

Abstract
Established cell lines are invaluable for studying cell and molecular biological questions. A variety of human ovarian cancer (OC) cell lines exist, however, most have acquired significant genetic alterations from their cells of origin, including deletion of important cell cycle regulatory genes. In order to analyze signaling events related to cell cycle control in human OC, we have modified existing protocols for isolating and culturing OC cells from patient ascites fluid and normal ovarian surface epithelial (OSE) cells from benign ovarian tissue sections. These cells maintain an epithelial phenotype and can be manipulated experimentally for several passages before cellular senescence. An example using TGFb1 treatment of OC cells to examine signaling and target gene activation is presented.
Keywords: ovarian neoplasms; blotting, northern; ovary
Introduction
Primary cells isolated from patients are often considerably different from established cell lines of similar origin. The requirement to identify a method for isolation and growth of normal OSE and OC cells from human patients partially stems from the known genetic alterations in existing established cell lines, and the absence of animal models for epithelial OC [with the exception of aging chickens (1). Several methods for primary culture of OC cells isolated from patient ascites have been described in the literature, however these methods often require multistep procedures prior to seeding culture vessels with the isolated cells (2-5). We have found that simply mixing ascites fluid 1:1 with growth medium typically results in cultures of predominantly epithelial OC cells. This technique provides a simple, reliable procedure for establishing OC cell cultures. In addition, our modification of the original Auersperg technique (2) combined with an enzymatic digestion protocol (6) allows for easy isolation and culture of normal OSE. These cultures can be subcultured several times before cellular senescence occurs, thus making them useful for a variety of experimental manipulations.
Alteration in mRNA expression is commonly used to analyze transcriptional responses of genes to extracellular stimuli. Our laboratory used Northern analysis to measure transcriptional responses in normal OSE and OC cells treated with TGFb1 (7). Signal transduction by TGFb involves a cascade of molecules resulting in modification of gene transcription to elicit the cellular response [reviewed in (8)]. TGFb directly modulates the cell cycle by up-regulation of the cyclin dependent kinase inhibitor, p15Ink4B. We found that Northern analysis using cDNA probes was insensitive or produced high background signal for analyzing p15Ink4B mRNA expression, whereas complementary RNA (cRNA) probes worked very effectively. In addition, higher specific activity cRNA probes allowed us to use a small amount of RNA on our Northern blots, which is invaluable for procedures where the volume of tissue or cells is a limiting factor.
Materials and Methods
Primary human ovarian tissue and ascites
Institutional approval for research with human materials was received prior to the initiation of these studies (QEII Health Sciences Centre, Research Ethics Committee, #QE-RS-99-016; IWK/Grace Hospital Research Ethics). Normal OSE was retrieved from ovarian specimens excised from patients undergoing oophorectomy for benign conditions. Patient ascites was obtained from epithelial OC patients at the time of diagnostic laparotomy. All ascites samples were obtained from chemotherapeutically naïve patients with Stage III ovarian adenocarcinoma. These tumors were diagnosed by Dr. Shawn Murray (gynecologic pathologist at the QEII Hospital, Halifax, NS) as serous, clear cell, mucinous, and poorly differentiated.
Normal human OSE isolation and culture
The normal surface epithelial cells from solid ovarian specimens were isolated by a method combining two protocols (2,6). We typically receive surgical samples of ovarian tissue, which includes OSE, in sterile saline that range in size from 1-2 cm3. Processing of the ovarian tissue is conducted using aseptic techniques in a cell culture cabinet. The tissue was removed from the saline and immediately incubated in a 6 well tissue culture plate, OSE side down, in 2 ml Dispase II (Roche Molecular Biochemicals) for 30 minutes at 37 °C with occasional agitation. Sheets of OSE cell fragments were very gently scraped with a scalpel blade directly into complete growth medium [MCDB105 (Sigma)/M199 (GibCo) supplemented with 10% fetal bovine serum (FBS) and 100 units/ml penicillin, 100 μg/ml streptomycin] and allowed to adhere and grow for 4 days. All experiments using normal primary OSE were performed at culture passages 3-4.
Ovarian cancer cell isolation and culture
Primary human OC cells were isolated from patient ascites; we usually receive 2-4 litres of ascites fluid in sterile vacuum containers. For each flask of cells 20 ml of ascitic fluid was mixed with 20 ml growth medium (MCDB105/M199 supplemented with 10% FBS and 100 units/ml penicillin, 100 μg/ml streptomycin) and plated in T75 flasks (5-10 flasks per sample). Ascitic fluid usually contains a large number of red blood cells, however, they do not interfere with cell plating. Ovarian tumor cell clumps or grape-like clusters will be apparent in the ascitic fluid, which will eventually adhere to the cell culture plate surface. After 3-4 days the supernatant was removed and attached cells were re-fed growth medium. All experiments using primary OC cells were performed at culture passages 1-8.
RNA isolation and Northern Blot analysis
Total cellular RNA was isolated using the Genelute Mammalian Total RNA kit (Sigma-Aldrich, Oakville, ON, Canada). We have found commercial kits to be convenient and consistently produce high quality RNA. Two 10 cm plates of subconfluent (70-80%) OC cells were used for each treatment condition which typically yields 20-60 µg of total RNA. Ten micrograms of total RNA was separated on a 1.5% agarose formaldehyde gel and transferred to BrightStar Plus membrane (Ambion Inc., Austin, Tx). Blots were incubated with 1x106 cpm/ml cRNA probe overnight at 60 ºC in hybridization buffer (400 mM sodium phosphate, 1 mM EDTA, 0.5% SDS, 1 mg/ml BSA, 0.2 mg/ml yeast tRNA, 50% formamide) and washed 2 times for 1 hour at 60 ºC, and one time for 15 min at 75 ºC in wash solution (0.1% SDS, 0.1 x SSC, 1 mM EDTA). Signals were visualized by autoradiography. To examine transcriptional modulation of TGFb target genes, cells were treated with 4 pM (0.1ng/ml) TGFb1 (Sigma-Aldrich, Oakville, ON, Canada) for 4 h prior to harvesting.
cRNA probe synthesis
cRNA probes can be synthesized from cDNA clones/fragments subcloned into plasmids containing T3, T7, or SP6 RNA polymerase promoters [originally described in (9)]. The orientation of the cDNA fragment must be known in order to synthesize the appropriate strand of RNA complementary to the target mRNA. cRNA probes were radiolabeled by including α-32P-UTP in the cRNA synthesis reaction. Probes were gel purified from denaturing polyacrylamide gels, eluted overnight at 37 ºC in probe elution buffer (500 mM ammonium acetate, 1 mM EDTA, 0.2% SDS), and the specific activity of the probe was determined. For the p15Ink4B probe we PCR amplified, subcloned, and sequence verified a cDNA fragment corresponding to nucleotides 70-450 (relative to the start of translation).
Results and Discussion
Choice of medium for culturing human OSE and ovarian cancer cells
The most commonly used medium for growing normal OSE or OC cells is the mixture of MCDB105/M199 in a 1:1 ratio supplemented with FBS [10-20%; (2)]. Previous formulations used a combination of MCDB202:M199 (1:1) supplemented with a variety of agents including epidermal growth factor (EGF; 20ng/ml) and hydrocortisone (0.4 µg/ml), which could increase the OSE growth rate and doubling populations, however these agents eventually cause an irreversible modulation of the OSE to an atypical, fibroblast phenotype (2). MCDB105 (a modification of medium F12) was found to enhance the growth potential of normal OSE while maintaining the epithelial phenotype. Another choice of medium used to grow OC cells is DMEM/F12 medium (without phenol red) supplemented with 3% FBS, 5 µg/ml insulin, 50 nM ethanolamine, 50 nM phosphoethanolamine, 5 ng/ml EGF, 10 µg/ml transferrin (10). We found, however, that cells grown in MCDB105/M199 supplemented with FBS maintained the desired epithelial phenotype and could be passaged a greater number of times compared to cells grown in the DMEM/F12 medium.
Normal human OSE
In general we find that our technique produces monolayer cultures of cobblestone epithelial cells that rarely contain stromal contaminants (Fig. 1A & B). A typical problem when isolating OSE is scraping the ovarian section too firmly, which can result in removal of underlying stromal cells. Observing sheets of cells lifting during the scraping procedure is an indication of scraping too firmly. If present, stromal cells quickly out grow the epithelial cells and are easily visualized by microscopy (Fig. 1C). Depending on the size of the ovarian biopsy, the yield of OSE cells can range from 1000-2000 cells at the time of isolation. Cells are subcultured using 0.06% trypsin in 1xPBS, 0.5 mM EDTA. Once the cells have reached 90-95% confluence (approximately 7 days), they can be lifted and plated onto a single 60 mm dish; subsequent subculturing (every 2-3 days) will produce four 10 cm plates by passage 4 (2 x106 cells per plate). After passage 4, cells typically take on the flattened morphology of cells in senescence and fail to divide further (Fig. 1D).
Fig. 1.
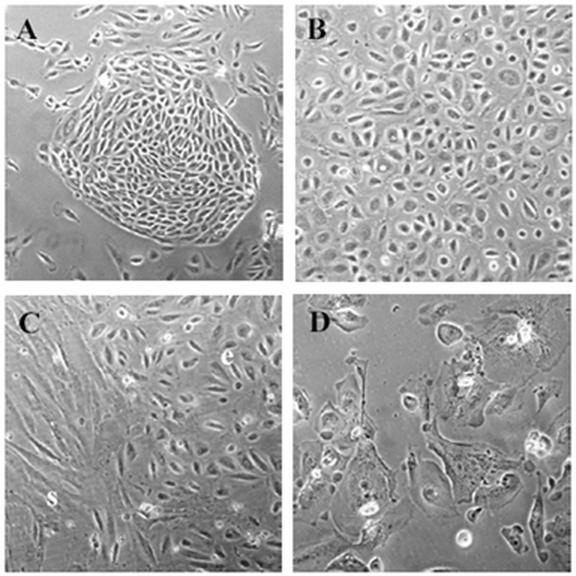
Primary human ovarian surface epithelium (OSE) morphology. A) Nest of OSE cells 4 days after initial plating. B) Monolayer of ‘cobblestone’ OSE at passage 1. C) Stromal cell (s) contamination of OSE (e) culture. D) Senescent OSE cells at passage 4. A = 100X magnification, B-D = 200X.
Ovarian cancer cell culture
Our method to grow OC cells isolated from ascites provides a rapid, convenient alternative to other multi-step procedures. We initially seed between 5-10 T75 flasks (100-200 ml ascites fluid), which, upon gaining confluence, provides us with a large number of cells for future experiments. We store several vials of low passage cells in liquid nitrogen, which will maintain the same growth characteristics as cells that remain unfrozen. One 100% confluent T75 flask of OC cells contains between 1-3 x 106 cells, and we would usually split one T75 flask to six 10 cm plates. These plates will reach 70-80% confluence within the next few days.
Tumor cells in ascitic fluid samples (n=34) were examined and diagnosed as serous, clear cell, mucinous, and poorly differentiated. The majority of the samples we have received were serous adenocarcinoma of the ovary and we have been able to establish primary cultures from 17 of 18 samples. Three additional samples that were diagnosed as serous adenocarcinoma were determined to be primary peritoneal tumors (tumors that did not originate in the ovary), however all three cultures grew with similar morphology and growth rate, and to a similar number of passages as the serous tumors originating in the ovary. Furthermore, we established cultures from cells in ascites diagnosed as clear cell adenocarcinoma (4 of 4 cultures), mucinous (2 of 3), poorly differentiated (2/2 ovarian and 3/3 primary peritoneal), and 1 malignant mixed Müllerian carcinosarcoma. In our experience the majority of the cells display a similar morphology in culture, grow at a similar rate, and can be passaged a similar number of times before becoming senescent.
The most common morphology of our primary ovarian cancer cell cultures is a cobblestone epithelial monolayer (Fig. 2A), however, other morphologies do occur, including a larger, flattened epithelial morphology (Fig. 2B). If stromal contamination occurs (Fig. 2C) these cells will eventually over grow any epithelial cells present.
Fig. 2.
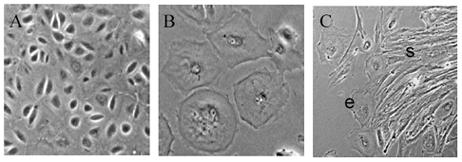
Primary ovarian cancer cell morphology. A) Typical cultures yield a cobblestone epithelial morphology (400X). B) Another common morphology of OC cultures are large, flattened epithelial cells (400X). C) Stromal cell (s) contamination can also occur, and these cells will eventually overtake the epithelial cells (e) (100X).
In our experience, the cobblestone epithelial cells grow well and are most useful for experimentation. Ovarian cancer cells have a limited growth potential (6-8 passages) and will eventually exhibit the flattened appearance of senescent cells that fail to divide (even after 30 days in culture). Therefore the optimal time to conduct most experiments is when the cells are at a low passage number. Although we usually isolate sufficient cells to conduct experiments in triplicate, the fact that the cells have limited cell doubling capacity suggests that more flasks of cells could be plated initially to generate more cell stocks. It can not be predicted, however, whether a given sample will yield a viable culture. Some of our ascites samples did not contain cells that grew in culture. Moreover, if a sample does contain a significant number of contaminating stromal cells, these cells will quickly outgrow the OC cells. Based on our experience, therefore, we balance the cost of initial plating with the potential for producing viable cultures that are useful for experimentation.
Other methods to isolate OC cells from ascites have utilized Percoll (Amersham Biosciences) density gradients and anti-CD45 immunomagnetic beads to separate red blood cells and leukocytes from individual OC cells and cell clusters (5). Our initial trials to isolate OC cells from ascites utilized a Percoll gradient (50-10%), however, we determined that despite our initial yield of total cells being higher, these cells had a lower plating efficiency and reduced capacity to adapt to growth in culture than if we simply plated the cells as described in this paper. We suggest that this may be due to a 'culture shock' when the cells are switched from the ascitic fluid environment to a defined medium. This problem could probably be circumvented if clarified ascitic fluid were used to supplement the growth medium. Furthermore, we did not find the presence of red blood cells to adversely affect the establishment of primary OC cell cultures. Indeed, we have often received samples of ascites fluid that was extremely bloody making it difficult to easily visualize cells or cell clusters upon initial seeding of the flasks. After 3-4 days, however, the medium was changed to reveal a sub-confluent monolayer of OC cells. Leukocytes are unlikely to adapt to growth conditions required for OC cells. Thus, special procedures to reduce the number of red blood cells or leukocytes can be conducted, however in our experience they are not necessary.
Markers to distinguish human OSE and ovarian cancer cells
There are no definitive markers for OSE or OC cells and thus a combination of markers can be used to distinguish these cells from potential contaminating cells, i.e. stromal fibroblasts, mesothelial cells, endothelial cells, leukocytes. Leukocytes are the least likely cells to cause concern due to the fact that they are unlikely to thrive in the OSE culture conditions or survive first passage of the cells. Endothelial cells can be easily distinguished immunocytochemically using anti-factor VIII. Stromal and/or pelvic mesothelial cells may be more difficult to distinguish because OSE or OC cells can take on a fibroblastic morphology when the cells are subconfluent, and normal OSE and pelvic mesothelium are quite similar morphologically and express similar epithelial and mesenchymal proteins. Examination of ascites cell spreads showed that some of our samples contained reactive or benign mesothelial cells, however the clusters of OC cells present in the ascites fluid vastly outnumbered these cells. The pathologic criteria we used to distinguish mesothelial from OC cells included a combination of the following criteria, i) high nuclear to cytoplasmic ratio, ii) large nucleolus (OC cells usually have macronucleoli), iii) cell size (OC cells were usually 10 times larger than reactive mesothelial cells, iv) mucin vacuoles (distinguished histochemically; not present in mesothelial cells), v) nuclear chromatin (sometimes more granular in OC cells), and vi) irregular nuclear membrane. In addition to these cellular features, a cocktail of markers can be used to immunocytochemically distinguish the desired cells; this information is detailed extensively in (11). It should also be noted that OSE cells could lose epithelial markers over time in culture (4).
Assessing mRNA expression
Our laboratory uses molecular and cellular biology techniques to assess various aspects of normal OSE and OC biology, including mRNA expression. We have found, however, that several of the target genes that we wished to examine by Northern analysis produced low levels of mRNA and detection using radiolabelled cDNA probes proved insensitive or presented a high background signal. Our attempts to produce Northerns having a specific signal with low background included increasing the amount of total RNA per lane (up to 40 µg), altering the stringency for hybridization or washing by varying the temperature or formulation of the washing buffer, or washing for longer periods of time. This usually resulted in either a higher background or loss of the signal. As an alternative approach we turned to the use of cRNA probes. Because the cRNA probe is synthesized with a-32P-UTP the probes have a higher specific activity than nick translated or random primed cDNA probes, and form more stable complexes with the target mRNA than similar cDNA:mRNA hybrids, allowing a higher level of stringency in the hybridization (60 oC with 50% formamide in the hybridization buffer) and wash (up to 75 oC, wash solution 0.1% SDS, 0.1 x SSC, 1 mM EDTA) procedures.
To conduct our experiments using Northern analysis we found that two 10 cm plates of subconfluent (70-80%) cells should be used for each treatment condition; our typical yield is 20-60 µg of total RNA. Total cellular RNA was isolated using the Genelute Mammalian Total RNA kit (Sigma-Aldrich, Oakville, ON, Canada), however, more traditional methods of RNA isolation, such as the single step guanidinium-thiocyanate-phenol-chloroform extraction method also work well (12). In addition, although we transfer our total RNA to BrightStar Plus membrane (Ambion Inc., Austin, TX), nitrocellulose or other charged nylon backed membranes can also be used.
We chose the cyclin dependent kinase inhibitor p15Ink4B gene to analyze the response of TGFb target genes involved in cell cycle regulation. Many established human OC cell lines have deleted the p15Ink4B gene and thus primary cell cultures are required to analyze the p15Ink4B response (7,13). In order to test whether OC cells could up-regulate p15Ink4B in response to TGFb1, we treated the cells with 4 pM TGFb1 for 4 h prior to harvesting total RNA. Using the p15Ink4B cDNA as a probe resulted in a very poor quality signal with high background (Fig. 3A). Our results using cRNA probes, however, clearly show that human OC cells transmit the TGFb signal to cause an up-regulation in the p15Ink4B mRNA (p15Ink4B generates 3.2 and 2.2kb mRNA signals; Fig. 3B).
Fig. 3.
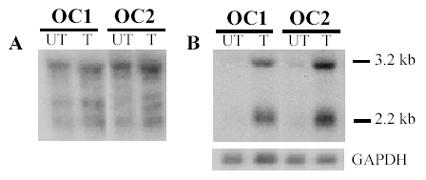
p15Ink4B mRNA expression increases in response to TGFβ1. Primary OC cells (OC1 & OC2) were untreated (UT) or treated (T) with 4 pM TGFβ1. A) Northern blot to detect p15Ink4B using cDNA probes produced poor quality signal with background, making results difficult to interpret. B) The same blot was re-probed using radiolabeled p15Ink4B cRNA which clearly improved results, providing a high quality signal and easily interpretable results. GAPDH mRNA is used as a loading control.
We found that the use of cRNA probes was crucial to examine mRNA expression of certain TGFb responsive genes. Thus we would advise the use of cRNA probes if initial attempts to detect mRNA with cDNA probes produces equivocal results. Because our studies involve investigation of cell cycle regulatory genes and established OC cell lines are of limited use for these studies, it was necessary to establish a method to consistently produce epithelial OC cells from patients. We use a modified protocol to isolate primary OC cells from ascites that involves a single step to reliably produce OC cultures. To compare events that occur in OC cells versus normal OSE, we also developed a modified technique to establish normal OSE cultures. Thus, we found that the combination of primary cell culture and cRNA probes for Northern analyses provided us with a effective experimental system for analyzing alterations in gene transcription in response to external stimuli.
Acknowledgments
The authors wish to acknowledge E.J. Campbell Dwyer for assistance with cRNA Northern analysis, Drs. R. Grimshaw and J. Bentley (QEII Health Science Centre) for providing human ovarian tumor samples, and Dr. T. F. Baskett and the staff of the QEII Ob/Gyn Unit for providing normal human ovarian tissue.
Appendix
Protocols
Isolation of Normal Ovarian Surface Epithelium
Working in laminar flow hood, place ovarian tissue OSE side down into 2 ml of Dispase II (Roche Biochemical) in a 35 mm well of a 6-well dish.
Place into incubator for 30 minutes, swirling the dish for 5-10 seconds every 10 minutes.
Add 2 ml of complete growth medium (MCDB/M199, 10% FBS, 100 U penicillin, 100 µg streptomycin) into a separate well of the same dish.
Transfer the ovarian tissue to the well containing the medium and gently rub the OSE surface with a sterile scalpel; leave these OSE cells in the well.
Draw off Dispase II from the other well and transfer to a sterile 15 ml tube and top up with complete medium.
Spin at 100 xg for 5 minutes at room temperature using a tabletop centrifuge.
Carefully aspirate the supernatant and resuspend pellet with complete medium and transfer to a separate well of the 6-well culture dish.
Place in incubator for 3-4 days before changing medium.
Freeze the remaining ovary tissue rapidly in liquid nitrogen and store at -80 oC.
Isolation of Ovarian Cancer Cells from Ascites
Aliquot 20 ml of ascitic fluid into a T75 flask containing 20 ml growth medium (MCDB105/M199 supplemented with 10% FBS and 100 units/ml penicillin, 100 mg/ml streptomycin). Ovarian tumor cell clumps or grape-like clusters will be apparent in the ascitic fluid, which will eventually adhere to the cell culture plate surface.
After 3-4 days remove supernatant, wash with 1X PBS, and feed with growth medium.
32P cRNA Probe Generation
(reaction is set up at room temp so spermidine in the transcription buffer does not precipitate DNA.)
Use sterile screw cap tubes for the reaction to reduce possible evaporation.
1 µg DNA template
2.0 µl 10 mM NTP stock: ATP, CTP, GTP
4.0 µl 5X transcription buffer (MBI)
0.8 µl RNase inhibitor
5.0 µl a-32P-UTP
1.0 µl RNA polymerase: Add last.
20 µl final volume (use sterile H2O to make up the reaction volume to 20 µl)
NOTE: GAPDH can be used as a control to normalize loading of the Northern blot. Since it is a highly abundant message, increase the amount of cold UTP and decrease the amount of a-32P-UTP, eg. 1.5-2.0 µl of 32P UTP and 3 µl of 0.1 mM cold UTP.
Heat the reaction for 1 h at 37 oC.
Add 2 µl of 1U/µl Dnase (RNase free) and incubate for 15 min at 37 oC.
Inactivate the reaction by heating to 95oC for 5 minutes.
For best results, the probe should be gel purified (7M urea/6% polyacrylamide gel). Add 20 µl of gel load buffer (95% formamide, 18 mM EDTA, 0.025% SDS, 0.025% bromophenol blue, 0.025% xylene cyanol) to probe. Put on ice until gel is ready.
Pre-run gel approximately 1 h at 35 mA before loading probe. Heat probe 90-95oC for 4 min and immediately place on ice.
Rinse away urea in gel wells. Load sample. Run at 35 mA for 1-1.5 h. The light blue xylene cyanol in the loading buffer runs at approximately 130 bases. When gel is done carefully take plates apart. Wrap plate with gel in plastic wrap being careful to avoid wrinkles where sample lane is. In a darkroom lay film, intensifying screen and plexiglass over gel. Leave approximately 5-10 min Develop film and outline where the transcript is on film. Cut flap in the outline with razor blade. Line up film and gel; cut out gel band containing full length transcript with razor blade. Transfer gel to 1.5 ml screw cap tube, add 400-500 µl of probe elution buffer (500 mM ammonium acetate, 1 mM EDTA, 0.2% SDS). Elute probe overnight at 37 oC. Pellet gel fragments by microcentrifugation and transfer supernatant to a new 1.5 ml tube. Count 1 µl. We use 1x106 cpm of probe per ml of hybridization solution.
Block the blot by prehyridizing in hybridization buffer (400 mM sodium phosphate, 1 mM EDTA, 0.5% SDS, 1 mg/ml BSA, 0.2 mg/ml yeast tRNA, 50% formamide) for at least 30 min prior to adding cRNA probe. Heat cRNA probe for 5 min at 90-95 oC. Immediately add the probe to the blot and hybridize overnight (or at least 4 h) at 60oC. After hybridization blots are washed at least 3 time 15 min in wash buffer (0.1 X SSC, 0.1% SDS, 1 mM EDTA) at 60oC. Washes can be for longer periods of time and at increasing temperatures up to 75 oC.
References
- Fredrickson TN. Ovarian tumors of the hen. Environ Health Perspect. 1987;73:35–51. doi: 10.1289/ehp.877335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kruk PA, Maines-Bandiera SL, Auersperg N. A simplified method to culture human ovarian surface epithelium. Lab Invest. 1990;63:132–136. [PubMed] [Google Scholar]
- Hirte HW, Clark DA, Mazurka J, O'Connell G, Rusthoven J. A rapid and simple method for the purification of tumor cells from ascitic fluid of ovarian carcinoma. Gynecol Oncol. 1992;44:223–226. doi: 10.1016/0090-8258(92)90046-l. [DOI] [PubMed] [Google Scholar]
- Auersperg N, Maines-Bandiera SL, Dyck HG, Kruk PA. Characterization of cultured human ovarian surface epithelial cells: phenotypic plasticity and premalignant changes. Lab Invest. 1994;71:510–518. [PubMed] [Google Scholar]
- Hurteau JA, Rodriguez GC, Whitaker RS, Shah S, Mills G, Bast RC, Berchuk A. Transforming growth factor-ß inhibits proliferation of human ovarian cancer cells obtained from ascites. Cancer. 1994;74:93–99. doi: 10.1002/1097-0142(19940701)74:1<93::aid-cncr2820740117>3.0.co;2-p. [DOI] [PubMed] [Google Scholar]
- Quirk SM, Cowan RG, Huber SH. Fas antigen-mediated apoptosis of ovarian surface epithelial cells. Endocrinology. 1997;138:4558–4566. doi: 10.1210/endo.138.11.5508. [DOI] [PubMed] [Google Scholar]
- Dunfield LD, Campbell Dwyer EJ, Nachtigal MW. TGFb-induced Smad signaling remains intact in primary human ovarian cancer cells. Endocrinology. 2002;143:1174–1181. doi: 10.1210/endo.143.4.8733. [DOI] [PubMed] [Google Scholar]
- Massague J, Blain SW, Lo RS. TGFbeta signaling in growth control, cancer, and heritable disorders. Cell. 2000;103:295–309. doi: 10.1016/s0092-8674(00)00121-5. [DOI] [PubMed] [Google Scholar]
- Krieg PA, Melton DA. Functional messenger RNAs are produced by SP6 in vitro transcription of cloned cDNAs. Nucleic Acids Res. 1984;12:7057–7070. doi: 10.1093/nar/12.18.7057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Evangelou A, Jindal SK, Brown TJ, Letarte M. Down-regulation of transforming growth factor beta receptors by androgen in ovarian cancer cells. Cancer Res. 2000;60:929–935. [PubMed] [Google Scholar]
- Auersperg N, Wong AS, Choi KC, Leung PC. Ovarian surface epithelium: biology, endocrinology, and pathology. Endocrine Reviews. 2001;22:255–288. doi: 10.1210/edrv.22.2.0422. [DOI] [PubMed] [Google Scholar]
- Chomczynski P, Sacchi N. Single-step method of RNA isolation by acid guanidinium thiocyanate- phenol-chloroform extraction. Anal Biochem. 1987;162:156–159. doi: 10.1006/abio.1987.9999. [DOI] [PubMed] [Google Scholar]
- Watson JE, Gabra H, Taylor KJ, Rabiasz GJ, Morrison H, Perry P, Smyth JF, Porteous DJ. Identification and characterization of a homozygous deletion found in ovarian ascites by representational difference analysis. Genome Res. 1999;9:226–233. [PubMed] [Google Scholar]