

Abstract
The tumor suppressor p53 is activated in response to many types of cellular and environmental insults via mechanisms involving post-translational modification. Here we demonstrate that, unlike phosphorylation, p53 invariably undergoes acetylation in cells exposed to a variety of stress-inducing agents including hypoxia, anti-metabolites, nuclear export inhibitor and actinomycin D treatment. In vivo, p53 acetylation is mediated by the p300 and CBP acetyltransferases. Overexpression of either p300 or CBP, but not an acetyltransferase-deficient mutant, efficiently induces specific p53 acetylation. In contrast, MDM2, a negative regulator of p53, actively suppresses p300/CBP-mediated p53 acetylation in vivo and in vitro. This inhibitory activity of MDM2 on p53 acetylation is in turn abrogated by tumor suppressor p19ARF, indicating that regulation of acetylation is a central target of the p53–MDM2–p19ARF feedback loop. Functionally, inhibition of deacetylation promotes p53 stability, suggesting that acetylation plays a positive role in the accumulation of p53 protein in stress response. Our results provide evidence that p300/CBP-mediated acetylation may be a universal and critical modifi cation for p53 function.
Keywords: acetylation/CBP/MDM2/p300/p53
Introduction
The tumor suppressor p53 plays a critical role in human cancer formation. In response to a variety of stress signals, often associated with the progression of neoplastic diseases, p53 becomes activated and induces cell cycle arrest and/or programmed cell death (apoptosis). By eliminating damaged and potentially dangerous cells that might otherwise become cancerous, p53 suppresses tumor formation. In unstressed cells, p53 is latent and is maintained at low levels by targeted degradation mediated by its negative regulator, MDM2 (reviewed in Freedman et al., 1999). The critical role of MDM2 in regulating p53 is best illustrated by a study carried out in mice where inactivation of p53 was shown to completely rescue the embryonic lethality caused by the loss of MDM2 function (Montes de Oca Luna et al., 1995). MDM2 counteracts p53 tumor suppressor activity by physically binding to p53 and suppressing its transcriptional activity. MDM2 also functions as the p53 ubiquitin ligase and triggers its degradation (reviewed in Freedman et al., 1999). This latter activity requires the Ring finger domain located at the C-terminus of MDM2 (Fang et al., 2000), and may also involve the acetyltransferase p300, which binds both MDM2 and p53 (Grossman et al., 1998). Therefore, MDM2 negatively regulates p53 by at least two independent mechanisms.
The activation and stabilization of p53 are thought to be mediated by specific protein modifications, with phosphorylation being the major focus of earlier studies (reviewed in Giaccia and Kastan, 1998; Appella and Anderson, 2000). Although the exact functions of specific phosphorylation events remain controversial, evidence indicates that they probably contribute to both the stabilization and activation of p53. For example, DNA-damaging agents activate phosphorylation at serine (Ser) 15 and Ser37, likely by a family of protein kinases including ATM and ATR (Canman et al., 1998; Tibbetts et al., 1999), and Ser20 by the Chk2 kinase (Hirao et al., 2000; Shieh et al., 2000). These phosphorylation events are believed to contribute to p53 stabilization by preventing the binding of MDM2 and rendering p53 more resistant to MDM2 (Shieh et al., 1997; Unger et al., 1999).
In addition to potentially regulating MDM2 binding, phosphorylation was also shown to modulate the transcriptional activity of p53. For example, phosphorylation at Ser15 stimulates p53 interaction with its transcriptional co-activators p300 and CBP, and a mutation that eliminates this phosphorylation leads to p53 transcriptional defects (Lambert et al., 1998; Dumaz and Meek, 1999). However, the requirement for the aforementioned phosphorylation is probably not universal for p53 stabilization or activation. For example, inhibition of RNA polymerase II by actinomycin D leads to p53 stabilization and activation without invoking either Ser15 or Ser20 phosphorylation (Ashcroft et al., 2000). Similarly, viral oncoprotein E1A-induced p53 acti vation is not accompanied by Ser15 phosphorylation (de Stanchina et al., 1998). These results suggest that alternative pathways and/or modifications exist and play important roles in modulating p53 activation. One such possible pathway involves the tumor suppressor p19ARF. Inappropriate expression of E1A and other cellular oncogenes, such as c-myc, leads to p53 activation through a p19ARF-dependent pathway (de Stanchina et al., 1998; Zindy et al., 1998). p19ARF functions, at least in part, by binding to MDM2 and neutralizing its activity (Pomerantz et al., 1998; Zhang et al., 1998). p19ARF inhibits the p53 ubiquitin ligase activity of MDM2 in vitro (Honda and Yasuda, 1999), and sequesters MDM2 into nucleoli, thereby preventing its nuclear export in vivo (Weber et al., 1999). Because the ubiquitin ligase activity and the nuclear export of MDM2 appear to be essential for the degradation of p53 (Tao and Levine, 1999a,b), it is possible that by directly binding and inactivating MDM2, p19ARF bypasses the need for phosphorylation in p53 activation.
Another potential mechanism that may play a critical role in p53 activation is acetylation. Multiple lysine (Lys) residues in p53 are reported to be acetylated. In vitro, Lys320 can be acetylated by P/CAF (p300/CBP associated factor) (Liu et al., 1999) and CBP (A.Ito and T.P.Yao, unpublished result), while Lys373 and Lys382 are acetylated by p300 and CBP (Sakaguchi et al., 1998; Liu et al., 1999). At least two additional lysine residues (Lys370 and Lys381) are acetylated by CBP (A.Ito and T.P.Yao, unpublished result). In vivo studies show that some of these sites are acetylated in response to DNA-damaging agents, demonstrating that acetylation is a bona fide modification for p53 (Sakaguchi et al., 1998; Liu et al., 1999). However, despite the observation that acetylation can stimulate p53 DNA binding activity in vitro (Gu and Roeder, 1997; Liu et al., 1999), the exact function of acetylation and the identities of the p53 acetylases that modify these sites in vivo remain to be established.
p300 and its family member CBP are the candidate in vivo p53 acetylases. p300 and CBP were originally discovered as transcriptional co-activators that play critical roles in integrating multiple signal-dependent transcription events (reviewed in Goodman and Smolik, 2000). In vivo, genetic experiments have clearly demonstrated essential roles for p300 and CBP in normal embryonic development (Tanaka et al., 1997; Yao et al., 1998; Kung et al., 2000). More recent analyses have indicated that p300 and CBP may have specific roles in tumor suppression pathways. p300 mutations were recently found in many types of tumor (Gayther et al., 2000) and mutation of human CBP causes Rubinstein–Taybi syndrome (RTS), which leads to an increased risk of cancers (reviewed in Giles et al., 1998). The human genetic evidence was further substantiated by the analysis of CBP knockout mice, which also display a higher risk of tumors of hematopoietic origin (Gayther et al., 2000; Kung et al., 2000). Interestingly, many of the p300 mutations identified from tumors actually result in the loss of acetyltransferase activity (Gayther et al., 2000), suggesting that the ability of p300 and CBP to acetylate one or more cellular proteins may be critical for their functions in growth control. The fact that p300 and CBP play important roles in p53 transcriptional activity (Gu et al., 1997; Lill et al., 1997) suggests that p53 might be a critical substrate of p300/CBP in mediating tumor suppression.
In this report, we present evidence that acetylation is a common modification associated with p53 activation in response to all p53-activating agents tested. We also establish that, in vivo, p300 and CBP can function as p53 acetylases and positively regulate p53 acetylation status, while MDM2 suppresses p53 acetylation. Consistent with p53 acetylation being a critical target of MDM2, we show that the tumor suppressor p19ARF can specifically inhibit the ability of MDM2 to negatively regulate p53 acetylation. Lastly, we provide evidence that inhibition of deacetylation increases the half-life of p53, suggesting that acetylation plays a role in p53 stability. Our results provide strong evidence that acetylation is a tightly regulated event and may be a universal and critical modification for p53 function.
Results
To initially address the potential importance of acetylation, we first determined whether p53 becomes acetylated in response to various environmental or cellular insults that are known to activate and stabilize p53. We used an antibody that specifically recognizes acetylated p53 at Lys382 (Sakaguchi et al., 1998) or an antibody that recognizes a cluster of acetylated lysine residues (pan-acetylated p53, including lysines 370, 372, 373, 381 and 382) to confirm specific acetylation. Because in most cases both antibodies give very similar results in assessing p53 acetylation in vivo (for example, see Figure 2), the majority of results in this report are based on the analysis of Lys382 acetylation.
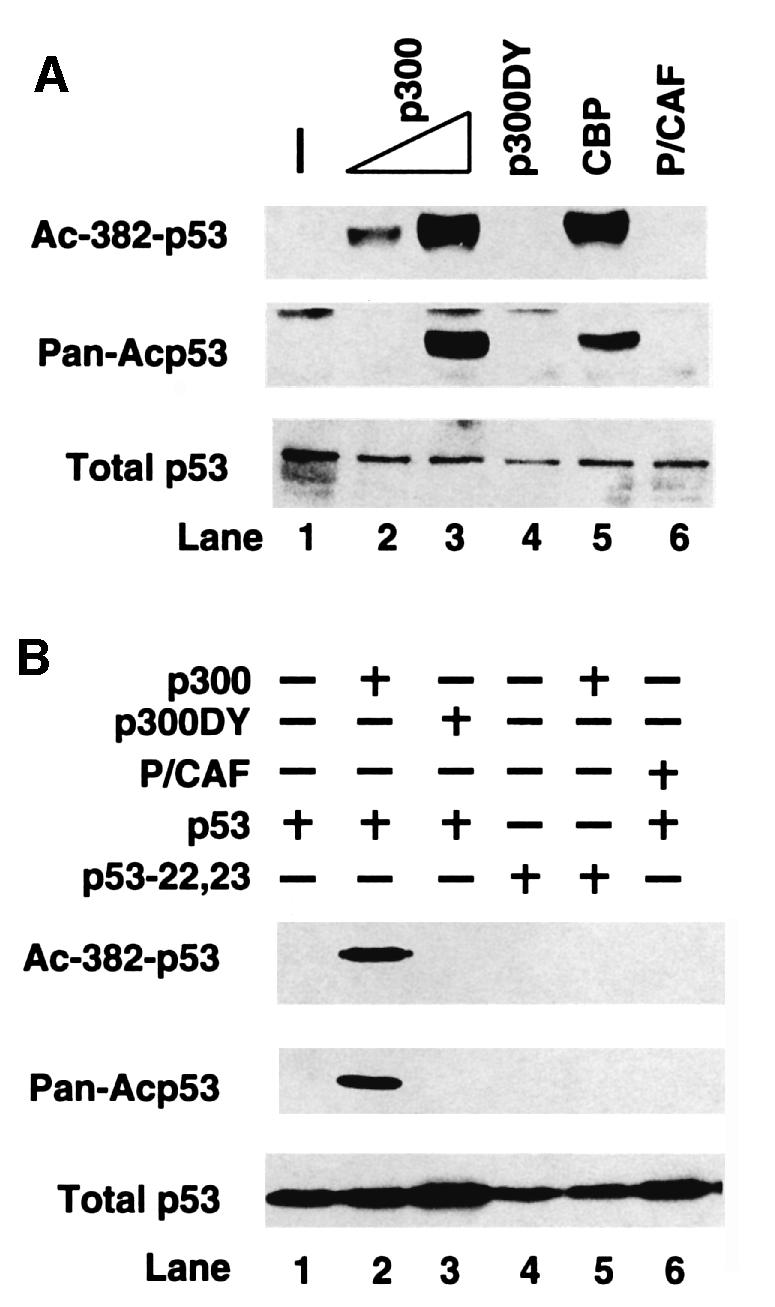
Fig. 2. Acetylation of p53 by p300 and CBP in vivo. (A) 293T cells were transfected with p300 (lanes 2 and 3), acetyltransferase-deficient p300 DY mutant (lane 4), CBP (lane 5) or PCAF (lane 6), and levels of endogenous acetylated p53 were assessed by either antibody specific for acetylated Lys382 (Ac-382-p53) or antibody against a cluster of acetylated lysines (Pan-Acp53; see text for details). The protein levels of p300, p300 DY, CBP and P/CAF were all comparable (data not shown). (B) H1299 cells (p53–/–) were transfected with expression plasmid for wild-type p53 alone (lane 1), or co-transfected with either p300 (lane 2), p300 DY mutant (lane 3) or PCAF (lane 6). H1299 cells were also transfected with an expression plasmid for p53(22,23) p300 binding mutant alone (lane 4) or co-transfected with p300 (lane 5). For (A) and (B), cell extracts were prepared (36 h post-transfection) and the detection of acetylated p53 (Ac-382-p53 and Pan-Acp53) or total p53 was determined as described in the legend to Figure 1.
p53 acetylation is commonly induced by multiple p53-activating agents
Consistent with earlier reports, DNA damaging agents, such as UV irradiation (Figure 1A) and the DNA strand breakers camptothecin and cis-platinum (data not shown), all efficiently induce p53 acetylation. However, in the earlier reports the deacetylase inhibitor trichostatin A (TSA) was added during treatment to enhance the acetylation signal. This treatment prevents analysis of the kinetics of p53 acetylation (Sakaguchi et al., 1998). To address this issue, we carried out the experiment in the absence of TSA. As shown in Figure 1A, p53 acetylation is a transient event and, after an initial increase, the abundance of acetylated p53 decreased due to the activity of a putative p53 deacetylase. Importantly, the kinetics of p53 acetylation paralleled that of its stabilization, suggesting that acetylation may play a role in p53 activation (Figure 1A).

Fig. 1. p53 acetylation induced by multiple p53-activating agents. A549 cells were treated with (A) UV-B (100 J/m2), (C) H2O2 (1 mM) or proteasome inhibitor LLnV (LL, 10 µM) for 3 h, (D) LMB (10 ng/ml) or (F) actinomycin D (5 nM). A549 and MCF7 cells (data not shown) were exposed to (B) deferoxamine mesylate (DFX) to mimic hypoxia (100 µM) or proteasome inhibitor LLnV (LL, 10 µM) for 12 h. WI-38 cells were exposed to (E) PALA (100 µM). (A–F) All cells were harvested at the times indicated. All cells contain wild-type p53. Total p53, acetylated p53 and the internal control α-tubulin levels were assessed by western blotting with α-p53 monoclonal antibody (middle panel), α-acetylated p53 (Lys382) (top panel) and α-tubulin monoclonal antibody (bottom panel), respectively. All treatments were carried out without the use of TSA, except for the DFX experiment where 5 µM of TSA was added to cells.
To investigate further the involvement of acetylation in p53 activation, we examined whether p53-activating agents other than DNA damaging treatment can induce p53 acetylation. Many different types of cellular and environmental insult are capable of activating p53. Here we tested hypoxia, oxidative stress, blocking of nuclear export by leptomycin B (LMB) and depletion of ribonucleotides pools by n-phosphonacetyl-L-aspartate (PALA) (reviewed in Giaccia and Kastan, 1998; Freedman et al., 1999). All of these treatments are capable of activating and stabilizing p53. As shown in Figure 1B–E, these agents stabilized p53 and, in every single case, p53 became acetylated. Importantly, treatment with the proteasome inhibitor LLnV, despite its ability to increase total p53 levels, did not result in increased acetylation, demonstrating that the acetylation signals detected were specific and not simply a consequence of higher protein levels (LL in Figure 1B and C). Inhibition of RNA polymerase II by actinomycin D is unique and different from DNA damaging or hypoxia treatment as it activates p53 without triggering phosphorylation of Ser15 or Ser20 (Ashcroft et al., 2000, and data not shown). Figure 1F shows that actinomycin D still efficiently induced p53 acetylation, distinguishing acetylation from phosphorylation during p53 activation. Altogether, these results demonstrate that p53 becomes acetylated in response to all p53-activating agents tested in this study, and further indicate that acetylation is a common modification associated with p53 activation.
p300 and CBP function as p53 acetylases in vivo
Prime candidates for the p53 acetylases are p300 and its family member CBP. Both p300 and CBP can acetylate p53 in vitro (Gu and Roeder, 1997; Sakaguchi et al., 1998; Liu et al., 1999; Figure 4A). However, it is not known whether these acetyltransferases can function as p53 acetylases in vivo. To address this issue, we determined whether overexpression of p300 or CBP can induce the specific acetylation of endogenous p53. As shown in Figure 2A, overexpression of wild-type p300 in human 293T cells significantly induced p53 acetylation levels as illustrated by antibodies against acetylated Lys382 (top panel), pan-acetylated p53 (middle panel) or acetylated Lys373 (data not shown). The acetylation of p53 depends on the acetyltransferase activity of p300, as an acetylase-deficient point mutant (DY mutant) derived from a human tumor mutation (C.-H.Lai and T.-P.Yao, manuscript in preparation) failed to induce p53 acetylation. In contrast to p300 or CBP, the expression of P/CAF, which acetylated p53 at Lys320 in vitro, did not result in acetylation detectable by the antibodies used in this study (Figure 2A). In p53-null H1299 cells, co-expression of wild-type p53 and p300 also led to specific acetylation of the transfected p53 species similar to that observed for the endogenous p53 (Figure 2B, lane 2). Again, the acetylation of the transfected p53 required wild-type p300 acetyltransferase activity (Figure 2B, lane 3). Importantly, a p300-binding-deficient p53 mutant could not be acetylated when co-expressed with p300 (lanes 4–5). This result indicates that direct binding between p53 and p300 is necessary for efficient acetylation and provides further evidence that p53 acetylation is mediated directly by p300 in vivo. Identical results were observed when CBP was evaluated for its role in p53 acetylation (Figure 2A, lane 5, and data not shown).

Fig. 4. Suppression of CBP acetyltransferase activity by MDM2 in vitro. (A and B) GST–p53 (A) or core histones (B) were acetylated by recombinant CBP in the presence of the indicated amounts of MDM2 or BSA, and analyzed by SDS–PAGE followed by autoradiography. Film was exposed (A) overnight and (B) for 3 h. Acetylated p53 and histone are indicated with arrows. Note that the level of acetylated p53 and acetylated histone decreases in the presence of MDM2. Acetylated MDM2 is marked with an arrowhead (A). (C and D) The intensity of the acetylated GST–p53 (C) or histones (D) was quantified by phosphoimager analysis and plotted. The intensity of acetylated GST–p53 or histones in the absence of MDM2 or BSA was set as 1. (C) reflects the average of three experiments, while (D) reflects the average of two experiments.
MDM2 suppresses p300/CBP-dependent p53 acetylation in vivo
The results presented thus far provide strong evidence that p53 is acetylated by its positive regulators p300 and CBP in response to a variety of signals. If acetylation plays a critical role in p53 function, it is likely that factors that negatively regulate p53 activity might interfere with this process. MDM2 is the most important p53 negative regulator and it also interacts with p300 (Grossman et al., 1998). These observations prompted us to ask whether MDM2 has the capacity to regulate the acetylation status of p53. As shown in Figure 3A, overexpression of MDM2 effectively reduced p300-dependent p53 acetylation in a dose-dependent manner (lanes 4–6). Of note, MDM2 overexpression does not affect the protein levels of transfected p300 (Figure 3A, top panel), supporting a direct effect of MDM2 on p53 acetylation. To rule out the possibility that the decrease in acetylation was caused by a corresponding decrease in p53 protein levels triggered by MDM2, the proteasome inhibitor LLnV was added to the culture to block p53 degradation. This treatment led to the stabilization of p53. Despite high protein levels, p53 remained non-acetylated in the presence of MDM2 (Figure 3A, lane 8). This result demonstrates that MDM2 can reverse the p53 acetylation induced by p300. In contrast to LLnV treatment, the deacetylase inhibitor TSA effectively abrogated the effect of MDM2 and restored p53 acetylation (Figure 3A, compare lanes 6 and 7), providing further evidence that MDM2 specifically modulated p53 acetylation. Interestingly, TSA treatment also increased p53 protein levels, suggesting the possibility that inhibition of p53 deacetylation promoted p53 stability (see below).

Fig. 3. Suppression of p300-dependent p53 acetylation by MDM2. (A) H1299 cells were transfected with expression plasmid for wild-type p53 and internal control GFP (lane 1), and co-transfected with either MDM2 (lanes 2 and 3), c-myc-tagged p300 (lane 4), or MDM2 and c-myc-tagged p300 (lanes 5–8). Cells were also treated 24 h post-transfection with either the deacetylase inhibitor TSA (5 µM) (lane 7) or the proteasome inhibitor LLnV (10 µM) (lane 8) for 12 h. Cell extracts were prepared (36 h post-transfection) and the level of acetylation (third panel) and total p53 protein (fourth panel) were determined by western blotting as described for Figure 1. (B) Schematic diagram of MDM2 deletion mutants used in (C). (C) H1299 cells were transfected with p53 wild-type and internal control GFP (lane 1), or co-transfected with c-myc-tagged p300 (lane 2), or c-myc-tagged p300 and the indicated amounts of MDM2 wild type (lanes 3 and 4), Δ58–92 mutant (lanes 5 and 6), Δ4 mutant (lanes 7 and 8) or ΔR mutant (lanes 9 and 10). p53 protein and acetylation levels were determined as described in (A). p300 levels were determined by either anti-myc (A14, Santa Cruz) (A) or by anti-p300 (RW128) (C). Both antibodies yielded similar results.
To study further how MDM2 suppresses p53 acetylation, we analyzed a series of MDM2 mutants with specific functional domains deleted (Figure 3B). Specifically, we tested MDM2 mutants that are deficient in p53 binding (Δ58–92) (Chen et al., 1993), p300 binding (Δ4, amino acids 192–222) (Grossman et al., 1998), or defective in ubiquitin ligase activity (ΔR, deletion of the Ring domain). As shown in Figure 3C, after transfection into H1299 cells, all these MDM2 variants were expressed (second panel). However, when compared with wild-type MDM2 (lanes 3–4), both the p53 binding mutant (Δ58–92, lanes 5–6) and the p300 binding mutant (Δ4, lanes 7–8) were defective as they only weakly suppressed p53 acetylation even when expressed at higher levels (Figure 3C, Ac-382). Importantly, both mutants are also deficient in degrading p53, further suggesting a functional link between p53 acetylation and stability. In contrast, the Ring domain mutant inhibited p53 acetylation to a level similar to that of wild-type MDM2 (ΔR, lanes 9–10). These results indicate that physical binding to both p53 and p300 is required for full activity of MDM2 to repress p53 acetylation. The Ring domain, which is essential for degrading p53 (Fang et al., 2000), is dispensable for this function. From this set of experiments, we conclude that MDM2 can actively repress p300-mediated p53 acetylation in vivo and that this activity requires physical binding to both p53 and p300.
MDM2 suppresses CBP acetyltransferase activity in vitro
In principle, MDM2 could repress p53 acetylation either by directly suppressing p53 acetylation or by promoting p53 deacetylation. To address these possibilities, we first determined whether MDM2 could directly inhibit p300/CBP-mediated p53 acetylation in vitro. As shown in Figure 4A, although recombinant MDM2 had no effect on CBP auto-acetylation, it efficiently inhibited p53 acetylation in a dose-dependent manner (Figure 4A and C). As this inhibition was not sensitive to the deacetylase inhibitor TSA (data not shown), MDM2 likely interfered with the acetyltransferase activity of CBP rather than functioning as a p53 deacetylase. Interestingly, while suppressing p53 acetylation, MDM2 itself became acetylated by CBP (Figure 4A, arrowhead). The functional importance of this acetylation is not yet clear. To determine whether the effect of MDM2 on CBP was specific to p53 acetylation, we tested whether MDM2 could suppress CBP-mediated histone acetylation. As shown in Figure 4B and D, MDM2 was able to suppress the acetylase activity of CBP towards core histones as well. In contrast, under the same experimental conditions, MDM2 had no apparent suppressive effect on another acetyltransferase, P/CAF (data not shown). Thus, MDM2 can specifically suppress CBP-mediated p53 and core histone acetylation in vitro. These observations suggest that the ability of MDM2 to repress p53 acetylation in vivo works, at least in part, by suppressing the acetyltransferase activity of p300 and CBP.
Tumor suppressor p19ARF reverses the inhibition of p53 acetylation by MDM2
p19ARF induces p53 activation by negatively regulating MDM2. This activity is proposed to be mediated by inactivating p53 E3 ligase activity of MDM2. Analysis of the role of MDM2 in p53 acetylation suggests an altern ative possibility that p19ARF might function by antagonizing the activity of MDM2 toward p53 acetylation. To examine this possibility, we determined whether co-expression of p19ARF and MDM2 could neutralize the latter’s ability to repress p300-dependent p53 acetylation. As shown in Figure 5, in the absence of p19ARF, co-transfection of MDM2 efficiently repressed p53 acetylation and induced its degradation in H1299 cells (compare lanes 2 and 3). However, upon co-expression of p19ARF, both p53 acetylation and protein levels were restored (lanes 4–5). Importantly, a p19ARF mutant, which does not bind MDM2 (C65, Zhang and Xiong, 1999) failed to suppress MDM2 in this assay (lane 6). Altogether, these results demonstrate that p19ARF can abrogate the ability of MDM2 to suppress p53 acetylation. The correlation between p53 protein level and acetylation level in response to MDM2 and p19ARF, however, does suggest that acetylation might influence p53 stability.
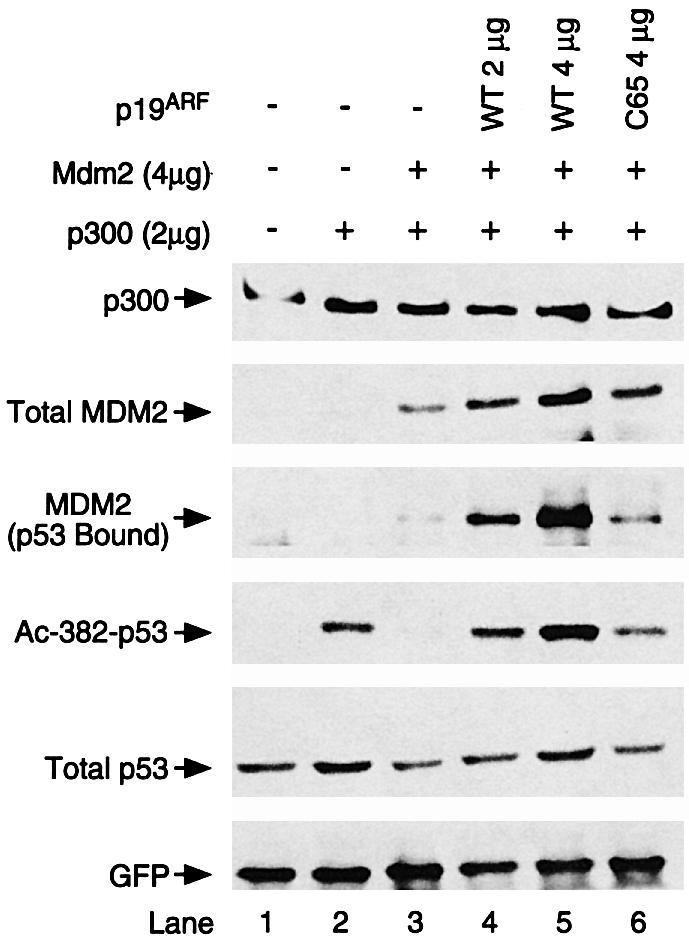
Fig. 5. p19ARF reverses the inhibition of p53 acetylation by MDM2. H1229 cells were transfected with either expression plasmid for p53 wild-type and internal control GFP (lane 1) or co-transfected with c-myc-tagged p300 (lanes 2–6), in combination with MDM2 and p19 expression plasmid as indicated. Analysis of p53 protein and acetylation levels was carried out as described for Figures 1 and 2. Note that expression of p19 effectively neutralized the effects of MDM2 on p53 acetylation (lanes 4 and 5). p300 levels were determined by RW128.
An increased level of total and p53-bound MDM2 was also observed when p19ARF was co-expressed (Figure 5, MDM2 panels, lanes 4–5). This might be due to the inhibition of MDM2 auto-ubiquitylation (Fang et al., 2000) and, consequently, the stabilization of MDM2. Importantly, despite the high levels of MDM2 associated with p53, MDM2 in this complex did not show appreciable repression toward p53 acetylation, supporting the idea that p19ARF dominantly inhibits the activity of MDM2 in this assay. This observation also suggests that p19ARF restores p53 acetylation and protein levels without dissociating MDM2 from p53. This set of results demonstrates that, in addition to inhibiting MDM2 as a p53 ubiquitin ligase, p19ARF is also capable of inactivating MDM2 in suppressing p53 acetylation, providing further evidence that acetylation is a modification regulated by a p300/CBP–MDM2–p19ARF feedback loop in the p53 network.
Inhibition of p53 deacetylation promotes p53 stability
The results presented so far support the idea that acetylation is a common modification regulated by a network of critical regulators of p53 function. In principle, acetylation could contribute to p53 stabilization and/or p53 activity. Several observations from our study suggest the possibility that acetylation may regulate p53 stability. First, there was a positive correlation between the kinetics of p53 protein levels and its acetylation levels in response to DNA damage (Figure 1A). Secondly, p19ARF concomitantly restored p53 protein and acetylation levels, which were negatively regulated by MDM2 (Figures 3 and 5). Lastly, treatment with the deacetylase inhibitor TSA seemed to result in higher p53 protein levels (Figure 3A). If acetylation were important for p53 stabilization, one would predict that TSA treatment should delay the normal rate of degradation by preventing p53 deacetylation. To test this hypothesis, p53 stability was determined following its activation by UV irradiation. As shown in Figure 6A (top panel), TSA treatment effectively inhibited the p53 deacetylase and increased the levels of acetylated p53 in A549 cells. Importantly, the apparent half-life of p53 was dramatically increased in the presence of TSA, suggesting that acetylated p53 is more stable (Figure 6A, middle panel, and B). In contrast, the same treatment did not affect the half-life of actin (Figure 6A, bottom panel), indicating that TSA did not have a general positive effect on protein stability. This result indicates that one function of specific p53 acetylation is to increase its stability.

Fig. 6. Inhibition of deacetylase promotes p53 stabilization. (A) A549 cells were exposed to UV-B (50 J/m2) in the presence (+) or absence (–) of TSA (5 µM). Four hours post-irradiation, cyclohexamide (10 µg/ml) was added to inhibit new p53 protein synthesis (designated 0 h). Cells were harvested at the time-points indicated after cyclohexamide treatment. Acetylated p53 (top panel) and total p53 (middle panel) were determined. Note that total p53 level and acetylation levels are significantly higher in the presence of TSA. As a control, direct western blotting with an α-actin polyclonal antibody also assessed actin levels (lower panel). (B) The band intensity of p53 protein levels was measured by NIH imaging software and calculated against the amount of p53 present at time point 0, which was set at 100%. Results are given in the presence (filled circles) or absence (empty circles) of TSA treatment.
Discussion
In this report, we show that p53 becomes acetylated in response to all p53-activating agents tested (Figure 1). Together with the recent report that p53 acetylation increases as fibroblasts senesce (Pearson et al., 2000), these results clearly establish acetylation as a common modification that invariably accompanies p53 activation. This is in contrast to the two well studied phosphorylation events on Ser15 and Ser20, which are activated only by a select few agents (Ashcroft et al., 2000), and further illustrates a unique requirement for acetylation in p53 activation. Although the complete function of p53 acetylation remains to be firmly established, we provide evidence that acetylation may at least contribute to p53 stability. Two recent reports have suggested that acetylation is important for p53 to suppress oncogenic ras-induced transformation (Pearson et al., 2000) and to induce metaphase chromosome fragility (Yu et al., 2000), adding more evidence for the functional significance of p53 acetylation. The findings that p300/CBP acetyltransferases and p19ARF promote p53 acetylation in vivo, while MDM2 inhibits acetylation, lend support to the idea that acetylation is an important modification targeted by both positive and negative regulators critical to p53 tumor suppressor activity.
Reversible acetylation was originally identified in histones and was thought to be important for transcriptional activity (Wade et al., 1997). However, a growing number of non-histone proteins are now being reported as targets of acetylation (reviewed in Kouzarides, 2000). Although in most cases the function of acetylation remains to be firmly established, analysis of E2F1 and myoD indicates that P/CAF-mediated acetylation appears to increase E2F1 stability (Martinez-Balbas et al., 2000) and contribute to myoD activity (Sartorelli et al., 1999). Similarly, p300/CBP-dependent GATA-1 acetylation has been shown to be critical for GATA-1 function (Boyes et al., 1998). In this report, we further show that MDM2 may be an acetylated protein as well (Figure 4A). Together with the demonstration that acetylation of p53 is tightly regulated and is important for p53 stability, these various lines of evidence support the hypothesis that acetylation is a prominent and likely general regulatory modification used to modulate protein function.
We have presented evidence that p300 and CBP are able to acetylate p53 and are likely to be the key p53 acetylases in vivo. Biochemical and genetic experiments indicate that p300 and CBP levels are limited in cells (reviewed in Goodman and Smolik, 2000), and apparently, they cannot support endogenous p53 acetylation under normal conditions. In theory, high levels of p300/CBP in the transfection setting should increase the probability of complex formation with p53. Moreover, transfection itself probably triggers some DNA damage response. These two factors together may contribute to p53 acetylation upon p300/CBP overexpression (Figure 2). Consistent with this idea, we have shown that a direct interaction between p300/CBP and p53 is necessary for efficient p53 acetylation (Figure 2B). Under normal physiological settings, it is likely that p53 and p300/CBP complexes are induced in response to activating signals. Consistent with this idea, we have found that mutations that eliminate phosphorylation at Ser15 but not Ser20 significantly reduced p53 acetylation in vivo (our unpublished observation). As Ser15 phosphorylation stimulates p53 binding to p300/CBP (Lambert et al., 1998), this result provides evidence that specific phosphorylation on Ser15 could be one activation step leading to p53–p300/CBP complex formation and subsequent p53 acetylation by p300/CBP. Ser15 phosphorylation, however, is not the only mechanism that can lead to p53 acetylation. Actinomycin D does not induce Ser15 phosphorylation (Ashcroft et al., 2000), yet it is a powerful agent in triggering p53 acetylation (Figure 1F). This result suggests a more general and unique requirement for acetylation than some specific phosphorylation events during p53 activation. The mechanism by which actinomycin D induces p53 acetylation without Ser15 phosphorylation, however, remains unknown.
Our results show clearly that MDM2 can suppress p300/CBP-mediated p53 acetylation in vitro and in vivo. There are at least four possible mechanisms that may explain this observation. First, MDM2 binds and inactivates p300/CBP acetyltransferase activity. This possibility is supported by our result that a p300-binding-deficient MDM2 mutant is defective in this activity. Secondly, p300/CBP and MDM2 bind to non-identical but overlapping regions at the N-terminus of p53. It is possible that high levels of MDM2 bind p53 and displace p300/CBP, thereby inhibiting p53 acetylation. This mechanism, however, may not explain how MDM2 suppresses histone acetylation, as there is no evidence that MDM2 binds histones. The observation that p19ARF restores p53 acetylation without dissociating MDM2 from p53 is also inconsistent with this model (Figure 5). Thirdly, MDM2 can interact directly with p300/CBP and itself becomes acetylated (Figure 4A). It is possible that MDM2 serves as a substrate competitor and thereby suppresses p53 acetylation. Further studies will be needed to verify the acetylation of MDM2 in vivo and the importance of this acetylation. Lastly, although MDM2 inhibits p53 acetylation by CBP directly in vitro, we could not eliminate the possibility that other mechanisms may also contribute to the suppression of p53 acetylation in vivo. For instance, MDM2 could stimulate deacetylation by recruiting a p53 deacetylase. The observation that TSA can completely abrogate the inhibitory effect of MDM2 on p53 acetylation (Figure 3A) and that MDM2 interacts with a specific deacetylase (A.Ito and T.P.Yao, unpublished result) is consistent with this possibility. Regardless of which mechanism is correct, our results clearly demonstrate that MDM2 is able to suppress p53 acetylation in vivo and in vitro.
Our analysis of MDM2 also reveals that MDM2 suppresses the core histone acetylation induced by p300/CBP. It has been hypothesized that p300 and CBP activate transcription by acetylating histones. The inhibitory activity of MDM2 on histone acetylation provides a biochemical mechanism to explain how MDM2 can inhibit p53 transactivation potency. In this scenario, the recruitment of MDM2 to the p53–p300 or p53–CBP complexes on target chromatin inhibits histone acetylation and thereby represses p53-dependent transcription.
By binding to MDM2, p19ARF plays a critical role in p53 activation. This activity of p19ARF was attributed, at least in part, to its ability to suppress the MDM2 E3 ligase activity toward p53 ubiquitylation (Honda and Yasuda, 1999). Our study now shows that p19ARF can also abrogate the inhibitory effect of MDM2 toward p53 acetylation in vivo (Figure 5). In fact, overexpression of p19ARF alone is sufficient to induce p53 acetylation (A.Ito, unpublished result). This observation adds a novel mechanism through which p19ARF regulates MDM2 activity and participates in tumor suppression. Two alternative hypotheses have been put forward to explain how p19ARF inhibits MDM2 activity. One proposes that p19ARF sequesters MDM2 in nucleoli and dissociates MDM2 from p53 (Weber et al., 1999), while the other shows that p53–MDM2–p19ARF forms a tripartite complex in the nucleoplasm, where MDM2 is not active (Zhang and Xiong, 1999). Unexpectedly, we found that upon p19ARF expression, a dramatic increase in MDM2 was found to complex with p53. However, the MDM2 in this complex is not active in suppressing p53 acetylation (Figure 5, lanes 4 and 5). These observations are more consistent with the possibility of a tripartite complex formation wherein p19ARF dominantly inhibits the activity of MDM2 toward p53 acetylation. However, we have found that recombinant p19ARF does not interfere with the ability of MDM2 to suppress CBP-mediated p53 acetylation in vitro (A.Ito and T.P.Yao, unpublished observation). Further studies will be required to determine how p19ARF suppresses MDM2 activity in this ternary complex. Regardless, our study demonstrates that p19ARF, in addition to regulating MDM2 ubiquitin ligase activity, can also suppress the activity of MDM2 towards p53 acetylation. These results suggest that all major regulators of p53 activity, including p300/CBP, MDM2 and p19ARF, integrate different extracellular and intracellular signals to modulate p53 acetylation level and thereby its stability and activity.
What is the importance of p53 acetylation in relation to p53 function? It was first reported that acetylation increases p53 DNA binding activity in vitro (Gu and Roeder, 1997). However, analyses of p53 mutants that can not be acetylated do not reveal obvious defects in DNA binding in vivo (data not shown), suggesting that acetylation might have other functions. Three lines of evidence derived from this study suggest that acetylation functions, at least in part, by modulating p53 stability. First, there is positive correlation between endogenous p53 protein and acetylation levels upon normal p53 activation (Figure 1). Secondly, in analyzing the ability of various MDM2 mutants and p19ARF to regulate p53 acetylation (Figures 3 and 5), we found a similar correlation between p53 protein and acetylation levels. This conclusion is further supported by the observation that TSA can efficiently reverse the degradation of p53 induced by MDM2 (Figure 3A and data not shown). Thirdly, prevention of p53 deacetylation leads to a more stable p53 species (Figure 6). Given that acetylation is always accompanied by p53 stabilization (Figure 1), this correlative evidence strongly suggests that acetylation may be a modification that contributes to p53 stabilization. How does acetylation stabilize p53? Since both the acetyltransferase and ubiquitin-conjugating system through which p53 is targeted for degradation modify lysine, it is possible that acetylation protects lysine residues from being ubiquitylated. It was reported recently that several lysine residues located at the C-terminus target p53 for ubiquitylation and degradation (Rodriguez et al., 2000). Importantly, these are the same lysine residues that can be acetylated by p300/CBP. These observations suggest the possibility that acetylation renders lysines unavailable for the ubiquitin-conjugating machinery, and thereby promotes p53 stability. If acetylation functions, at least in part, to inhibit ubiquitylation, reversible acetylation might have a more general role in regulating protein stability.
In conclusion, we propose that in response to cellular stresses, p53 becomes acetylated by the p300/CBP acetyltransferases. This modification requires either specific phosphorylation, such as at Ser15, or the activation of tumor suppressor p19ARF. Acetylation leads to p53 stabilization and the subsequent induction of MDM2. MDM2 then in turn triggers p53 deacetylation followed by p53 inactivation and destruction. The modulation of p53 acetylation by CBP/p300, MDM2 and p19ARF suggests the existence of an intricate pathway regulating the acetylation equilibrium that is crucial to the tumor suppressor activity of p53. Further characterization of the function of p53 acetylation will be critical for understanding the regulation of p53 tumor suppressor activity.
Materials and methods
Cell lines and transfection
A549, WI38, 293T and H1299 human cells were maintained in Dulbecco’s modified Eagle’s medium (DMEM). All cells were grown at 37°C in the presence of 10% fetal bovine serum and penicillin/streptomycin in a humidified atmosphere of 5% CO2. A549 and WI38 cells have wild-type p53, while H1299 cells are devoid of any p53 expression. All transfections were performed by the calcium phosphate method as described previously (Yao et al., 1992).
Plasmids
Wild-type human p53 cDNA was cloned into the BamHI–XhoI site of pCDNA3. The mutant p53(22,33), which can not bind to p300 has been described previously (Gu et al., 1997). The human MDM2 wild-type cDNA was cloned into the BamHI–EcoRI site of pCDNA3. The MDM2 ΔR mutant cDNA was made by digesting wild-type pCDNA3-MDM2 with SalI to delete the Ring domain (amino acids 442–491). The MDM2 Δ4 and Δ58–92 mutants were described previously (Chen et al., 1993; Grossman et al., 1998). The human p300-DY (Lys1399 converted to tyrosine) mutant was generated by site-direct mutagenesis and cloned into the pCMV vector. The full length mouse p19ARF and p19ARF N-terminal fragment (C65) (MDM2-binding-deficient mutant) were described previously (Zhang and Xiong, 1999).
Pulse–chase
A549 cells at 80–90% confluence were exposed to a 310 nm wavelength UV source. The deacetylase inhibitor TSA (Sigma) was added at a final concentration of 5 µM immediately after UV irradiation. Four hours after irradiation, cells were treated with 10 µg/ml of cyclohexamide to stop new p53 protein synthesis, and cells were then harvested at the indicated time points as described in Figure 6.
Immunoprecipitation and immunoblotting
Cells were lysed in buffer [20 mM Tris–HCl pH 7.6, 170 mM NaCl, 1 mM EDTA, 0.5% NP-40, 1 mM dithiothreitol (DTT)] supplemented with 5 µM TSA and protease inhibitors. For immunoprecipitation with anti-p53 antibody, equal amounts of lysate (containing 200–300 µg of total cellular protein) were incubated with 1 µg of goat anti-p53 antibody (Santa Cruz) and protein G–Sepharose (Pharmacia) for 3 h at 4°C. The use of goat antibody eliminates the heavy chain signal that co-migrates with p53 in subsequent immunoblotting. For immunoprecipitation with anti-p300 antibody, equal amounts of lysate (containing 100–150 µg of total cellular protein) were incubated with anti-p300 antibody (RW128) and protein G–Sepharose (Pharmacia) for 3 h at 4°C. When immunoprecipitation was not performed, 20–30 µg of total extracts were analyzed. Proteins were detected by chemiluminescent ECL kit (Amersham) with one of the following antibodies: anti-human p53 antibody (Ab-6, Calbiochem), anti-human acetylated (Lys382) p53 antibody (Calbiochem), anti-human MDM2 antibody (SMP14, Santa Cruz), anti-α-tubulin antibody (DM1A, Sigma), anti-c-myc antibody (A14, Santa Cruz), anti-p300 antibody (RW128, Eckner et al., 1994), anti-green fluorescent protein (GFP) antibody (Boehringer Mannheim) or anti-actin antibody (C-11, Santa Cruz).
In vitro acetyltransferase assay
Recombinant CBP protein (1 µg) purified from baculovirus was pre-incubated with the indicated amounts of purified bacterially expressed MDM2 protein or bovine serum albumin (BSA) for 10 min at room temperature. After pre-incubation, substrates [1 µg of glutathione-S-transferase (GST)–p53 or histone] were added and incubated with 50 nCi [14C]acetyl-coenzyme A in 30 µl of reaction buffer (50 mM Tris–HCl pH 8.0, 10% glycerol, 1 mM DTT, 100 µM EDTA, 1 mM phenylmethylsulfonyl fluoride) for another 45 min at 37°C. Acetylation was analyzed by SDS–PAGE followed by autoradiography, or by a phosphoimager.
Acknowledgments
Acknowledgements
We thank Dr W.Gu for his generous gift of the pan-acetylated p53-specific antibody and for sharing unpublished results, Dr S.Grossman for the MDM2 and p53 reagents, Drs E.Hura, J.Nevins and Y.Xiong for the p19ARF reagents and Dr J.Direnzo for CBP baculovirus. We thank Dr K.Sakaguchi for advice on the acetylated p53-specific antibody. We are grateful to Drs C.Anderson, A.R.Means, B.Harvat, Ms C.Hubbert, A.Guardiola and Mr T.Bolger for critically reading the manuscript. M.H. is supported by DAMD17-98-1-8072 and DK50494 from the NIH to Dr D.McDonnell. This work is supported partially by funding from the Damon Runyon–Walter Wichell Cancer Foundation to T.-P.Y. (DRS 20) and the National Institutes of Health (CA85676-01A1).
References
- Appella E. and Anderson,C.W. (2000) Signaling to p53: breaking the posttranslational modification code. Pathol. Biol. (Paris), 48, 227–245. [PubMed] [Google Scholar]
- Ashcroft M., Taya,Y. and Vousden,K.H. (2000) Stress signals utilize multiple pathways to stabilize p53. Mol. Cell. Biol., 20, 3224–3233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boyes J., Byfield,P., Nakatani,Y. and Ogryzko,V. (1998) Regulation of activity of the transcription factor GATA-1 by acetylation. Nature, 396, 594–598. [DOI] [PubMed] [Google Scholar]
- Canman C.E., Lim,D.S., Cimprich,K.A., Taya,Y., Tamai,K., Sakaguchi,K., Appella,E., Kastan,M.B. and Siliciano,J.D. (1998) Activation of the ATM kinase by ionizing radiation and phosphorylation of p53. Science, 281, 1677–1679. [DOI] [PubMed] [Google Scholar]
- Chen J., Marechal,V. and Levine,A.J. (1993) Mapping of the p53 and mdm-2 interaction domains. Mol. Cell. Biol., 13, 4107–4114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Stanchina E. et al. (1998) E1A signaling to p53 involves the p19(ARF) tumor suppressor. Genes Dev., 12, 2434–2442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dumaz N. and Meek,D.W. (1999) Serine 15 phosphorylation stimulates p53 transactivation but does not directly influence interaction with HDM2. EMBO J., 18, 7002–7010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eckner R., Ewen,M.E., Newsome,D., Gerdes,M., DeCaprio,J.A., Lawrence,J.B. and Livingston,D.M. (1994) Molecular cloning and functional analysis of the adenovirus E1A-associated 300-KD protein (p300) reveals a protein with properties of a transcriptional adaptor. Genes Dev., 8, 869–884. [DOI] [PubMed] [Google Scholar]
- Fang S., Jensen,J.P., Ludwig,R.L., Vousden,K.H. and Weissman,A.M. (2000) Mdm2 is a RING finger-dependent ubiquitin protein ligase for itself and p53. J. Biol. Chem., 275, 8945–8951. [DOI] [PubMed] [Google Scholar]
- Freedman D.A., Wu,L. and Levine,A.J. (1999) Functions of the MDM2 oncoprotein. Cell. Mol. Life Sci., 55, 96–107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gayther S.A. et al. (2000) Mutations truncating the EP300 acetylase in human cancers. Nature Genet., 24, 300–303. [DOI] [PubMed] [Google Scholar]
- Giaccia A.J. and Kastan,M.B. (1998) The complexity of p53 modulation: emerging patterns from divergent signals. Genes Dev., 12, 2973–2983. [DOI] [PubMed] [Google Scholar]
- Giles R.H., Peters,D.J. and Breuning,M.H. (1998) Conjunction dysfunction: CBP/p300 in human disease. Trends Genet., 14, 178–183. [DOI] [PubMed] [Google Scholar]
- Goodman R.H. and Smolik,S. (2000) CBP/p300 in cell growth, transformation and development. Genes Dev., 14, 1553–1577. [PubMed] [Google Scholar]
- Grossman S.R., Perez,M., Kung,A.L., Joseph,M., Mansur,C., Xiao,Z.X., Kumar,S., Howley,P.M. and Livingston,D.M. (1998) p300/MDM2 complexes participate in MDM2-mediated p53 degradation. Mol. Cell, 2, 405–415. [DOI] [PubMed] [Google Scholar]
- Gu W. and Roeder,R.G. (1997) Activation of p53 sequence-specific DNA binding by acetylation of the p53 C-terminal domain. Cell, 90, 595–606. [DOI] [PubMed] [Google Scholar]
- Gu W., Shi,X.L. and Roeder,R.G. (1997) Synergistic activation of transcription by CBP and p53. Nature, 387, 819–823. [DOI] [PubMed] [Google Scholar]
- Hirao A., Kong,Y.Y., Matsuoka,S., Wakeham,A., Ruland,J., Yoshida,H., Liu,D., Elledge,S.J. and Mak,T.W. (2000) DNA damage-induced activation of p53 by the checkpoint kinase Chk2. Science, 287, 1824–1827. [DOI] [PubMed] [Google Scholar]
- Honda R. and Yasuda,H. (1999) Association of p19(ARF) with Mdm2 inhibits ubiquitin ligase activity of Mdm2 for tumor suppressor p53. EMBO J., 18, 22–27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kouzarides T. (2000) Acetylation: a regulatory modification to rival phosphorylation? EMBO J., 19, 1176–1179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kung A.L., Rebel,V.I., Bronson,R.T., Ch’ng,L.E., Sieff,C.A., Livingston,D.M. and Yao,T.P. (2000) Gene dose-dependent control of hematopoiesis and hematologic tumor suppression by CBP. Genes Dev., 14, 272–277. [PMC free article] [PubMed] [Google Scholar]
- Lambert P.F., Kashanchi,F., Radonovich,M.F., Shiekhattar,R. and Brady,J.N. (1998) Phosphorylation of p53 serine 15 increases interaction with CBP. J. Biol. Chem., 273, 33048–33053. [DOI] [PubMed] [Google Scholar]
- Lill N.L., Grossman,S.R., Ginsberg,D., DeCaprio,J. and Livingston,D.M. (1997) Binding and modulation of p53 by p300/CBP coactivators. Nature, 387, 823–827. [DOI] [PubMed] [Google Scholar]
- Liu L., Scolnick,D.M., Trievel,R.C., Zhang,H.B., Marmorstein,R., Halazonetis,T.D. and Berger,S.L. (1999) p53 sites acetylated in vitro by PCAF and p300 are acetylated in vivo in response to DNA damage. Mol. Cell. Biol., 19, 1202–1209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martinez-Balbas M.A., Bauer,U.M., Nielsen,S.J., Brehm,A. and Kouzarides,T. (2000) Regulation of E2F1 activity by acetylation. EMBO J., 19, 662–671. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Montes de Oca Luna R., Wagner,D.S. and Lozano,G. (1995) Rescue of early embryonic lethality in mdm2-deficient mice by deletion of p53. Nature, 378, 203–206. [DOI] [PubMed] [Google Scholar]
- Pearson M. et al. (2000) PML regulates p53 acetylation and premature senescence induced by oncogenic Ras. Nature, 406, 207–210. [DOI] [PubMed] [Google Scholar]
- Pomerantz J. et al. (1998) The Ink4a tumor suppressor gene product, p19Arf, interacts with MDM2 and neutralizes MDM2’s inhibition of p53. Cell, 92, 713–723. [DOI] [PubMed] [Google Scholar]
- Rodriguez M.S., Desterro,J.M., Lain,S., Lane,D.P. and Hay,R.T. (2000) Multiple C-terminal lysine residues target p53 for ubiquitin– proteasome-mediated degradation. Mol. Cell. Biol., 20, 8458–8467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sakaguchi K., Herrera,J.E., Saito,S., Miki,T., Bustin,M., Vassilev,A., Anderson,C.W. and Appella,E. (1998) DNA damage activates p53 through a phosphorylation–acetylation cascade. Genes Dev., 12, 2831–2841. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sartorelli V., Puri,P.L., Hamamori,Y., Ogryzko,V., Chung,G., Nakatani,Y., Wang,J.Y. and Kedes,L. (1999) Acetylation of MyoD directed by PCAF is necessary for the execution of the muscle program. Mol. Cell, 4, 725–734. [DOI] [PubMed] [Google Scholar]
- Shieh S.Y., Ikeda,M., Taya,Y. and Prives,C. (1997) DNA damage-induced phosphorylation of p53 alleviates inhibition by MDM2. Cell, 91, 325–334. [DOI] [PubMed] [Google Scholar]
- Shieh S.Y., Ahn,J., Tamai,K., Taya,Y. and Prives,C. (2000) The human homologs of checkpoint kinases Chk1 and Cds1 (Chk2) phosphorylate p53 at multiple DNA damage-inducible sites. Genes Dev., 14, 289–300. [PMC free article] [PubMed] [Google Scholar]
- Tanaka Y., Naruse,I., Maekawa,T., Masuya,H., Shiroishi,T. and Ishii,S. (1997) Abnormal skeletal patterning in embryos lacking a single Cbp allele: a partial similarity with Rubinstein–Taybi syndrome. Proc. Natl Acad. Sci. USA, 94, 10215–10220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tao W. and Levine,A.J. (1999a) Nucleocytoplasmic shuttling of oncoprotein Hdm2 is required for Hdm2-mediated degradation of p53. Proc. Natl Acad. Sci. USA, 96, 3077–3080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tao W. and Levine,A.J. (1999b) P19(ARF) stabilizes p53 by blocking nucleo-cytoplasmic shuttling of Mdm2. Proc. Natl Acad. Sci. USA, 96, 6937–6941. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tibbetts R.S., Brumbaugh,K.M., Williams,J.M., Sarkaria,J.N., Cliby,W.A., Shieh,S.Y., Taya,Y., Prives,C. and Abraham,R.T. (1999) A role for ATR in the DNA damage-induced phosphorylation of p53. Genes Dev., 13, 152–157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Unger T., Juven-Gershon,T., Moallem,E., Berger,M., Vogt Sionov,R., Lozano,G., Oren,M. and Haupt,Y. (1999) Critical role for Ser20 of human p53 in the negative regulation of p53 by Mdm2. EMBO J., 18, 1805–1814. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wade P.A., Pruss,D. and Wolffe,A.P. (1997) Histone acetylation: chromatin in action. Trends Biochem. Sci., 22, 128–132. [DOI] [PubMed] [Google Scholar]
- Weber J.D., Taylor,L.J., Roussel,M.F., Sherr,C.J. and Bar-Sagi,D. (1999) Nucleolar Arf sequesters Mdm2 and activates p53. Nature Cell Biol., 1, 20–26. [DOI] [PubMed] [Google Scholar]
- Yao T.P., Segraves,W.A., Oro,A.E., McKeown,M. and Evans,R.M. (1992) Drosophila ultraspiracle modulates ecdysone receptor function via heterodimer formation. Cell, 71, 63–72. [DOI] [PubMed] [Google Scholar]
- Yao T.P. et al. (1998) Gene dosage-dependent embryonic development and proliferation defects in mice lacking the transcriptional integrator p300. Cell, 93, 361–372. [DOI] [PubMed] [Google Scholar]
- Yu A., Fan,H.Y., Liao,D., Bailey,A.D. and Weiner,A.M. (2000) Activation of p53 or loss of the Cockayne syndrome group B repair protein causes metaphase fragility of human U1, U2 and 5S genes. Mol. Cell, 5, 801–810. [DOI] [PubMed] [Google Scholar]
- Zhang Y. and Xiong,Y. (1999) Mutations in human ARF exon 2 disrupt its nucleolar localization and impair its ability to block nuclear export of MDM2 and p53. Mol. Cell, 3, 579–591. [DOI] [PubMed] [Google Scholar]
- Zhang Y., Xiong,Y. and Yarbrough,W.G. (1998) ARF promotes MDM2 degradation and stabilizes p53: ARF-INK4a locus deletion impairs both the Rb and p53 tumor suppression pathways. Cell, 92, 725–734. [DOI] [PubMed] [Google Scholar]
- Zindy F., Eischen,C.M., Randle,D.H., Kamijo,T., Cleveland,J.L., Sherr,C.J. and Roussel,M.F. (1998) Myc signaling via the ARF tumor suppressor regulates p53-dependent apoptosis and immortalization. Genes Dev., 12, 2424–2433. [DOI] [PMC free article] [PubMed] [Google Scholar]