

Abstract
The Chromatin Immunoprecipiation (ChIP) provides a powerful technique for identifying the in vivo association of transcription factors with regulatory elements. However, obtaining meaningful information for promoter interactions is extremely challenging when the promoter is a member of a class of highly homologous elements. Use of PCR primers with small numbers of mutations can limit cross-hybridization with non-targeted sequences and distinguish a pattern of binding for factors with the regulatory element of interest. In this report, we demonstrate the selective in vivo association of NF-κB, p300 and CREB with the human Iγ1 promoter located in the intronic region upstream of the Cγ1 exons in the immunoglobulin heavy chain locus. These methods have the ability to extend ChIP analysis to promoters with a high degree of homology.
Keywords: Gene Expression Regulation, Transcription Factors, Chromatin Immunoprecipitation, B cells
Introduction
The association of cellular proteins with chromatin can elicit a range of cellular responses involving transcriptional modulations, initiation or attenuation of DNA replication and in specific cases, the induction of non-homologous recombination during lymphocyte development and activation. The Chromatin Immunoprecipitation (ChIP) assay has proved to be an invaluable tool for identifying transcription factors associated with promoters at distinct stages of gene activation. This assay, for the most part, has refined in vitro and transfection studies that evaluate the capacity of a distinct factor to bind an identified sequence within a known promoter by demonstrating an in vivo association with the same binding site in chromatin reviewed in (1, 2). Accordingly, promoters that have the appropriate factor binding sites may be restricted, via chromatin organization, from interacting with a transcription factor under specific cellular conditions.
In a humoral immune response, multiple genes go through a cycle of transcriptional activation and attenuation in the course of B cell differentiation into antibody-producing cells (reviewed in (3, 4). A critical set of genes known to be directly regulated by activation signals at the transcriptional level are the immunoglobulin (Ig) constant region genes within the Ig heavy chain (CH) locus. In the human, there are nine Ig heavy chain constant region genes, Cμ, Cδd, Cγ1, Cγ2, Cγ3, Cγ4, Cα1, Cα2 and Cε, that encode the heavy chain polypeptides for the IgM, IgD, IgG1-4, IgA1-2 and IgE antibodies, respectively. The Cγ, Cα and Cε heavy chains are generated via class switch recombination (CSR) after antigen and / or T cell stimulation and thus contribute to the antibody diversity required for a comprehensive humoral response (reviewed in (3)). Upon B cell activation and prior to undergoing CSR, this group of genes becomes transcriptionally active due to the presence of intronic, or I region promoters located upstream of the different CH exons. The I region promoters within a subclass are highly homologous, yet have unique responses to diverse stimuli and thus transcriptional activation of the different I region promoters can greatly influence the antibody profile of a specific response (5-11).
Our laboratory’s focus has been to understand the basis for Iγ promoter activity in response to signals through the CD40 pathway (12-14). This pathway is triggered by cognate interactions with CD40 ligand (CD40L or CD154) expressed on activated CD4+ T cells and has been shown to be highly dependent on NF-κB (15-17). We, and others, have observed that the different Iγ promoters respond distinctly differently to CD40 signals both in B cell lines and in primary human B cells (12, 18-20). In particular, there is a very strong Iγ1 transcriptional response and a very restricted Iγ4 response. Iγ3 and Iγ2 promoters give variable responses depending on the cell line and stimulus. These observations are unexpected given the fact that the proximal promoters are approximately 97% identical within the subclass. We have analyzed the different Iγ promoters in transfection and reporter assays and found discrete differences in transcriptional responses that are sequence-specific (13). In particular, we identified a 36bp region in the Iγ promoter that contained CREB/ATF binding sites and an adjacent putative NF-κB binding site (κB6 site) that was absent in the Iγ3 promoter. We previously demonstrated that the CREB/ATF binding sites function as an “amplifier” element such that its insertion into the Iγ3 promoter induces a response greater than that seen with the Iγ3 promoter alone (13). Our recent work extended this finding and demonstrated a critical role for the NF-κin Iγ1 promoter activity in the Ramos B cell line (14). Taken together, our transfection and reporter data revealed important differences in Iγ promoter responses to CD40 signaling, however it was evident from the different responses obtained with the reporter constructs compared to the endogenous promoters, that a major form of Iγ promoter regulation was at the chromatin level.
Our recent experimental goal focused on determining whether binding at the different κB sites in the Iγ1 promoter was critical for in vivo expression. Also, we wished to determine whether the interaction between NF-κB binding at the κB6 site, with proteins bound at the adjacent CREB/ATF site and/or upstream κB3-5 sites, was necessary for Iγ1 expression in both Ramos B cells and as an extension, primary B cells. In order to study the CD40 regulation of Iγ1 transcription at the chromatin level several obstacles had to be overcome. First, it was clear that ChIP assays would give the most accurate pattern of both NF-κB and co-activator binding in the Iγ1 promoter, however these experiments have inherent problems that needed to be solved empirically. Second, only a small number of cells in the Ramos B cell line actually respond to exogenous signals and express Iγ transcripts, thus in order to obtain meaningful results, we had to use peripheral B cells from isolated blood for our ChIP studies. Finally, we had to utilize PCR protocols that would distinguish the Iγ1 promoter from the other three highly homologous Iγ subclass promoters in order to be able to convincingly ascribe binding to the Iγ1 promoter. Here, we describe, in-depth, our methods for identifying the in vivo association of specific transcription factors with a single promoter region that is highly homologous to three other promoters. Furthermore, we outline our procedure for carrying out ChIP on primary human B cells.
Materials and Methods
Isolation and activation of human CD19+ B cells
Blood products were prepared by the NBAH Blood Center at Robert Wood Johnson University Medical Center or the New York Blood Center from peripheral blood samples healthy donors. Mononuclear cells (PBMCs) were separated by Ficoll-Hypaque gradient centrifugation. Blood products were diluted 1:1 in RPMI 1640 media layered over Ficoll-Hypaque, and spun for 30 min at 2000 RPM. Buffy coat (the leukocyte layer) was aspirated from the gradient and washed for 30 min at 1500 RPM in RPMI 1640 media. Total leukocytes were resuspended in 1X PBS containing 2% FBS and B cells were removed via biomagnetic separation by incubating for 1 hr at 4°C with anti-CD19-conjugated superparamagnetic beads provided by Dynal (Lake Success, NY). Beads were washed five times with 1X PBS containing 0.1% BSA to remove non-specifically bound cells, and plated in 10 mls RPMI 1640 media supplemented with 10% heat inactivated fetal calf serum (FCS), 50U/ml penicillin, 50 μg/ml streptomycin and 1mM L-glutamine at 37°C / 5% CO2 for 12 hr. Media was collected and cells were detached from beads using 30 μl Dynal anti-CD19 DETACHabead (diluted 100-fold in RPMI containing 1% FBS) for 30 min at 25°C. Beads were washed and the supernatant containing CD19+ B cells was collected. To deplete the population of “activated” B cells, IgG+ B cells were removed from the total CD19+ B cell population by negative selection. Specifically, anti-IgG coated 100 mM plates were prepared with 5 mls coating buffer (0.5M Tris-Cl pH 9.5, 0.15M NaCl) containing 25 g/ml human anti-IgG (catalog # 2040-07, Southern Biotech) overnight at 4°C. CD19+ B cells from multiple units (up to 1 × 108 cells) were pooled and plated in 10 mls of complete RPMI media, on the anti-IgG coated petri dishes for 30 min at 25°C. Following incubation, IgG- B cells were analyzed by flow cytometry to verify the success of the isolation procedure.
Flow cytometry
To analyze lymphocyte populations relative to purity at different stages of the isolation procedure, cells were incubated with specific antibodies and surface expression monitored by flow cytometry. Specifically, 1 × 105 B cells were washed in 3% FCS/0.1% NaN3 /1X PBS followed by incubation for 10 min at 4°C with 5 g heat aggregated IgG to inhibit non-specific binding. Cells were incubated for 45 min at 4°C with saturating amounts of fluorescein isothiocyanated (FITC)-conjugated or biotin-conjugated mAbs against human CD20 (cat# 169-020) using 80 μof 10 μg/ml (Ancell, Bayport MN) IgM (cat# 9020-08) using 80 μl of 20 μg/ml, IgD (cat# 2030-02) using 80 μl of 10μg/ml, or IgG (cat# 2040-01) using 80 μl of 20μg/ml (Southern Biotechnology Associates, Birmingham, AL). Biotin-conjugated samples were further incubated with phycoerythrin-conjugated streptavidin (cat# 253-050) using 20 μl of a 1:10 dilution for 45 min at 4°C. Cells were washed and fixed with 500 μl of 1% paraformaldehyde in 1X PBS. Cells were analyzed using FACScan (Becton Dickinson, Mountain View, CA).
Primary B cell stimulation
The total population of CD19+/IgG- B cells collected from blood isolations were divided equally and plated in 2-5 ml of complete RPMI in a 6-well dish. The volume of media used for cultures was determined by the number of cells obtained with each isolation, with a final concentration of approximately 1 × 107 /ml. Cultures were either unstimulated or supplemented with 0.5 μ/ml of sCD40L for 2hr, and harvested for chromatin immnoprecipitation assays. To obtain sufficient CD19+/IgG- B cells for chromatin immunoprecipitations, multiple blood isolations were performed and the CD19+/IgG- B cells were pooled. In the event that multiple blood units could not be obtained from blood sources within a 24 hr period, individual blood units were processed. Isolated CD19+/IgG- B cells were stimulated as described above, harvested for formaldehyde crosslinking, and snap-frozen after washing with PBS. To continue with chromatin immunoprecipitation assays, these cell pellets were thawed and pooled to obtain a final cell number of 5 × 107 primary B cells per experiment.
EMSA to confirm B cell activation
To determine whether the stimulation conditions were sufficient for primary B cell activation, 5 × 106 primary IgM+/IgG- B cells were removed from culture at 2 hr and prepared for nuclear extracts using a modification of Dignam’s method (21). Cells were washed once in 1X PBS, and resuspended in 400 μl ice-cold buffer A (10 mM Hepes, pH 7.9, 10 mM KCl, 0.1 mM EDTA, 0.1 mM EGTA, 1 mM DTT, 0.5 mM PMSF, 2 μM aprotinin, 2 μM pepstatin A). After incubation on ice for 10 min NP-40 was added to a final concentration of 0.6%, and nuclei isolated by centrifugation at 14,000 rpm for 30 s. Nuclei were resuspended in 50 μl ice- cold buffer C (20 mM Hepes, pH 7.9, 0.4 M NaCl, 1 mM EDTA, 1 mM EGTA, 1 mM DTT, 0.5 mM PMSF, 2 μM aprotinin, 2 μM pepstatin A). Samples were kept on ice for 30 min, with brief vortexing every 5 min and centrifuged at 14,000 rpm for 10 min. Protein concentration was determined using the Bradford Assay (BioRad).
CD40 signaling in B cells results in an influx of NF-κB into the nucleus upon activation, therefore the nuclear extracts were incubated with a double-stranded NF-κB 5’agt tga ggg gac ttt ccc agg c 3’ competitor consensus DNA fragment purchased from Promega Corporation and end labeled with [γ-32P] ATP, to visualize activation of NF-κB. Binding reactions were prepared using 4 μextract, 1 μg poly dI-dC in binding buffer (10mM Tris Cl pH 7.5, 50mM NaCl, 1mM DTT, 1mM EDTA, 5% glycerol). 3-4 × 104 cpm was added to reactions and incubated 20 min at 25°C. In order to confirm the presence of NF-κB, a supershift assay was performed using 1 μl of anti-p50 sera (gift of Dr. N. Rice, National Cancer Institute, Bethesda, MD), and incubating the above mix an additional 1.5 h at 25°C prior to addition of the probe. Samples were loaded on a 6% acrylamide gel and visualized by autoradiography.
ChIP assay using CD19+ B cells
The chromatin immunoprecipitation (ChIP) assay that we devised for our work was an extension of previous protocols (22, 23) with some specific modifications. Briefly, 5 × 107 primary B cell cultures were harvested and diluted to 100 ml with 37°C media and cross-linked by the addition of one-tenth volume of 11% formaldehyde in 0.1 M NaCl, 1 mM EDTA, 0.5 mM EGTA and 50 mM Hepes, pH 8.0 in growth medium for 5 min at RT, before addition of glycine to a final concentration of 0.125 M. After washing 2X with ice-cold PBS containing 1X protease inhibitor cocktail (PIC) (Sigma) cells were resuspended in 25 mls ice-cold cell lysis buffer 1 (10mM Tris-CL pH 8.0, 10mM EDTA, 10mM Na-Butyrate, 0.5mM EGTA, 0.25% Triton X-100, 1X PIC, 1 mM PMSF) and incubated on ice 10 min. Nuclei were recovered by centrifugation and resuspended in lysis buffer 2 (0.2M NaCl, 10mM Tris-CL pH 8.0, 1mM EDTA, 0.5mM EGTA, 1X PIC, 1 mM PMSF) and incubated 10 min. Samples were centrifuged and resuspended in 2 ml sonication buffer (10 mM Tris-Cl, pH 8.0, 1 mM EDTA, 0.5 mM EGTA). Chromatin was sonicated 12 rounds for 20 s using a 250 Branson Sonifier (30% output) alternating with 30 s incubations on ETOH/ice. Chromatin was centrifuged 10 min at 14,000 RPM to pellet debris, and stored at -80°C. Chromatin samples were pre-cleared for 1-2 h at 4°C by adding 50 μl Protein A/ssDNA-agarose beads (Upstate Biotechnology), followed by incubation with antibody in a 10-fold dilution of 1 X RIPA buffer (140mM NaCl, 1% Triton X-100, 0.1% deoxycholate, and 1mM PMSF) at 4°C overnight. Immune complexes were recovered at 4°C for 1 h using 60 μl Protein A/ss DNA agarose beads. Complexes were washed five times with IP1 (0.1% SDS, 1% Triton X-100, 2 mM EDTA, 20 mM Tris, pH 8.0, 150 mM NaCl), once with IP2 (0.1% SDS, 1% Triton X-100, 2 mM EDTA, 20 mM Tris, pH 8.0, 500mM NaCl), and twice with Tris-EDTA pH 8.0. Immunoprecipitation reactions and input chromatin were digested with 200 μg/ml RNase A for 1 h and 200 μg/ml proteinase K in TE with 0.5% SDS for 2 h at 55°C. Crosslinks were reversed overnight at 65°C. Samples were extracted once with phenol/chloroform and once with chloroform/isoamyl alcohol, ETOH precipitated and resuspended in 12 μl TE.
Sequence-specific PCR with ChIP products
Due to the high sequence conservation of the four Iγ subclass promoters it was difficult to selectively amplify only the Iγl promoter region using standard PCR techniques. Therefore, in order to distinguish Iγ1 from the remaining subclasses, PCR was carried out with primers that contained a minimum number of 4 naturally occurring base alterations and 3-4 additional changes that were intentionally introduced into the primer sequence. This level of mispriming still allowed amplification of the Iγ1 promoter sequence. To test the specificity of primers, PCR was performed using either 0.2 ng purified HindIII-BamHI Iγ subclass promoter sequence, 1 μl of 1:200 dilution of input chromatin, 3 μl of undiluted or 10-fold serial diluted immunoprecipitant in a 50 μl reaction containing 10 mM Tris Cl (pH 8.1), 1.5 mM MgCl2, 50 mM KCl, 0.1% Triton X-100, 5% DMSO, 0.2 mM dNTPs, 2.5 U Taq (Promega) and 100 ng of 5’ and 3’ primers: Iγ non-specific- 5’gcc ctc tga ccc aga aac c 3’; 5’cct tcc tgt tct ggc gag gt 3’; Iγ1 specific- 5’ tcc atg tag tgc cgg aca cga ccc cat 3’; Iγ1 WT 5’ tcc atg tgg ggc cgg cct cga ccc cat 3’; Iγ1 Δ 1bp 5’ tcc atg tgg tgc cgg cct cga ccc cat 3’; Iγ1 Δ 2bp 5’ tcc atg tgg tgc cgg cca cga ccc cat 3’; Iγ1 Δ 3bp 5’ tcc atg tgg tgc cgg aca cga ccc cat 3; IgHG1- 5’ctc cac caa ggg ccc atc ggt 3’; 5’caa atc ttg tga caa aac tca cac at 3’. Amplification of all reactions was for 35 cycles, with a hot-start at 3 min at 92°C, 30 s at 92°C, 45 s at 56°C, 45 s at 72°C, and 3 min at 72°C. Final PCR amplifications were subjected to gel electrophoresis and bands were quantified using Kodak imaging software.
Results and Discussion
Isolation and characterization of CD19+ human B cells from blood
Successful ChIP is highly dependent on having sufficient starting material to carry out a number of immunoprecipitation reactions using chromatin from the same source of cells. Therefore, using primary human B cells in this assay is highly challenging given the relatively low numbers of isolated B cells from peripheral blood. Also, since our goal was to analyze factor binding before and after activation through CD40, it was critical that the isolated B cells were non-activated prior to isolation. We therefore set out to isolate CD19+ IgG- B cells to obtain a population of B cells that were predominantly IgM+/IgD+. From 500 ml/unit of human blood, we were able to isolate an average of 3.4 × 107 B cells/unit or 4.9% of the total peripheral blood mononuclear cells (PBMCs) (Table 1).
Table 1.
Leukocyte and B cell yields from human blood.
| Cell population | Range of cells obtained (total units = 10) | Average cell number | % yield |
| PBMCs | 2.5 × 108 - 1.5 × 109 | 7.0 × 108 | 100% |
| CD19+ | 1.5 × 107 - 9.5 × 107 | 4.0 × 107 | 5.7% |
| CD19+/IgG- | ND | 3.4 × 107 | 4.9% |
The positively selected population was shown to be approximately 98% CD20+ (pan B cell marker). We selected to use anti-CD20 antibody to measure successful B cell isolation, since use of the CD19+ magnetic beads may cause CD19+ epitopes to be obscured. Further purification of this population by negatively selecting IgG+ B cells produced cells that were 87% IgM+ and 89% IgD+ (Fig. 1).
Fig. 1. Purification of a IgM+/IgD+ population of primary B cells.
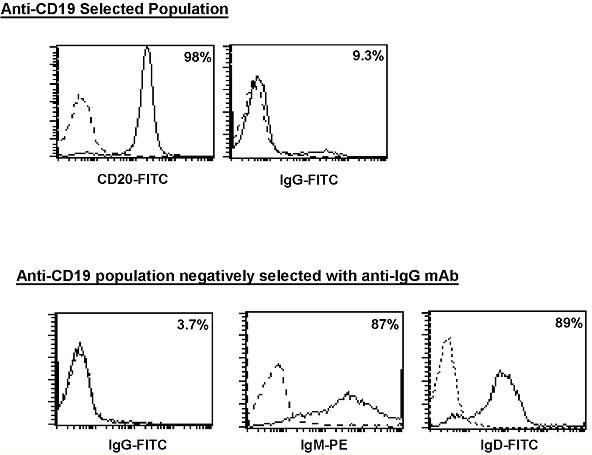
CD19+ isolated IgM+/IgG- peripheral B cells were analyzed for surface expression of CD20 and IgG (upper panel). IgG, IgM, and IgD expression was determined following subsequent negative selection using anti-IgG antibody (lower panel). Percentages of positively stained cells are indicated in the right corner of each histogram. The stippled line represents the isotype control for each antibody.
The percentage of IgG+ B cells left in the final population was below that observed with the isotype control and supported out assertion that the starting population was predominantly IgM+/IgD+ B cells (Table 2).
Table 2.
Phenotypic analysis of B cell populations.
| Surface maker | Average expression |
| CD20 | 98% |
| IgG- pre selection | 9.3% |
| IgG- post selection | 3.7% |
| Isotype control | 5.3% |
To obtain sufficient numbers of CD19+/IgM+ B cells for each ChIP assay, units were pooled prior to stimulation. Purified CD19+/IgG- B cells were stimulated with soluble, trimerized CD154 for 2 hr. Because of our interest in analyzing the interaction of Rel/NF-κB subunits with chromatin, it was necessary to confirm that the stimulation conditions were, in fact, inducing NF-κB activity. To this end, EMSAs were carried out with nuclear extracts from resting and activated B cells with and without antibody to the p50 NF-κB subunit. As seen in Figure 2, prior to activation there is a measurable level of NF-κB activity in the B cells (lane 1). However, upon activation there is a significant increase of binding activity (lane 2) and a considerable amount of the binding activity is super-shifted with anti-p50 antibodies (lane 3). This result indicated that under the stated conditions of CD40 activation, there was a marked increase in NF-κB activity in the primary CD19+/IgG- B cells.
Fig. 2. NF-κB activity is upregulated in response to CD40 signaling.
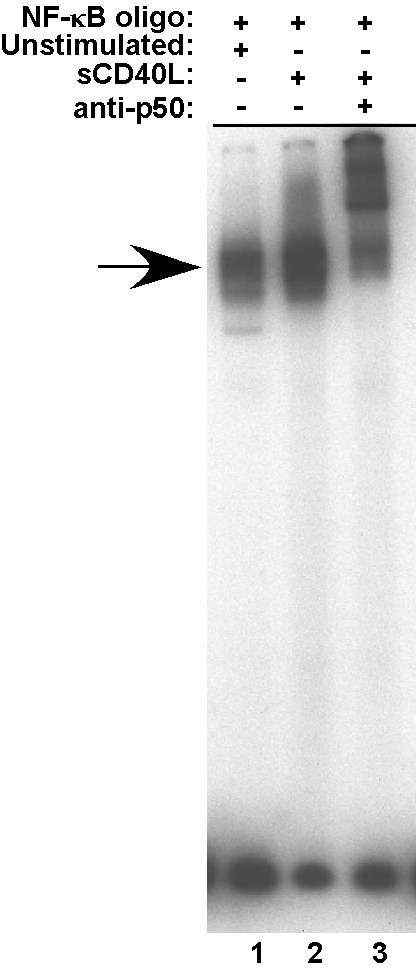
NF-κB consensus oligonucleotide was incubated with extracts from IgM+/IgG- peripheral B cells either unstimulated (lane 1) or stimulated for 2 h with 0.5 μg/ml sCD40L (lanes 2 and 3). Supershifting was performed by subsequent incubation with a p50 antibody (lane 3). NF-κB complex is indicated by the arrow.
In addition to the intrinsic challenges of the ChIP protocol (reviewed in (24, 25)) our experiments had the added difficulty of establishing binding specificity to a single promoter within a class of four highly homologous gene promoters. As shown in Figure 3, these promoters all contain multiple NF-κB sites; four of them (sites 3-6) have been shown to be active in regulating transcription in transfection studies in both mouse and human B cells (15, 26, 27). It was critical for the questions that we were asking, i.e. whether binding at the Iγ1 locus occurred prior to, or after activation, to be able to attribute in vivo NF-κB/rel, CREB, and p300 binding to the Iγ1 and not the Iγ2-4 promoters. Therefore, once the DNA was isolated from chromatin we carried out PCR with primers designed to distinguish between Iγ1 and Iγ4 based on size and between Iγ1 and Iγ3 based on differential amplification. This protocol was adapted from the mutagenically separated PCR (MS-PCR) technique in which variable length allele-specific primers are used to identify single allelic point mutations (14, 28, 29). In addition to size differences in the Iγ1 and Iγ4 3’ primers, an additional base change was introduced to reduce potential cross-reactions between near identical sequences thereby increasing the specificity of the reaction. We generated a 5’ primer that recognized only the Iγ1, Iγ3, and Iγ4 promoter sequences (I-1/4) sequences and two variable length 3’ primers (I-1 and I-4) that amplified different sized Iγ sequences corresponding to Iγ1 or Iγ4 (Fig. 3B). This approach proved to be unfeasible for quantitation-based experiments since the efficiency of amplification of the two reactions was found to be markedly different (arrows, Fig. 3B). Additional primer pairs were tested and they too presented quantification problems due to the preferential amplification of individual products (data not shown).
Fig. 3. Nucleotide sequence of the four human Iγ proximal promoter regions. (A).

Alignment of the four Iγ subclass full length proximal promoters is shown, with nucleotide variances indicated in bold. NF-κB binding sites are denoted by shading. Initial primer sequences used for ChIP PCR amplification are indicated by arrows. Numbering is relative to the Iγ1 transcriptional start site defined by Sideras et al. (30). (B) Total chromatin from CD19+ B cells was amplified using the 5’-I-1/4 primer and either the 3’-1-1 primer (lane 1), the 3’I-4 primer (lane 2) or both the 3’ I-1 and the 3’I-4 primers. Products representing the Iγ1 and Iγ4 promoters were separated on a 1.5% TBE gel.
To address the issues presented with MS-PCR, additional primer sets were generated, for which each Iγ subclass was amplified in a separate reaction using the same 5’ primer (ChIP 5’), but distinct 3’ primers that hybridized to a region of each promoter that had a minimum of three base pair differences among the sequences (ChIP 3’ Iγ1 and ChIP 3’ Iγ4) (Fig. 4A). Again, additional non-templated base changes were added within the primer sequences to limit cross-reactivity and amplification of multiple subclasses (28). As shown in Figure 4B, The number of induced changes was determined by testing various 3’ primers, denoted Iγ1Δ3, Δ2, and Δ1 bp, in PCR reactions containing each Iγ subclass promoter region as a cloned DNA template. In order to eliminate amplification of all other subclasses, a 3’ primer, containing at least 2 additional bp changes, was required (upper panel). However, to eliminate potential primer-dimer products and reduce the GC-stretches, another 3’ primer was generated that contained 4 induced alterations. Using this Iγ1 promoter-specific primer the promoter sequences of the other three Iγ subclasses failed to amplify (Fig. 4C). The size of the fragment generated with this set of Iγ1-specific primers (~380bp) precluded us from using these primers effectively in qPCR. Thus, we had to expand our PCR analysis by carrying out dilution PCR to determine the amount of enrichment with specific antibodies under distinct conditions of stimulation.
Fig. 4. The Iγ1 proximal promoter is selectively amplified with PCR. (A).
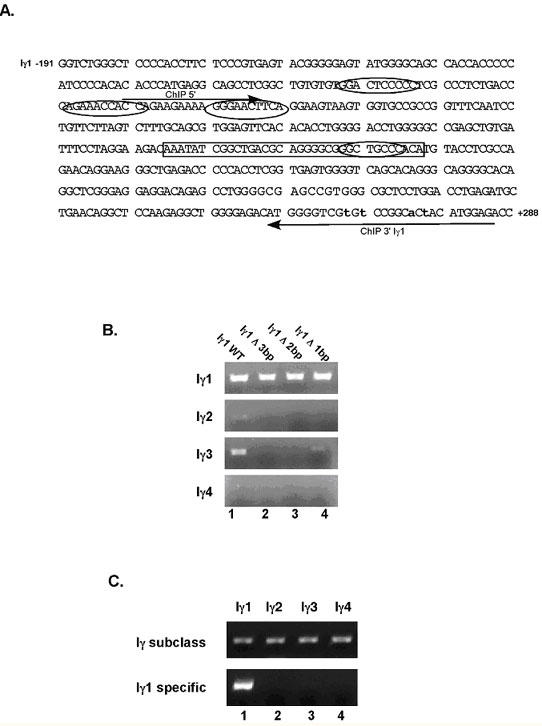
Shown is a schematic of the Iγ1 proximal promoter sequence from -191 to +288 (see Fig. 2), with arrows indicating the forward and reverse primers used to amplify ChIP products. Non-templated nucleotide substitutions, introduced into the 3’ primer sequence, are denoted in lower case. NF-κB sites 3 through 6 are indicated by ovals, whereas the 36 bp sequence is enclosed by a rectangle. (B) PCR amplification of cloned DNA containing the specific Iγ promoter sequences was performed using Iγ1-specific primers with exchange of 0 (lane 1), 3 (lane 2), 2 (lane 3) and 1 (lane 4) non-templated nucleotides in the Iγ1 sequence. (C) PCR amplification of cloned DNA sequences of Iγ1, Iγ2, Iγ3 and Iγ4 promoter sequences using the ChIP Iγ1-5’ and Iγ1-3’ primers (Fig. 3C used with permission, The Journal of Immunology 2005; 175:4499-4507).
The objective of our experiments, using the selective primers, was to analyze NF-κB, CREB and p300 binding specifically to the Iγ1 promoter in resting and CD40-activated primary human B cells. Because the strength of the Iγ1 promoter appears to be higher than the other Iγ subclass promoters in both primary B cells and a number of B cell lines, and the fact that we observed transcription factor binding in vitro, led us to question whether there was selective binding to the Iγ1 promoter in vivo. Using the ChIP assay with chromatin purified from resting and CD40-stimulated CD19+IgG- B cells we were able to see a distinct difference in binding of CREB and p300 in unstimulated versus stimulated B cells (Fig. 5).
Fig. 5. Co-activator binding is enhanced in CD40-activated B cells.
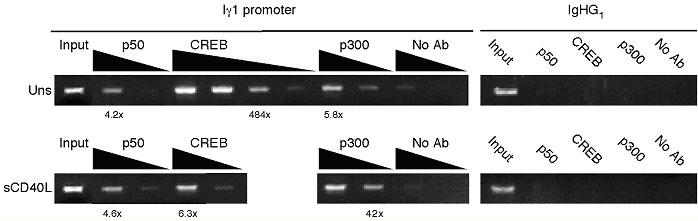
Chromatin was prepared from resting IgM+/IgG- peripheral B cells that was either unstimulated (upper panel) or stimulated for 2 h with 0.5 μg/ml sCD40L. Immunoprecipitation was carried out with antibodies specific for p50, CREB and p300. Eluted DNA was amplified with Iγ1-specific primers (left panels) or control IgHG1 primers (left panels). 10-fold serial dilutions of the chromatin (starting with a 1:10 dilution of the immunoprecipitated product) were used to demonstrate linearity of the PCR reaction. Products were quantified by determining the fold increase signal over the “no antibody” signal (used with permission, The Journal of Immunology 2005; 175:4499-4507).
Specifically, we observed that CREB binding was more pronounced in unstimulated B cells compared to stimulated B cells suggesting that CREB binds on this promoter prior to and after CD40 activation. With CD40 signaling there is a distinct loss of CREB signal but an enhancement of co-activator p300 binding indicating a recruitment of this factor to the Iγ1 promoter (compare upper and lower panels). As noted above, quantitation of the bands were carried out using dilution PCR to assess the enrichment of the Iγ1 band in the different samples.
In conclusion, using a modified ChIP protocol with primary human B cells we were able to show selective binding of transcription factors and co-activators at the Iγ promoter. Importantly, with this assay we also demonstrated an in vivo change in factor binding upon stimulation of naïve B cells upon activation through the CD40 signal transduction pathway.
Acknowledgments
We thank Dr. Nancy Rice (National Cancer institute, Bethesda, MD) for the NF-κB antibodies. Also, Dr. Celíne Gelínas (University of Medicine and Dentistry of New Jersey, Piscataway, NJ), Ralph Bernstein (CBER, FDA, Bethesday, MD) and Caroline Woo (Albert Einstein college of Medicine, Bronx, NY) are gratefully acknowledged for their input regarding technical issues related to ChIP. Finally, the help and insightful comments of Frank Sinquett and other members of the Covey lab were greatly appreciated during the entire course of this study. This work was supported by a National Institutes of Health Grant RO1AI3708 (to L. R. C.) and a National Institutes of Health Training Fellowship T32AI07403 (to R. L. D.). The authors have no conflicts of interest to declare related to this publication.
Appendix
Protocols
Transfer 5 × 107 cells and dilute to 100 ml volume with 37°C pre-warmed media, add 10 ml of 11% formaldehyde solution while swirling.
Incubate at 37°C 7-10 min, stop crosslinking by adding glycine to a final concentration of 0.125M, incubate for 5 minutes on ice.
Harvest cells at 1250 RPM for 5 minutes and wash twice with cold 1X PBS.
Pellet cells and resuspend in 25 ml of Lysis solution 1, incubate on ice 10 minutes, with periodic mixing.
Pellet cells at 2500 RPM and resuspend pellet in 25 ml lysis solution 2, to ensure complete resuspension, add 1 ml to pellet first and pipet up and down, add remaining 24 ml and place at 4°C on rocker.
Pellet and discard supernatant, resuspend pellet in 2 mls sonication buffer.
Add 200 mg of acid washed glass beads for sonication.
Place tube in ethanol/ice beaker to keep cold during sonication, position tube so that probe is near the bottom, but not touching. This will minimize frothing of samples.
Sonicate for 8-12 rounds, for 20 secs at 30% output, ice for 1 minute between each interval. Change ice frequently to ensure proper cooling of samples.
Spin chromatin at 14K RPM for 10 minutes to pellet debris.
Aliquot samples 200 μl and and continue with assay, or store at -80°C. Set aside 20 μl to use as input.
Thaw samples and dilute 1:10 with 1X RIPA buffer, add 50 μl of Protein A/ssDNA and pre-clear chromatin for 1 hour on a rotator at 4°C.
Spin at 1200RPM to remove beads and transfer supernatant to a fresh tube.
Add 2-4 μg of the desired antibody, rotate overnight at 4°C.
Add 60 μl Protein A/ssDNA slurry to each sample and rotate 1 hr.
Spin at 1200 RPM for 3 minutes to pellet beads.
Wash 5 times with IP1, with 5 minute rotation at RT.
Wash once with IP2, with a 5 minute rotation, and twice with TE.
To reverse crosslinke, bring bead pellet up to 400 with TE and add 8 μl RNAse A (10mg/ml) to final of 200 μg/ml, incubate 1 hour at 55°C. *Remove TC input samples from -80°C and reverse X-links at this time.
Add 20 μl of 10% SDS (FC 0.5%) and 8 μl Proteinase K (10mg/ml) and incubate 2-3 hours at 55°C, move to 65°C overnight.
Phenol chloroform samples once, chloroform/isoamyl once, and ethanol precipitate sample.
Resuspend final IP product in 12 TE, use 3μl per 50 μl PCR reaction.
Chromatin immunoprecipitation assay
Formaldehyde solution
11% formaldehyde
0.1M NaCl
1mM EDTA
0.5mM EGTA
50mM HEPES, pH 8.0
Lysis solution 1
10mM Tris-CL pH 8.0 10mM EDTA
10mM Na-Butyrate, 0.5mM EGTA
0.25% Triton X-100
1X PIC
1 mM PMSF
Lysis solution 2
0.2 M NaCl
10 mM Tris-Cl, pH 8.0
1 mM EDTA
0.5 mM EGTA
1X PIC
1 mM PMSF
Sonication buffer
10 mM Tris Cl, pH 8.0
1 mM EDTA
0.5 mM EGTA
2X RIPA
280 mM NaCl
2% Triton X-100
0.2% Deoxycholate
2 mM PMSF
IP Wash 1
0.1% SDS
1% Triton X-100
2 mM EDTA
20 mM Tris, pH 8.0
150 mM NaCl
IP Wash 2
0.1% SDS
1% Triton X-100
2 mM EDTA
20 mM Tris, pH 8.0
500mM NaCl
References
- Kuo MH, Allis CD. In vivo cross-linking and immunoprecipitation for studying dynamic Protein:DNA associations in a chromatin environment. Methods. 1999;19:425–433. doi: 10.1006/meth.1999.0879. [DOI] [PubMed] [Google Scholar]
- Wells J, Farnham PJ. Characterizing transcription factor binding sites using formaldehyde crosslinking and immunoprecipitation. Methods. 2002;26:48–56. doi: 10.1016/S1046-2023(02)00007-5. [DOI] [PubMed] [Google Scholar]
- Stavnezer J. Molecular processes that regulate class switching. Curr Top Microbiol Immunol. 2000;245:127–168. doi: 10.1007/978-3-642-59641-4_6. [DOI] [PubMed] [Google Scholar]
- Stavnezer J, Amemiya CT. Evolution of isotype switching. Semin Immunol. 2004;16:257–275. doi: 10.1016/j.smim.2004.08.005. [DOI] [PubMed] [Google Scholar]
- Gaff C, Gerondakis S. RNA splicing generates alternate forms of germline immunoglobulin alpha heavy chain transcripts. Int Immunol. 1990;2:1143–1148. doi: 10.1093/intimm/2.12.1143. [DOI] [PubMed] [Google Scholar]
- Gerondakis S. Structure and expression of murine germ-line immunoglobulin epsilon heavy chain transcripts induced by interleukin 4. Proc Natl Acad Sci USA. 1990;87:1581–1585. doi: 10.1073/pnas.87.4.1581. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lebman DA, Nomura DY, Coffman RL, Lee FD. Molecular characterization of germ-line immunoglobulin A transcripts produced during transforming growth factor type beta-induced isotype switching. Proc Natl Acad Sci USA. 1990;87:3962–3966. doi: 10.1073/pnas.87.10.3962. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lutzker S, Rothman P, Pollock R, Coffman R, Alt FW. Mitogen and IL-4 regulated expression of germline Ig γ2b transcripts: evidence for directed heavy chain class switching. Cell. 1988;53:177–184. doi: 10.1016/0092-8674(88)90379-0. [DOI] [PubMed] [Google Scholar]
- Radcliffe G, Lin YC, Julius M, Marcu KB, Stavnezer J. Structure of germ line immunoglobulin alpha heavy-chain RNA and its location on polysomes. Mol Cell Biol. 1990;10:382–386. doi: 10.1128/mcb.10.1.382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rothman P, Chen YY, Lutzker S, Li SC, Stewart V, Coffman R, Alt FW. Structure and expression of germline immunoglobuin heavy-chain epsilon transcripts: interleukin-4 plus lipopolysaccharide-directed switching to C epsilon. Mol Cell Biol. 1990;10:1672–1679. doi: 10.1128/mcb.10.4.1672. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rothman P, Lutzker S, Gorham B, Stewart V, Coffman R, Alt FW. Structure and expression of germline immunoglobulin γ3 heavy chain gene transcripts: implications for mitogen and lymphokine directed class switching. Int Immunol. 1990;2:621–627. doi: 10.1093/intimm/2.7.621. [DOI] [PubMed] [Google Scholar]
- Ford GS, Yin CH, Barnhart B, Sztam K, Covey LR. CD40 ligand exerts differential effects on the expression of Iγ transcripts in subclones of an IgM+ human B cell lymphoma line. J Immunol. 1998;160:595–605. [PubMed] [Google Scholar]
- Bhushan A, Covey LR. CREB/ATF proteins enhance the basal and CD154- and IL-4-induced transcriptional activity of the human Igamma1 proximal promoter. Eur J Immunol. 2001;31:653–664. doi: 10.1002/1521-4141(200102)31:2<653::AID-IMMU653>3.0.CO;2-D. [DOI] [PubMed] [Google Scholar]
- Dryer RL, Covey LR. A Novel NF-{kappa}B-Regulated Site within the Human I{gamma}1 Promoter Requires p300 for Optimal Transcriptional Activity. J Immunol. 2005;175:4499–4507. doi: 10.4049/jimmunol.175.7.4499. [DOI] [PubMed] [Google Scholar]
- Lin S-C, Stavnezer J. Activation of NF-κB/Rel by CD40 engagement induces the mouse germ line immunoglobulin Cγ1 promoter. Mol Cell Biol. 1996;16:4591–4603. doi: 10.1128/mcb.16.9.4591. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin S-C, Wortis HH, Stavnezer J. The ability of CD40L, but not lipopolysaccharide, to initiate immunoglobulin switching to immunoglobulin G1 is explained by differential induction of NF-κB/Rel Proteins. Mol Cell Biol. 1998;18:5523–5532. doi: 10.1128/mcb.18.9.5523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Snapper CM, Rosas FR, Zelazowski P, Moorman MA, Kehry MR, Bravo R, Weih F. B cells lacking relB are decfective in proliferative responses, but undergo normal B cell maturation to Ig secretion and Ig class switching. J Exp Med. 1996;184:1537–1541. doi: 10.1084/jem.184.4.1537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jumper MD, Nishioka U, Davis LS, Lipsky PE, Meek K. Regulation of human B cell function by recombinant CD40 ligand and other TNF-related ligands. J Immunol. 1995;155:2369–2378. [PubMed] [Google Scholar]
- Jumper MD, Splawski JB, Lipsky PE, Meek K. Ligation of CD40 induces sterile transcripts of multiple Ig H chain isotypes in human B cells. J Immunol. 1994;152:438–445. [PubMed] [Google Scholar]
- Fujieda S, Zhang K, Saxon A. IL-4 plus CD40 monoclonal antibody induces human B cells γ subclass-specific isotype switch: Switching to γ1, γ3, and γ4, but not γ2. J Immunol. 1995;155:2318–2328. [PubMed] [Google Scholar]
- Dignam J, Lebovitz RM, Roeder RD. Accurate transcription initiation by RNA polymerase II in a soluble extract from isolated mammalian nuclei. Nucl Acids Res. 1983;11:1475–1489. doi: 10.1093/nar/11.5.1475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Orlando V, Strutt H, Paro R. Analysis of chromatin structure by in vivo formaldehyde cross-linking. Methods. 1997;11:205–214. doi: 10.1006/meth.1996.0407. [DOI] [PubMed] [Google Scholar]
- Parekh BS, Maniatis T. Virus infection leads to localized hyperacetylation of histones H3 and H4 at the IFN-beta promoter. Mol Cell. 1999;3:125–129. doi: 10.1016/S1097-2765(00)80181-1. [DOI] [PubMed] [Google Scholar]
- Orlando V. Mapping chromosomal proteins in vivo by formaldehyde-crosslinked-chromatin immunoprecipitation. Trends Biochem Sci. 2000;25:99–104. doi: 10.1016/S0968-0004(99)01535-2. [DOI] [PubMed] [Google Scholar]
- Weinmann AS. Novel ChIP-based strategies to uncover transcription factor target genes in the immune system. Nat Rev Immunol. 2004;4:381–386. doi: 10.1038/nri1353. [DOI] [PubMed] [Google Scholar]
- Schaffer A, Cerutti A, Shah S, Zan H, Casali P. The evolutionarily conserved sequence upstream of the human Ig heavy chain S gamma 3 region is an inducible promoter: synergistic activation by CD40 ligand and IL-4 via cooperative NF-kappa B and STAT-6 binding sites. J Immunol. 1999;162:5327–5236. [PubMed] [Google Scholar]
- Warren WD, Roberts KL, Linehan LA, Berton MT. Regulation of the germline immunoglobulin Cgamma1 promoter by CD40 ligand and IL-4: dual role for tandem NF-kappaB binding sites. Mol Immunol. 1999;36:31–44. doi: 10.1016/S0161-5890(98)00114-X. [DOI] [PubMed] [Google Scholar]
- Rust S, Funke H, Assmann G. Mutagenically separated PCR (MS-PCR): a highly specific one step procedure for easy mutation detection. Nucl Acids Res. 1993;21:3623–3629. doi: 10.1093/nar/21.16.3623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schluter B, Erren M, Schotte H, Junker R, Rust S, Assmann G. The mutagenically separated polymerase chain reaction is a rapid and reliable method for genotyping of the tumour necrosis factor-alpha promoter polymorphism (-308 G/A). Clin Chim Acta. 2002;320:135–138. doi: 10.1016/s0009-8981(02)00054-2. [DOI] [PubMed] [Google Scholar]
- Sideras P, Mizuta TR, Kanamori H, Suzuki N, Okamoto M, Kuze K, Ohno H, Doi S, Fukuhara S, Hassan MS, et al . Production of sterile transcripts of C gamma genes in an IgM-producing human neoplastic B cell line that switches to IgG-producing cells. Int Immunol. 1989;1:631–642. doi: 10.1093/intimm/1.6.631. [DOI] [PubMed] [Google Scholar]