

Abstract
Protein-tyrosine phosphatases (PTPases) have a catalytic cysteine residue whose reduced state is integral to the reaction mechanism. Since exposure to air can artifactually oxidize this highly reactive thiol, PTPase assays have typically used potent reducing agents to reactivate the enzymes present; however, this approach does not allow for the measurement of the endogenous PTPase activity directly isolated from the in vivo cellular environment. Here we provide a method for using an anaerobic chamber to preserve the activity of the total PTPase complement in a tissue lysate or of an immunoprecipitated PTPase homolog to characterize their endogenous activation state. Comparison with a sample treated with biochemical reducing agents allows the determination of the activatable (reducible) fraction of the endogenous PTPase pool.
Keywords: Signal Transduction, Cysteine, protein-tyrosine-phosphatase, oxidation-reduction
Introduction
Dozens of enzyme homologs in the protein-tyrosine phosphatase (PTPase) superfamily have been cloned and characterized over the past decade (1). The unifying structural feature among PTPases, is a conserved ~230 amino acid domain that contains the characteristic sequence motif, (I/V)H C XAGXGR(S/T)G, with the cysteine residue that catalyzes protein phosphotyrosine hydrolysis by the formation of a cysteinyl-phosphate intermediate (2). The catalytic cysteine residue must be present in an unconjugated, unoxidized state to carry out phosphotyrosine hydrolysis. Thus, cellular and environmental reactions that lead to the oxidation or modification of this site will effectively block the enzyme's catalytic activity and reduce the specific activity of the cellular PTPase pool (2,3). Since alterations in the oxidation state of the catalytic cysteine within the cellular environment has profound effects on the PTPase specific activity, attention has recently been focused on how the redox state of the catalytic thiol is regulated in vivo. Crystallographic studies have shown that this thiol moiety is in spatial proximity to amino acid side chains adjacent to the enzyme active site that strongly affect its ionization (4). These interactions enhance the reactivity of the cysteine thiol group and effectively lowers its pKa to more than 3 units below that found in a typical free cysteine thiol or protein cysteine side chains. Thus, at physiological pH, the catalytic cysteine is ionized as a thiolate anion which is easily derivatized or oxidized in preference to other neighboring sulfhydryls (3,5).
One method to directly assess the reversible oxidation of thiol-dependent cellular enzymes, including PTPases, is to work under a neutral atmosphere. We have recently demonstrated that from a variety of tissue sources, PTPases are sensitive to oxidation by exposure to air when working with cell lysates on the benchtop (6). Frequently, reduction of the PTPases present in a cell lysate by biochemical treatment with strong reducing agents is incomplete, preventing an accurate assessment of both the endogenous level of PTPase activity, as well as the fraction of the PTPase pool that is reversibly oxidized or conjugated. Working within an anaerobic chamber allows for the measurement of endogenous PTPase levels, and including a biochemical reductant in the assay buffer allows for the measurement of the reducible pool of PTPase in the sample. This technique is useful for assay of cellular lysates or immunoprecipitated enzymes where specific antibodies are available.
Materials and Methods
Materials
Cell culture media and sera were obtained from Cellgro (Herndon, VA). Para-nitrophenylphosphate (pNPP) and reduced, carboxymethylated-maleylated (RCM)-lysozyme were obtained from Sigma. γ-[32P]-ATP was from Amersham Pharmacia Biotech (Piscataway, NJ). Monoclonal PTP1B antibody (Ab-2) was from Calbiochem/Oncogene Sciences (La Jolla, CA). Trisacryl protein G was obtained from Pierce (Rockford, IL). The BBL GasPak Disposable Anaerobic Indicator, #4370504 was purchased from Becton Dickinson, (Sparks, MD). All other reagents were obtained from Sigma Chemical (St. Louis, MO) or Fisher Scientific (Pittsburgh, PA).
Cell Culture
3T3-L1 pre-adipocytes were maintained in Dulbecco's modified Eagle's medium (DMEM) / Ham's F12 medium (1:1) supplemented with 10% (v/v) fetal calf serum. Adipocyte differentiation was induced at 2 days post confluence with medium supplemented with 100 nM insulin, 0.25 μM dexamethasone, and 0.5 mM isobutylmethylxanthine (IBMX). After 2 days, dexamethasone and IBMX were removed from the medium and the cells were allowed to differentiate further for an additional 6 days. Prior to use, the cells were serum-starved in medium containing 0.1% (w/v) bovine serum albumin (BSA) for 16 hours. Human HepG2 cells were maintained in Eagle's Minimal Essential Medium (EMEM) supplemented with 10% (v/v) fetal calf serum and 1X each of non-essential amino acids and L-glutamine (Cellgro). Cells were used at 80% confluence, after 16 hours of serum starvation in medium containing 0.5% (w/v) BSA.
Cell and Tissue Fractionation
Cells are lysed by homogenization in ice-cold homogenization buffer (150 mM NaCl, 5 mM EDTA, 5 mM EGTA, in 50 mM Hepes, pH 7.5, containing a protease inhibitor cocktail (Sigma), with brief sonication. Prior to use, all buffer solutions are deoxygenated within the chamber by stirring overnight.
Anaerobic Chamber and Experimental Conditions
An enclosed anaerobic work station (Forma Scientific, model # 901024) was used to provide an oxygen-free environment for cell and tissue homogenization and anaerobic PTPase assay. The use of this chamber is detailed in the Protocol section. This chamber uses palladium catalyst wafers and desiccant wafers to maintain strict anaerobiosis to less than 10 ppm O2 (according to the specifications provided by the manufacturer). High purity N2 is used for purging the chamber initially and the working anaerobic gas mixture was N2:H2:CO2 proportioned at 85:10:5. Monitoring the O2 concentration to identify possible air leaks while working in the chamber is performed using a BBL GasPak Disposable Anaerobic Indicator which changes color when the O2 concentration reaches 0.5% (~ 5000 ppm). For anaerobic conditions, the tissue samples and cultured cells were introduced into the chamber in a frozen state and disrupted as described above into deoxygenated homogenization buffers.
Following appropriate cell treatment with various hormones, cytokines or other reagents, the dish is snap-frozen by pouring on a layer of liquid nitrogen. Once the nitrogen has evaporated, the dishes are introduced into the chamber in a frozen state according to the detailed protocol discussed below.
PTPase Enzyme Activity Using pNPP as Substrate
Aliquots containing an appropriate amount of cell fraction protein are incubated in a final volume of 100 μl at 30°C for cell lysates or 37°C for immunoprecipitated enzymes, for 10-120 min in reaction buffer containing 10 mM pNPP and 2 mM EDTA in 20 mM 2-(N-morpholino)ethanesulfonic acid (MES) at pH 6.0, with and without 2 mM DTT. The reaction was stopped by the addition of 900 μl of 1 M NaOH and the absorption was determined at 410 nm (7). The initial rate of pNPP hydrolysis was estimated from the linear portion of the earliest time points of the enzymatic reaction.
PTPase Enzyme Activity Using [32P]-RCM-Lysozyme as Substrate
Alternatively, RCM-lysozyme can be used as a more specific PTPase substrate. Recombinant human insulin receptors from transfected CHO cells (8) or commercially available were partially purified on wheat germ lectin-agarose (Vector Laboratories, Burlingame, CA) as described (9). RCM-lysozyme was radioactively labeled on tyrosine by phosphorylating with the insulin receptor preparation and γ-[32P]-ATP (10). The reaction was initiated with the addition of 0.5 mg of RCM-lysozyme and incubated at 25°C for 16 hrs. The reaction was terminated with the addition of trichloroacetic acid (TCA) to a final concentration of 20% (w/v) and centrifuged at 30,000 x g for 15 min at 4°C. The pellet was washed 3 times with 20% TCA and dialyzed against 50 mM imidazole-HCl, pH 7.2.
PTPase activity was assayed using the indicated amount of cell fraction protein at 30°C for cell lysates or 37°C for immunoprecipitated enzymes, for 10-120 min in reaction buffer containing 50 mM HEPES, pH 7.0 and 2 mM EDTA, with and without 2 mM DTT. The reaction was initiated by the addition of 20 μl of [32P]-phosphotyrosyl RCM-lysozyme (~20,000 dpm) and terminated by the addition of 0.9 ml of acidic charcoal mixture, consisting of 0.9 M NaCl, 90 mM sodium pyrophosphate, 2 mM NaH2PO4 and 4% (w/v) Norit A activated charcoal (11). After centrifugation in a microfuge, the amount of radioactivity in 0.4 ml of supernatant was measured by Cerenkov counting in a liquid scintillation counter. The initial rate of RCM-lysozyme hydrolysis was estimated from the linear portion of the earliest time points of the enzymatic reaction where less than 20% of the RCM-lysozyme was hydrolyzed during the 30 minute reaction period.
Results
Assay of overall PTPase activity in cell or tissue lysates. We have evaluated this method using differentiated 3T3-L1 adipocytes. Cells were snap frozen with liquid nitrogen and transferred into the chamber for assay. Cells were lysed within the chamber environment into deoxygenated buffers and lysates were withdrawn from the chamber using tightly-capped sealed tubes for centrifugation. The enzyme assay was performed within the chamber using pNPP as substrate.
Cell lysis on the benchtop under aerobic conditions resulted in a 27% reduction in PTPase activity (P<0.001) in the crude homogenates measured in the absence of added reducing agents (Fig. 1). When PTPase activity was measured in the presence of DTT, we detected no significant change in the activity as measured in the chamber. However, the activity that had been inhibited by air exposure was restored by treatment with DTT to the level found when samples were maintained in the anaerobic chamber (Fig. 1).
Fig. 1.
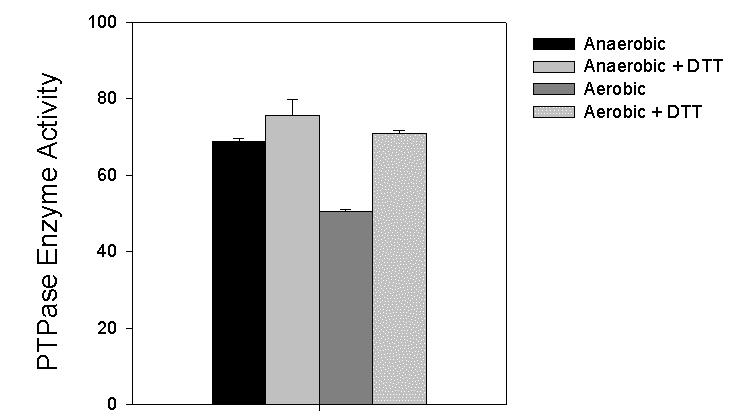
Effect of air exposure on PTPase activity in lysates of 3T3-L1 adipocytes and reversibility with DTT. 3T3-L1 adipocytes were differentiated as described in Methods and individual 10-cm dishes were snap-frozen with liquid nitrogen. Replicate dishes were then introduced into the anaerobic chamber in the frozen state and disrupted into deoxygenated buffers or homogenized into the same buffer on the bench top in air. Samples containing 30 μg protein were then subjected to the pNPP assay in the presence or absence of 2 mM DTT as described. PTPase activity (μmol min-1 mg-1) is calculated from the A410 of the samples following a 10 min incubation using a molar extinction coefficient of 1.78 x 104 M-1 cm-1(7). Modified from reference 6 and reproduced with permission.
We also evaluated whether the catalytic activity of PTP1B, a specific cellular PTPase enzyme immunoprecipitated from HepG2 cells was altered by exposure to air (Fig. 2). The measurement of PTP1B-specific activity was facilitated by using a monoclonal antibody to PTP1B that can immunoadsorb the enzyme in a catalytically active state. To maintain the endogenous reduced state of the enzyme thiol, all stages of the immunoprecipitation process, including centrifugation, washing of the pellets, and the assay itself were performed under strict nonoxidizing conditions. The activity of PTP1B was reduced dramatically by air exposure to 36% of the level observed in the samples maintained under anaerobic conditions throughout the experiment within the chamber environment (P< 0.001).
Fig. 2.
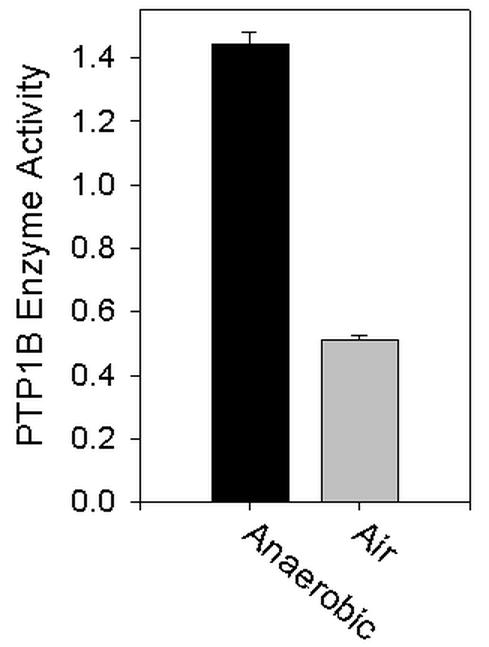
Effect of air exposure and in vitro oxidation and reduction reactions on the catalytic activity of PTP1B. Following a preclearing step, PTP1B was immunoprecipitated from lysates of snap-frozen HepG2 cells either within the anaerobic chamber environment or on the bench top in air, as described in Materials and Methods. The immunoprecipitated samples were washed, and PTPase activity was assayed by using pNPP as substrate. A comparison between PTP1B activity under aerobic vs. anaerobic conditions is shown. PTPase activity is presented as μmol min-1 mg-1 immunoprecipitated cell lysate. Modified from reference 6 and reproduced with permission.
Discussion
PTPases are highly active enzymes with high turnover rates, and their activity in the cell must be attenuated for signal transduction mechanisms involving protein tyrosine phosphorylation to proceed in a balanced manner (12). Recently, several laboratories have provided evidence for novel regulatory mechanisms involving reactive oxygen species, especially H2O2, which can oxidize and inactivate PTPase enzymes in a stepwise fashion (13-15). The regulation of PTPase catalytic activity by reactions involving the catalytic cysteine is emerging as a key mode for suppressed of these enzymes in signal transduction by hormones and cytokines in the cell (16,17).
This methodology can be used to assay the total PTPase activity in lysates of various cells and tissues from different sources as well as following treatments with hormones or cytokines. Cells are stabilized by snap-freezing with liquid nitrogen and transferred into the chamber for assay. Handling of the samples within the chamber can maintain the state of enzyme reduction and activation from the time of isolation. Samples can also be withdrawn from and reintroduced into the chamber keeping the samples within the confines of tightly-capped O-ring sealed tubes for various experimental manipulations, such as centrifugation or incubations with reagents that require refrigerated temperatures.
Sample assay in the absence or presence of added dithiothreitol provides a means of directly evaluating the proportion of endogenous enzyme activity that can be reactivated by biochemical reduction. The reactivated activity fraction represents enzyme forms that are partially oxidized by intracellular reactions with reactive oxygen species as well as artifactually by air exposure (Fig. 3).
Fig. 3. Some oxidative structural modifications of the PTPase catalytic site that occur in vivo.
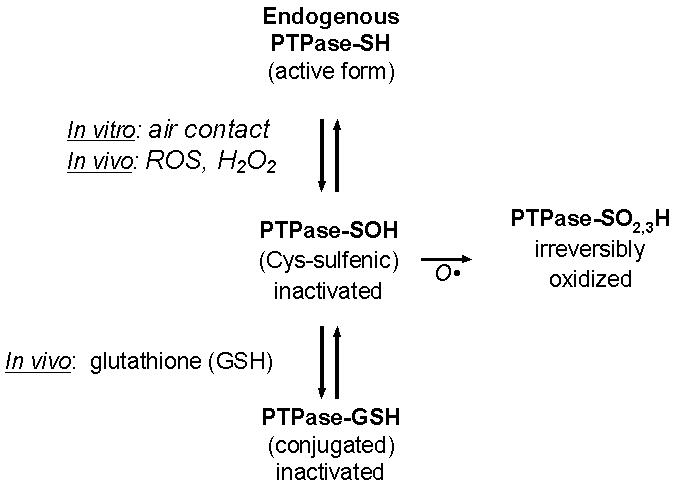
The catalytic thiol of PTPases is susceptible to a variety of inactivating modifications. These include oxidation in a stepwise fashion to progressively more inert forms (13-15). The catalytic cysteine thiol is first oxidized to the sulfenic (-SOH) form, which is amenable to reduction by cellular enzymatic mechanisms or with reducing agents in vitro, followed by further sequential steps of oxidation, to sulfinic (-SO2H) and sulfonic (-SO3H) forms, leading to irreversible PTPase inactivation. Also, the reduced, ionized cysteine thiol, or a partially oxidized form is susceptible to conjugation with glutathione, which provides an inactive, but potentially reactivatable form of the enzyme (22).
Our laboratory has been interested in studying PTPases, in particular characterizing their role in the regulation of the insulin action pathway (18). The intracellular PTPase PTP1B, has emerged as a key regulator of insulin signaling and is a target of inhibition for the potential treatment of type 2 diabetes mellitus (19). PTP1B is the target of an oxidant signal generated by the action of EGF and other growth factors (17). Insulin also generates a cellular oxidant burst that appears to be coupled to the oxidative inhibition of PTP1B in insulin-sensitive cell types (20,21).
In summary, we describe the use of a commercially available anaerobic chamber system to evaluate the endogenous level of PTPase activity from various cell and tissue sources (6). Since cellular PTPases, as well as other enzyme systems that may be susceptible to oxidation-reduction reactions, are susceptible to oxidative inhibition by air exposure, the use of the anaerobic chamber methodology provides an important advantage for enzyme characterization. Evaluation of the changes in the activation state of cellular PTPases from endogenous sources has recently provided additional evidence for modulation of the activation state of this important class of regulatory enzymes within the cellular milieu in response to changes in the abundance of cellular reactive oxygen species and other thiol side-chain reactions, including glutathione conjugation. This approach is widely adaptable to the characterization of regulation of thiol reactivity in determining the catalytic activity of cellular PTPases and how this process impacts on signal transduction by reversible protein-tyrosine phosphorylation.
Acknowledgments
We appreciate helpful discussions during the initiation of this work with Dr. Sue Goo Rhee, Chief, Laboratory of Cell Signaling, NHLBI, NIH. This work was supported by grants DK43396 and DK53388 from the NIH and a Mentor-based postdoctoral fellowship in support of Dr. Li Zhu to Dr. Goldstein.
Appendix
Protocols
Annotated from Model 1O25/1O29 Anaerobic System Installation and Operation Manual, Manual No. 7011O25, Revision 2, Forma Scientific, Inc., PO Box 649, Marietta, OH 45750.
Anaerobic manipulation procedures
General information
The anaerobic work station (model #901O24) was purchased from Forma Scientific, Marietta, OH.
High-purity N2 for purging and the working anaerobic gas mixture (N2:H2:CO2=85:10:5) from a reliable commercial source.
The anaerobic environment inside the chamber should be monitored with O2 indicator strips (BBL strips, Becton Dickinson and company) prior to and during experiments. The strips will react if the ambient O2 concentration is more than 0.5% in the chamber.
Buffer solutions and equipment introduced into the chamber need to be deoxygenized overnight prior to use in experiments.
Installation of charcoal, catalyst and desiccant activated wafers, and the chamber relief bubbler
Open the air filter system door in the top center of the right side wall.
Slide the wafers into the correct slots, charcoal on top, palladium catalyst wafer in the middle and desiccant wafer at the bottom.
The palladium catalyst wafers and desiccant wafers need to be regenerated by heating 160°C for two hours weekly, and can be reused for one year. The charcoal activated filter needs to be replaced every 6 months.
To prepare the chamber relief bubbler, fill the glass bottle shipped with the unit, with mineral oil to a level of approximately 1-1/2 inches. Clip the bottle into the chamber relief with the tubing extending approximately 1/8 inches in the mineral oil.
Change the mineral oil once a month and clean the bottle. Check the bottle daily to ensure that the tube extends into the mineral oil.
Preparation of anaerobic environment for the first use, operation of the interchange device both automatically and manually, are performed exactly as described in the manual.
Initial settings:
Gas system switch → on.
Catalyst fan switch → on.
Light switch → on.
Vacuum, nitrogen, and equalize switch → auto.
Manual fill knob → off.
Deoxygenation of solutions:
Open the outer door with the inner door closed.
Put the solution in a baker with stir bar and other related material into the exchanger.
Close the outer door.
Press the start cycle button to run one or more degas cycles automatically or run manually.
Open the inner door and transfer the items into the glove chamber.
Close the inner door.
Put the baker on a stir plate overnight to equalize the solution.
Check for anaerobiosis with a BBL strip.
If the solution is anaerobic, aliquot in bottles with sealable cap, and transfer out of chamber.
Store the anaerobic solution in 4°C.
Measurement of activities of PTP1B and PTPase of cultured cells and human adipose tissue under anaerobic environment
Cell Culture and cell differentiation
3T3-L1 cell culture and differentiation:
3T3-L1 pre-adipocytes are maintained in Dulbecco's modified Eagle's medium (DMEM) / Ham's F12 medium (1:1) supplemented with 10% (v/v) fetal calf serum. Never let the cells grow to confluence for passing or seeding.
Cells are seeded in 10cm culture dishes and the medium is changed every other day.
When cells become confluent, change medium and let cells grow two more days.
Cell differentiation is induced at 2 days post confluence with medium supplemented with 100 nM insulin, 0.25 μM dexamethasone, and 0.5 mM isobutylmethylxanthine (IBMX).
After 2 days, change medium with 100 nM insulin every other day for an additional 6 days.
Prior to use, the cells are serum-starved in medium containing 0.1% (w/v) bovine serum albumin (BSA) for 16 hours.
Human HepG2 cell culture:
Human HepG2 cells are maintained in Eagle's Minimal Essential Medium (EMEM) supplemented with 10% (v/v) fetal calf serum and 1x each of non-essential amino acids and L-glutamine (Cellgro).
Cells are used at 80% confluence, after 16 hours of serum starvation in medium containing 0.5% (w/v) BSA.
Cell and Tissue Fractionation
Solutions:
-
Homogenization buffer (deoxygenized)
- 150 mM NaCl
- 5 mM EDTA
- 5 mM EGTA
- 50 mM Hepes, pH 7.5
- protease inhibitor cocktail (Sigma)
-
Solubilization buffer (deoxygenized)
- Homogenization buffer +1% (v/v) Triton X-100
Preparation of lysates:
For cultured cells
Pour liquid N2 in each of 10 cm culture dishes to pulverize cells after serum-starvation.
Transfer cells into the interchange and run an automatic transfer cycle.
Move dishes into the anaerobic chamber and add 1 ml of ice-cold homogenization buffer for each 10 cm culture dish after liquid N2 is evaporated.
Harvest cells with cell lifter and transfer cells in 1.5 ml Eppendorf tube.
Cells were sonicated briefly inside the anaerobic chamber.
Fractionation procedures
The cell and tissue lysates were transferred into centrifuge tubes with air-tight lids and centrifuged at 100,000 x g for 45 min at 4°C.
Transfer tubes into the chamber and save the supernatants as the cytosol fractions.
The pellets were incubated in solubilization buffer on ice for 45 min.
Centrifuge at 15,000 x g for 20 min at 4°C.
Transfer tubes into the chamber and save the supernatants as the soluble particulate fractions.
Protein was measured using the method of Bradford.
Immunoprecipitation of PTP1B
Working within the chamber, take 500 μg protein of whole cell lysates or fractionations and normalize into 1μg/μl in 0.5 ml in 2 ml tubes with air-tight O-ring sealed lids (Fisher Scientific, catalog #05-669-4).
Add 2 μl of non-immune IgG and 20 μl of trisacryl protein G (50% beads) for pre-clearing and incubate at 4°C with rolling for 1 hour.
Briefly spin down the beads and transfer the samples back into the chamber.
Transfer the supernatants into a set of fresh tubes and add 2 μg of anti-PTP1B antibody (Ab-2, Oncogene Sciences), a monoclonal antibody directed at a C-terminal epitope of the enzyme that preserves its enzymatic activity.
Incubate at 4°C with rolling overnight.
Transfer samples into the chamber and add 50 μl of trisacryl protein G.
Continue to incubate at 4°C with rolling for two hours.
Centrifuge briefly and remove supernatants in the chamber.
Wash beads with ice-cold PBS three times and the sample are ready for PTP-1B activity assay by pNPP method.
PTPase Enzyme Activity Using pNPP as Substrate
Solution:
Reaction buffer (deoxygenized):
10 mM of para-nitrophenylphosphate (pNPP, sigma)
2 mM EDTA
20 mM 2-(N-Morpholino) ethanesulfonic acid (MES) at pH 6.0
1-2 mM DTT
No DTT for non-reducing reaction buffer
Stopping buffer:
0.2 M NaOH
Procedures:
Add reaction buffer to sample for total volume of 100 μl and mix.
For cell lysates, incubate at 30°C incubator or water bath for 10 min to 2 hours inside the chamber. For immunoprecipitated enzymes, incubate at 37°C incubator or water bath for 10 min to 2 hours inside the chamber
Terminate the reaction by adding 900 μl of stopping buffer.
The absorption was determined at 410 nm
The initial rate of pNPP hydrolysis was estimated from the linear portion of the earliest time points of the enzymatic reaction.
Enzyme activity is calculated from the molar extinction coefficient for phenyl phosphate = 1.78 x 104 M-1 cm-1 (Pot et al. J. Biol. Chem. 1991;266:19688-19696).
PTPase Enzyme Activity Using [32P]-RCM-Lysozyme as Substrate:
Reagents:
Reduced carboxamidomethylated and maleylated (RCM)-lysozyme
partial purified recombinant human insulin receptor
[γ-32P]ATP (~200 cpm/pmol)
Solutions:
- RCM-lysozyme phosphorylation buffer:
- 40 mM imidazole hydrochloride, pH 7.6
- 50 mM NaCl
- 12 mM magnesium acetate
- 4 mM MnCl2
- 100 uM ammonium molybdate
- 0.1 mM Na Vanadate
- 30 mM N-acetylglucosamine
- 0.2% deoxycholate
- 0.2 mM EGTA
- 0.05% Triton X-100
- 3% glycerol
- 2 mM dithiothreitol (DTT)
- 300 nM insulin
- Phosphatase assay buffer (deoxygenized):
- 50 mM HEPES, pH 7.0
- 2 mM EDTA,
- 25 mM imidazole hydrochloride, pH 7.2
- 1 mg/ml fatty acid- and globulin-free BSA
- Buffer is prepared with and without added 1-2 mM DTT
- Acidic charcoal mixture:
- 0.9 M NaCl
- 90 mM sodium pyrophosphate
- 2 mM NaH2PO4
- 4% (v/v) Norit A activated charcoal
Procedures:
- RCM-lysozyme phosphorylation (preparation of substrate):
- Add 0.5 ml of RCM-lysozyme phosphorylation buffer to an 1.5 ml tube
- Add 400 μg partial purified recombinant human insulin receptor
- Add 4 mM [γ-32P]ATP (~200 cpm/pmol)
- Add 1 mg RCM-lysozyme to initiate the reaction
- Incubate at 30°C overnight
- Add 100% Trichloroacetic acid (TCA) to a final concentration of 10% to terminate reaction
- Incubate on ice for 30 min
- Centrifuge at 27,000 x g for 15 min at 4°C
- Discard the supernatant
- Wash the protein pellet three times further with 20% TCA to remove [γ-32P]ATP
- Resuspend the pellet in 1 ml of 2 M Tris base, using a glass rod.
- Leave the resuspended solution on ice overnight
- Dialyze the sample against 50 mM imidazole hydrochloride, pH 7.2
- Count 10 μl of sample by Cerenkov counting in a liquid scintillation counter.
-
Assay of PTPases:
The assay is based on the release of [32P]Pi from phosphorylated protein substrate (labeled RCM-lysozyme) and is performed in the anaerobic chamber. The initial rate of RCM-lysozyme hydrolysis was estimated from the linear portion of the earliest time points of the enzymatic reaction where less than 20% of the RCM-lysozyme was hydrolyzed. For blank control PTPases sample is replaced by phosphatase assay buffer and the counts are less than 2% of total radioactivity in the assay.
- Transfer samples, phosphatase assay buffer, substrate in a radioisotope-protective rack into the anaerobic chamber
- Add 0.02> ml samples containing 30 μg proteins to 1.5 ml centrifuge tubes
- Add 0.02 ml of phosphatase assay buffer
- Preincubate at 30°C for 5 min
- Add the substrate (>10,000 dpm)
- For cell lysates, incubate at 30°C for 10 min to 2 hours inside the chamber. For immunoprecipitated enzymes, incubate at 37°C for 10 min to 2 hours.
- Add 0.9 ml acidic charcoal mixture to terminate reactions
- Transfer the reaction out of the chamber
- Freeze briefly to facilitate the precipitation of protein
- Thaw and centrifuge at 12,500 g for 5 min
- The amount of radioactivity in 0.4 ml of supernatant was measured by Cerenkov counting in 5 ml liquid scintillation counter.
References
- Tonks NK, Neel BG. Combinatorial control of the specificity of protein tyrosine phosphatases. Curr Opin Cell Biol. 2001;13:182–195. doi: 10.1016/s0955-0674(00)00196-4. [DOI] [PubMed] [Google Scholar]
- Zhang ZY. Protein-tyrosine phosphatases - biological function, structural characteristics, and mechanism of catalysis. Crit Revs Bioch Mol Biol. 1998;33:1–52. doi: 10.1080/10409239891204161. [DOI] [PubMed] [Google Scholar]
- Den JM, Dixon JE. Protein tyrosine phosphatases - mechanisms of catalysis and regulation. Curr Opin Chem Biol. 1998;2:633–641. doi: 10.1016/s1367-5931(98)80095-1. [DOI] [PubMed] [Google Scholar]
- Barford D, Das AK, Egloff MP. The structure and mechanism of protein phosphatases - insights into catalysis and regulation. Ann Rev Biophys Biomol Struct. 1998;27:133–164. doi: 10.1146/annurev.biophys.27.1.133. [DOI] [PubMed] [Google Scholar]
- Lohse DL, Denu JM, Santoro N, Dixon JE. Roles of aspartic acid 181 and serine-222 in intermediate formation and hydrolysis of the mammalian protein-tyrosine-phosphatase ptp1. Biochemistry. 1997;36:4568–4575. doi: 10.1021/bi963094r. [DOI] [PubMed] [Google Scholar]
- Zhu L, Zilbering A, Wu X, Mahadev K, Joseph JI, Jabbour S, Deeb W, Goldstein BJ. Use of an anaerobic environment to preserve the endogenous activity of protein-tyrosine phosphatases isolated from intact cells. Published online as The FASEB Journal express article 10 1096/fj 00-0795fje FASEB J. 2001;15:1637–1639. doi: 10.1096/fj.00-0795fje. [DOI] [PubMed] [Google Scholar]
- Pot DA, Woodford TA, Remboutsika E, Haun RS, Dixon JE. Cloning, bacterial expression, purification, and characterization of the cytoplasmic domain of rat LAR, a receptor-like protein tyrosine phosphatase. J Biol Chem. 1991;266:19688–19696. [PubMed] [Google Scholar]
- Hashimoto N, Feener EP, Zhang WR, Goldstein BJ. Insulin receptor protein-tyrosine phosphatases - Leukocyte common antigen-related phosphatase rapidly deactivates the insulin receptor kinase by preferential dephosphorylation of the receptor regulatory domain. J Biol Chem. 1992;267:13811–13814. [PubMed] [Google Scholar]
- Pike LJ, Kuenzel EA, Casnellie JE, Krebs EG. A comparison of the insulin- and epidermal growth factor-stimulated protein kinases from human placenta. J Biol Chem. 1984;259:9913–9921. [PubMed] [Google Scholar]
- Tonks NK, Diltz CD, Fischer EH. Purification and assay of CD45: An integral membrane protein-tyrosine phosphatase. Meth Enzymol. 1991;201:442–451. doi: 10.1016/0076-6879(91)01040-9. [DOI] [PubMed] [Google Scholar]
- Streuli M, Krueger NX, Thai T, Tang M, Saito H. Distinct functional roles of the two intracellular phosphatase like domains of the receptor-linked protein tyrosine phosphatases LCA and LAR. EMBO J. 1990;9:2399–2407. doi: 10.1002/j.1460-2075.1990.tb07415.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hunter T. Protein kinases and phosphatases: the yin and yang of protein phosphorylation and signaling. Cell. 1995;80:225–236. doi: 10.1016/0092-8674(95)90405-0. [DOI] [PubMed] [Google Scholar]
- Barrett WC, DeGnore JP, Keng YF, Zhang ZY, Yim MB, Chock PB. Roles of superoxide radical anion in signal transduction mediated by reversible regulation of protein-tyrosine phosphatase 1B. J Biol Chem. 1999;274:34543–34546. doi: 10.1074/jbc.274.49.34543. [DOI] [PubMed] [Google Scholar]
- Claiborne A, Yeh JI., Mallett TC, Luba J, Crane EJ, Charrier V, Parsonage D. Protein-sulfenic acids: Diverse roles for an unlikely player in enzyme catalysis and redox regulation. Biochemistry. 1999;38:15407–15416. doi: 10.1021/bi992025k. [DOI] [PubMed] [Google Scholar]
- Barrett WC, DeGnore JP, Konig S, Fales HM, Keng YF, Zhang ZY, Yim MB, Chock PB. Regulation of PTP1B via glutathionylation of the active site cysteine 215. Biochemistry. 1999;38:6699–6705. doi: 10.1021/bi990240v. [DOI] [PubMed] [Google Scholar]
- Denu JM, Tanner KG. Specific and reversible inactivation of protein tyrosine phosphatases by hydrogen peroxide - evidence for a sulfenic acid intermediate and implications for redox regulation. Biochemistry. 1998;37:5633–5642. doi: 10.1021/bi973035t. [DOI] [PubMed] [Google Scholar]
- Lee SR, Kwon KS, Kim SR, Rhee SG. Reversible inactivation of protein-tyrosine phosphatase 1B in A431 cells stimulated with epidermal growth factor. J Biol Chem. 1998;273:15366–15372. doi: 10.1074/jbc.273.25.15366. [DOI] [PubMed] [Google Scholar]
- Goldstein BJ. Protein-tyrosine phosphatases and the regulation of insulin action. In: LeRoith D, Olefsky JM, Taylor SI, editors. Diabetes mellitus: A fundamental and clinical text. 2nd edition., Philadelphia: Lippincott Press; 2000. p. 206-217.
- Goldstein BJ. Protein-Tyrosine Phosphatase 1B (PTP1B): A Novel Therapeutic Target for Type 2 Diabetes Mellitus, Obesity and Related States of Insulin Resistance. Curr Drug Targets - Immune, Endocrine Metab Dis. 2001;1:265–275. doi: 10.2174/1568008013341163. [DOI] [PubMed] [Google Scholar]
- Mahadev K, Zilbering A, Zhu L, Goldstein BJ. Insulin-stimulated hydrogen peroxide reversibly inhibits protein-tyrosine phosphatase 1B in vivo and enhances the early insulin action cascade. J Biol Chem. 2001;276:21938–21942. doi: 10.1074/jbc.C100109200. [DOI] [PubMed] [Google Scholar]
- Mahadev K, Wu X, Zilbering A, Zhu L, Lawrence JTR, Goldstein BJ. Hydrogen peroxide generated during cellular insulin stimulation is integral to activation of the distal insulin signaling cascade in 3T3-L1 adipocytes. J Biol Chem. 2001;276:48662–48669. doi: 10.1074/jbc.M105061200. [DOI] [PubMed] [Google Scholar]
- Sullivan DM, Wehr NB, Fergusson MM, Levine RL, Finkel T. Identification of oxidant-sensitive proteins: TNF-alpha induces protein glutathiolation. Biochemistry. 2000;39:11121–11128. doi: 10.1021/bi0007674. [DOI] [PubMed] [Google Scholar]