

Abstract
The cell cycle machinery consists of regulatory proteins that control the progression through the cell cycle ensuring that DNA replication alternates with DNA segregation in mitosis to maintain cell integrity. Some of these key regulators have to be degraded at each cell cycle to prevent cellular dysfunction. Mitotic exit requires the inactivation of cyclin dependent kinase1 (cdk1) and it is the degradation of the cyclin subunit that inactivates the kinase. Cyclin degradation has been well characterized and it was shown that it is ubiquitin proteasome pathway that leads to the elimination of cyclins. By now, many other regulatory proteins were shown to be degraded by the same pathway, among them members of the aurora kinase family, degraded many other regulatory proteins. Aurora kinases are involved in mitotic spindle formation as well as in cytokinesis. The abundance and activity of the kinase is precisely regulated during the cell cycle. To understand how proteolysis regulates transitions through the cell cycle we describe two assays for ubiquitination and degradation of xenopus aurora kinase A using extracts from xenopus eggs or somatic cell lines.
Keywords: Xenopus, ubiquitin
Introduction
The ubiquitin/proteasome pathway is the major proteolytic system in the eukaryotic cell for selective degradation of short lived regulatory proteins (1,2). This pathway consists of a ubiquitin activating enzyme (E1) a ubiquitin conjugating enzyme (E2) and a ubiquitin ligase (E3). The 26S proteasome then degrades the ubiquitinated proteins (3,4).
The common feature of proteasome mediated protein degradation is the covalent attachment of ubiquitin, a 76 amino acids protein, to an epsilon amino-group of lysin residues of proteins targeted for degradation. Repeated addition of ubiquitin molecules to the substrate leads to the formation of polyubiquitin chains that are recognized by the 26S Proteasome (5). In contrast to the E1 and E2 enzymes the E3 ubiquitin ligases display substrate specificity. The E3 enzyme for the ubiquitination of several mitotic proteins has been purified from Xenopus egg extracts and called APC/C (Anaphase Promoting Complex/Cyclosome) (6). The activity of APC is regulated by two additional proteins, cdc20 and cdh1. While cdc20 is a mitosis specific activator, cdh1 maintains APC activity in G1 phase (7,8). Most known substrates of APC contain a destruction box (D box) and are degraded in mitosis or G1 phase of the cell cycle. The Cdc20 dependent pathway targets proteins that possess a cyclin-like “destruction box” which sequence contains two invariable conserved residues, an arginine and a leucine, separated by two amino-acids (9). The minimal required sequence RxxL is generally localized in the N-terminal domain of the protein (9,10). The “D boxes” have been also found to be important for several non-cyclin proteins that are mitosis-specifically ubiquitinated and degradated (11-13).
For example, the mitotic aurora-A kinase whose over-expression is detected in tumour cells undergoes cell cycle dependent regulation. Aurora must also be inactivated or degraded when the cell proceeds in G1 phase. The over-expression of aurora in cultured cells produces a transformed phenotype, and transformed cells induce tumour formation when implanted in nude mice, indicating that the cell cannot function with a high level of aurora kinase activity (14). Expression of mammalian aurora proteins is cell cycle regulated, being maximal in G2/M phases (15-18).
When cells exit from metaphase many proteins involved in the regulation of mitotic events are degraded via anaphase promoting complexes (APC) through a ubiquitin dependent pathway (7,9,19). Human aurora-A has been found to be associated with APC/C (16), but although the kinase is ubiquitinated in vivo and degraded through a mechanism sensitive to APC/C inhibitors, a functional destruction box has now been identified.
We report an assay that permits the ubiquitination analysis of a specific protein. Additonally, a degradation assay is performed to analyze the in vitro behaviour of this protein. This method analysis of any cell cycle regulated protein.
Materials and Methods
Reagents
1C1 and 6E3 monoclonal antibodies were directed against pAurora-A (20,21). 1C1 detects both the histidine-tagged and the endogenous aurora-A whereas 6E3 detects only the histidine-tagged protein (20). Bromodeoxyuridine (BrdU), primary monoclonal anti-BrdU antibody, aphidicolin, and trypsin-EDTA solution were obtained from Sigma. A secondary Texas-red conjugated antibody was obtained from Interchim. Leibovitz 15 cell culture medium (L-15), antibiotic-antimycotic solution (penicillin-streptomycin-amphotericin) was supplied by GIBCO BRL laboratories. Foetal calf serum was obtained from Biotimes.
Cell culture
Xenopus XL2 cells were cultured and synchronized as previously described (22). Xenopus cells were cultured at a density of 105 cells in 75 cm2 flasks in L-15 medium with 10% FCS until cell attachment. The cells were then washed three times with PBS and cultured in serum-free medium for 24 h. Flow cytometry was used to determine the degree of cell synchronization. Cells were synchronized for each phase as described above. For each step of synchronization, the cells were incubated in 40 µM BrdU, trypsinized and cell suspensions were centrifuged at 3,000 x g for 5 min, followed by fixation in ethanol at -20°C for 1 h. Fixed cells were treated with the anti BrdU antibody for 1 h (1/200) at room temperature and then with Texas-red conjugated immunoglobulin antimouse (1/50) for 1 h. Samples were stored at -20°C until analysis. Cells were stained with 10 µg/ml propidium iodide (PI) and DNA content was analyzed on a FACS (Becton Dickinson) with two fluorescence photomultipliers equipped with an argon laser operating at 15 mW, generating a light of 488 nm wavelength. PI emission was measured at wavelengths between 620 and 655 nm. Cell cycle analysis was performed with multicycle program.
Recombinant proteins
The aurora-A encoding sequence was inserted in the bacterial expression vector pET21 (Novagen) as previously described (21). In this study we used a construct in which the two cyclin-like “destruction boxes” were deleted by restriction enzyme digestions (11). All histidine-tagged recombinant proteins were purified from BL21(DE3)pLysS by Ni-NTA-agarose affinity chromatography following manufacturer's instructions (Qiagen S.A.). All the purified proteins proved to be soluble and were conserved in a buffer containing 20 mM Tris, pH7.5, 500 mM NaCl, and 50% glycerol at -20°C.
In vitro ubiquitination assays
The DNA coding for aurora-A wt (wild type) inserted in Bluescript was in vitro transcribed/translated in reticulocyte lysate at 30°C for 2 hours using a TNT kit (ex: Promega T7 quick translation kit). 2 µl of the translation reaction was added to a ubiquitination mix containing 40 mM Tris pH 7.5, 5 mM MgCl2, 1 mM DTT, 10% glycerol, 10 mM creatine phosphate, 0.1 µg/ml creatine kinase, 0.5 mM ATP, 1 µM okadaic acid, 1 µM ubiquitin aldehyde (Calbiochem), 1 mg/ml methylated ubiquitin (Sigma Chemicals) and 3.5 µl of Xenopus egg mitotic extract prepared according to Murray (1991) (23) in a total volume of 10 µl. The reaction was incubated for 60 minutes at room temperature, stopped by the addition of Laemmli sample buffer and heated at 90°C for 10 min. Reaction products were separated by SDS-polyacrylamide gel electrophoresis and electrotransferred onto nitrocellulose membrane. Aurora-A (wt) and aurora-A ubiquitin conjugates were visualized by Western blots using the 6E3 monoclonal antibody.
Cell extract preparation
Cell extracts were prepared as described in Bastians et al (1999) (24) with some modifications. Cells were harvested by trypsin treatment and washed twice in ice cold PBS (136 mM NaCl, 26 mM KCl, 2 mM Na2HPO4, 2 mM KH2PO4, pH 7.2) and once in low salt buffer (LSB: 50 mM Hepes-NaOH pH7.5, 5 mM KCl, 1.5 mM MgCl2, 1 mM DTT) supplemented with protease inhibitors mixture from Sigma Chemicals. The pellet was then resuspended in a lysis solution (50mM Hepes-NaOH pH7.4, 5mM KCl, 1.5mM MgCl2, 1mM DTT, 5 mM NaF, 5 µg/ml leupeptin, 5 µg/ml pepstatin, 5 µg/ml chymostatin). The cells were homogenized in the buffer, incubated on ice for 20 min and lysed by sonication at 4°C. Cellular debris was removed by centrifugation at 14,000 x g for 30 min. The supernatant was aliquoted and frozen in liquid nitrogen and stored at -80°C.
In vitro degradation assay
2 µl of thawed cell extract were supplemented with an ATP regenerating system (1.5 mM ATP, 40 mM phospho-creatine, 80 µg/ml creatine kinase from Sigma Chemicals) and incubated with the recombinant purified protein in 7.4 mM Hepes pH 7.4, 5 mM KCl, 1.5 mM MgCl2, 1 mM DTT. The amount of recombinant protein added to the extract never exceeded the amount of endogenous protein to avoid any saturation of the degradation machinery. The reaction was incubated at 30°C and aliquots were taken at various time points. Protein degradation products were mixed with Laemmli sample buffers and separated on a 12.5% SDS-polyacrylamide gel electrophoresis (7 cm x 12 cm), The gels were then electrotransferred on nitrocellulose membrane, in Tris-glycine 100mM, methanol 20% buffer. The degradation product was then detected by Western Blot using the appropriate antibody followed by chemoluminescence and Quantification with the Image Quant software. Inhibition of the 26S proteasome was achieved by preincubating the cell extract with 40 µM ALLN (acetyl-leucyl-leucyl-norleucinal) during 30 min at 4°C (Calbiochem, San Diego, CA).
Western blot analysis
After electrophoresis and transfer onto the nitrocellulose membrane (Amersham Pharmacia Biotech) by standard methods, the membranes were blocked in TBST (20 mM Tris, pH 7.6, 137 mM NaCl, 0.1% Tween) containing 5% skimmed milk for 2 hours at 4°C. The membranes were incubated with specific monoclonal antibodies (1C1 or 6E3) for 1 hour at 4°C in TBST containing 2.5% skimmed milk and washed for 1 hour at 20°C in TBST containing 2.5% skimmed milk. The membranes were then incubated with peroxydase conjugated secondary antibody (Sigma Chemicals) for 2 hours in TBST with 2.5% skimmed milk. The immunocomplexes were revealed using peroxydase chemoluminescent substrates according to the manufacturer's instructions (NEN Life Sciences) and analyzed on a FluoroImager (Molecular Dynamics) in order to quantify the signal.
Results and Discussion
We attempted to determine if the overall level of the aurora-A kinase is constant throughout the cell cycle. According to the protocol describe previously, the XL2 cells were synchronized at various stages of the cell cycle (20). Sequential use of serum deprivation, aphidicolin treatment, release from S block and incubation of the cells with ALLN (acetyl leucyl leucyl norleucinal-proteasome inhibitor) arrested the cells in G0, G1/S, G2 and M phase respectively. After release, the cells started another cell cycle and the degree of synchronization was determined by FACS analysis. The protein levels of the corresponding cell cycle phases were analyzed by western blotting.
Using the anti aurora-A antibody 1C1, we analyzed aurora-A protein levels at the different stages of the cell cycle (Fig. 1A). In extracts from cells released from serum starvation, the protein was poorly detected suggesting that the protein is absent from the G1 cells. In the extract prepared from cells blocked in S phase by aphidicholin treatment, the western blot revealed that aurora-A increases in this stage. Accumulation of the aurora-A occurred 6 hours after release from the aphidicholin block. At this time, a large percentage of the cells are in G2 phase. Even higher amount of aurora-A is detected when cells were blocked in M phase with ALLN (acetyl-leucyl-leucyl norleucinal). When cells were released from the ALLN block and progressed into another cell cycle, the amount of aurora-A declined rapidly. Six hours after release from the ALLN block, 85% of the cells were in G1 phase and the amount of pAurora-A was hardly detectable. For quantification, protein levels were normalized with γ-tubulin whose protein levels do not vary during the cell cycle (Fig. 1B).
Fig. 1.
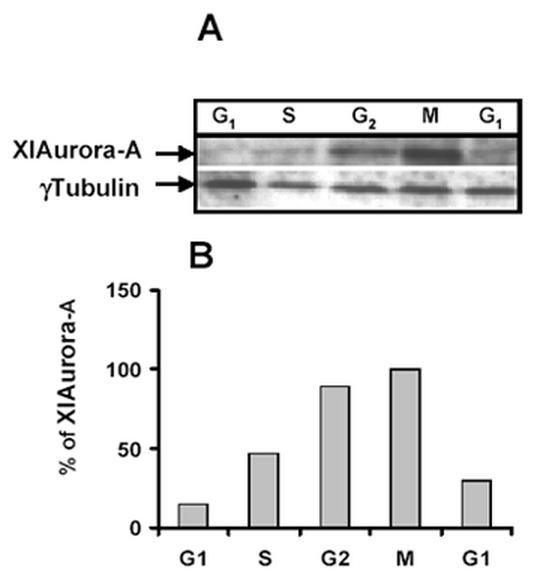
Cell cycle-dependent expression of aurora A protein. XL2 cells were blocked at various stages of the cell cycle by serum starvation or treatment with the different drugs as described in material and methods. After release from the block, cells were harvested at different stages of the cell cycle. The cell lysates prepared from 5x105 cells were separated by SDS PAGE and analyzed by Western blot with XlAurora A antibody and anti γ tubulin as a control. The appropriate portion of a blot (according to the molecular weight of proteins to be analyzed) was separately probed with anti X1Aurora-A (dilution: 1/500) and anti γ tubulin (dilution: 1/200) for the internal control of quantity of protein (Fig. 1A). The cell cycle distribution was analyzed by BrdU analysis. Percentage of cells in different phases of the cell cycle was calculated (Fig. 1B). The membrane has been scanned on a fluoroimager (Molecular Dynamics) and quantification of the protein has been realized with Image Quant.
Aurora-A is degraded in G1 phase by the ubiquitin-proteasome pathway
The above results indicate that pAurora-A starts to accumulate in S phase reaching its maximum in G2/M and disappears as cells enter in G1 of the next cell cycle. This leads to the suggestion that the decrease in aurora-A protein levels is due to an increase in its degradation rate. Sequence analysis of aurora-A revealed the presence of a putative destruction box similar to that found in mitotic cyclin implicating that aurora-A is also degraded by the ubiquitin /proteasome pathway via the APC complex (Fig. 2).
Fig. 2.
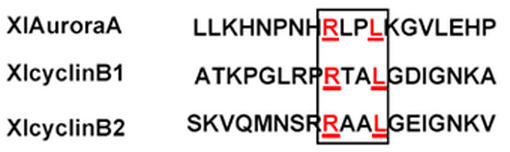
Diagram to compare the amino acid sequences of the destruction boxes and flanking sequences of cyclin B1, cyclin B2 and aurora-A. The destruction boxes are marked by a rectangle and the conserved amino acids are indicated in bold.
We therefore investigated if aurora-A is ubiquitinated like the cyclins during M phase. We established an ubiqutination assay in a metaphase arrested Xenopus egg extracts (CSF extract). This extract is known to contain all the enzymes required for ubiquitination (described in the introduction) necessary for mitotic degradation. The ubiquitination reaction was performed with in vitro translated aurora-A the “wild type” or the “destruction box mutant” (data not shown) in the presence of ubiquitin aldehyde and methylated ubiquitin. Ubiquitin aldehyde was added to the reaction to compete with isopeptidases, which remove ubiquitin molecules from its substrates. Methylated ubiquitin was used to prevent the formation of polyubiquitin chains that would result in distinct higher molecular weight bands rather then a higher molecular weight smear. Although the presence of the CSF extract was not absolutely required for the ubiquitination its presence significantly stimulated the reaction (compare Fig. 3, Lanes 1 and 4). Ubiquitination of aurora-A could not be detected in the absence of ubiquitin aldehyde and methylated ubiquitin (Fig. 3, Lane 2) or in the in vitro mock-translated reaction (Fig. 3, Lane 3). In contrast, when ubiquitin aldehyde and methylated ubiquitin was added to aurora-A and incubated in the egg extract, ubiquitinated aurora-A conjugates were detected (Fig. 3, Lane 1).
Fig. 3.
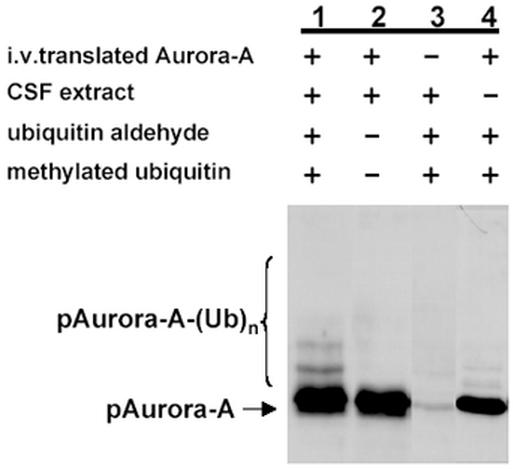
Ubiquitination of p Aurora A in a xenopus cell extract. In vitro-translated pAurora A was incubated in Xenopus CSF arrested eggs extracts (35 µg of protein) in the presence of 1µM ubiquitin aldehyde and 1mg/ml of methylated ubiquitin (Lane 1). Lane 2: ubiquitin aldehyde and methylated ubiquitin were omitted from the reaction. Lane 3: without in vitro translation of pAurora-A. Lane 4: without CSF extract the reactions were stopped, analyzed on SDS-PAGE and transferred onto nitrocellulose membrane. Western blot analysis was performed with the monoclonal antibody ICI (dilution 1:500) in order to detect the pAurora-A-ubiquitin conjugates (Published in FEBS Lett, 508: 149-152, 2001 and used with permission).
Degradation assay in cell extract
We demonstrated that Aurora A is ubiquitinated and therefore asked whether we could also show that is degraded in a cell free extract. As the protein disappears in G1 phase, we could not use metaphase arrested CSF even if the extract was released into G1 by the addition of calcium as aurora-A is not degraded in xenopus egg (data not shown). We decided to use a cellular extract from xenopus XL2 cells arrested in G1 phase. For this assay, we used purified proteins expressed in bacteria as it was easier to adjust the protein concentration than would be possible with the in vitro translated proteins.
The amount of recombinant pAurora-A (wt and DB mutant) added to the extract was carefully estimated to avoid any saturation of the degradation machinery (Fig. 4A). Xenopus cells were synchronized in G1 phase by serum deprivation and cell extracts were prepared as described in materials and methods. The recombinant pAurora-A (wt and BD mutants) was incubated in the G1 cell extract. Degradation of recombinant proteins was followed at different time points by western blot analysis. Fig. 4B shows that pAurora-A wild type was degraded more rapidly than the destruction box mutant (pAurora-A DD1D2). When incubated in the G1 cell extract, 50% of the added wt pAurora-A was degraded within the first 30 minutes (Fig. 4B, first panel). Two hours after incubation, the protein level was reduced to 20% while there was no decrease in the amount of the destruction box mutant.
Fig. 4.
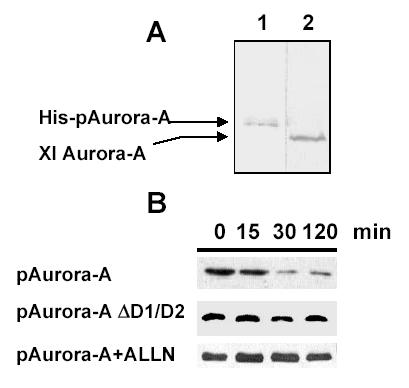
In vitro degradation assay of the ubiquitinated protein pAuroraA. A specific amount of the recombinant protein (Lane 1) was analyzed on a 12.5% polyacrylamide gel and compared with the endogenous protein from a G1 cell extract (Lane 2). Western blot analysis was performed with the monoclonal antibody 1C1 (dilution 1:500) in order to detect the recombinant pAurora-A and the endogenous protein.Fig. 4B: XL2 cells were arrested in G1 phase by serum starvation. Cell extracts were prepared with lysis buffer complemented with an ATP regenerating system and incubated in the presence of pAurora A wild type or pAurora A mdb (mutated destruction box). Samples were analysed on a 12.5% SDS-PAGE followed by a western blot performed with the 1C1 monoclonal antibody. Degradation kinetics of the various recombinant aurora-A proteins containing potential destruction box deletions were performed in XL2 cell extracts prepared from G1 cells. The reaction was incubated at 30°C. The specificity of the degradation was controlled by addition of the APC/C inhibitor ALLN. Reaction products were separated by 12.5% SDS-polyacrylamide gel electrophoresis and analysed by western blot using the 1C1 monoclonal antibody (1:200). Aliquots of the reaction were taken at 0, 15, 30 and 120 minutes. Western blots were quantified with Imag Quant and the amount of protein detected at various time points was expressed as a percentage of the protein detected at t = 0 min (input).
In order to prove that the observed degradation was due to the proteasome activity, the reaction was also performed in the presence of the proteasome inhibitor ALLN (acetyl leucyl leucyl norleucinal). Under these conditions pAurora-A was no longer degraded (Fig. 4B, third panel).
In summary, the paper describes two in vitro functional assays to analyse the ubiquitination / degradation pathway of a cell cycle regulated protein. The ubiquitination assay is performed with xenopus egg extract and allows the analysis of the ubiquitination of in vitro translated proteins. Although xenopus extracts are adequate to study the mechanism of ubiquitination, somatic cell extracts are more applicable to analyze the mechanism of degradation of a cell cycle regulated protein as one can prepare a cell extract at any specific stage of the cell cycle.
Acknowledgments
This research work was supported by the Centre National de la Recherche Scientifique, the Association pour la Recherche contre le Cancer, the Ligue Nationale contre le Cancer, and the Conseil Régional Bretagne. G.P. is a fellow of the French Government.
Appendix
Protocols
Cell culture
Xenopus cells were cultured in 100 x 20 mm culture dish, (Falcon, BD Science) in L-15 medium (Leibovitz 15 –GIBCO BRL) 10% FCS (Biological Industries - European grade filtered on 0.0.07 µM) antibiotic and antimycotic (penicillin 10000 units /ml; streptomycin 10000 µg/ml; amphotericineB 25 µg/ml) from GIBCO-BRL Lifes biotechnologies.
The cells were then washed three times with phosphate buffered saline PBS Dulbeccos without Ca2+and Mg 2+.
-
XL2 cells in the different phases of the cell cycle were obtained as follows:
- Wash the cells four times with PBS.
- Incubate the cells for 24h hours at 25°C in serum free medium L-15 to obtain cells in G1 phase.
- Wash the cell culture three times with PBS and incubate the cells at 25°C with full medium and aphidicholin (Sigma-Aldrich) at 2µg/ml for 30 h to obtain cells in S phase.
- Wash the cells three times with PBS and incubate them for 8 hours at 25°C in full medium to obtain cells in G2 phase.
At the end, incubate the cells for 7 hours at 25°C in the presence of full medium added with ALLN 40µg/ml to accumulate cells in M phase.
FACS analysis
For each step of synchronization, the cells were incubated in 40 µM BrdU, trypsinized and cell suspensions were centrifuged at 3,000xg for 5 min, fixed in cold ethanol 70°at -20°C for 1 h.
All the experiments are performed in 12 x 75 mm tubes (Falcon recommended for flow cytometers manufactured by Becton Dickinson). The cells were washed three times with ice cold PBS, and treated with HCl 4N for 30 min at room temperature (RT) and washed ten times with ice cold PBS in order to eliminate the HCl until the pH of the solution is 7.5. Then incubate the cells with PBT (PBS with 0; 2% Tween 20 (Sigma) and 1% BSA (Sigma)) during 30 min at room temperature.
The antibody anti-BrdU was diluted in PBT (1/30) and incubated with the cells 30 min at RT. The secondary antibody, texas-red conjugated immunoglobulin antimouse diluted at 1/40 was added to the mixture during 1 h at RT.
The cells were then washed with PBS and treated with propidium iodide (IP) to a final concentration of 10µg/ml. DNA content was analysed on a FACS (Becton Dickinson) with two fluorescence photomultipliers equipped with an argon laser operating at 15 mW, generating a light of 488 nm wavelength. PI emission was measured at wavelengths between 620 and 655 nm. Cell cycle analysis was performed with multicycle program.
Recombinant protein production
All the proteins are produced in bacteria as the following procedure.
The DNA sequence was inserted in the bacterial expression vector pET21 (Novagen). The transformed BL21(DE3)pLysS bacteria were cultivated for 2 hours at 37°C in 2XTY medium (2XTY = bacto Agar 1.6%; bacto yeast extract 1%; and NaCl 85 mM pH 7.5) and 1% glycerol until the DO600 reached 0.7 units. Then the mixture was cooled to 25°C and the induction was performed under IPTG (Isoprpyl-bthiogalactopyranoside - Euromedex) at a final concentration of 1 mM at 25°C for 2 hours.
At the end of incubation the culture medium was centrifuged at 4°C for 30 min at 5000 rpm (JA20-Beckman). The pellet was washed twice with PBS and was frozen at -80°C for 24 hours.
The pellet was then resuspended in IMAC 5 (IMAC = Tris/Hcl 20 mM , Nacl 500mM , Glycerol 10%) and Imidazol (sigma) 5mM containing lysozyme (Sigam) 1mg/ml and PMSF (phenylMethylSulfonylFluoride-Sigma) at a final concentration of 1 mM. The solution was sonicated and centrifuged 30 min at 9000 rpm in 50 ml Oakridge tube (JA-20 Beckman).
The final supernatant containing the histidine-tagged recombinant protein was purified from by Ni-NTA-agarose affinity chromatography (Qiagen S.A.). 100µl of agarose was loaded in a seringue and washed three times with the IMAC 5 buffer.
The protein supernatant was loaded on the column. The column was then washed twice with 20 volumes of IMAC 5 and IMAC 20 (IMAC + 20 mM imidazol). The histidine tagged protein was eluted with small volumes of IMAC 250 (IMAC + 250 mM imidazol). All the purified proteins prooved to be soluble were conserved in a buffer containing 20 mM Tris, pH7.5, 500 mM NaCl, and 50 % glycerol at -20°C.
In vitro ubiquitination assay
The DNA coding for the proteins was inserted in Bluescript and was in vitro transcribed/translated in reticulocyte lysate at 30°C for 2 hours using a TNT kit (ex: Promega T7 quick translation kit). The translation has to be realised immediatly before the ubiquitination assay and cannot be stored at minus -80°C for another ubiquitination assay.
2 µl of the translation reaction were added to a ubiquitination mix containing 40 mM Tris pH 7.5, 5 mM MgCl2, 1 mM DTT, 10% glycerol, 10 mM creatine phosphate, 0.1 µg/ml creatine kinase, 0.5 mM ATP, 1 µM okadaic acid, 1 µM ubiquitin aldehyde (Calbiochem), 1 mg/ml methylated ubiquitin (Sigma Chemicals) and 3.5 µl of Xenopus egg mitotic extract (Murray et al., Nature, 339: 280-286, 1989).
The reaction was incubated for 60 minutes at room temperature, stopped by the addition of Laemmli sample buffer and heated at 90°C for 10 min. Reaction products were separated by SDS-polyacrylamide gel electrophoresis and electrotransferred onto nitrocellulose membrane.
Cell extract preparation
Cells synchronized as previously described were harvested by trypsin treatment and washed twice in ice cold PBS (136 mM NaCl, 26 mM KCl, 2 mM Na2HPO4, 2 mM KH2PO4, pH 7.2).
The cells were resuspended in low salt buffer (LSB: 50 mM Hepes-NaOH pH7.5, 5 mM KCl, 1.5 mM MgCl2, 1 mM DTT) supplemented with protease inhibitors mixture from Sigma Chemicals The cells were homogenised in the buffer, incubated on ice for 20 min and lysed by sonication at 4°C. The mixture was centrifuged for 30 min at 15000 rpm, 4°C in a microfuge (Hearaus).
The pellet was then resuspended in a lysis solution (50 mM Hepes-NaOH pH7.4, 5mM KCl, 1.5mM MgCl2, 1mM DTT, 5 mM NaF, 5 µg/ml leupeptin, 5 µg/ml pepstatin, 5 µg/ml chymostatin). The supernatant was aliquoted and frozen in liquid nitrogen and stored at -80°C.
Degradation assay
2 µl of thawed XL2 cell extract were supplemented with an ATP regenerating system (1.5 mM ATP, 40 mM phospho-creatine, 80 µg/ml creatine kinase) (Sigma Chemicals) and incubated with the recombinant purified protein in 7.4 mM Hepes pH 7.4, 5 mM KCl, 1.5 mM MgCl2, 1 mM DTT. The amount of recombinant protein added to the extract has to be controlled comparing to the amount of endogenous protein in order to avoid any saturation of the degradation machinery.
The reaction was incubated at 30°C and aliquots were taken at various time points. Protein degradation products were mixed with Laemmli sample buffers and separated on a 12.5% SDS-polyacrylamide gel electrophoresis (7cm x 12 cm).
References
- Ciechanover A, Schwartz AL. The ubiquitin-proteasome pathway: the complexity and myriad functions of proteins death. Proc Natl Acad Sci USA. 1998;95:2727–2730. doi: 10.1073/pnas.95.6.2727. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ciechanover A. The ubiquitin-proteasome pathway: on protein death and cell life. Embo J. 1998;17:7151–7160. doi: 10.1093/emboj/17.24.7151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Finley D, Sadis S, Monia BP, Boucher P, Ecker DJ, Crooke ST, Chau V. Inhibition of proteolysis and cell cycle progression in a multiubiquitination-deficient yeast mutant. Mol Cell Biol. 1994;14:5501–5509. doi: 10.1128/mcb.14.8.5501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hershko A, Ciechanover A. The ubiquitin system for protein degradation. Annu Rev Biochem. 1992;61:761–807. doi: 10.1146/annurev.biochem.61.1.761. [DOI] [PubMed] [Google Scholar]
- Hershko A, Ciechanover A. The ubiquitin system. Annu Rev Biochem. 1998;67:425–479. doi: 10.1146/annurev.biochem.67.1.425. [DOI] [PubMed] [Google Scholar]
- Yu H, King RW, Peters JM, Kirschner MW. Identification of a novel ubiquitin-conjugating enzyme involved in mitotic cyclin degradation. Curr Biol. 1996;6:455–466. doi: 10.1016/s0960-9822(02)00513-4. [DOI] [PubMed] [Google Scholar]
- Peters JM. Subunits and substrates of the anaphase-promoting complex. Exp Cell Res. 1999;248:339–349. doi: 10.1006/excr.1999.4443. [DOI] [PubMed] [Google Scholar]
- Visintin R, Prinz S, Amon A. CDC20 and CDH1: a family of substrate-specific activators of APC-dependent proteolysis. Science. 1997;278:460–463. doi: 10.1126/science.278.5337.460. [DOI] [PubMed] [Google Scholar]
- Glotzer M, Murray AW, Kirschner MW. Cyclin is degraded by the ubiquitin pathway. Nature. 1991;349:132–138. doi: 10.1038/349132a0. [DOI] [PubMed] [Google Scholar]
- Murray AW, Solomon MJ, Kirschner MW. The role of cyclin synthesis and degradation in the control of maturation promoting factor activity. Nature. 1989;339:280–286. doi: 10.1038/339280a0. [DOI] [PubMed] [Google Scholar]
- Arlot-Bonnemains Y, Klotzbucher A, Giet R, Uzbekov R, Bihan R, Prigent C. Identification of a functional destruction box in the Xenopus laevis aurora-A kinase pEg2. FEBS Lett. 2001. pp. 149–152. [DOI] [PubMed]
- Honda K, Mihara H, Kato Y, Yamaguchi A, Tanaka H, Yasuda H, Furukawa K, Urano T. Degradation of human Aurora2 protein kinase by the anaphase-promoting complex-ubiquitin-proteasome pathway. Oncogene. 2000;19:2812–2819. doi: 10.1038/sj.onc.1203609. [DOI] [PubMed] [Google Scholar]
- Walter AO, Seghezzi W, Korver W, Sheung J, Lees E. The mitotic serine/threonine kinase Aurora2/AIK is regulated by phosphorylation and degradation. Oncogene. 2000;19:4906–4916. doi: 10.1038/sj.onc.1203847. [DOI] [PubMed] [Google Scholar]
- Bischoff JR, Anderson L, Zhu Y, Mossie K, Ng L, Souza B, Schryver B, Flanagan P, Clairvoyant F, Ginther C, Chan CS, Novotny M, Slamon DJ, Plowman GD. A homologue of Drosophila aurora kinase is oncogenic and amplified in human colorectal cancers. Embo J. 1998;17:3052–3065. doi: 10.1093/emboj/17.11.3052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kimura M, Matsuda Y, Yoshioka T, Sumi N, Okano Y. Identification and characterization of STK12/Aik2: a human gene related to aurora of Drosophila and yeast IPL1. Cytogenet Cell Genet. 1998;82:147–152. doi: 10.1159/000015089. [DOI] [PubMed] [Google Scholar]
- Shindo M, Nakano H, Kuroyanagi H, Shirasawa T, Mihara M, Gilbert DJ, Jenkins NA, Copeland NG, Yagita H, Okumura K. cDNA cloning, expression, subcellular localization, and chromosomal assignment of mammalian aurora homologues, aurora-related kinase (ARK) 1 and 2. Biochem Biophys Res Commun. 1998;244:285–292. doi: 10.1006/bbrc.1998.8250. [DOI] [PubMed] [Google Scholar]
- Terada Y, Tatsuka M, Suzuki F, Yasuda Y, Fujita S, Otsu M. AIM-1: a mammalian midbody-associated protein required for cytokinesis. Embo J. 1998;17:667–676. doi: 10.1093/emboj/17.3.667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gopalan G, Chan CS, Donovan PJ. A novel mammalian, mitotic spindle-associated kinase is related to yeast and fly chromosome segregation regulators. J Cell Biol. 1997;138:643–656. doi: 10.1083/jcb.138.3.643. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ciechanover A. The ubiquitin-proteasome proteolytic pathway. Cell. 1994;79:13–21. doi: 10.1016/0092-8674(94)90396-4. [DOI] [PubMed] [Google Scholar]
- Giet R, Uzbekov R, Cubizolles F, Le Guellec K, Prigent C. The Xenopus laevis aurora-related protein kinase pEg2 associates with and phosphorylates the kinesin-related protein XlEg5. J Biol Chem. 1999;274:15005–15013. doi: 10.1074/jbc.274.21.15005. [DOI] [PubMed] [Google Scholar]
- Roghi C, Giet R, Uzbekov R, Morin N, Chartrain I, Le Guellec R, Couturier A, Doree M, Philippe M, Prigent C. The Xenopus protein kinase pEg2 associates with the centrosome in a cell cycle-dependent manner, binds to the spindle microtubules and is involved in bipolar mitotic spindle assembly. 1998; 111: 557-572. J Cell Sci. 1998;111:557–572. doi: 10.1242/jcs.111.5.557. [DOI] [PubMed] [Google Scholar]
- Uzbekov R, Chartrain I, Philippe M, Arlot-Bonnemains Y. Cell cycle analysis and synchronization of the Xenopus cell line XL2. Exp Cell Res. 1998;242:60–68. doi: 10.1006/excr.1998.4097. [DOI] [PubMed] [Google Scholar]
- Murray AW. Cell cycle extracts. Methods Cell Biol. 1991;36:581–605. [PubMed] [Google Scholar]
- Bastians H, Topper LM, Gorbsky GL, Ruderman JV. Cell cycle-regulated proteolysis of mitotic target proteins. Mol Biol Cell. 1999;10:3927–3941. doi: 10.1091/mbc.10.11.3927. [DOI] [PMC free article] [PubMed] [Google Scholar]