

Abstract
Recent evidence indicates novel role for matrix metalloproteinases (MMPs), in particular gelatinase A (MMP-2), in the regulation of vascular biology that are unrelated to their well-known proteolytic breakdown of matrix proteins. We have previously reported that MMP-2 can modulate vascular reactivity by cleavage of the Gly32-Leu33 bound in big endothelin-1 (ET-1) yielding a novel vasoactive peptide ET-1[1-32]. These studies were conducted to investigate whether gelatinolytic MMPs could affect neutrophil-endothelial cell attachment. ET-1[1-32] produced by MMP-2 up-regulated CD11b/CD18 expression on human neutrophils, thereby promoted their adhesion to cultured endothelial cells. ET-1[1-32] evoked release of gelatinase B (MMP-9), which in turn cleaved big ET-1 to yield ET-1[1-32], thus revealing a self-amplifying loop for ET-1[1-32] generation. ET-1[1-32] was rather resistant to cleavage by neutrophil proteases and further metabolism of ET-1[1-32] was not a prerequisite for its biological actions on neutrophils. The neutrophil responses to ET-1[1-32] were mediated via activation of ETAreceptors through activation of the Ras/Raf-1/MEK/ERK signaling pathway. These results suggest a novel role for gelatinase A and B in the regulation of neutrophil functions and their interactions with endothelial cells. Here we describe the methods in detail as they relate to our previously published work.
Keywords: matrix metalloproteinases, endothelin-1
Introduction
Matrix metalloproteinases (MMPs) are a class of secreted enzymes with major functions in the degradation and remodeling of all components of the extracellular matrix (1). Gelatinase A (MMP-2) and gelatinase B (MMP-9) cleave denaturated collagens and type IV collagen, which is present in the basement membranes. This latter action is important for the mobilization of stem cells (2) and migration of lymphocytes and tumor cells (3). An increasing body of evidence indicates novel roles for gelatinases in the innate and adaptive immunity that are unrelated to matrix remodeling. For instance, gelatinase B was found to process cytokines and chemokines, resulting in skewed immune functions (3,4); while gelatinase A was reported to mediate platelet aggregation (5). Gelatinase A can also modulate vascular reactivity by reducing the vasodilatory potency of calcitonin gene-related peptide through cleavage of the Gly14-Leu15 bound (6) and by cleavage of the Gly32-Leu33 bound in big endothelin-1 (ET-1) to yield a novel vasoconstrictor peptide ET-1[1-32] (7). On a molar basis, ET-1[1-32] appears to be a more active peptide than the 21-amino acid ET-1 (7). Of particular interest, increased levels of gelatinase activity can often be detected with simultaneous increases in big ET-1 at sites of inflammation associated with myocardial ischemia (8,9), neointima formation (10) and atherosclerosis (11,12). Since these pathological conditions are also characterized by increased adhesiveness of leukocytes to the vascular endothelium (13) and ET-1 may function as an autocrine/paracrine modulator of leukocyte functions (14-17), we investigated whether ET-1[1-32] formed by gelatinase A and gelatinase B could affect neutrophil adhesion to endothelial cells and studied the underlying molecular mechanisms.
Materials and Methods
Activation of MMPs
Highly purified human gelatinases A or B are commercially available as pro-enzymes as well as active enzymes (Chemicon International, Mississauga, ON, Canada). Preparations of pro-MMP are often contaminated with small quantities of the corresponding active MMP. Larger quantities of active MMP can be obtained in the laboratory by incubating the pro-MMP (140 nM) with 4-aminophenylmercuric acetate (APMA, Sigma, 1 mM). APMA was prepared freshly by dilution from a 10 mM stock in NaOH with 1M Tris-HCl, pH 7.5. APMA activates pro-MMPs by disrupting a cystein switch. The activation reaction was allowed to proceed for 2 hrs at room temperature. If needed, unreacted APMA was scavenged by addition of BSA to a final concentration of 50 µM. Activity of MMP-2 and MMP-9 was tested against an extracellular matrix protein (e.g., Collagen type IV, Calbiochem) and by zymography (see below). Activated MMPs were also able to cleave some small vasoactive hormone peptides. We have shown that both MMP-2 and MMP-9 can cleave big endothelin-1[1-38] to yield two peptides, ET-1[1-32] and ET-1[33-38] (7,18). Only ET-1[1-32] has known biological activities, which were first discovered using an in vitro arterial system upon the preparation of the peptide in vitro (7).
In vitro preparation and characterization of ET-1[1-32]
ET-1[1-32] was prepared as described previously by cleaving synthetic human big ET-1 (Sigma-Aldrich, Oakville, ON, Canada) with activated MMPs. The cleavage reaction was conducted for16 h at 37°Cin HEPES-phosphate saline solution (in mM: NaCl 142, KCl 4.7, MgSO41.17, CaCl21.56, HEPES 10, KH2PO41.18; pH 7.4).
HPLC analysis: The incubation mixture was separated on an HPLC-chromatograph (Waters) using a 12.5 cm x 4 mm C-18 column (LiChrospher, Merck) with a (1% per minute)-gradient of 5% CH3CN in 0.1% aqueous TFA against 0.1% TFA in acetonitrile at 0.5 ml/min flow rate.
Mass analysis: HPLC resolved peaks were collected and the mass of peptides was determined with a Voyager Elite matrix assisted laser desorption ionization (MALDI) mass spectrometer (Applied Biosystems, Framingham, MA) equipped with delayed extraction and a reflectron. The instrument was run in the reflectron mode using 20 kV acceleration. External calibrations were completed using a mixture of known peptides.Samples were analysed using a two-layer preparation method (19). The first layer was 0.7 µL of 5 mg/ml sinapinic acid (Aldrich) in 3:2 acetone: methanol. The second layer was 0.4 µl of a 1:1 mixture of the sample solution and sinapinic acid solution. The second layer sinapinic acid solution was prepared at a concentration of 10 mg/ml in 1:1 acetonitrile: 0.1% trifluoroacetic acid (aq.) (Supelco). After the second layer dried, the sample spot was rinsed to remove excess salts by placing a 5 µl drop of 0.1% TFA on top of the sample spot and then removing the drop using a Kimwipe.
For the studies, ET-1[1-32] stock solution (10-4M) was diluted in phosphate buffered saline, containing 0.1% bovine serum albumin, pH 7.4 immediately before use. Endotoxin concentration of ET-1[1-32] preparations was below the detection limit (0.125 EU/ml) of the Limulus amebocyte lysate assay (E-Toxate, Sigma).
Isolation and treatment of neutrophil granulocytes
Neutrophil granulocytes were isolated from the peripheral venous blood obtained from non-smoking healthy volunteers by dextran sedimentation, centrifugation through Ficoll-Hypaque gradients (Pharmacia Diagnostics AB) and hypotonic lysis of red blood cells (17). Neutrophils (107cells/ml, purity >97%) were re-suspended in a modified Hanks’ balanced salt solution (in mM: NaCl 140, KCl 5, K2PO410, CaCl21.4, MgCl21.2, glucose 5, and 0.1% bovine serum albumin, pH 7.4), pre-incubated with enzyme inhibitors, as indicated, then challenged with ET-1[1-32] for the indicated times at 37ºC. The cells were then pelleted, and pellets and supernatants were collected for further analysis.
Flow cytometry analysis of adhesion molecule expression
Direct immunofluorescence labeling of control or treated neutrophils was performed as described previously (20). In brief, leukocytes were stained with a saturating concentration of FITC-labeled anti-human L-selectin monoclonal antibody (mAb) DREG-56 (IgG1, PharMingen) or R-phycoerythrin-labeled anti-human CD11b mAb Leu-185 (IgG1, BD Biosciences). Appropriately labeled, class-matched irrelevant mouse IgG1 was used as a negative control for each staining. Single or double-color immunofluorescence was analyzed by a FACScan flow cytometer (Becton Dickinson Immunocytometry Systems) with Lysis II software.
Activation of the Ras/Raf-1/MEK/ERK signaling pathway
Neutrophils were lysed in ice-cold lysis buffer (in mM: Tris 20, EGTA 1, Na3VO42, NF 25, 0.5% Triton X-100, PMSF 2, aprotinin 40 μg/ml and 10 μg/ml each of pepstatin A, chymostatin and leupeptin, pH 7.4) for 15 min on ice, and centrifuged at 4ºC for 10 min at 14,000 g. Western blot analysis of phosphorylated MEK and ERK 1/2 was performed using the Phospho Plus MEK 1/2 and ERK 1/2 MAP kinase antibody kits (New England Biolabs, Beverly, MA) as described (21). ERK 1/2 activity was assayed following immunoprecipitation of ERK 1/2 from the cell lysates with an immobilized anti-phospho-p44/p42 MAP kinase antibody (New England Biolabs) using Elk-1 fusion protein, as a specific target for ERK 1/2 (22). Phosphorylation of ERK 1/2 at Ser383 was quantified by densitometry following immunoblotting by using an affinity purified anti-phospho-Elk-1 polyclonal antibody (New England Biolabs) and chemiluminescence detection (Lumi-Light Western Detection Substrate, Roche Diagnostics).
Raf-1 kinase assay: Raf-1 activity was determined by using the method of Garner et al. (23) with modifications. In brief, Raf-1 was immunoprecipitated from the neutrophil lysates with an anti-Raf-1 antibody (C-12, Santa Cruz Biotechnology, Santa Cruz, CA), antigen-antibody complexes were then isolated by protein A-Sepharose CL-4B (Sigma), and Raf-1 activity was measured using the Raf-1 Kinase Cascade Assay kit (Upstate Biotechnology, Lake Placid, NY) in accordance with the manufacturer’s protocol.
Ras activation assay: Activated p21Ras(Ras-GTP) from neutrophil lysates was affinity purified by using GST-Ras binding domain of Raf-1 (residues 1-149) fusion protein conjugated to agarose beads (Upstate Biotechnology) (24). The beads were washed extensively and boiled in reducing sample buffer. The eluted proteins were resolved on a 10% SDS-polyacrylamide gel, transferred to a PVDF membrane, probed with a mouse anti-Ras mAb (clone RAS10, Upstate Biotechnology), and visualized using a goat anti-mouse secondary antibody conjugated to horseradish peroxidase (BioRad) and a chemiluminescence detection system (Lumi-Light Western Detection Substrate, Roche Diagnostics).
Metabolism of ET-1[1-32]
Measurements were performed as previously described for ET-1[1-38] and ET-1[1-21] (25). Briefly, ET-1[1-32] (100 pmol) was incubated with preparations of neutrophil cytosol (0.5 - 5mg protein) or membrane (0.2 - 2mg protein) at 37°C for 2 hrs. The incubation mixture was separated on HPLC and the resolved peaks were collected and further characterized on a MALDI mass spectrometer, as described above.
Measurement of gelatinase release
Gelatinolytic activity released from unchallenged and challenged neutrophils was determined using [3H]-acetylated gelatin as a substrate following activation of the latent gelatinase by 1.7 mM phenylmercuric acetate (26). Gelatinase released was expressed as the percentage of total enzyme units released from neutrophils treated with 0.1 % Triton X-100.
Zymographic analysis of MMPs
Thegelatinolytic activity of neutrophil MMPs was measured essentially as described before (7). The samples of conditioned supernatants and neutrophil homogenate were mixed with electrophoresis loading buffer (10:1, vol:vol, 20ml per well) and subjected to electrophoresis, which was carried out on a 7.5% SDS-PAGE co-polymerized with gelatin (2 mg/ml). Gelatin was added from a 10 fold concentrated stock containing pre-boiled gelatin type A (20 mg/ml, Sigma, Cat. # G 8150) and 1% SDS. Following electrophoresis, gels were washed with 2.5% Triton X-100 for 1 hr (3 times, 20 min each) and incubated for 48 hrs in enzyme assay buffer (25 mM Tris, pH 7.5, 5 mM CaCl2, 0.9% NaCl, 0.05% Na3N) for the development of enzyme activity bands. After incubation, the gels were stained with 0.05% Coomassie brilliant blue G-250 in a mixture of methanol: acetic acid: water (2.5: 1: 6.5) and de-stained in 4% methanol with 8% acetic acid. The gelatinolytic activities were detected as transparent bands against the background of Coomassie brilliant blue-stained gelatin. If more sensitivity was necessary, the gels were further destained in a solution of 1% Triton x-100 (1–2 hrs). This procedure increased the signal-to-noise ratio, allowing the visualization of faint gelatinase bands at 92, 125 and > 200 kDa. To measure the activities of the detected enzymes, zymograms were read using a ScanJet 3c scanner (Hewlett Packard, Boise, ID). The intensity of the bands was expressed as arbitrary units and analyzed using SigmaGel measurement software (Jandel Corporation, San Rafael, CA).
Neutrophil-endothelial cell adhesion assay
Culture of endothelial cells: Human coronary artery endothelial cells (HCAEC, cryopreserved in passage 3) obtained from Clonetics/BioWhittaker were cultured in T25 flasks (26). HCAEC (passages 4 to 6) seeded into 96-well microplates (Sarstedt) and grown to confluence were used in the adhesion assays.
Adhesion assay: Monolayers of HCAEC in 96-well microplates were stimulated with bacterial lipopolysaccharide (1 μg/ml, E. coli O155:B4, Sigma) for 4 h at 37ºC in a 5% CO2atmosphere. The cells were then washed three times with PBS, and 2x10551Cr-labeled neutrophils in 100 μl were added. In some experiments, ET-1[1-32] was added with neutrophils to activated HCAEC. After incubation for 30 min at 37ºC on an orbital shaker at 90 rpm, loosely adherent or unattached neutrophils were washed three times, and the endothelial monolayer plus the adherent neutrophils were lysed in 150 μl of 0.1% Triton X-100 in water. The number of adherent neutrophils in each experiment was estimated from the radioactivity of a control sample.
Statistical analysis
Results are reported as means ± SEM. Statistical comparisons were made by ANOVA using ranks (Kruskal-Wallis test), followed by Dunn’s multiple contrast hypothesis test to identify differences when various treatments were compared to the same control or by the Mann-Whitney test for unpaired observations. P values < 0.05 were considered significant for all tests.
Results and Discussion
Neutrophil gelatinase cleaves big ET-1 to yield ET-1[1-32]
Neutrophils can metabolize big ET-1 into smaller peptides (25).When big ET-1 was incubated with neutrophil cytosol and membrane fractions and the resultant incubates were analyzed using mass spectrometry or HPLC/mass spectrometry, fragments of ET-1[1-38] including ET-1[23-38], ET-1[1-22], ET-1[1-20], ET-1[1-19] and ET-1[1-18] were detected Fig. 1A.Gelatin-zymography revealed the presence of gelatinase B, but not gelatinase A, in both cytosol and membrane preparations of human neutrophils (18). Since gelatinase A can cleave big ET-1 to ET-1[1-32] (7); we tested whether gelatinase B also possess a similar activity. Indeed, APMA-activated gelatinase B cleaved big ET-1 to yield ET-1[1-32], as demonstrated using HPLC and MALDI-TOF mass spectroscopy Fig. 1B.
Fig. 1.
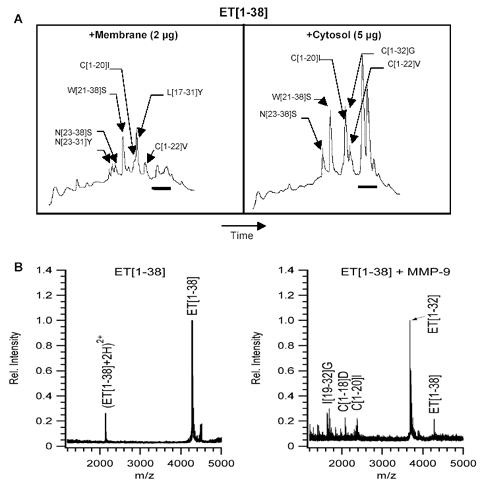
HPLC/MALDI-TOF analysis shows that big ET-1 (ET-1[1-38]) is cleaved to yield many smaller peptides by proteases in the cytosol and cell membrane of neutrophils. Based on their masses, these peptides were assigned the ET-sequences indicated in brackets. (A). Bar: peaks in the region of the HPLC chromatogram corresponding to neutrophil proteins, not ET peptides. Mass analysis on a MALDI TOF instrument reveals that MMP-9 (gelatinase B), cleaves big ET-1 to yield ET-1[1-32] (B).
It is not clear whether neutrophils are a major source of bioactive ET peptides, such as ET-1[1-21] and ET-1[1-32]. Neutrophils contain predominantly pro-MMP-9, not active MMP-9 (18). Therefore, the conversion of big ET-1 into ET-1[1-32] is likely regulated by a specific MMP-9 activation event (that occurs during or following release of granule contents) as well as by rapid peptide degradation. In line with this, only small quantities of ET-1 peptides were detected with our HPLC/mass spectrometry assays Fig. 1A.
Metabolism of ET-1[1-32] by neutrophils
We next investigated whether further cleavage of ET-1[1-32] was a requirement for its bioactivity. ET-1[1-32] (100 pmol) was incubated with preparations of neutrophil cytosol (0.5-5mg protein) or membrane (0.2-2mg protein) and assessed for conversion into smaller peptides. There was little conversion of ET-1[1-32] into other smaller peptides such as ET-1[1-22], ET-1[1-21], ET-1[1-20] and ET-1[1-19] using a combination of HPLC with mass spectroscopy (18). Consistent with these observations, the effects of ET-1[1-32] on adhesion molecule expression were insensitive to phosphoramidon, an inhibitor of the metal-dependent endothelin converting enzymes (L-selectin expression was 35±6% and 31±7% of control, CD11b expression 178±17% and 184±16% of control in response to 100 nM ET-1[1-32] in the absence or presence of phosphoramidon, respectively, n=5, P>0.1) (18).
To further confirm that the biological effects of ET-1[1-32] were due to the inherent bioactivity of this peptide, neutrophils were challenged with big ET-1 (100 nM), the inactive precursor of ET-1 peptides, in the presence of phosphoramidon (an endothelin converting enzyme inhibitor) and o-phenanthroline (an MMP inhibitor). There were no detectable changes in adhesion molecule expression on neutrophils under these conditions (L-selectin and CD11b/CD18 expression were 110±6% and 102±3% of control, respectively, n=4, P>0.1) (18).
ET-1[1-32] induces gelatinase release from neutrophils
Next, we investigated whether ET-1[1-32] is capable of inducing gelatinase release from human neutrophils. Indeed, ET-1[1-32] evoked a concentration-dependent release of gelatinase (Fig. 2) without affecting cell viability (data not shown). Since neutrophils contain only gelatinase B, but not gelatinase A, and both gelatinase A and B are capable of cleaving big ET-1 to yield ET-1[1-32], a self-amplifying loop may exist for stimulation of ET-1[1-32] formation and gelatinase B release and consequently for neutrophil activation. Intriguingly, ET-1[1-32] appears to function as a partial neutrophil agonist, for it did not induce superoxide formation (18).
Fig. 2.

ET-1[1-32] induces gelatinase release from human neutrophils. Isolated neutrophils were challenged with ET-1[1-32] for 30 min at 37°C. Gelatinase released into the culture medium was measured as described in Methods and is expressed as percentage of total cellular enzyme activity. N=4, P<0.05 vs unstimulated.
ET-1[1-32] induces L-selectin shedding and up-regulates CD11b/CD8 expression on neutrophils
ET-1[1-32] induced a rapid down-regulation of L-selectin and up-regulation of CD11b/CD18 on the surface of neutrophils (Fig. 3). These actions of ET-1[1-32] were concentration-dependent with EC50 values of 1 to 3 nM (18). Consistent with previous observations (7), on a molar basis ET-1[1-32] appeared to be a more potent neutrophil agonist than ET-1 (17,18). Our previous pharmacological and receptor binding studies showed that human neutrophils predominantly express ETAreceptors (17). Consistently, the actions of ET-1[1-32] on neutrophil expression of L-selectin and CD11b/CD18 as well as gelatinase release were almost completely inhibited by the selective ETAreceptor antagonist FR139317, but not by the ETBreceptor-selective antagonist BQ788 (18).
Fig. 3.

Effects of ET-1[1-32] on surface expression of adhesion molecules on human neutrophils. Neutrophils were challenged with 100 nM ET-1[1-32] for 30 min at 37°C, then stained with fluorescein-labeled anti-L-selectin and anti-CD11b monoclonal antibodies. C, negative control of immunostaining with irrelevant antibodies. Results are representative of 6 experiments.
ET-1[1-32] activates ERK through the Ras/Raf-1/MEK pathway
Experiments with PD98059, which inhibit the activation of MEK and consequently of ERK, revealed that neutrophil responses to ET-1[1-32] were ERK-dependent (18). However, PD98059 was more effective in reversing ET-1[1-32]-induced up-regulation of CD11b/CD18 expression and gelatinase release (72±5% and 80±5% inhibition, respectively), than down-regulation of L-selectin expression (59±4% inhibition) (18). To provide direct evidence for the involvement of the Ras/Raf-1/MEK/ERK signaling pathway, we studied activation of this cascade. ET-1[1-32] induced rapid (within 2 min) association of GTP-bound active Ras with the glutathione-S-transferase-Ras binding domain of Raf-1 Fig. 4A and increased Raf-1 kinase activity Fig. 4B. Consequently, enhanced phosphorylation of MEK and ERK was also detected within 5 min of addition of ET-1[1-32] Fig. 4C.
Fig. 4.
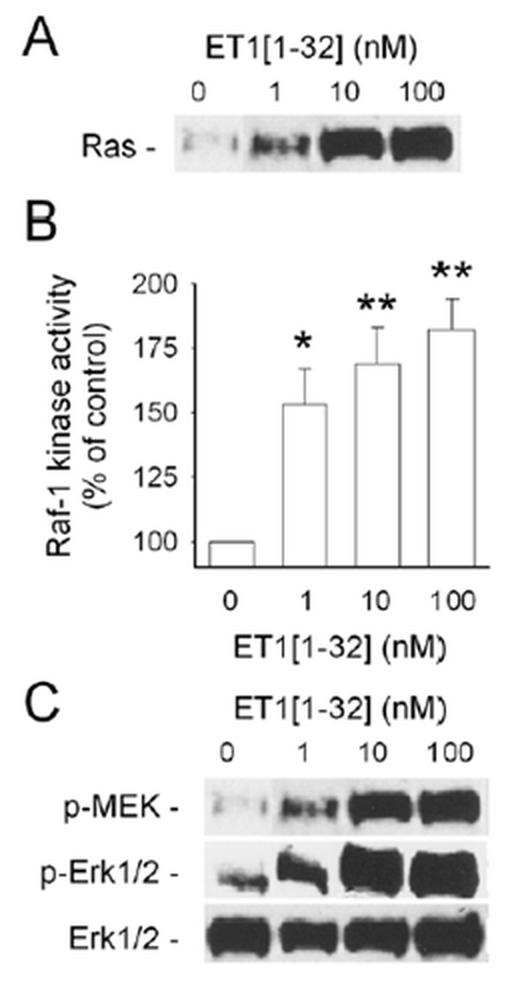
ET-1[1-32] activates the Ras/Raf-1 kinase/MEK/ERK 1/2 signaling pathway. A, Ras was detected by immunoblotting following affinity precipitation of GTP-bound active Ras from lysates of neutrophils. B, Raf-1 kinase activity was determined using a MAPK cascade system following immunoprecipitation of Raf-1. C, MEK and ERK 1/2 phosphorylation was detected by immunoblotting. Neutrophils were challenged with ET-1[1-32] for 2 min (A,B) or 5 min (C). Results are representative of 3 independent experiments.
These actions of ET-1[1-32] were concentration-dependent and was comparable to those evoked by 100 nM platelet-activating factor (18). ET-1[1-32]-induced ERK phosphorylation was inhibited by 75±6% with PD98059 (18). The similar degree of inhibition of ERK phosphorylation and CD11b/CD18 expression and gelatinase release observed with PD98059 indicates a tight correlation between ERK activation and neutrophil responses to ET-1[1-32]. It should be noted that the inhibition was incomplete, indicating the involvement of other signaling pathways.
ET-1[1-32] promotes neutrophil adhesion to human coronary artery cells in vitro
To assess the biological significance of ET-1[1-32]-induced changes in expression of adhesion molecules on the neutrophil surface, we performed a neutrophil-endothelial cell adhesion assay. To mimic blood flow, the assay was performed under non-static conditions (26). While ET-1[1-32] did not stimulate neutrophil adhesion to monolayers of unstimulated HCAEC, about 1.5-fold more neutrophils adhered to LPS-stimulated HCAEC when neutrophils were added to HCAEC together with ET-1[1-32] than in the absence of ET-1[1-32] (Fig. 5). Since neutrophils were incubated in the adhesion assays for 30 min with activated HCAEC, stimulation of neutrophil adhesion by ET-1[1-32] can be attributed primarily to the effects of this peptide on neutrophils. Indeed, much longer periods of culture of HCAEC with ET-1[1-32] were required to detect slight increases in expression of E-selectin and ICAM-1 on HCAEC (18).
Fig. 5.
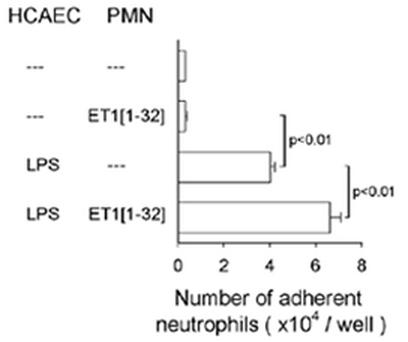
ET-1[1-32] enhances adhesion of neutrophils (PMN) to monolayers of human coronary artery endothelial cells (HCAEC). HCAEC were incubated with LPS (1 μg/ml) for 4 h, washed and PMN without or together with ET-1[1-32] (30 nM) were then added for 30 min at 37°C. Values are means ± SEM (n=5).
No adhesion experiments were performed with neutrophils alone pre-incubated with ET-1[1-32] since this peptide may induce neutrophil aggregation through up-regulation of CD11b/CD18 expression, thereby making interpretation of the results difficult. Our studies with function-blocking monoclonal antibodies revealed that ET-1[1-32]-induced neutrophil adherence to activated HCAEC were predominantly mediated through CD18 integrins and their endothelial counter-ligand, ICAM-1 (18).
Conclusions
Our results showed that two MMPs, gelatinase A and B, via cleavage of big ET-1 could affect expression of adhesion molecules on human neutrophils and consequently promote their adhesion to endothelial cell. Such actions may have relevance to pathological conditions where simultaneous over-expression of gelatinolytic MMPs and big ET-1 can be detected at the sites of tissue injury and inflammation. Thus, gelatinase A and B, often considered as pure effector molecules, may act as amplifiers of leukocyte recruitment, one of the key events of inflammation. Finally, gelatinases constitute a prototypic example of the regulation of innate immune reactions by proteolysis.
Acknowledgments
This work was supported by grants from the Medical Research Council of Canada/Canadian Institutes of Health Research (to J.G.F., J.S.D.C. and S.T.D.), the Heart and Stroke Foundation of Canada (to S.T.D., C.F.P. and J.G.F.) and the Natural Sciences and Engineering Council of Canada (to C.F.P.).
Appendix
Protocols
Protocol I: Zymography
Gelatin-SDS-zymography technique
Prepare a gel following a standard SDS-PAGE protocol with the exception that gelatin is added to the gel solution. For the detection of MMP-2 and MMP-9 gelatinolytic activity, the recommended composition is 7.5% acrylamide. Cast SDS-PAGE gel according to the procedure of Laemmli E.K. (Nature, 227, 1970, 680-685). An updated procedure is described in Current Protocols in Molecular Biology, Greene Publishing Associates and Wiley-Interscience, 1991, Vol. 2 pp 10.2.4-10.2.9. However, replace water in the procedure for gelatin (Gelatin, type A, from pork skin, electrophoresis reagent, Sigma cat # G-8150). Final concentration of gelatin should be between 0.5 and 2.0 mg/ml (recommended is 2 mg/ml).
Run the gel at 4ºC. Before starting the electrophoresis cool down the running buffer solution.
3.After the electrophoresis is complete, wash the gel with an aqueous solution of Triton X-100 (2.5%) (3 times 20 min) at room temperature.
4.Replace Triton for enzyme assay buffer (Tris-HCl, pH 7.6, supplemented with 0.15 M NaCl, 5 mM CaCl2, and 0.05% NaN3). Incubate the gel at 37ºC for 24 hrs. Low activity bands may require longer incubation (48 hrs).
5.Replace enzyme assay solution for Coomassie Blue stain (0.05% Coomassie Brilliant Blue, 10 % acetic acid, 30% methanol).
6. Gelatinase bands are detected as clear bands contrasting against the blue stained gel background. Sensitivity of this procedure is in the 50 to 100 pg range for MMP-2.
Protocol II: Analysis of metabolism of ET peptides by neutrophils
Peptide metabolism reactions
Solutions of ET-1[1-38] and ET-1[1-32] peptides (10-4M) in HEPES-phosphate saline solution (in mM: NaCl 142, KCl 4.7, MgSO41.17, CaCl21.56, HEPES 10, KH2PO41.18; pH 7.4) are incubated with neutrophil cytosol or membrane fractions for various times (from 30 minutes to 30 hrs) at 37ºC and in two dilutions containing a total of 2 µg and 0.2 µg protein. Control incubations are carried out with APMA-activated MMP-2 and MMP-9 to confirm cleavage of big ET-1 to ET-1[1-32].
Snap freeze the peptide digest until analysis by matrix assisted laser desorption ionization (MALDI)- time of flight (TOF) mass spectrometry (MS). MALDI-TOF MS is chosen for being relatively tolerant to the presence of salts and detergents in the peptide preparation.
Sample preparation for MALDI
Our Voyager Elite matrix assisted laser desorption ionization (MALDI) mass spectrometer (Applied Biosystems, Framingham, MA) is equipped with delayed extraction and a reflectron. The instrument is run in the reflectron mode using 20 kV acceleration.
Complete external calibration using a mixture of known peptides.
Analyse samples using a two-layer preparation method. The first layer is 0.7 µl of 5 mg/ml sinapinic acid (Aldrich) in 3:2 acetone: methanol. The second layer is 0.4 µl of a 1:1 mixture of the sample solution and sinapinic acid solution. The second layer sinapinic acid solution was prepared at a concentration of 10 mg/ml in 1:1 vol/vol acetonitrile: 0.1% trifluoroacetic acid (aq.) (Supelco). Wait until the second layer is completely dry. Rinse the sample spot to remove excess salts by placing a 5 µl drop of 0.1% TFA on top of the sample spot and then remove the drop using a Kimwipe.
HPLC analysis and HPLC/MS analysis
Mass analysis offers quick insight into the metabolism of peptides, reflecting possible cleavage by proteases and the mass of the resulting fragments. To confirm the results from MALDI-MS analysis, it is useful to fractionate the ET peptides and subcellular fractions on an HPLC provided with a C18 column and analyze each fraction separately.
Separation on a C18 column is largely based on the differential hydrophobic property of peptides and proteins. It is a method of choice for separation and analysis of peptides and soluble proteins. Upon developing a gradient of 0.1% TFA against and acetonitrile, ET peptides of different lengths are resolved in time. Elution of peptides is detected using an on-line UV absorbance detector (210 nm) as absorbance peaks.
The components that elute from the HPLC C18 column are collected in separate plastic vials and frozen for later analysis by MALDI-TOF MS.
MALDI analysis is used to assign a mass to every component after it has been resolved on the C18 column, thus facilitating component identification. Other alternative mass spectrometry methods such as liquid chromatography (LC) quadrupol time of flight (Q-TOF) MS might be employed for the on-line sequencing of LC peptides. The above described MALDI-TOF MS method is useful for most applications and alone has a high sensitivity of detection (in the low femto- to attomol levels). Importantly, HPLC with absorbance detection can be used for quantitative purposes. However, HPLC retention time does not allow identification. Protein and peptide identification is possible by coupling HPLC with spectrometry techniques. However, mass spectrometry does not easily allow for quantification. This is due to the fact that peptide ionization in the mass spectrometer is affected by the presence of other components of the sample, including other peptides. Nevertheless, mass spectrometry can be very powerful in the qualitative analysis as well as in following up the enzymatic conversion of proteins and peptides.
Protocol III: Quantification of gelatinase release from neutrophils
Preparation of [3H]-acetylated gelatin
Type I collagen (50 mg, Sigma) is dissolved in 25 ml 0.05% acetic acid. Gelatin is obtained from collagen by denaturation at 45ºC for 1 hr.
Dialyze extensively against 100 mM phosphate buffer containing 140 mM NaCl, 20 mM KCl and 50 mM sodium acetate.
To prepare [3H]-acetylated substrate, the dialyzed gelatin solution is mixed with 0.1 mmol [3H]-acetic anhydride (50 Ci/mmol, NET-018A, DuPont NEN) in 0.5 ml acetonitrile with stirring and kept at 4ºC for 60 min. Acetic anhydride is supplied in 5 μl benzene in a break seal glass tube. Immediately before use, the tube should be immersed in liquid nitrogen for 20 sec, then add 500 μl of acetonitrile to the upper compartment and break the seal. The flow of acetonitrile into the lower compartment is relatively slow (20-25 min).
The acetylated gelatin should be dialyzed against three changes of 50 mM Tris/HCl, pH 7.6, containing 50 mM NaCl. Determine incorporated radioactivity, and store the acetylated gelatin at 4ºC.
Gelatinase assay
Materials: di-isopropyl-phosphoro-fluoridate (DFP)-treated samples, [3H]-acetylated gelatin, buffer A: 50 mM Tris/HCl, pH 7.6 containing 50 mM NaCl, 0.064% Triton X-100, 12.8 mM CaCl2, 2.57 mM p-amino-phenylmercuric acetate and 0.13 mg/ml bovine serum albumin.
Following incubation of neutrophils with appropriate stimuli, prepare cell supernatant by centrifugation (400 g 15 min). For total neutrophil gelatinase content, lyse the cells with 0.1 % Triton X-100 and collect supernatant after centrifugation at 10,000 g for 15 min.
Mix 40 μl cell supernatant with equal volume of buffer B (100 mM Tris/HCl, pH 7.6 containing 6 mM DFP, 100 mM NaCl and 0.1% Triton X-100) and incubate for 60 min at 37ºC. Run all conditions in duplicate.
Stop the reaction by adding 4 μl of bovine serum albumin solution (2 mg/ml).
Mix 20 μl of [3H]-acetylated gelatin solution, 30 μl of DFP-treated sample and 175 μl buffer A in an Eppendorf tube.
Incubate mixture for 60 min at 37ºC.
To stop the reaction, add 25 μl BSA solution (100 mg/ml in 50 mM Tris/HCl, pH 7.6 containing 50 mM NaCl). Vortex.
Add 50 μl ice-cold TCA (90% weight/vol), vortex, and keep samples on ice for 30 min.
Centrifuge with 1250 g for 15 min at 4ºC.
Transfer 100 μl supernatant into liquid scintillation vials, overlay with scintillation fluid and read in scintillation counter.
Calculate gelatinase release as the percentage of total cellular gelatinase content.
Protocol IV: Measurement of Ras and Raf-1 kinase activity
Affinity precipitation and detection of activated Ras
Following challenge of neutrophils (107cells/ml, 400 μl) with stimuli for 1 to 5 min, centrifuge the cells with 400 g for 5 min at 4ºC. Discard supernatant.
Lyse the cells for 15 min on ice in 150 μl lysis buffer (25 mM HEPES, pH 7.5, 150 mM NaCl, 1% Igepal CA-630 (Sigma), 10 mM MgCl2, 1 mM EDTA and 2% glycerol).
Centrifuge with 14,000g for 10 min at 4ºC.
Transfer supernatant into new Eppendorf tubes. Determine protein concentration and adjust protein concentration to 1mg/ml with lysis buffer.
Add 1/10 volume of glutathione agarose, 50% slurry and incubate for 15 min at 4ºC on a rotator (Adams Nutator). Glutathione agarose should be prepared in advance by swelling glutathione immobilized on cross-linked 4% beaded agarose (Sigma, G-4510) in water at 200 ml/g for 2 hours. This suspension can be stored at 4ºC. Immediately before use, remove an aliquot, wash it twice with equal volume of water, then three times with equal volume of lysis buffer, and re-suspend in 0.5 volume of lysis buffer.
Add 10 μl of Raf-1 RBD agarose conjugate (Upstate Biotechnology) and incubate at 37ºC for 15 min with gentle agitation.
Centrifuge with 12,000 g for 5 sec. Discard supernatant.
Wash beads five times with 400 μl lysis buffer. Collect the agarose beads by 5 sec centrifugation in the microcentrifuge at 14,000 g, and drain off the supernatant.
Resuspend the agarose beads (approx. 15 μl) in 15 μl Laemmli loading buffer 2x and boil for 5 min.
Perform SDS-polyacrylamide gel electrophoresis on 20 μl supernatant and transfer the proteins to nitrocellulose. Perform western blotting using anti-Ras monoclonal antibody (clone RAS10, Upstate Biotechnology cat. No. 05-516) at 1 μg/ml final concentration.
Stimulation of cells will activate approximately 10-15% of the available Ras; in vitro GTP-γS protein loading will activate nearly 100% of the Ras that can be activated. To activate Ras with GTP-γS protein, incubate 1 ml of cell lysate with GTP-γS (final concentration 0.1 mM) in the presence (positive control) and absence of EDTA (10 mM) (negative control) at 30ºC for 15 min with agitation. Terminate the reaction by placing the tubes on ice and adding MgCl2to a final concentration of 60 mM. Then proceed as described under point 4.
Immunoprecipitation and detection of Raf-1
Following incubation with appropriate stimuli, pellet neutrophils by centrifugation. Lyse cells by adding 1/10th volume of RIPA buffer pH 7.2 (100 mM Na2PO4, 150 mM NaCl, 2 mM EDTA, 1% sodium deoxycholate, 1% Nonidet (NP-40), 0.1% SDS, 10 μg/ml aprotinin, 2 mM PMSF, 0.001% β-mercaptoethanol, and 0.8 mM sodium orthovanadate) for 15 min on ice.
Centrifuge in a microcentrifuge with 14,000 g for 15 min at 4ºC.
Transfer supernatant into new Eppendorf tubes. Add 1 μl anti-Raf antibody (C-12, Santa Cruz Biotechnology, Santa Cruz, CA) or monoclonal rabbit IgG (SC-133, Santa Cruz Biotechnology) per 100 μl supernatant. Incubate on a rotator for 60 min at 4ºC.
Add 4 μl protein A-Sepharose CL4B per 100 μl supernatant, and incubate on a rotator for 60 min at 4ºC. Protein A-Sepharose CL4B (Sigma, cat. # P-3391) should be prepared in advance. Suspend protein A-Sepharose CL4B at 50 mg/ml in PBS for 1 hr at 4ºC, centrifuge with 800 g for 10 min at 4ºC, wash 5 times with equal volume of RIPA buffer, and finally resuspend it in 0.5 volume of RIPA buffer and store at 4ºC.
Centrifuge with 800-900 g for 20 min at 4ºC.
Wash the pellets twice with 0.5 ml RIPA buffer, then twice with 0.5 ml PAN buffer (100 mM PIPES, pH 7.4, 100 mM NaCl) containing 30 μg/ml aprotinin, and once with 0.5 ml PAN buffer. Re-suspend pellets in 5 μl PAN buffer.
To assess Raf-1 kinase activity, we used the Raf-1 Kinase Cascade Assay kit from Upstate Biotechnology (Cat. No. 17-172). Use 4 μl of the samples in the assay and follow the manufacturer’s protocol.
Protocol V: Neutrophil-endothelial cell adhesion assay
Preparation of 51Cr-labeled neutrophils
Isolate polymorphonuclear leukocytes from peripheral blood.
Resuspend leukocytes at 2x107cells/ml in Hanks’ balanced salt solution, pH 7.2, 0.1% BSA.
Add 56 μCi51Cr (sodium chromate in saline, Cat. No. NEZ030, NEN) per ml cell suspension.
Incubate for 60 min at 37ºC.
Wash cells twice with 10 ml Hanks’ balanced salt solution without calcium and magnesium.
Resuspend neutrophils at 2.5x106cells/ml in RPMI 1640 medium containing 1% BSA. Check neutrophil viability using Trypan blue exclusion test, and determine chromium-51 incorporation into known number of neutrophils (e.g., 500,000 cells).
Adhesion assay
Endothelial cells should be grown into confluence in 96-well microplates (Sarstedt).
Change endothelial cell culture medium at least 16 hours before the experiments for RPMI 1640 medium containing 1% BSA.
Culture endothelial cell for 4-6 hours with LPS (1 μg/ml), TNF-α (50 ng/ml) or IL-1β (25 ng/ml) to induce near-maximum expression of E-selectin and ICAM-1.
Wash wells three times with 200 μl RPMI 1640, 1% FBS.
Add 100 μl radiolabeled neutrophils with or without a neutrophil agonist to endothelial cells.
Incubate for 30 min at 37ºC, 5% CO2on a horizontal rotator at 90 rpm.
Remove medium. Wash wells three times with phosphate buffered saline, pH 7.2 to remove unattached or loosely attached leukocytes.
Overlay wells with 150 μl 0.1% Triton X-100 solution, and incubate until cell lysis is complete.
Transfer 100 μl of cell lysate into counting vials, and determine radioactivity using a γ-counter.
The number of neutrophils adhered per well can be calculated by knowing the amount of chromium-51 incorporated into neutrophils.
References
- Nagase H, Woessner JF. Matrix metalloproteinases. J Biol Chem. 1999;274:21491–21494. doi: 10.1074/jbc.274.31.21491. [DOI] [PubMed] [Google Scholar]
- Pruijt et al JFM. Prevention of interleukin-8-induced mobilization of haematopoietic progenitor cells in rhesus monkeys by inhibitory antibodies against gelatinase B (MMP-9). Proc Natl Acad Sci USA. 1999;96:10863–10868. doi: 10.1073/pnas.96.19.10863. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Opdenakker G, Van den Steen PE, Van Damme J. Gelatinase B: a tuner and amplifier of immune functions. Trends Immunol. 2001;22:571–579. doi: 10.1016/s1471-4906(01)02023-3. [DOI] [PubMed] [Google Scholar]
- Van den Steen PE, Proost P, Wuyts A, Van Damme J, Opdenakker G. Neutrophil gelatinase B potentiates interleukin-8 tenfold by amino terminal processing, whereas it degrades CTAP-III, PF-4, and GRO-a and leaves RANTES and MCP-2 intact. Blood. 2000;96:2673–2681. [PubMed] [Google Scholar]
- Sawicki G, Salas E, Murat J, Miszta-Lane H, Radomski MW. Release of gelatinase A during platelet activation mediates aggregation. Nature. 1997;386:616–619. doi: 10.1038/386616a0. [DOI] [PubMed] [Google Scholar]
- Fernandez-Patron C, Stewart KG, Zhang Y, Koivunen E, Radomski MW, Davidge ST. Matrix metalloproteinase-2-dependent cleavage of calcitonin gene-related peptide promotes vasoconstriction. Circ Res. 2000;87:670–676. doi: 10.1161/01.res.87.8.670. [DOI] [PubMed] [Google Scholar]
- Fernandez-Patron C, Radomski MW, Davidge ST. Vascular matrix metalloproteinase-2 cleaves big endothelin-1 yielding a novel vasoconstrictor. Circ Res. 1999;85:906–911. doi: 10.1161/01.res.85.10.906. [DOI] [PubMed] [Google Scholar]
- Miyauchi T, Yanagisawa M, Tomizawa T, Sugoshita Y, Suzuki N, Fujino M, Ajisaka R, Goto K, Masaki T. Increased plasma concentration of endothelin-1 and big endothelin-1 in acute myocardial infarction. Lancet. 1989;2:53–54. doi: 10.1016/s0140-6736(89)90303-6. [DOI] [PubMed] [Google Scholar]
- Watanabe T, Suzuki N, Shimamoto N, Fujino M, Imada A. Contribution of endogenous endothelin to the extension of myocardial infarct size in rats. Circ Res. 1991;69:370–377. doi: 10.1161/01.res.69.2.370. [DOI] [PubMed] [Google Scholar]
- Jenkins GM, Crow MT, Bilato C, Gluzband Y, Ryu WS, Li Z, Stetler-Stevenson W, Nater C, Froehlich JP, Lakatta EG, Cheng L. Increased expression of membrane-type matrix metalloproteinase and preferential localization of matrix metalloproteinase-2 to the neointima of balloon-injured rat carotid arteries. Circulation. 1998;97:82–90. doi: 10.1161/01.cir.97.1.82. [DOI] [PubMed] [Google Scholar]
- Lerman A, Edwards BS, Hallett JW, Heublein DM, Sandberg SM, Burnett Jr JC. Circulating and tissue endothelin immunoreactivity in advanced atherosclerosis. N Engl J Med. 1991;325:997–1001. doi: 10.1056/NEJM199110033251404. [DOI] [PubMed] [Google Scholar]
- Newby AC, Southgate KM, Davies M. Extracellular matrix degrading metalloproteinases in the pathogenesis of arteriosclerosis. Basic Res Cardiol. 1993;89:59–70. doi: 10.1007/978-3-642-85660-0_6. [DOI] [PubMed] [Google Scholar]
- Lefer AM, Lefer DJ. Pharmacology of the endothelium in ischemia-reperfusion and circulatory shock. Ann Rev Pharmacol Toxicol. 1993;33:71–90. doi: 10.1146/annurev.pa.33.040193.000443. [DOI] [PubMed] [Google Scholar]
- López-Farré A, Riesco A, Espinosa G, Digiuni E, Cernadas MR, Alvarez V, Monton M, Rivas F, Gallego MJ, Egido J, Casado S, Caramelo C. Effect of endothelin-1 on neutrophil adhesion to endothelial cells and perfused hearts. Circulation. 1993;88:1166–1171. doi: 10.1161/01.cir.88.3.1166. [DOI] [PubMed] [Google Scholar]
- Filep JG, Fournier A, Földes-Filep E. Acute pro-inflammatory actions of endothelin-1 in the guinea pig lung: involvement of ETA and ETB receptors. Br J Pharmacol. 1995;115:227–236. doi: 10.1111/j.1476-5381.1995.tb15868.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Helset E, Lindal S, Olsen R, Hyklebust R, Jorgensen L. Endothelin-1 causes sequential trapping of platelets and neutrophils in pulmonary microcirculation in rats. Am J Physiol. 1996;271:L538–L546. doi: 10.1152/ajplung.1996.271.4.L538. [DOI] [PubMed] [Google Scholar]
- Zouki C, Baron C, Fournier A, Filep JG. Endothelin-1 enhances neutrophil adhesion to human coronary artery endothelial cells: role of ETA receptors and platelet-activating factor. Br J Pharmacol. 1999;127:969–979. doi: 10.1038/sj.bjp.0702593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fernandez-Patron C, Zouki C, Whittal R, Chan JSD, Davidge ST, Filep JG. Matrix metalloproteinases regulate neutrophil-endothelial cell adhesion through generation of ET-1[1-32]. FASEB J. 2001;15:2230–2240. doi: 10.1096/fj.01-0178com. [DOI] [PubMed] [Google Scholar]
- Dai Y, Whittal RM, Li L. Two-layer sample preparation: a method for MALDI-MS analysis of complex peptide and protein mixtures. Anal Chem. 1999;71:1087–1091. doi: 10.1021/ac980684h. [DOI] [PubMed] [Google Scholar]
- Filep JG, Delalandre A, Payette Y, Földes-Filep E. The glucocorticoid receptor regulates expression of L-selectin and CD11/Cd18 on human neutrophils. Circulation. 1997;96:295–301. doi: 10.1161/01.cir.96.1.295. [DOI] [PubMed] [Google Scholar]
- Zouki C, Zhang SL, Chan JSD, Filep JG. Peroxynitrite induces integrin-dependent adhesion of human neutrophils to endothelial cells via activation of the Raf-1/MEK/Erk pathway. FASEB J. 2001;15:25–27. doi: 10.1096/fj.00-0521fje. [DOI] [PubMed] [Google Scholar]
- Marias R, Wynne J, Treisman R. The SRF accessory protein Elk-1 contains a growth factor-regulated transcriptional activation domain. Cell. 1993;73:381–393. doi: 10.1016/0092-8674(93)90237-k. [DOI] [PubMed] [Google Scholar]
- Gardner AM, Lange-Carter CA, Vaillancourt RR, Johnson GL. Measuring activation of kinases in mitogen-activated protein kinase regulatory network. Meth Enzymol. 1994;238:258–270. doi: 10.1016/0076-6879(94)38024-4. [DOI] [PubMed] [Google Scholar]
- Taylor SJ, Shalloway D. Cell cycle-dependent activation of Ras. Curr Biol. 1996;6:1621–1627. doi: 10.1016/s0960-9822(02)70785-9. [DOI] [PubMed] [Google Scholar]
- Kaw S, Hecker M, Vane JR. The two step conversion of big endothelin 1 to endothelin 1 by subcellular fractions from human polymorphonuclear leukocytes. Proc Natl Acad Sci USA. 1992;89:6886–6890. doi: 10.1073/pnas.89.15.6886. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zouki C, Beauchamp M, Baron C, Filep JG. Prevention of in vitro neutrophil adhesion to endothelial cells through shedding of L-selectin by C-reactive protein and peptides derived from C-reactive protein. J Clin Invest. 1997;100:522–529. doi: 10.1172/JCI119561. [DOI] [PMC free article] [PubMed] [Google Scholar]