

Abstract
Isolated adult cardiac ventricular myocytes have been a useful model for cardiovascular research for more than 20 years. With the recent advances in cellular physiology and transgenic techniques, direct measurement of isolated ventricular myocyte mechanics is becoming an increasingly important technique in cardiac physiology that provides fundamental information on excitation-contraction coupling of the heart, either in drug intervention or pathological states. The goal of this article is to describe the isolation of ventricular myocytes from both rats and mice, and the use of real-time beat-to-beat simultaneous recording of both myocyte contraction and intracellular Ca2+ transient.
Keywords: myocardial contraction, research design, calcium
Introduction
The essential contractile unit of the heart is the ventricular myocyte. Measurement of ventricular contractile function provides the most direct information on cardiac contractile function either with drug intervention or under certain pathological conditions. Over the past two decades, a plethora of new techniques have been presented to the physiologist for studies of cardiac contractile function. Several techniques have been developed including video-based edge-detection systems, photodiode arrays, and spatial video-based edge imaging (1,2). The most commonly used approach is the edge-detection method, which measures changes in myocyte length (isotonic contraction).
The application of isolated myocytes provides unique information on cardiac physiology. The presence of heterogeneous cell types and nerve terminals often makes the results obtained from multicellular preparations such as papillary muscles difficult to interpret. Mechanical function of the myocardium may be affected by non-myocyte factors such as the coronary vasculature and/or interstitial connective tissue. For example, alterations in contractile performance under ethanol exposure may simply be due to enhanced interstitial fibrosis but not reduced function of individual myocytes. Therefore, it is imperative that there be a tool for physiologists to study the myocyte specifically, regardless of the function of its surrounding tissues. This is accomplished with video-based edge-detection of the isolated ventricular myocyte.
Materials and Methods
This section describes the typical methods used to ultimately obtain individual ventricular myocytes. Some of these methods are specific to the type of animal used, so we have noted in the text if the method is specific to either rat or mouse.
Preparation of the animals
Animals are usually injected with heparin (1000U/kg, i.p.) 20 mins. prior to the experimental protocol. To anesthetize the animal, ketamine/xylazine has been the drug of choice in both rats and mice (0.1ml/100g, i.p.) since it has little direct cardiac toxicity. Once the animal is anesthetized, a surgical incision is made posterior to the xiphoid process. Cut superiorly until reaching the xiphoid process and make another incision running down the chest cavity. Continue cutting through the sternum until the diaphragm is evident. Cut down along the diaphragm to avoid damaging the heart. The rib cage may be dissected and removed. To remove the heart, pick the organ up between the index finger and thumb and cut the aorta below. Place the heart in a weigh bath with some of the Ca2+-containing buffer and weigh the organ. While the heart is in the buffer, carefully remove all remnant tissue around the organ, specifically exposing the aorta. Make sure that there is enough aortic tissue to allow adequate perfusion superiorly to the carotid bifurcation. Holding the aorta with two forceps, lift the heart from the buffer and place on the perfusion cannula, which should have buffer running through at a slow rate. Clamp the aorta to the perfusion needle and tie it (below the branches) with a piece of surgical thread. Increase the buffer flow rate until the drops fall between 5 and 10 times per minute. If the procedure is performed successfully, the carotid arteries will fill with buffer and become clear, the heart will begin to beat rhythmically and the drops will become clear.
Isolation Procedure of Rat Ventricular Myocytes
Once the heart is successfully hung, it is perfused (at 37°C) with Krebs-Henseleit bicarbonate (KHB) buffer containing (in mM): 118 NaCl, 4.7 KCl, 1.25 CaCl2, 1.2 MgSO4, 1.2 KH2PO4, 25 NaHCO3, 10 N-[2-hydro-ethyl]-piperazine-N'-[2-ethanesulfonic acid] (HEPES) and 11.1 glucose, equilibrated with 5%CO2-95% O2. Hearts are subsequently perfused with a nominally Ca2+-free KHB buffer for 2-3 min until spontaneous contractions cease, followed by a 15-20 min perfusion (depending on the physiologic condition of the animal) with Ca2+-free KHB containing 223 U/ml collagenase (Worthington Biochemical Corp., Freehold, NJ) and 0.1 mg/ml hyaluronidase (Sigma Chemical, St. Louis, MO). After perfusion, ventricles are removed and minced, under sterile conditions, and incubated with the Ca2+-free KHB with collagenase solution for 3-5 min. The cells are further digested with 0.02 mg/ml trypsin (Sigma) before being filtered through a nylon mesh (300 μm) and subsequently separated from the collagenase-trypsin solution by centrifugation (60 x g for 30 sec). Myocytes are resuspended in a sterile-filtered, Ca2+-free Tyrode's buffer containing (in mM): 131 NaCl, 4 KCl, 1 MgCl2, 10 HEPES, and 10 glucose, supplemented with 2% bovine serum albumin, with a pH of 7.4 at 37oC. Cells are initially washed with Ca2+-free Tyrode's buffer to remove residual enzyme and extracellular Ca2+ is slowly added back up to 1.25 mM. Myocytes with obvious sarcolemmal blebs or spontaneous contractions are not used. Only rod-shaped myocytes with clear edges are selected for recording of mechanical properties or intracellular Ca2+ transients as previously described (3).
Isolation Procedure for Mouse Ventricular Myocytes
A similar hanging procedure is utilized in the mouse, however a light magnifier is helpful in isolating the aorta. Hearts are rapidly removed from anesthetized mice via cardiectomy and immediately mounted on a temperature controlled (37°C) perfusion system. After perfusion with modified Tyrode solution (Ca2+ free) for 2 min, the heart is digested for 15-20 min with 0.9 mg/ml collagenase D (Boehringer Mannheim Biochemicals) in modified Tyrode solution. The modified Tyrode solution (pH 7.4) contained the following (in mM): NaCl 135, KCl 4.0, MgCl2 1.0, HEPES 10, NaH2PO4 0.33, glucose 10, butanedione 10, and the solution was equilibrated with 5% CO2-95% O2. The digested heart is removed from the cannula, and the left ventricle is cut into small pieces in the modified Tyrode solution. These pieces are gently agitated and the pellet of cells is resuspended in modified Tyrode solution and allowed to settle for another 20 min at room temperature during which time extracellular Ca2+ is added incrementally back up to 1.20 mM. Isolated myocytes are always used for experiments within 8 h after isolation, however we have recently had some success with culturing mouse myocytes for up to 48 hours.
Fig. 1.
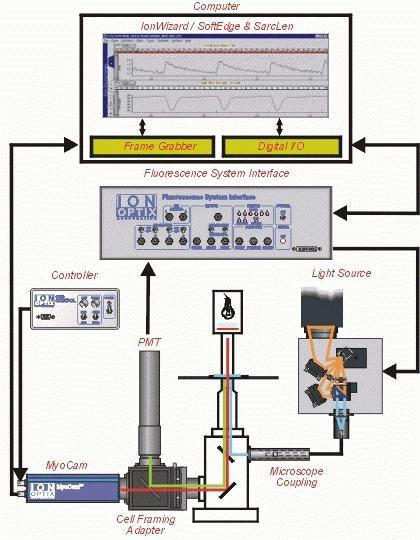
IonOptixTM video-based edge-detection cartoon. As the above panel illustrates, cell shortening and intracellular Ca2+ transients can be monitored simultaneously within the same cell. Figure provided courtesy of Dr. Doug Tillotson, IonOptixTM.
Cell Shortening/Relengthening
Mechanical properties of ventricular myocytes are assessed using a video-based edge-detection system (IonOptix Corporation, Milton, MA) (3). Cells are placed in a Warner chamber mounted on the stage of an inverted microscope (Olympus, X-70) and superfused (~1 ml/min at 25°C) with a buffer containing (in mM): 131 NaCl, 4 KCl, 1 CaCl2, 1 MgCl2, 10 glucose, 10 HEPES, at pH 7.4. The cells are field stimulated with a suprathreshold (50%) voltage and at a frequency of 0.5 Hz, 3 msec duration, using a pair of platinum wires placed on opposite sides of the chamber connected to a FHC stimulator (Brunswick, NE). The polarity of the stimulatory electrodes is reversed frequently to avoid possible build up of electrolyte by-products. The myocyte being studied is displayed on the computer monitor using an IonOptix MyoCam camera, which rapidly scans the image area every 8.3 msec such that the amplitude and velocity of shortening/relengthening is recorded with good fidelity. The soft-edge software (IonOptix) is used to capture changes in cell length during shortening and relengthening.
Intracellular Fluorescence Measurement
A separate cohort of myocytes is loaded with fura-2/AM (0.5 μM) for 15 min and fluorescence measurements are recorded with a dual-excitation fluorescence photomultiplier system (Ionoptix) as previously described (3). Myocytes are placed in a chamber on the stage of an Olympus X-70 inverted microscope and imaged through a Fluor 40x oil objective. Cells are exposed to light emitted by a 75W lamp and passed through either a 360 or a 380 nm filter (bandwidths are ± 15 nm), while being stimulated to contract at 0.5 Hz. Fluorescence emissions are detected between 480-520 nm by a photomultiplier tube after first illuminating cells at 360 nm for 0.5 sec then at 380 nm for the duration of the recording protocol (333 Hz sampling rate). The 360 nm excitation scan is repeated at the end of the protocol and qualitative changes in intracellular Ca2+ concentration ([Ca2+]i) are inferred from the ratio of the fluorescence intensity at the two wavelengths.
Results and Discussion
This section describes several problems that may be encountered during the isolation of single ventricular myocytes.
Myocyte Isolation and Yield:
The nature and quality of the myocyte isolation procedure may be the single most important factor in the success of mechanical assessment of the myocytes, and also for reliable comparison between the experimental and control groups. Although the cell culture medium may provide a favorable environment for the cells to recover from enzymatic trauma, the yield, survival and viability of the myocytes are largely dependent on the quality of isolation. Caution has to be taken when working with the mice myocytes in such variables as temperature, type of enzyme, and difficulty of culturing. A good isolation will yield approximately 70-80% viable cells or 4-5 million rod-shaped myocytes in both rats and mice.
Inadequate Heparinization:
Inadequate heparinization due to injection of heparin (i.p.) into the gut, gall bladder or other organs in the abdominal cavity may result in inadequate heparinization and blood clotting in the coronary arteries. This will significantly affect the outcome of enzymatic digestion and yield of myocytes. If after perfusion there are blood clots in the ventricles of the heart, increase the amount of heparin given and take particular caution to inject medial to the hindlimb in further studies. This will secure that the heparin is given intraperitoneally.
Air bubble accumulation
Air bubbles may build up and be trapped over time in the heart, causing a physical occlusion of the perfusion. Therefore, the perfusion system should be clear of any air bubbles at the start of the perfusion. Should any air bubbles develop during the perfusion, an air bubble trap device (usually a valve in the perfusion route or a 0.45-μm pore size cellulose acetate filter) may be inserted into the perfusion path. Make sure to replace the filter frequently as it may become plugged.
Cleaning of the perfusion system
It is essential to wash the entire perfusion system with 70% ethanol and distilled water before and after each isolation. Enzyme may stick to the glassware tubing due to inappropriate washing after perfusion or bacteria may grow within the system, contaminating the perfusion system.
Inadequate cell yield
The number one problem in myocyte isolation is inadequate cell yield. If the animal under study is older than average or substantially overweight (such as in an obesity study), the heart may need to be digested for a longer period of time. Also, the perfusion pressure can be increased to increase the digestion pressure within the heart. On the other hand, younger animals will require less perfusion pressure and shorter digestion times. All of this comes with practice in determining adequate perfusion and digestion.
Inability to stimulate myocyte
A particular problem normally encountered in myocyte studies is the inability to get the cells to beat. Make sure to clean the platinum wires routinely to remove any electrolyte by-product buildup. It is also helpful to allow the cells to recover for a short period of time (~15 mins.) once in the Warner chamber. It is essential that CaCl2 be added to the HEPES buffer in order to facilitate contraction.
Inability to obtain an adequate fluorescence signal
Fura-2/AM is cleaved into AM and fura-2, which is able to penetrate the cell membrane and bind to intracellular Ca2+. If the fura-2-loaded cell is excited, it will fluoresce during contraction and the signal will be displayed on the computer monitor. Make sure that the shutter is open, the emission filter is incorporated and the room is as dark as possible. We have found that the addition of 1 μl of fura-2/AM stock to the Warner chamber will provide a beautiful signal after 15 mins. loading and wash.
Applications
In this section, the authors present several different applications that have been used to provide models of compromised heart function in which myocytes can be studied. This is by no means an exhaustive list, simply several models that have benefited from video-based edge-detection.
(1). Drug intervention: Drugs are commonly used in an attempt to ameliorate the devastating effects of a particular disorder. We have used a model of fetal alcohol syndrome whereby dams are administered alcohol in liquidiet form during gestation, in concert with magnesium supplementation (4) (alcohol is known to contribute to hypomagnesemia). Isolation of ventricular myocytes from this model has allowed us to examine the effect of magnesium supplementation on the heart, specifically the functional unit or myocyte. We have also utilized video-based edge-detection in the study of leptin and cardiac contractile function. We found that leptin elicits a dose dependent decrease in myocyte shortening and intracellular Ca2+ transients that is attenuated by pretreatment with L-NAME, a NOS inhibitor (5).
(2). Disease models: Overt cardiac mechanical abnormalities are generally observed in diabetes of both chemical and genetic origin, with the severity of dysfunctions increasing with time (6-8). Characteristics of abnormal function include prolonged duration and reduced rate of ventricular contraction and relaxation, as measured in isolated ventricular myocytes (6,8). Diabetes is commonly studied by our laboratory and others and the heart is of particular interest due to the development of diabetic cardiomyopathy. Cardiomyopathy is a heart disorder that presents itself in the diabetic patient, regardless of arterial disease. Isolated myocytes from this model of diabetic cardiomyopathy have allowed the researcher to probe the effects of the disease on the heart. We have also used models of hypertension (9) and obesity (10) to examine cardiac specifics effects.
(3). Transgene: Transgenic animals are particularly important in the study of disease and its progression. We have examined the effect of metallothionein overexpression on the development of diabetic cardiomyopathy in the mouse (11). Myocytes isolated from this animal model has allowed us to probe the effect of transgenic alteration on the heart.
Summary
Isolation of ventricular myocytes is a powerful tool that allows the researcher the freedom to examine myogenic, cardiac specific effects, with all other tissues removed. The development of video-based edge-detection was an extremely important and useful tool that has expanded the level of understanding of cardiac specific effects of numerous disease states. We have included several helpful hints and techniques that will aid in proper isolation and maintenance of ventricular myocytes, however this technique is extremely touchy and requires patience beyond that of the beginning researcher. However, mastery of the technique will allow the researcher power beyond what was initially available to study cardiac specific problems under disease.
Acknowledgments
The work conducted in our laboratory has been supported by grants from the American Diabetes Association, the American Heart Association - Northland Affiliate, the Max Baer Fund and the North Dakota Experimental Program to Stimulate Competitive Research (ND EPSCoR). The authors are indebted to Dr. Doug Tillston and IonOptix Cooperation for all of their hard work over the past several years in helping us improve the video-based edge-detection system.
Appendix
Protocols
Adult rat ventricular myocyte isolation and culture procedures (Jun Ren)
Solutions
Krebs - Henseleit (K-H) Bicarbonate Buffer 100 ml 0.2 g D-glucose
Ca2+ - free K-H buffer 100 ml Leave out CaCl2, all else the same as K-H buffer. 0.2 g D-glucose
Enzyme Buffer 200 ml Ca2+ - free K-H buffer 0.11 g Collagenase CLS 2 (Worthington; 326U/mg) (170 U/ml) 0.02 g Hyaluronidase Type II (Sigma H-2126) 0.4 g D-glucose
Incubation Buffer - (Do not make up until after enzyme buffer is used). 50 ml Enzyme buffer 1 mg Trypsin IX (Sigma T-0134)
Sedimentation Buffer (sterile filter) 50 ml Ca2+ - free K-H buffer 1 g Albumin, bovine Fraction V (Sigma A- 7906) 0.09 g D-glucose
Keep solutions and tubing clean, but do not bother sterilizing until sedimentation buffer. Once cells are in sedimentation buffer, use sterile techniques in all subsequent steps. Bold items are not added until immediately before isolation.
Krebs - Henseleit Bicarbonate Buffer constituents:
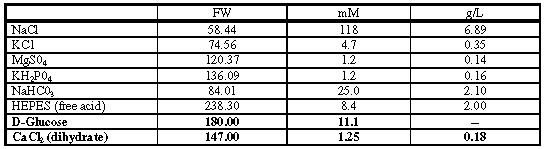 |
Add glucose to the working solutions each day (e.g. 0.24 g/120 ml buffer) Warm buffer to 37°C and gas with 95%O2 / 5% CO2 (no more than 30 minutes). (All chemicals are purchased from Sigma except where indicated).
Isolation Procedures:
Excise the heart and hang in Langendorf configuration (cannulate aorta and perfusion coronaries). Perfusion pressure is important (try 5 ml/min once heart is up and beating).
Clear blood from coronaries and tie aorta on cannula while perfusing with K-H buffer (Ca2+ containing). Make sure heart is beating quickly and rhythmically.
Cut slit in pulmonary vein in order to reduce afterload on coronaries.
Perfuse for 3-5 minutes.
Solution should be clear of blood and beating rhythmically before perfusion is stopped.
Switch perfusate to Ca2+-free K-H buffer - in order to stop beating.
Perfuse for about three minutes after cessation of beating.
Switch perfusate to enzyme buffer.
Recirculate enzyme buffer after enzyme solution has filled all tubes.
Perfuse for 15 minutes.
Stop perfusion. Mix 25 ml of enzyme buffer with 1mg of Trypsin (incubation buffer).
Remove the heart from the perfusion apparatus and trim off atria and large vessels.
Finely mince ventricular tissue (~one minute) in about 25 ml incubation buffer.
Cut tip off of plastic transfer pipette (or 5 ml Eppendorf pipette tip) to be used for gently tritriating tissue. Purpose is to release cells from tissue matrix. Spend about two minutes and be very gentle.
Filter through nylon mesh (about 300 μm mesh); size is not too critical. Purpose is to separate cells from tissue mass.
Wash procedures:
-
Add an equal volume of sedimentation buffer to cells and centrifuge at 600 rpms (slow) for 60 seconds; aspirate off supernatant and resuspend cells in sedimentation buffer up to 10 mls. Purpose of centrifuging is to separate cells from enzymes quickly.
Sedimentation buffer is used for washing cells (removes excess enzymes, separates dead cells and increases extracellular Ca2+ slowly so that more cells survive.)
Add Ca2+ back in increments with subsequent washes e.g. after resuspending cells in sedimentation buffer, add 10 μm Ca2+ (we make up a 100 mM stock CaCl2, sterile filtered; and add 10 μl stock/10 ml sedimentation buffer).
Allow cells to settle for 10 minutes (live cells settle fast, dead cells tend to float in the BSA (albumin)).
Remove supernatant, resuspend in sedimentation buffer, add 25 μl Ca2+ stock/10 ml buffer
Settle 10 minutes. Resuspend 50 μl/10 ml; Settle 10 minutes. Resuspend 75 μl/10 ml; Settle 10 minutes. Resuspend in normal culture medium.
NOTE: We add Ca2+ back slowly because our diabetic cells do not tolerate Ca2+ well. Normal cells do not need to be treated as gently, but we do it none-the-less.
Culturing myocytes
-
Precoat culture dishes/glass cover slips with laminin. Be sure to use culture dishes and not petri dishes; myocytes will not stick to the surface of a petri dish. Make up laminin stock (10 μg/ml) in sterile culture medium.
Laminin, mouse (Collaborative Biomedical Products, cat# 40232; tel (617) 275-0004)
1 mg/bottle
Add contents of bottle to 100 ml culture medium (keep under sterile conditions).
Aliquot and store at -70°C
(If stored at -20°C, it only lasts about one month).
Allow laminin to sit on the coverslip surface for 30-60 minutes (do not let it dry out). A thin surface coating is all you need. Before plating out cells, aspirate off excess laminin.
Resuspend myocytes in culture medium. Typically we get 4-5 million rod shaped cells/heart. We resuspend cells from one heart in 50 ml medium. Plate out 5 ml per 100 mm culture dish.
Allow myocytes to settle and stick for one hour (at 370C). Gently aspirate off dead cells and add fresh medium.
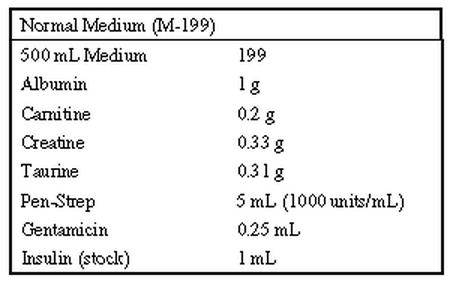
Recipe for Serum-free culture medium, for adult rat ventricular myocytes
(Ellingsen et al., Am J Physiol, 265:H747-H754, 1993)
For details, see next page.
 |
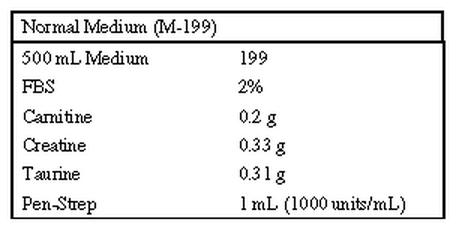
Recipe for culture medium for adult mouse ventricular myocytes (see protocol below)
 |
Protocol for Rat Culture Medium preparation.
1. Weigh out Albumin, Carnitine, Creatine and Taurine and place in a 250 ml sterile bottle (or flask).
2. Dissolve with 100 ml of M 199 (from the 500 ml bottle).
3. Add insulin and Pen-Strep
4. Sterile filter and add back to 500 ml bottle.
Protocol for Mouse Culture Medium preparation.
1. Weigh out Carnitine, Creatine and Taurine and place in a 250 ml sterile bottle (or flask).
2. Dissolve with 100 ml of M 199 (from the 500 ml bottle).
3. Add Pen-Strep
4. Sterile filter and add back to 500 ml bottle.
Medium 199 with Earle's salts, 25 mM HEPES and bicarbonate, without glutamine (Sigma M-7528)
Store at 4°C.
Albumin, bovine (Sigma A-7906)
Store at 4°C.
L-Carnitine (Sigma C-0283), FW 197.7
Store desiccated at room temp.
Final concentration 2 mM
Creatine (Sigma C-3630), FW 131.1
Store at room temp.
Final concentration 5 mM
Insulin (Sigma I-5500), FW 5733.5
Final concentration: 10-7M
Dissolve 0.29 mg/ml (e.g. 15 mg in 50 ml) in 0.01 N (41 μl stock HCl (12 N)/50 ml H2O) and store at 4oC for up to 1 month.
Use 1 ml per 500 ml medium
Taurine (Sigma T-7146), FW 125.1
Store at room temp.
Final concentration 5 mM
3, 3', 5-Triiodo-D-Thyronine (Sigma T-2502), FW 651
Final concentration: 10-10M
Dissolve 10-3 M (0.65 mg/ml, e.g. 2 mg in 3 ml) in 0.05 N NaOH (30 μl 5 N NaOH in 3 ml H2O) and dilute to 10-6 M (50 μl in 50 ml) with water. Freeze in aliquots at -20o C and neutral pH for up to 4 months.
Use 50 ul of 10-6 M per 500 ml medium.
Penicillin/Streptomycin (Gibco 600-5070 AG)
Final concentration 100 IU/ml, 100 mg/ml
Use 5 ml of 10,000 IU/ml, 10,000 mg/ml for 500 ml medium
Store at -20°C
Gentamicin (Sigma 10 mg/ml; G1272)
Withdraw from bottle with 1 ml syringe and 25 g needle
HEPES Buffer constituents (to be used for cell wash):
Bold items are not added until the day of isolation
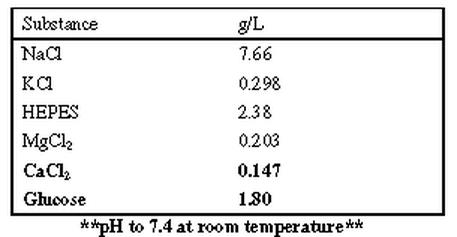 |
Fura-2 AM:
4 ml Medium 199 (Sigma Chemical) 2 μl Fura-2 (Molecular Probes)
Fura-2 is kept in the freezer and can be thawed and refrozen numerous times. Make sure to vortex the above solution vigorously before applying to the cells.
Place 5-6 drops of fura-2 into the cell bath and incubate for 5 minutes. Wash with 1 mM Ca2+-containing HEPES for 15 minutes before starting fluorescence recording.
Adult Mouse Myocyte Isolation Protocol
Solutions
Buffer 1:
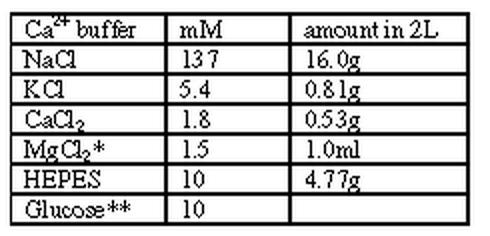 |
* = from 1M stock solution ** = 0.4507g per 250ml of above volume pH = 7.4 @ 37 C; 7.55 @ RT
Buffer 2:
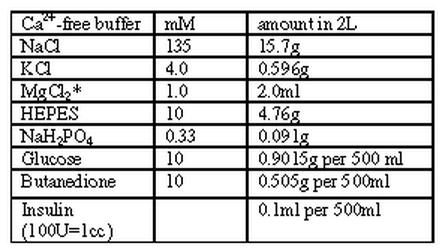 |
pH = 7.4 @ RT
Buffer 3:
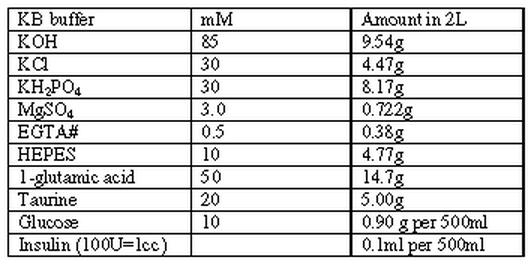 |
Buffer 4:
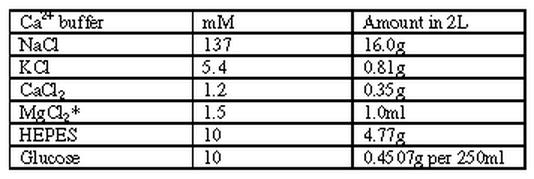 |
* = from 1M stock solution pH = 7.4 @ 37 C; 7.55 @ RT
IMPORTANT: Make sure to filter all the above buffers. They may be made up beforehand. No not add glucose, butanedione or insulin until the day of the isolation.
Ca2+-addition recipe
0.06mM Ca2+
1ml of buffer 4
19ml of buffer 2
0.24mM Ca2+
4ml of buffer 4
16ml of buffer 2
0.6mM Ca2+
10ml of buffer 4
10ml of buffer 2
1.2mM Ca2+
20ml of buffer 4
General information:
Enzyme
Protease (3-4mg/35ml Ca-free)
Collagenase D (10-13mg/35ml Ca-free; older mouse, 16mg/35ml)
1.8mM Ca2+ buffer
This is the first buffer used for perfusion. Note the difference in molarity than that used in adding back calcium. To these solution add Glucose (10mM; 0.4507g/250ml)
1.2mM Ca2+ buffer
This buffer is made the same as the 1.8mM Ca2+ buffer, however the amount of Ca2+ added is different. For this buffer, we add 0.35g of CaCl2 to 2L of buffer 1.
Collagenase D
Boehringer Mannheim
Lot #84612223
Protease
Sigma
Lot #87H0534
Protocol
Mice myocytes are extremely sensitive to any air bubbles within the tubing of the perfusion system. Make sure that there are absolutely no bubbles anywhere within the tubing. Pipetting up and down is helpful to remove any bubbles found. It is also extremely critical that only the times listed below for perfusion of the buffers are used. Therefore, it is imperative that three Langendorf perfusion tubes be set up in series so that the buffers can be switched quickly.
Prepare the buffers as listed on the buffers page. Add glucose, insulin and butanedione on the day of isolation. Place about 25-30ml buffer 2 in two weigh boats and refrigerate. Weigh out the enzymes and place in a sterile 25ml Erlenmeyer flask and refrigerate. Fill two small culture plates with buffer 3 and place at RT near the perfusion system.
Fill the perfusion system with the buffer 1 and buffer 2. Make sure that the perfusion lines are filled and the temperature is calibrated to keep them at 37 °C. It may be necessary to have the water bath at a higher temperature to maintain this 37°C. Fill the third perfusion system with buffer 2 (this will be used for the enzyme buffer) and remove any air bubbles. Mouse myocytes require 100% O2 perfusion throughout the entire procedure.
Set up thermometer, light and timer.
Heparinize the mouse with 0.2cc of heparin
Anesthetize with 0.22cc of ketamine/xylazine (15 mg/kg and 150 mg/kg, respectively).
Weigh the mouse.
Quickly remove the heart and place in cold buffer 2 (from the fridge) to cold arrest the heart. The trick to removing the heart is to remove some of the lung with it in order to have a large aorta to work with. While in the cold buffer, trim off any excess debris or tissue.
Hang the heart by cannulating the aorta. It is helpful to use two fine point tweezers and to place one of the tweezers within the aorta. Tie the aorta onto the needle and remove the clip.
DO NOT CUT ANY PART OF THE HEART. Perfuse with buffer 1 for 4.5 minutes. The heart should start to beat rhythmically. Maintain the temperature of the heart at 37° C by adjusting the intensity of the light. Also, maintain the drop rate of all solution at 4-5 drops/10 secs.
Perfuse with buffer 2 for 4 minutes. Once this is started, mix the two enzymes with 35 ml of buffer 2 that was placed in the incubator (at 37°C). Filter the enzyme and then replace the third perfusion tube with the enzyme buffer. One can do this by simply sucking out buffer 2 with a syringe.
Perfuse the enzyme solution for 6-7 minutes (contingent on the age of the animal). For a normal adult mouse, use only 6 minutes.
Trim off the atria and the right ventricle while the heart is still hanging. Place the left ventricle into buffer 3 and remove the pericardium.
Transfer the left ventricle into buffer 3 containing culture dish. Simply pull the tissue apart, watching for the myocytes to release. The solution will become milky, contingent on a proper isolation procedure. After mincing the tissue, slowly triturate the solution.
Filter #14 to get rid of the tissue.
Let the solution settle for 15 minutes. DO NOT CENTRIFUGE.
Begin adding Ca2+ back incrementally from 0.06mM to 1.2mM. When transferring between two different molarities of Ca2+ solution, simply remove the myocyte pellet by a pipet and place into the bottom of the next buffer. Tap the side of the tube to facilitate the formation of the pellet.
Let myocytes settle for approximately 5 minutes between each Ca2+ concentration.
The myocytes are now ready for the experiment. Mouse myocytes do not culture well. Cells are good for up to 8 hours.
Note: We have had some success in culturing mouse myocytes per the recipe above. It is important that laminin-coated coverslips be used for all experiments involving these myocytes. Follow the same protocol for coating laminin coverslips as described under culturing rat myocytes detailed above. The medium (note changes from rat culture medium) should be replaced every 6-8 hours and the myocytes should be maintained at 37°C with 100% CO2. Mouse myocytes can be cultured for 24-48 hours with limited decrease in viability, however after 48 hours viability is drastically decreased.
References
- Wang Z, Mukherjee R, Lam CF, Spinale FG. Spatial characterization of contracting cardiac myocytes by computer-assisted, video-based image processing. Am J Physiol. 1996;39:H769–H779. doi: 10.1152/ajpheart.1996.270.2.H769. [DOI] [PubMed] [Google Scholar]
- Mukherjee R, Crawford FA, Hewett KW, Spinale FG. Cell and sarcomere contractile performance from the same cardiocyte using video microscopy. J Appl Physiol. 1993;74:2023–2033. doi: 10.1152/jappl.1993.74.4.2023. [DOI] [PubMed] [Google Scholar]
- Ren J, Bode AM. Altered cardiac excitation-contraction coupling in ventricular myocytes from spontaneously diabetic BB rats. Am J Physiol. 2000;279:H238–244. doi: 10.1152/ajpheart.2000.279.1.H238. [DOI] [PubMed] [Google Scholar]
- Wold LE, Ren J. Prenatal alcohol exposure alters cardiac myocyte contractile function in offspring: influence of maternal Mg2+ supplementation. Cardiovasc Tox, in revision. [DOI] [PubMed]
- Nickola MW, Wold LE, Colligan PB, Wang G-J, Samson WK, Ren J. Leptin attenuates cardiac contraction in adult rat ventricular myocytes: Role of nitric oxide. Hypertension. 2000;36:501–505. doi: 10.1161/01.hyp.36.4.501. [DOI] [PubMed] [Google Scholar]
- Ren J, Davidoff AJ. Diabetes rapidly induces contractile dysfunctions in isolated ventricular myocytes. Am J Physiol. 1997;272:H148–H158. doi: 10.1152/ajpheart.1997.272.1.H148. [DOI] [PubMed] [Google Scholar]
- Ren J, Walsh MF, Hamaty M, Sowers JR, Brown RA. Altered inotropic response to IGF-I in diabetic rat heart: influence of intracellular Ca2+ and NO. Am J Physiol. 1998;275:H823–H830. doi: 10.1152/ajpheart.1998.275.3.H823. [DOI] [PubMed] [Google Scholar]
- Ren J, Bode AM. Altered cardiac excitation-contraction coupling in ventricular myocytes from spontaneously diabetic BB rats. Am J Physiol. 2000;279:H238–H244. doi: 10.1152/ajpheart.2000.279.1.H238. [DOI] [PubMed] [Google Scholar]
- Ren J, Jefferson L, Sowers JR, Brown RA. Influence of age on contractile response to insulin-like growth factor I on ventricular myocytes from spontaneously hypertensive rats. Hypertension. 1999;34:1215–1222. doi: 10.1161/01.hyp.34.6.1215. [DOI] [PubMed] [Google Scholar]
- Ren J, Sowers JR, Walsh MF, Brown RA. Reduced contractile response to insulin and IGF-1 in ventricular myocytes from genetically obese Zucker rats. Am J Physiol. 2000;279:H1708–H1714. doi: 10.1152/ajpheart.2000.279.4.H1708. [DOI] [PubMed] [Google Scholar]
- Wold LE, Duan J, Epstein PN, Ren J. Cardiac-targeted overexpression of metallothionein prevents the development of diabetic cardiomyopathy in streptozotocin-induced diabetes. Circ Res, submitted.