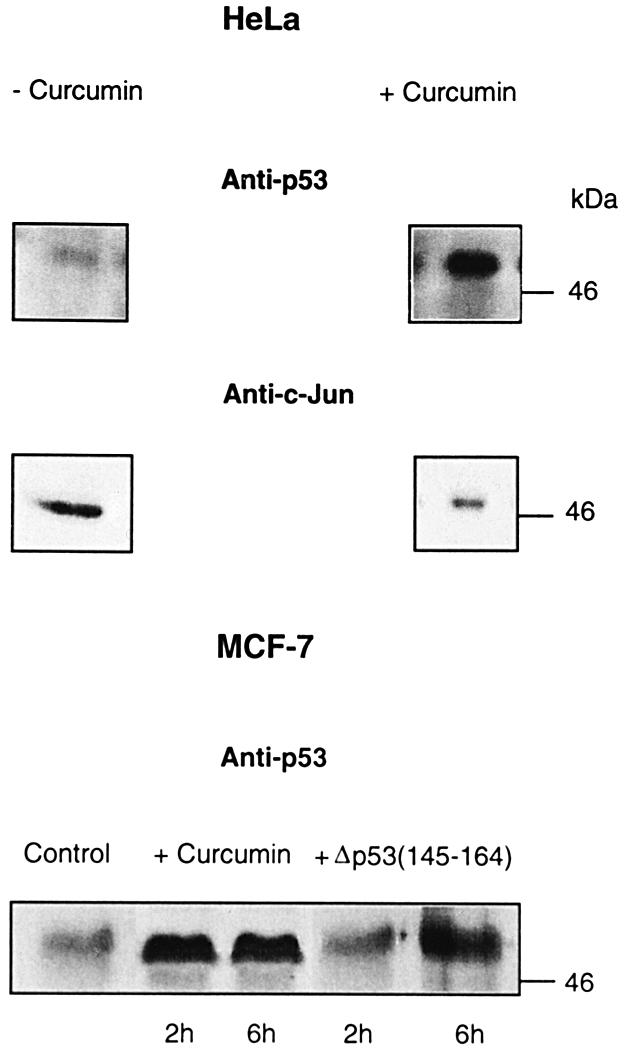
Fig. 7. Inhibition of CSN-specific phosphorylation leads to accumulation of endogenous p53 in HeLa and MCF-7 cells. At 6 h after curcumin treatment (+Curcumin), endogenous p53 increased dramatically in HeLa cells as compared with cells incubated without curcumin (–Curcumin). At the same time, the transcription factor c-Jun (Anti-c-Jun) was degraded, indicating an intact Ub–26S proteasome system. In MCF-7 cells, endogenous p53 is stabilized in the presence of curcumin or Δp53(145–164) as compared with untreated cells (control).
MCF-7 cells possess p53wt and degrade the tumor suppressor exclusively by the Mdm2-dependent pathway. In MCF-7 cells, a substantial increase in endogenous p53 was detected already after 2 h of curcumin treatment (Figure 7). After 12 h, we observed a decrease in the amount of p53 accompanied by a massive cell death, which might be due to apoptosis (data not shown). An increase in endogenous p53 was also obtained by incubating MCF-7 cells with Δp53(145–164), indicating a specific involvement of the CSN.
Discussion
CSN-specific phosphorylation of the human tumor suppressor p53 depends on its binding to subunit CSN5/Jab1
The data presented here clearly show that full-length p53 is phosphorylated by CSN. The reaction is sensitive to curcumin, an effective inhibitor of CSN kinase (Henke et al., 1999). Interestingly, the fragment Δp53(1–154) is not phosphorylated although it contains CSN-specific phosphorylation sites. This observation is equivalent to the situation for c-Jun. As in p53, the oligomerization domain of c-Jun is localized in the C-terminal region, and N-terminal fragments cannot be phosphorylated, implying that dimerization is a prerequisite for phosphorylation via CSN (Seeger et al., 1998; Naumann et al., 1999). Oligomerized p53 binds to CSN as shown by density gradient centrifugation in which most free p53 sediments at ∼200 kDa (Figure 2A). Electron microscopy studies clearly support these data and show that oligomeric p53–gold clusters bind with high affinity to CSN. As several proteins cover one gold colloid in protein–gold conjugates prepared by the technique used (Slot and Geuze, 1985), the actual number of p53 molecules bound to the CSN is either equal to or higher than the number of gold particles seen. The predilection for tetrameric and also trimeric gold clusters suggests that the CSN preferentially binds tetrameric p53. However, the exact structure of p53 oligomers that bind to the CSN cannot be deduced from the present data. An excess of unlabeled p53 did not change the ratio between different oligomeric forms bound to the CSN (data not shown). Also, the occurrence of only one CSN5/Jab1 subunit in the CSN complex is very likely (Kapelari et al., 2000). Nevertheless, we cannot exclude the possibility that there is more than one p53-binding site on the CSN complex.
Although amino acids 1–154 of p53 are sufficient for binding to CSN subunit CSN5/Jab1, as demonstrated by far-western blots (Figure 3B) and pull-down assays (Figure 3C), phosphorylation by CSN requires full-length or perhaps tetramerized p53. On the other hand, binding to CSN5/Jab1 is essential for phosphorylation, as shown by competition with Δp53(1–154) (Figure 1B), indicating that the process is CSN specific.
Whether amino acids 82–105 form the interacting surface with CSN5/Jab1, as indicated by sequence alignment (Figure 3C), has yet to be tested. Interestingly, CSN5/Jab1 is a receptor for many different cellular regulators such as p53 (this study), c-Jun (Claret et al., 1996), p27 (Tomoda et al., 1999), Bcl-3 (Dechend et al., 1999), rLHR (Li et al., 2000), progesterone receptor (Chauchereau et al., 2000) and integrin LFA-1 (Bianchi et al., 2000). This implies competition for the binding site and, in fact, our preliminary in vitro data show that p53 inhibits CSN-specific phosphorylation of c-Jun (data not shown).
CSN-specific phosphorylation of Thr155 is sufficient to target p53 to degradation by the Ub–26S proteasome system
Peptide sequencing and mass spectrometry revealed novel p53 phosphorylation sites specifically modified by CSN kinase. A phosphorylated peptide encompassing the CSN-specific phosphorylation sites [Δp53(145–164)] reduces CSN-dependent modification of p53. The phosphorylation sites are localized within the core domain that is responsible for specific promoter DNA binding of p53. Since evidence is increasing for a cooperation of CSN and the Ub–26S proteasome pathway in regulating the stability of important cellular proteins, the most interesting question was whether CSN-specific phosphorylation of p53 has any impact on the degradation of the tumor suppressor. Therefore, the effects of CSN-dependent phosphorylation on the two major degradation mechanisms of p53, the HPV E6- and the Mdm2-dependent pathways, were tested. Curcumin or Δp53(145–164) almost completely inhibited CSN-specific phosphorylation and abolished E6-dependent degradation of p53 in reticulocyte lysate. Endogenous p53 was stabilized by curcumin in HeLa cells possessing the E6-dependent degradation route (Beer-Romero et al., 1997). MCF-7 cells, which degrade p53 in a Mdm2-dependent manner, accumulate endogenous p53 in the presence of curcumin or Δp53(145–164). Similar effects were obtained by solely mutating Thr155 to valine, one of the CSN-specific phosphorylation sites. The mutant p53T155V transfected into cells was dramatically stabilized in HeLa cells, as well as in HL 60 cells where p53 is degraded via the Mdm2-dependent pathway. In HL 60 cells, stabilization of p53 is most probably due to reduced binding of p53T155V to Mdm2. Mutations of additional CSN-specific phosphorylation sites had no impact on p53 stability. Perhaps the additional phosphorylation sites, Ser149 and Thr150, and the putative sites on the chymotryptic fragment 94–103 are involved in other functions such as releasing p53 from the DNA or nuclear–cytoplasmatic transport, which has to be tested further.
p53 conformation seems to correlate with the susceptibility of the tumor suppressor to the Ub–26S proteasome system. Using the conformation-specific antibodies PAb 1620 and 240, we found that proteasome-dependent degradation of p53wt and p53S149A, T150V is associated with wild-type conformation, i.e. PAb 1620 positive (PAb 1620+). On the other hand, stabilization of p53 by preventing CSN-specific phosphorylation of T155 via mutation (p53T155V or p53S149A, T150V, T155V) or by curcumin is associated with a so-called mutant conformation (PAb 240+). Interestingly, DNA-damaging agents lead to stabilization of p53 (Chehab et al., 2000) and induce the mutant conformation (PAb 240+) (Webley et al., 2000). All our p53 mutants and p53wt were able to induce the inhibitor of G1 cyclin-dependent kinase, p21. This is in agreement with recent data demonstrating that both wild-type (PAb 1620+) and mutant (PAb 240+) conformations can induce p21 (Webley et al., 2000).
Interestingly, Δp53(1–154) is not degraded by the E6-dependent pathway, indicating that binding to CSN5/Jab1 is not sufficient for Ub-dependent degradation. Importantly, the competition of the N-terminal fragment with full-length p53 might explain the induction of apoptosis in HeLa cells by a Δp53(1–214) fragment (Haupt et al., 1995). In this case, endogenous p53 might be stabilized and cause apoptosis. The finding implies tumor therapeutic possibilities at least for cervical carcinomas.
On the basis of the present data, we conclude that CSN-specific phosphorylation of Thr155 is an important prerequisite for degradation of the tumor suppressor by the Ub–26S proteasome system. The question of why CSN phosphorylation accelerates p53 degradation via both pathways, the E6- and Mdm2-dependent mechanisms, cannot be answered at the moment. Perhaps CSN-specific phosphorylation of p53 leads to a conformation with increased affinity for both E6 and Mdm2, with the consequence of ubiquitylation and degradation by the 26S proteasome.
Models of functional cooperation between CSN and the Ub–26S proteasome system in the regulation of p53 stability
The data presented here demonstrate a clear connection between CSN-dependent phosphorylation and Ub–26S proteasome-dependent degradation of p53. We assume that CSN kinase is constitutively active, since the purified complex possesses kinase activity (Seeger et al., 1998). Thus, CSN is a likely candidate responsible for maintenance of low p53 levels under normal cell growth conditions by phosphorylating p53 with the consequence of Ub-dependent degradation. This is supported by the fact that low p53 levels in HeLa as well as in MCF-7 cells depend on CSN activity sensitive to curcumin or Δp53(145–164). Under conditions of DNA damage and genomic instability, the CSN-dependent mechanism of p53 destabilization should be switched off. There are many possible ways to prevent CSN-specific phosphorylation of p53. The recently described phosphorylation of p53 at Ser20 by Chk2 (Chehab et al., 2000; Hirao et al., 2000) might lead to a conformation of the tumor suppressor that is no longer phosphorylated by CSN. Perhaps direct modification by kinases such as ATM or Chk2 might cause inactivation of CSN kinase. Future investigations will show the exact links between p53 stabilizing and destabilizing mechanisms, which might be a matter of p53 conformation.
Examination of the core domain–DNA complex crystal structure (Cho et al., 1994) shows that the CSN-specific phosphorylation sites (Ser149, Thr150 and Thr155) are localized on a loop between two β-strands distal from the protein–DNA interface. Therefore, the phosphorylation sites should be accessible for CSN, and an impact on p53 DNA binding by CSN-specific phosphorylation is possible. The CSN-dependent release from the DNA might result in a nuclear export connected with Ub–26S proteasome-dependent degradation, as demonstrated for p27Kip1 (Tomoda et al., 1999).
The findings described here indicate that high CSN kinase activity might propagate tumor growth whereas low activity might drive cells into apoptosis. This is in agreement with the induction of p53-dependent apoptosis in human basal cell carcinoma cells by curcumin described recently (Jee et al., 1998). The fact that CSN-specific phosphorylation of p53 targets the tumor suppressor to degradation might explain the role of curcumin as an anti-tumorigenic substance. Thus, the function of CSN as a destabilizing factor of p53 makes the CSN complex an interesting target for future tumor therapy.
Materials and methods
Purification of human CSN and kinase assay
The CSN was isolated from human red blood cells and its kinase activity was determined with [γ-32P]ATP as described before (Seeger et al., 1998; Henke et al., 1999). Chymotryptic phospho-peptide analyses of His6-tagged p53wt and p53 mutants were performed as outlined previously for c-Jun (Seeger et al., 1998). The peptide Δp53(145–164) encompassing the amino acids (C)145LWVDSTPPPGTRVRAMAIYK-CONH2164 was synthesized with phosphorylated serine and threonine residues using the FMOC strategy on a 433A peptide synthesizer (ABI).
Wild-type p53, site-directed mutagenesis, recombinant Mdm2 and recombinant CSN subunits
Wild-type p53 (p53wt) cDNA was obtained by RT–PCR using total RNA from human monocytes isolated by standard methods. The cDNA was cloned into pBluescript (pSK) between BamHI and HindIII sites and verified by DNA sequencing. To obtain Δp53(1–154) cDNA, the construct was cleaved with BanI and re-ligated. p53wt and Δp53(1–154) cDNAs were subcloned into pQE expression vectors coding for the His6 tag at the N-terminus (Qiagen). Expressions were carried out in Escherichia coli and His-tagged proteins were isolated using the Ni-NTA purification kit (Qiagen). Site-directed mutagenesis was performed with p53 cDNA in pBluescript using the QuikChange Site-Directed Mutagenesis Kit (Stratagene) following the manufacturer’s instructions. DNA sequencing verified mutated cDNAs of p53. For expression of recombinant p53 mutants, cDNAs were subcloned into pQE vectors (Qiagen) and then as described for p53wt. For cell experiments, p53wt and p53 mutant cDNAs were subcloned into pcDNA3.1 (Invitrogen).
Mdm2 cDNA was obtained by RT–PCR using total RNA of LNCAP cells as template. To obtain recombinant Mdm2, the pQE cloning strategy was used. Recombinant CSN subunits were produced using the pQE strategy (Qiagen) as in Kapelari et al. (2000).
Glycerol gradients, western and far-western blots
To test binding between purified CSN and recombinant p53wt, 10–30% glycerol gradients were used as described in Seeger et al. (1998). A 40 µg aliquot of CSN and 5 µg of recombinant His6-tagged p53 were mixed, pre-incubated for 30 min at 37°C and then loaded on a 12 ml gradient. The gradients were calibrated with the 11S regulator (∼200 kDa) and the 20S proteasome (∼700 kDa). Fractions of 600 µl were collected, protein was precipitated with trichloroacetic acid, resuspended in 15 µl of 1× protein sample buffer and every second fraction was loaded on a 10% SDS–polyacrylamide gel. Following SDS–PAGE, proteins were subjected to western blot analysis with the indicated antibodies. The anti-CSN2 antibody was obtained as described (Seeger et al., 1998).
Far-western blots with recombinant CSN subunits were performed as outlined previously (Kapelari et al., 2000). In brief, ∼0.5 µg of recombinant His6-tagged CSN subunits or p53wt and Δp53(1–154) were separated on a 10% SDS–polyacrylamide gel and blotted to nitrocellulose. Immobilized CSN subunits were incubated for 2 h at room temperature with 1.5 µg/ml recombinant p53wt in phosphate-buffered saline (PBS). Immobilized p53wt and Δp53(1–154) were incubated with 1.5 µg/ml recombinant CSN5 in PBS. After washing, the blots were probed with monoclonal anti-p53 (IC Chemikalien) or anti-CSN5 (Gene Tex) antibodies.
Western blot analysis with cell lysates was performed using the same monoclonal anti-p53 antibody described above and a monoclonal anti-p21 antibody (Santa Cruz). All blots were developed by the ECL technique (Amersham).
Electron microscopy with purified CSN and gold-labeled p53
The preparation of colloidal gold was adapted from Slot and Geuze (1985) with modifications according to Hölzl et al. (2000). In brief, formation of ∼3.5 nm colloidal gold particles was achieved by rapid mixing of an aqueous solution of 0.012% NaAuCl4 with an aqueous solution of 0.25% tannic acid, 0.2% sodium citrate and 1 mM potassium carbonate at 60°C.
A 7 µl aliquot of recombinant p53 (0.8 mg/ml) was incubated with 40 µl of colloidal gold solution for 15 h at 4°C. After centrifugation for 10 min at 80 000 g (Beckman, Airfuge), the supernatant with the unlabeled p53 was removed and the soft pellet containing the stable conjugate, covalently connected via gold–sulfur bonds, was resuspended in 5 µl of distilled water.
A 0.6 µl aliquot of gold-labeled p53 was incubated in 30 µl of 20 mM Tris buffer pH 7.2, 30 mM NaCl for 5 min at 4°C in the absence (p53 alone) or presence of 6 µl of purified CSN (0.3 mg/ml) (p53+CSN). Droplets (5 µl) of p53 alone or p53+CSN were applied to 100 × 400 mesh copper grids that had been coated with carbon and glow discharged in a plasma cleaner for 45 s. After blotting and washing with 20 mM Tris pH 7.2 and 30 mM NaCl, negative staining and electron microscopy were performed as described previously (Kapelari et al., 2000). Images were recorded at 2 µm defocus by means of a slow scan CCD camera (Photometrix; 2048 × 2048 pixels) at a total magnification of ×47 660 (corresponding to a pixel size of 2.94 Å).
HPV E6-dependent degradation of p53 and p53 mutants in reticulocyte lysate
In vitro transcription/translation was performed using the TNT Coupled Reticulocyte Lysate System (Promega). A 1 µg aliquot of cDNA in pBluescript was used for in vitro transcription/translation. The reaction was carried out for 90 min at 30°C. Since in vitro translated p53 is stable in reticulocyte lysate, the HPV type 16 E6 protein (E6-16) was added. The degradation assay was carried out as described by Scheffner et al. (1990). A 10 µl aliquot of lysate containing translated 35S-labeled p53wt or p53 mutants was mixed with 5 µl of lysate with translated, unlabeled E6-16 in a final volume of 40 µl containing 25 mM Tris–HCl pH 7.5, 100 mM NaCl, 3 mM dithiothreitol (DTT) and 7.5 µl of untreated reticulocyte lysate (Promega). The mixture was incubated at 30°C and, after the indicated times, the reaction of 10 µl aliquots was stopped by adding 2 µl of 4× SDS sample buffer and boiling for 4 min. Total reaction mixtures of 12 µl were electrophoresed on SDS–polyacrylamide gels and the radioactive proteins were visualized by autoradiography. Lactacystin was added to the degradation assay as indicated at a final concentration of 20 µM. Curcumin and Δp53(145–164) were added to the transcription/translation reaction and to the degradation assay at final concentrations of 50 and 200 µM, respectively.
Pull-down assays
Binding of p53wt and Δp53(1–154) to CSN5/Jab1 and of p53wt and p53 mutants to Mdm2 was assayed using Ni-NTA magnetic agarose beads according to the manufacturer’s instructions (Qiagen). In brief, 10 µg of recombinant p53, Δp53(1–154) or Mdm2 were incubated with Ni-NTA magnetic agarose. In vitro translated 35S-labeled CSN5/Jab1 or p53wt or p53 mutants were added to p53– or Mdm2–magnetic agarose and incubated according to the manufacturer’s protocol. Magnetic agarose-bound proteins were eluted, separated by SDS–PAGE and visualized by autoradiography.
Immunoprecipitation
Immunoprecipitations of p53wt in vitro translated with and without 50 µM curcumin and of p53 mutants were performed with 2 µg of conformation-specific anti-p53 antibodies PAb 1620 and PAb 240 (Calbiochem) according to standard protocols. 35S-labeled p53wt and p53 mutants were obtained by in vitro translation as described above, and 25 µl of reticulocyte lysates were used for each reaction. Immuno precipitates were separated by SDS–PAGE and 35S-labeled proteins visualized by autoradiography.
Transient transfection of p53wt and p53 mutants into HeLa and HL 60 cells
HeLa, HL 60 and MCF-7 cells were cultured in RPMI 1640 medium supplemented with 10% fetal calf serum, 2 mM glutamine (Life Technologies Inc.), penicillin (100 U/ml) and streptomycin (100 µg/ml) in a humidified 5% CO2 atmosphere. For transient expression experiments, HL 60 cells were transfected with 20 µg of expression constructs of p53wt or p53 mutant cDNAs in pcDNA3.1 vector or with vector alone by electroporation as described (Melkonyan et al., 1996). HeLa cells were transfected with 10 µg of expression construct DNAs or with vector alone using the PerFect™ Transfection kit (Invitrogen) according to the manufacturer’s instructions.
Cells were harvested 24 h after transfection and assayed for gene expression by western blotting. Cell extracts were prepared using standard procedures. HL 60 cells were collected by centrifugation and the pellet was washed with PBS. Ice-cold triple-detergent lysis buffer (50 mM Tris–HCl pH 8.0, 150 mM NaCl, 0.02% sodium azide, 0.1% SDS, 1 µg/ml aprotinin, 1% NP-40, 0.5% sodium deoxycholate) with freshly prepared phenylmethylsulfonyl fluoride (PMSF; 1 mg/ml) was added to the pellet. In the case of HeLa cells, ice-cold triple-detergent lysis buffer was added to cell culture plates on ice and the cells were scraped from the plates with a cell scraper. Cells were disrupted by repeated aspiration through a 21-gauge needle. After centrifugation at 15 000 g for 30 min at 4°C, 20 µl of the total lysate (supernatant) were used for western blotting.
For inhibition by curcumin, 4 × 106 HeLa cells were incubated with 50 µM curcumin in 0.25% dimethylsulfoxide (DMSO). Control cells were treated with 0.25% DMSO. Six hours after curcumin treatment, cells were harvested and lysates were assayed for p53 and c-Jun (anti-c-Jun antibody, Amersham) levels by western blotting. MCF-7 cells were incubated with 50 µM curcumin or 200 µM Δp53(145–164).
Acknowledgments
Acknowledgements
We are grateful to Dr O.Medalia for preparing the colloidal gold, Dr W.Henke for the generous gift of RNA from LNCAP cells, Gene Tex for the anti-Jab1 antibody, and Dr M.Kania for critical reading of the manuscript. This work was supported by a grant DU 229/5-1 from the Deutsche Forschungsgemeinschaft to W.D.
References
- Arbiser J.L., Klauber,N., Rohan,R., van Leeuwen,R., Huang,M.T., Fisher,C., Flynn,E. and Byers,H.R. (1998) Curcumin is an in vivo inhibitor of angiogenesis. Mol. Med., 4, 376–383. [PMC free article] [PubMed] [Google Scholar]
- Banin S. et al. (1998) Enhanced phosphorylation of p53 by ATM in response to DNA damage. Science, 281, 1674–1677. [DOI] [PubMed] [Google Scholar]
- Beer-Romero P., Glass,S. and Rolfe,M. (1997) Antisense targeting of E6AP elevates p53 in HPV-infected but not in normal cells. Oncogene, 14, 595–602. [DOI] [PubMed] [Google Scholar]
- Bianchi E., Denti,S., Granata,A., Bossi,G., Geginat,J., Villa,A., Rogge,L. and Pardi,R. (2000) Integrin LFA-1 interacts with the transcriptional co-activator JAB1 to modulate AP-1 activity. Nature, 404, 617–621. [DOI] [PubMed] [Google Scholar]
- Carr A.M. (2000) Piecing together the p53 puzzle. Science, 287, 1765–1766. [DOI] [PubMed] [Google Scholar]
- Chauchereau A., Georgiakaki,M., Perrin-Wolff,M., Milgrom,E. and Loosfelt,H. (2000) JAB1 interacts with both the progesterone receptor and SRC-1. J. Biol. Chem., 275, 8540–8548. [DOI] [PubMed] [Google Scholar]
- Chehab N.H., Malikzay,A., Appel,M. and Halazonetis,T.D. (2000) Chk2/hCds1 functions as a DNA damage checkpoint in G1 by stabilizing p53. Genes Dev., 14, 278–288. [PMC free article] [PubMed] [Google Scholar]
- Cho Y., Gorina,S., Jeffrey,P.D. and Pavletich,N.P. (1994) Crystal structure of a p53 tumor suppressor–DNA complex: understanding tumorigenic mutations. Science, 265, 346–355. [DOI] [PubMed] [Google Scholar]
- Claret F.X., Hibi,M., Dhut,S., Toda,T. and Karin,M. (1996) A new group of conserved coactivators that increase the specificity of AP-1 transcription factors. Nature, 383, 453–457. [DOI] [PubMed] [Google Scholar]
- Dechend R., Hirano,F., Lehmann,K., Heissmeyer,V., Ansieau,S., Wulczyn,F.G., Scheidereit,C. and Leutz,A. (1999) The Bcl-3 oncoprotein acts as a bridging factor between NF-κB/Rel and nuclear co-regulators. Oncogene, 18, 3316–3323. [DOI] [PubMed] [Google Scholar]
- Deng X.-W. et al. (2000) Unified nomenclature for the COP9 signalosome and its subunits: an essential regulator of development. Trends Genet., 16, 202–203. [DOI] [PubMed] [Google Scholar]
- Elsea S.H., Mykytyn,K., Ferrell,K., Coulter,K.L., Das,P., Dubiel,W., Patel,P.I. and Metherall,J.E. (1999) Hemizygosity for the COP9 signalosome subunit gene, SGN3, in the Smith–Magenis syndrome. Am. J. Med. Genet., 87, 342–348. [PubMed] [Google Scholar]
- Ferrell K., Wilkinson,C.R.M., Dubiel,W. and Gordon,C. (2000) Regulatory subunit interactions of the 26S proteasome, a complex problem. Trends Biochem. Sci., 25, 83–88. [DOI] [PubMed] [Google Scholar]
- Freedman D. and Levine,A.J. (1998) Nuclear export is required for degradation of endogenous p53 by Mdm2 and human papillomavirus E6. Mol. Cell. Biol., 18, 7288–7293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Freilich S., Oron,E., Kapp,Y., Nevo-Caspi,Y., Orgad,S., Segal,D. and Chamovitz,D.A. (1999) The COP9 signalosome is essential for development of Drosophila melanogaster. Curr. Biol., 9, 1187–1190. [DOI] [PubMed] [Google Scholar]
- Glickman M.H., Rubin,D.M., Coux,O., Wefes,I., Pfeifer,G., Cjeka,Z., Baumeister,W., Fried,V.A. and Finley,D. (1998) A subcomplex of the proteasome regulatory particle required for ubiquitin-conjugate degradation and related to the COP9-signalosome and eIF3. Cell, 94, 615–623. [DOI] [PubMed] [Google Scholar]
- Haupt Y., Rowan,S., Shaulian,E., Vousden,K.H. and Oren,M. (1995) Induction of apoptosis in HeLa cells by trans-activation-deficient p53. Genes Dev., 9, 2170–2183. [DOI] [PubMed] [Google Scholar]
- Henke W., Ferrell,K., Bech-Otschir,D., Seeger,M., Schade,R., Jungblut,P., Naumann,M. and Dubiel,W. (1999) Comparison of human COP9 signalsome and 26S proteasome lid. Mol. Biol. Rep., 26, 29–34. [DOI] [PubMed] [Google Scholar]
- Hershko A. and Ciechanover,A. (1998) The ubiquitin system. Annu. Rev. Biochem., 67, 425–479. [DOI] [PubMed] [Google Scholar]
- Hirao A., Kong,Y.Y., Matsuoka,S., Wakeham,A., Ruland,J., Yoshida,H., Liu,D., Elledge,S.J. and Mak,T.W. (2000) DNA damage-induced activation of p53 by the checkpoint kinase Chk2. Science, 287, 1824–1827. [DOI] [PubMed] [Google Scholar]
- Hölzl H. et al. (2000) The regulatory complex of Drosophila melanogaster 26S proteasomes: subunit composition and localization of a deubiquitylating enzyme. J. Cell Biol., 150, 119–130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Honda R. and Yasuda,H. (1999) Association of p19ARF with Mdm2 inhibits ubiquitin ligase activity of Mdm2 for tumor suppressor p53. EMBO J., 18, 22–27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang M.T., Ma,W., Lu,Y.P., Chang,R.L., Fisher,C., Manchand,P.S., Newmark,H.L. and Conney,A. (1995) Effects of curcumin, demethoxycurcumin, bisdemethoxycurcumin and tetrahydrocurcumin on 12-O-tetradecanoylphorbol-13-acetate-induced tumor promotion. Carcinogenesis, 16, 2493–2497. [DOI] [PubMed] [Google Scholar]
- Jee S.-H., Shen,S.-Ch., Tseng,Ch.-R., Chiu,H.-Ch. and Kuo,M.-L. (1998) Curcumin induces a p53-dependent apoptosis in human basal cell carcinoma cells. J. Invest. Dermatol., 111, 656–661. [DOI] [PubMed] [Google Scholar]
- Kapelari B., Bech-Otschir,D., Hegerl,R., Schade,R., Dumdey,R. and Dubiel,W. (2000) Electron microscopy and subunit–subunit interaction studies reveal a first architecture of COP9 signalosome. J. Mol. Biol., 300, 1169–1178. [DOI] [PubMed] [Google Scholar]
- Li S., Liu,X. and Ascoli,M. (2000) p38JAB1 binds to the intracellular precursor of the lutropin/choriogonadotropin receptor and promotes its degradation. J. Biol. Chem., 275, 13386–13393. [DOI] [PubMed] [Google Scholar]
- Maki C.G., Huibregtse,J.M. and Howley,P.M. (1996) In vivo ubiquitination and proteasome-mediated degradation of p53. Cancer Res., 56, 2649–2654. [PubMed] [Google Scholar]
- Melkonyan H., Sorg,C. and Klempt,M. (1996) Electroporation efficiency in mammalian cells is increased by dimethyl sulfoxide (DMSO). Nucleic Acids Res., 24, 4356–4357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mundt K.E. et al. (1999) The COP9/signalosome complex is conserved in fission yeast and has a role in S phase. Curr. Biol., 9, 1427–1430. [DOI] [PubMed] [Google Scholar]
- Musti A.M., Treier,M. and Bohmann,D. (1997) Reduced ubiquitin-dependent degradation of c-Jun after phosphorylation by MAP kinases. Science, 275, 400–402. [DOI] [PubMed] [Google Scholar]
- Naumann M., Bech-Otschir,D., Huang,X., Ferrell,K. and Dubiel,W. (1999) COP9 signalosome-directed c-Jun activation/stabilization is independent of JNK. J. Biol. Chem., 274, 35297–35300. [DOI] [PubMed] [Google Scholar]
- Pagano M., Tam,S.W., Theodoras,A.M., Beer-Romero,P., Del Sal,G., Chau,V., Yew,P.R., Draetta,G.F. and Rolfe,M. (1995) Role of the ubiquitin–proteasome pathway in regulating abundance of the cyclin-dependent kinase inhibitor p27. Science, 269, 682–685. [DOI] [PubMed] [Google Scholar]
- Potocki L. et al. (1999) Molecular mechanism for duplication 17p11.2—the homologous recombination reciprocal of the Smith–Magenis microdeletion. Nature Genet., 24, 84–87. [DOI] [PubMed] [Google Scholar]
- Rapp L. and Chen,J.J. (1998) The papillomavirus E6 protein. Biochim. Biophys. Acta, 1378, F1–F19. [DOI] [PubMed] [Google Scholar]
- Scheffner M., Werness,B.A., Huibregtse,J.M., Levine,A.J. and Howley,P.M. (1990) The E6 oncoprotein encoded by human papillomavirus types 16 and 18 promotes the degradation of p53. Cell, 63, 1129–1136. [DOI] [PubMed] [Google Scholar]
- Scheffner M., Huibregtse,J.M., Vierstra,R.D. and Howley,P.M. (1993) The HPV-16 E6 and E6–Ap complex functions as a ubiquitin–protein ligase complex in the ubiquitination of p53. Cell, 75, 495–505. [DOI] [PubMed] [Google Scholar]
- Seeger M., Kraft,R., Ferrell,K., Bech-Otschir,D., Dumdey,R., Schade,R., Gordon,C., Naumann,M. and Dubiel,W. (1998) A novel protein complex involved in signal transduction possessing similarities to 26S proteasome subunits. FASEB J., 12, 469–478. [PubMed] [Google Scholar]
- Slot J.W. and Geuze,H.J. (1985) A new method of preparing gold probes for multiple-labeling cytochemistry. Eur. J. Cell Biol., 38, 87–93. [PubMed] [Google Scholar]
- Tomoda K., Kubota,Y. and Kato,J. (1999) Degradation of the cyclin-dependent-kinase inhibitor p27Kip1 is instigated by Jab1. Nature, 398, 160–165. [DOI] [PubMed] [Google Scholar]
- Webley K.M., Shorthouse,A.J. and Royds,J.A. (2000) Effect of mutation and conformation on the function of p53 in colorectal cancer. J. Pathol., 191, 361–367. [DOI] [PubMed] [Google Scholar]
- Wei N. and Deng,X.W. (1999) Making sense of the COP9 signalosome. A regulatory protein complex conserved from Arabidopsis to human. Trends Genet., 15, 98–103. [DOI] [PubMed] [Google Scholar]
- Wei N., Chamovitz,D.A. and Deng,X.W. (1994) Arabidopsis COP9 is a component of a novel signaling complex mediating light control of development. Cell, 78, 117–124. [DOI] [PubMed] [Google Scholar]
- Wei N., Tsuge,T., Serino,G., Dohmae,N., Takio,K., Matsui,M. and Deng,X.W. (1998) The COP9 complex is conserved between plants and mammals and is related to the 26S proteasome regulatory complex. Curr. Biol., 8, 919–922. [DOI] [PubMed] [Google Scholar]