

Abstract
There is accumulating evidence that membrane-bound receptors interact with many intracellular proteins. Multiprotein complexes associated with ionotropic receptors have been extensively characterized, but the identification of proteins interacting with G protein-coupled receptors (GPCRs) has so far only been achieved in a piecemeal fashion, focusing on one or two protein species. We describe a method based on peptide affinity chromatography, two-dimensional electrophoresis, mass spectrometry and immunoblotting to identify the components of multiprotein complexes interacting directly or indirectly with intracellular domains of GPCRs or, more generally, any other membrane-bound receptor. Using this global approach, we have characterized multiprotein complexes that bind to the carboxy-terminal tail of the 5-hydroxytryptamine type 2C receptor and are important for its subcellular localization in CNS cells (Bécamel et al., EMBO J., 21(10): 2332, 2002).
Keywords: proteomics; spectrum analysis, mass
Introduction
Many cellular processes implicate large protein networks. The global identification of the components of these networks is essential to study their functions. There is accumulating evidence that membrane-bound receptors such as ionotropic receptors or tyrosine kinases are associated with large multiprotein complexes (1, 2). G-protein coupled receptors (GPCRs), which constitute one of the largest known categories of proteins, also interact with several intracellular proteins in addition to G proteins (3-5). These proteins are important for some of the GPCR functions that do not implicate G-proteins (targeting, clustering, desensitization and resensitization, signaling not mediated by G-proteins) (6). Although proteins interacting with the intracellular loops of GPCRs have already been described, most of the proteins identified as binding partners of GPCRs associate with their cytosolic carboxy (C)-terminal tail (5). Many GPCRs express a PDZ (PSD95/DiscLarge/ZO-1) domain recognition motif at their C-terminal extremity (5, 6). PDZ domains have emerged in the last few years as important modular protein interaction domains found in a new class of structurally related adaptor proteins (7). These proteins often form molecular scaffolds that link proteins into large signaling networks. This suggests that GPCRs can interact with multiprotein complexes, organized around PDZ domain based scaffolds, via their C-terminal extremity. Several studies, often based on two-hybrid screens, have focused on the identification of proteins interacting with the C-terminal tail of a particular GPCR (8-10). However, this approach provides the identification of only one or two protein species that directly interact with the receptor. We have recently developed a proteomic approach, based on peptide affinity chromatography, associated with high resolution 2-dimensional (2-D) electrophoresis, a combination of Matrix-Assisted Laser Desorption Ionization-Time Of Flight Mass Spectrometry (MALDI-TOF MS) and tandem mass spectrometry (MS/MS), and immunoblotting, to identify multiprotein complexes interacting with the 5-hydroxytryptamine type 2C (5-HT2C) receptor. This GPCR expresses a C-terminal PDZ ligand (SSV), suggesting that it interacts with a PDZ-based multiprotein complex (11). Using this approach, we have identified 15 proteins that bind directly or indirectly to the C-terminal tail of the 5-HT2C receptor (12). These include synapse-enriched multi domain proteins containing one or several PDZ domains and proteins devoid of PDZ domains. This method, which provides a global characterization of multiprotein complexes, can thus be used for systematic identification of proteins interacting with the intracellular domains of membrane-bound receptors and of protein networks associated with any protein.
Materials and Methods
Antibodies and Plasmids
The rabbit polyclonal anti-Veli3 antibody was purchased from Zymed Laboratories (San Francisco, CA), mouse monoclonal anti-CASK and anti-Mint1 antibodies from Becton Dinckinson Biosciences. The Veli2 antibody was obtained by immunization of rabbits with a synthetic peptide derived from the Veli2 sequence (QHHSYSSLESRG) as previously described (13). This antibody recognized a single polypeptide around 24 kDa in Western blots of mouse brain extracts (12). Horseradish peroxidase-coupled donkey anti-rabbit and antimouse secondary antibodies were from Amersham Biosciences (Upsalla, Sweeden). The expression vectors encoding the wild type and mutated C-terminal tail of the human 5-HT2C receptor (residues 368-458) fused to GST (pGEX2C90SSV and pGEX902CSSA, respectively) have been described elsewhere (14).
Membrane preparations and GST pull-down
Mice brains were homogenized with a polytron in 20 ml phosphate buffered saline (PBS) and centrifuged at 200 x g for 5 min. Pellets were resuspended in ice-cold lysis buffer containing Tris-HCl (50 mM, pH 7.4), EDTA (1 mM), and a protease inhibitor cocktail (Roche), homogenized 20 times on ice with a glass-Teflon homogenizer and centrifuged at 10,000 x g for 30 min. The membrane pellets were resuspended in CHAPS extraction buffer (Tris-HCl, 50 mM pH 7.4; EDTA, 0.05 mM; CHAPS, 10 mM and protease inhibitors) for 3 h in rotation at 4°C. Then, samples were centrifuged for 1 h at 10,000 x g and solubilized proteins (supernatants, 20 mg protein per condition) were incubated with immobilized GST-fusion proteins overnight at 4°C. GST and GST-fusion proteins were expressed in Escherichia coli strain BL21 and immobilized (50 mg each) on glutathione-sepharose 4B beads (Amersham Biosciences). Samples were washed five times with 150 mM NaCl and eluted with 10 mM reduced glutathione. Samples were then precipitated with 10% ice-cold trichloroacetic acid (TCA) for 2 h and precipitates were washed three times with diethyl ether.
Two-dimensional electrophoresis
TCA precipitates were resuspended in 350 ml isoelectrofocusing medium containing urea (7 M), thiourea (2 M), CHAPS (4% w/v), ampholines (preblended, pI 3.5-9.5, 8 mg/ml, Amersham Biosciences), dithiothreitol (DTT, 100 mM), Tergitol NP7 (0.2% v/v, Sigma) and traces of bromophenol blue (15). Proteins were first separated according to their isoelectric point along linear immobilized pH-gradient (IPG) strips (pH 3-10, 18 cm long) using the IPGphor apparatus (Amersham Biosciences). Sample loading for the first dimension was performed by passive in-gel re-swelling. After the first dimension, the IPG strips were equilibrated for 10 min in a buffer containing urea (6M), Tris-HCl (50 mM, pH 6.8), glycerol (30% v/v), SDS (2% w/v), DTT (10 mg/ml) and bromophenol blue and then for 15 min in the same buffer containing 15 mg/ml iodoacetamide instead of DTT. For the second dimension, the strips were loaded onto vertical 12.5% SDS polyacrylamide gels. The gels were silver stained according to the procedure of Shevchenko et al. (16).
Image acquisition and 2-D gel spot pattern analysis
Gels to be compared were always processed and stained in parallel. Gels were scanned using a computer-assisted densitometer (Amersham Biosciences). Spot detection, gel alignment and spot quantification were performed using the Image Master 2-D Elite software (Amersham Biosciences). Quantitative variations of proteins were expressed as volumes of spots. To correct for variability resulting from silver staining, results were expressed as relative volumes of all spots in each gel. Data are the means of values from four gels originating from different pull-down experiments.
Protein identification by MALDI-TOF mass spectrometry
Proteins of interest were excised and digested in gel using trypsin (sequencing grade, Promega, Madison, WI), as previously described (16, 17). Digest products were completely dehydrated in a vacuum centrifuge and resuspended in 10 ml formic acid (2% v/v), desalted using Zip Tips C18 (Millipore, Bedford, MA), eluted with 10 ml acetonitrile:trifluoroacetic acid, (80:0.1%) and concentrated to 2 ml. Aliquots of analyte solutions were mixed with the same volume of α-cyano-4-hydroxy-trans-cinnamic acid (10 mg/ml in acetonitrile:trifluoroacetic acid, 50:0.1%) and loaded on the target of a BIFLEX III MALDI-TOF mass spectrometer (Bruker-Franzen Analytik, Bremen, Germany) using the Dry-droplet procedure (18). Analysis was performed in reflectron mode with an accelerating voltage of 20 kV and a delayed extraction of 400 ns. Mass spectra were acquired as the sum of the ion signals that were generated by irradiation of the target with a mean of 300 laser pulses. Spectra were analyzed using the XTOF software (Bruker-Franzen Analytik) and autoproteolysis products of trypsin (m/z 842.51, 1045.56, 2211.10) were used as internal calibrates. Peptides were selected in the mass range of 800-3500 Da. Identification of proteins was performed using both Mascot and PeptIdent softwares, available online at http://www.matrixscience.com and http://www.expasy.org/tools/peptident.html, respectively. A mass deviation of 100 ppm was allowed for data base interrogation, but the mass accuracy of our analyses was usually better than 50 ppm. Coverage of the full-length protein exceeding 15% was considered to be sufficient unless there were some obvious conflicts between the experimental molecular weight or isoelectric point and those of the identified protein (19). Matching peptides with missed cleavages were considered as pertinent only when there were two consecutive basic residues or when arginine and lysine residues were followed by a proline or acidic residues inside the peptide amino acid sequence.
Protein identification by tandem mass spectrometry
Nanoelectrospray mass spectrometry was performed on a quadrupole time-of-flight (Q-TOF) mass spectrometer (QSTAR; Sciex, Toronto, Canada) equipped with a nanospray source (Protana Inc., Odense, Denmark). The needle containing 1 ml peptide mixtures, purified by using POROS R2 micro columns (Protana), was briefly touched against the interface plate and then centered in front of the mass spectrometer orifice. A potential of 900 V at the needle started the nanospray. Peptide fragmentation was performed in the collision cell using nitrogen gas on the doubly charged peptide ions detected, with a collision energy profile optimized individually for each peptide (30-55 V). Tandem mass spectra were interpreted using the BioAnalyst software (Applied BioSystems). Identification of proteins was performed by matching the experimental MS/MS spectra of multiple peptides from the same protein against calculated spectra for all peptides in a protein sequence database, using the Mascot software (http://www.matrixscience.com). For Mascot searches, a positive score was defined to be greater than 30 for each peptide ion. The identification was systematically confirmed by manual interpretation of the MS/MS spectra by the “peptide sequence tag” method using the BioAnalyst and PeptideSearch softwares (www.narrador.emblheidelberg.de) (20, 21).
Immunoblotting
Proteins, resolved on 1-D or 2-D gels, were transferred electrophoretically onto nitrocellulose membranes (Hybond-C, Amersham Biosciences). Membranes were incubated overnight with primary antibodies. Immunoreactivity was detected with an enhanced chemiluminescence method (Renaissance Plus, NEN DuPont, Boston, MA).
Results and Discussion
Proteins interacting with the cytosolic C-terminal tail of the 5-HT2C receptor were purified by peptide affinity chromatography using the entire C-terminal tail of the receptor (90 amino acids) fused to GST (GST902CSSV) as bait. The bait (50 mg per assay) was immobilized onto glutathione sepharose beads. Whole brain extracts were used in this purification step because the 5-HT2C receptor displays a wide and discrete distribution in the central nervous system. The bound proteins were eluted, separated by 2-D electrophoresis and stained with silver. For silver staining, we used a procedure allowing further micro characterization of proteins by mass spectrometry (16). This method omits the sensitization with glutaraldehyde, which is known to make covalent bounds with proteins through Schiff base formation with amino groups, and minimizes oxidation of proteins. Fig. 1 illustrates a typical 2-D gel obtained with the GST902CSSV bait. These gels exhibited numerous protein spots, probably due to the high sensitivity of the silver staining procedure used, which detects proteins in the nanogramme range (Fig. 1). Among these proteins, several major spots were identified as GST and/or GST fusion protein by MALDI-TOF mass spectrometry (Fig. 1).
Fig. 1.
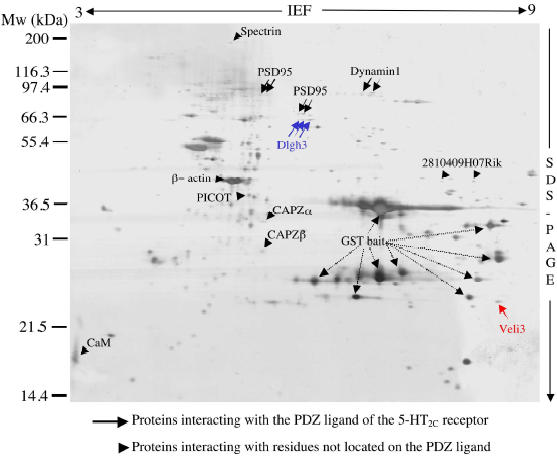
Protein 2-D map of multiprotein complexes interacting with the C-terminal extremity of the 5-HT2C receptor. Proteins interacting with the C-terminal tail of the 5-HT2C receptor were purified by peptide affinity chromatography on glutathione sepharose beads using the C-terminal 90 amino acid residues of the receptor (GST902CSSV) as bait. Proteins, recovered with reduced glutathione, were resolved on 2-D gels and stained with silver. A representative gel is illustrated. Arrows and arrowheads indicate protein spots that were not detectable (or much less represented) in gels obtained using the GST902CSSA and GST baits, respectively. The unlabelled spots were found in equal amounts in gels performed with all baits. Some of them were identified as E. coli proteins or major brain proteins. MALDI-TOF MS and MS/MS analyses of Veli3 (red) and Dlgh3 (blue) are described in detail on Figs. 2 and 3, respectively.
To detect proteins that specifically interact with the C-terminal tail of the 5-HT2C receptor, we performed differential analyses of 2-D gel protein patterns obtained with the GST902CSSV bait and two control baits: 1) GST alone and 2) GST fused to the C-terminal tail of the 5-HT2C receptor which was mutated on the C-terminal residue of the PDZ ligand (GST902CSSA). This residue is critical for the interaction with target PDZ domains (14). A comparison of protein patterns in the 2-D gels obtained with the GST902CSSV and GST902CSSA baits, indicated a selective recruitment of 5 spots or groups of spots (indicated by arrows in Fig. 1) by the wild type bait. These spots were not detectable on gels performed with the GST902CSSA bait (n = 4 gels originating from 4 pull-down experiments, see Figs. 2 and 3). These proteins were unambiguously identified by MALDI-TOF and/or MS/MS as four distinct proteins (Figs. 1-3). Three of them are proteins that contain one or several PDZ domains. The fourth protein that bound selectively to the wild type bait (dynamin 1) was devoid of PDZ domains, but its association with proteins containing PDZ domains has already been suggested. This protein may thus be indirectly recruited by the C-terminal tail of the 5-HT2C receptor through a PDZ domain-based scaffold. Altogether, these results validate the method used to purify and detect proteins interacting specifically with PDZ domain recognition motifs located at the C-terminal extremity of membrane-bound receptors.
Fig. 2.
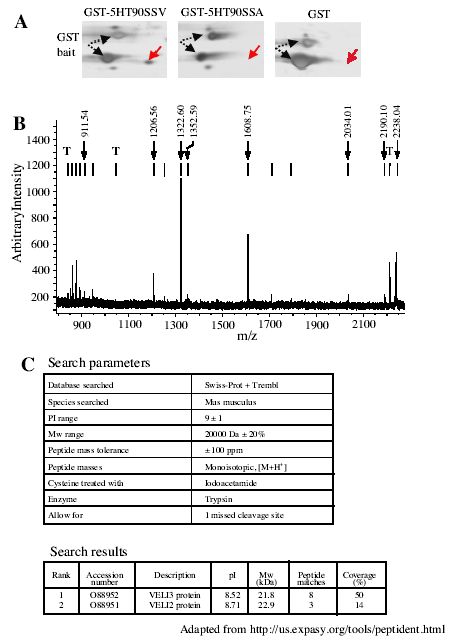
MALDI-TOF mass spectrometry identification of Veli3 as a binding partner of the PDZ ligand of the 5-HT2C receptor. A. Areas of interest of silver-stained 2-D gels obtained in experiments using the GST902CSSV, GST902CSSA and GST baits, respectively, and showing the specific recruitment of one protein spot by the PDZ ligand of the 5-HT2C receptor. B. MALDI-TOF peptide mass map obtained from in-gel trypsin digestion of this spot. Ion signals with measured masses that matched calculated masses of protonated tryptic peptides of mouse Veli3 are indicated. T indicates the ion signals corresponding to the autolysis products of trypsin that were used for internal calibration of spectra (mol wt: 842.51, 1045.56 and 2211.10, respectively). C. Parameters and results of SwissProt and Trembl database search.
Fig. 3.
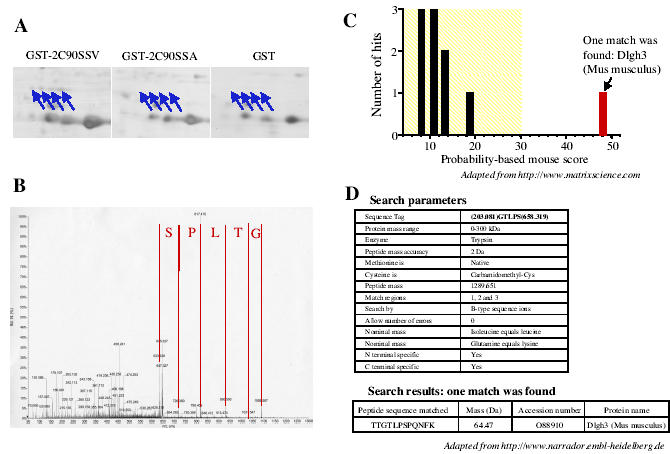
Tandem mass spectrometry identification of Dlgh3 as a binding partner of the PDZ ligand of the 5-HT2C receptor. A. Areas of interest of silver-stained 2-D gels obtained in experiments using the GST902CSSV, GST902CSSA and GST baits, respectively, and showing the specific recruitment of four protein spots (indicated by arrows) by the PDZ ligand of the 5-HT2C receptor. These spots were identified as a single protein by MALDI-TOF MS (not shown). B. Fragmentation spectrum of a double charged ion at m/z = 645.80 detected on the nanoelectrospray mass spectrum of the tryptic digestion product of one of these spots. A sequence of five amino acids is apparent in the higher mass part of the spectrum. C. Graphical representation of hits obtained by matching the fragmentation spectrum illustrated in B. and the calculated spectra of all the peptides in the NCBr database. D. Parameters and results of Non-Redundant Database search using the peptide sequence tag method. Both methods yielded a single match with a mouse Dlgh3 peptide.
We also detected an enrichment of several protein species in 2-D gels obtained in pull down experiments performed with the GST902CSSV and GST902CSSA baits, when compared with those obtained with GST alone (four gels performed in each experimental condition). These proteins are indicated by arrowheads on Fig. 1. Identical amounts of these proteins were retained by the wild type and mutated baits. These proteins that include cytoskeleton proteins as well as signaling proteins, lack obvious PDZ domains. This indicates that they interact with residues that are not located in the PDZ ligand of the receptor.
The data illustrated in Fig. 1 demonstrate that the resolution provided by 2-D gel electrophoresis combined with the high sensitivity of the silver staining procedure was required to detect the proteins specifically recruited by the 5-HT2C receptor C-terminal tail. As previously mentioned, the 2-D protein pattern recovered from the GST pull down assays exhibited numerous protein spots. Some of these spots were identified as either bacterial proteins that bind to GST and/or fusion peptide during bait production in bacteria, or major proteins from mice brain. The gels also showed a large enrichment of GST baits, compared with most of the proteins identified as specific binding partners of the receptor. Separating proteins by 1-D electrophoresis would have probably precluded the detection of proteins with a molecular weight similar to that of the bait, such as the Veli3 protein (Figs. 1 and 2). Moreover, identification of proteins by MALDI-TOF mass spectrometry, which only provides peptide mass fingerprints but not sequence information, requires an efficient purification procedure such as that provided by 2-D electrophoresis. However, GST proteins or any other contaminating protein may mask specific binding partners of the receptor, even when proteins are separated by 2-D electrophoresis. Moreover, several limitations inherent to 2-D electrophoresis make difficult the detection and the identification of some of the proteins purified during the affinity chromatography step such as hydrophobic, basic (pI > 9), large (> 200 kDa) and small (< 10 kDa) proteins. An alternative approach, that combines efficient separation of biological materials and high sensitivity, associates 1-D electrophoresis and liquid chromatography (LC) coupled to MS/MS (LC-MS/MS) (21). This approach, based on LC separation of tryptic peptides prior to MS/MS, can theoretically analyze complicated mixtures containing up to hundreds of proteins (21), and has already been used to identify proteins physically associated to the N-methyl-D-aspartate receptor and separated by 1-D electrophoresis (1). This methodology largely removes the difficulty to identify proteins containing several trans-membrane domains and basic proteins. A procedure which do not require any gel separation and staining and based on direct trypsin digestion of proteins purified by affinity chromatography followed by LC-MS/MS analysis, would also resolve the issue of identifying large proteins as well as peptides that bind to the C-terminal tail of the receptors. This strategy has been successfully used by Ficarro et al. (22) to characterize the yeast phosphoproteome purified by affinity chromatography. However, the large variations in protein concentration in our samples (e.g. GST bait and Veli3) may render uncertain the identification of minor proteins by these alternative approaches. Moreover, our strategy based on 2-D gels remains a method of choice to perform differential analyses of protein expression. In our experience, MALDI-TOF mass spectrometry yielded non-ambiguous identification of most of the proteins examined. Mean sequence coverage of 40% was obtained, which is a much higher score than that generally admitted (15-20% of protein coverage) (19). Moreover, the identity of the protein was always confirmed in 2-D immunoblotting when antibodies against the identified proteins were available. The high success rate in these analyses was linked to the high mass accuracy achieved. Indeed, the deviation between experimental peptide masses and those resulting from theoretical digestion of the protein was generally inferior to 50 ppm using trypsin autolysis peptides as internal calibrates. Another important characteristic of the MALDI-TOF technology is its ability to discriminate between closely related proteins, even when these proteins share high degrees of identity. Veli3, one of the proteins that was recruited by the C-terminal tail of the 5-HT2C receptor, belongs to a family of related proteins containing one PDZ domain and named Veli1, Veli 2 and Veli3 (for Vertebrate homologues of LIN-7) (23). Veli3 and Veli2 amino acid sequences show 81% identity. Moreover, both proteins display similar molecular weights and isoelectric points, indicating a possible overlap of both proteins on 2-D gels. The peptide masses deduced from the MALDI-TOF mass spectrum illustrated in Fig. 2B only matched the theoretical peptide masses calculated for Veli3 according to the criteria defined in “Materials and Methods.” Eight tryptic peptides, corresponding to the coverage of almost 50% of the sequence, were assigned to the sequence (Fig. 2C). The second ranked protein was Veli2, but only 3 peptides corresponding to 14% sequence coverage were assigned to the sequence. This specific identification of Veli3 versus Veli2 as a binding partner of the 5-HT2C receptor was further confirmed by immunoblotting using specific antibodies raised against both proteins (data not shown). The identity of several proteins recruited in our in vitro binding assay was confirmed by MS/MS. Because MS/MS spectra contain sequence information on the peptides rather than only their mass, this approach is certainly more discriminating. MS/MS analysis was systematically performed when no antibody against the protein of interest was available. It is noteworthy that in our analyses MS/MS data always confirmed the results obtained by MALDI-TOF MS. Two methods that generally provided identical results were used to identify proteins by MS/MS: 1) the match of the experimental fragmentation spectrum against the theoretical spectra of all peptides in protein databanks and 2) the sequence tag method. Peptide sequence tag combines a short sequence stretch with mass information that specifies the location of the identified sequence inside the tryptic peptide (21). These two approaches generally yielded identical results. Fig. 3 illustrates the MS/MS analysis of one of the double charged tryptic peptides (m/z = 645.83) of Dlgh3, a protein that was identified as one of the binding partners of the PDZ ligand of the 5-HT2C receptor by MALDI-TOF MS. Comparing the experimental fragmentation spectrum (Fig. 3B) with a calculated spectrum for all peptides in the NCBI database yielded a single match with a high score and corresponding to a Dlgh3 peptide (Fig. 3C). We obtained the same unique peptide match by the peptide sequence tag (determined by interpretation of the MS/MS spectrum with the BioAnalyst software) method and Non-Redundant Protein Database interrogation using the PeptideSearch algorithm (Fig. 3D).
Our procedure using silver staining of 2-D gels allows the detection of proteins recruited in relatively low amounts (a few ng) in the affinity chromatography step. The sensitivity of silver staining allows full use of the sensitivity of current MALDI-TOF and Quadrupole-TOF mass spectrometers like those used in our study. In this regard, the intensities of ion signals detected by MALDI-TOF mass spectrometry in digests of small protein spots (such as those identified as Dlgh3) were in the same range as those obtained with a mixture of external peptide calibrates (50-100 fmol each). However, the low dynamic range and the rapid saturation of silver staining only allow semi-quantitative measurements of proteins detected on 2-D gels. Other staining procedures using fluorescent dyes (e.g. Sypro Ruby), that are just as sensitive as silver staining, would certainly provide more quantitative information without precluding further identification of proteins by mass spectrometry. Despite the reasonably high sensitivity of silver staining, we failed to detect some of the binding partners of the receptor, in particular those that indirectly interact with its C-terminal extremity. This critical limitation for a global characterization of multiprotein complexes cannot be overcome by increasing the amount of proteins loaded on the 2-D gels. Indeed, due the high amount of GST baits in the samples, compared with the other proteins, overloading the gels would alter the electrofocusing of proteins during the first dimension. To identify additional protein components of the complexes purified by affinity chromatography, we combined mass spectrometry with immunoblotting screens. These immunoblotting screens were based on the data of the literature on the known partners of the proteins previously identified by mass spectrometry analyzes. For example, the Veli proteins are known to be part of ternary protein complexes also composed of CASK, a membrane-associated guanylate kinase that contains one PDZ domain, and Mint1, a modular protein that contains two PDZ domains (24, 25). Immunoblotting experiments performed with a CASK and a Mint1 antibody after the affinity chromatography step showed that both CASK and Mint1 were recruited by the C-terminal extremity of the 5-HT2C receptor 12. This suggests that the 5-HT2C receptor interacts with the entire ternary complex Veli3-CASK-Mint1. CASK is known to interact with both Veli proteins and Mint1. These interactions do not involve PDZ domains, which are free to interact with additional protein partners 24. Several pieces of evidence indicate that the ternary complex Veli3-CASK-Mint1 interacts with the 5-HT2C receptor through Veli3 and, thus, that CASK and Mint1 are indirect partners. 1) Only Veli3 was detected by silver staining, indicating that lower amounts of CASK and Mint1 were recruited. 2) Veli3 and the 5-HT2C receptor interacted in heterologous cells 12. 3) The PDZ domain of Veli3 is class I, which determines optimal binding to the PDZ ligand of the 5-HT2C receptor. In contrast, the PDZ domain of CASK is class II, which renders improbable its direct binding to the PDZ ligand of the 5-HT2C receptor 7. This example illustrates how immunoblotting associated with mass spectrometry may be useful to detect low amounts of proteins in a complex such as proteins indirectly associated with a receptor. One important issue concerns the specificity of the protein complexes identified by our fishing approach. Co-immunoprecipitation experiments performed with some of the binding partners of the 5-HT2C receptor indicated for all the proteins tested that the receptor interacts with them in the mouse CNS 12. Moreover, immunohistochemistry and electron microscopy experiments revealed a similar distribution of these proteins and the 5-HT2C receptor in CNS cells. Finally, we have used a similar approach to identify the protein complexes interacting with the C-terminal extremities of three other 5-HT receptors that express PDZ-like ligands (namely the 5-HT2A receptor and two splice variants of the 5-HT4 receptor). The four complexes examined showed different protein composition (unpublished results).
Conclusions
We have established a specific and sensitive method to purify and identify the proteins that interact directly or indirectly with the C-terminal extremity of GPCRs. This approach may be used to characterize the multiprotein complexes interacting with the intracellular domains of any membrane-bound receptors and, more generally, the protein complexes associated with any intracellular protein.
Acknowledgments
This work was supported by grants from the CNRS, the Génopole de Montpellier and the Région Languedoc-Roussillon. C. Bécamel was a recipient of a fellowship from the Association pour la Recherche contre le Cancer.
Appendix
Protocols
Production and immobilization of GST fusion proteins
Solutions and media
SOC medium: 2% tryptone, 0.5% yeast extract, 10 mM NaCl, 2.5 mM KCl, 10 mM MgCl2 and 20 mM glucose
SOB-ampicillin medium: 2% tryptone, 0.5% yeast extract, 10 mM NaCl, 2.5 mM KCl, 10 mM MgCl2 and 100 mg/ml ampicillin
LB-ampicillin plate: 1% tryptone, 0.5% yeast extract, 1% NaCl, 10 mM MgCl2, 100 mg/ml ampicillin and 1.3% agar
Cell lysis solution: phosphate buffered saline (PBS), 0.5 mM DTT and a protease inhibitor cocktail (Roche)
Protocol
Transfer 1 ng of plasmid DNA in 25 ml of BL21 competent cell suspension. Stir gently to mix. Incubate successively at 0°C for 30 min, at 42°C for 30 sec and then at 0°C for 2 min
Add 500 ml of SOC medium. Incubate at 37°C for 1 h with shaking
Plate 200 ml of BL21 cell suspension on LB-ampicillin. Incubate overnight at 37°C
Remove a single colony and culture in 50 ml of SOB-ampicillin medium overnight at 37°C
Add 450 ml of SOB-ampicillin medium and culture at 37°C until OD600nm = 0.5
Induce synthesis of recombinant proteins by adding 0.25 mM isopropyl-ß-D-thiogalactopyranoside for 3 h at 37°C
Harvest cells by centrifugation at 1,000 x g for 15 min at 4°C
Suspend completely the cell pellet in 25 ml cell lysis solution and sonicate on ice 5 times for 1 min
Add 1% Triton X100 and leave samples in rotation at 4°C for 1 h
Centrifuge at 12,000 x g for 10 min at 4°C. Keep the surpernatant. The amount of recombinant protein can be estimated by SDS-PAGE of an aliquot using bovine serum albumin as standard.
Incubate 50 mg of each GST-fusion protein with 500 ml of glutathione Sepharose beads for 1 h in rotation at 4°C
Wash beads 3 times with PBS
Preparation of CHAPS-soluble brain extracts
Solutions
Lysis buffer: 50 mM Tris-HCl, pH 7.4, 1 mM EDTA and the protease inhibitor cocktail
Solubilization buffer: 50 mM Tris-HCl, pH 7.4, 0.05 mM EDTA, 10 mM CHAPS and the protease inhibitor cocktail
Protocol
Dissect 2 mice brains per condition and homogenize with a polytron in 10 ml PBS. Centrifuge at 200 x g for 5 min
Suspend the pellet in 2 ml ice-cold lysis buffer/2 mice brains and homogenize 20 times on ice with a glass-Teflon homogenizer. Centrifuge at 10,000 x g for 30 min at 4°C
Suspend membrane pellet in 2 ml solubilization buffer and homogenize 5 times on ice with a glass-Teflon homogenizer. Leave samples on rotation for 3 h at 4°C
Centrifuge at 10,000 x g for 1 h at 4°C. Keep the supernatant (solubilized proteins)
GST pull-down
Incubate solubilized proteins of 2 mice brains with immobilized GST fusion proteins overnight in rotation at 4°C
Wash beads 5 times with 0.5 M NaCl
Incubate beads with 10 mM reduced glutathione for 1 h in rotation at 4°C
Centrifuge at 1,000 x g for 10 min at 4°C. Keep the supernatant
Precipitate proteins with 10% ice-cold TCA for 2 h on ice
Centrifuge at 10,000 x g for 15 min at 4°C
Wash the TCA precipitates 3 times with 1 ml diethyl ether
Two-dimensional electrophoresis
Materials
First dimension: IPGPhor + strip holders (18 cm long, Amersham Biosciences)
Second dimension: Etan-Dalt II (Amersham Biosciences)
Solutions
Isoelectrofocusing medium: 7 M urea, 2 M thiourea, 4% CHAPS, ampholines (preblended, pI 3.5-9.5, 8 mg/ml), 100 mM DTT, 0.2% Tergitol NP7 and traces of bromophenol blue
Equilibration medium with DTT: 50 mM Tris-HCl, pH 6.8, 30% glycerol, 2% SDS, 10 mg/ml DTT and traces of bromophenol blue
Equilibation medium with iodoacetamide: 50 mM Tris-HCl, pH 8.8, 30% glycerol, 2% SDS, 15 mg/ml iodoacetamide and traces of bromophenol blue
Polycrylamide gel mixture: 12.5% acrylamide (Plus One, Amersham Biosciences), 0.16% bisacrylamide (Plus One), 375 mM Tris-HCl, pH 8.8, 0.1% SDS, 0.015% ammonium persulfate and 0.05% TEMED
Agarose solution: 1% agarose (low-melting), 150 mM Tris-HCl, pH 6.8, 0.2% SDS
Protocol
Suspend the TCA precipitates in 350 ml isoelectrofocusing medium.
Apply the sample between the electrodes of a strip holder.
Add a pre-casted IPG dry strip (pH 3-10, linear, 18 cm long, Amersham Biosciences).
Recover with 1 ml mineral oil.
Perform the isoelectrofocusing with the following parameters and steps: 20°C, 50 mA per strip, rehydration period, 12 h; gradient 0-300 V, 1 min; step-and-hold 300 V, 30 min; gradient 300-5000 V, 3 h; step-and-hold 5000 V, 40,000 V/h. The strips can be kept for several months at –80°C after isoelectrofocusing.
Incubate the strip for 10 min with 6 ml equilibration medium with DTT.
Remove the excess of DTT on the strip.
Incubate the strip for 15 min with 6 ml equilibration medium with iodoacetamide.
Load the strip on the top of a 12.5% polyacrylamide gel (20 x 20 cm, 1 mm thick).
Seal the strip with 1-2 ml agarose solution (heated at 60°C).
Perform the second dimension in two steps: 1) 1 h at 60 V and 45 mA per gel (an initial low-voltage step reduces electroendosmosis); 2) 5 h at 300 V, 45 mA per gel.
Silver staining
Fix the gel in ethanol:acetic acid (50:5% in distilled water) for 20 min.
Incubate the gel in 50% ethanol for 10 min.
Wash the gel with distilled water for 10 min.
Incubate the gel with sodium thiosulfate (0.02%) for 1 min.
Wash the gel for 10 sec twice with distilled water.
Stain the gel with AgNO3 (1 g/l) for 20 min at 4°C.
Wash the gel for 10 sec twice with distilled water.
Incubate the gel with the developing solution (40 g/l Na2CO3, 0.1% formaldehyde). Change rapidly the developing solution after a yellow precipitate appears and incubate the gel with the new solution until appropriate staining is achieved.
Stop the revelation by replacing the developing solution by 5% acetic acid.
Store the gel in 1% acetic acid at 4°C (the gels can be stored for a few weeks before digestion).
Image acquisition and two-dimensional protein pattern analysis
Scan the gels with a computer-assisted densitometer (such as the ImageScanner, Amersham Biosciences, etc.).
Detect protein spots in the gels to be processed with a 2-D gel analysis software (such as Image Master 2D Elite, Amersham Biosciences, etc.).
Suppress dark noise.
Match the corresponding spots in the gels to be processed and perform gel alignment.
Perform spot quantification (expressed as spot volume relative to either the volume of all spots in each gel or the volume of a single spot found in equal amount in all experimental conditions, to correct for variability resulting from silver staining).
Calculate mean values from at least 4 gels performed from different pull down experiments and perform the appropriate statistical tests.
In-gel protein digestion with trypsin
Excise carefully pieces of gels including the spots to be processed, cut them in 1-mm large pieces and transfer them to 0.5 ml polypropylene tubes. Cut a control piece of gel from a blank region of the gel and process it in parallel with the samples.
Shrink the gel pieces by dehydration with acetonitrile for 10 min (in this step as in the following ones, use the minimal solution volume required to recover the gel pieces) and remove the liquid phase.
Swell the gel pieces by rehydration with 100 mM NH4HCO3 for 10 min.
Dehydrate the gel pieces with acetonitrile and remove the liquid phase.
Dry the gel pieces in a vacuum centrifuge for 5 min.
Swell the gel pieces with the digestion buffer (100 mM NH4HCO3, 5 mM CaCl2 and 150 ng trypsin per 2-100 ng protein spot) on ice for 45 min.
Add the same volume of the digestion buffer, but without trypsin, to keep the gel pieces wet during the digestion period (25°C, overnight).
Transfer the liquid phase to a 1.5 ml polypropylene tube.
Dehydrate the gel pieces with acetonitrile for 10 min and transfer the liquid phase to the same 1.5 ml tube.
Swell the gel pieces by rehydration with 100 mM NH4HCO3 for 10 min.
Dehydrate the gel pieces with acetonitrile for 10 min and transfer the liquid phase to the same 1.5 ml tube.
Renew 10 and 11.
Swell the gel pieces by rehydration with 5% formic acid for 10 min.
Dehydrate the gel pieces with acetonitrile for 10 min and transfer the liquid phase to the same 1.5 ml tube.
Renew 13 and 14.
Dry peptide samples with a vacuum centrifuge.
Sample preparation for MALDI-TOF analysis
Dissolve the peptide mixture in 10 ml formic acid (2%).
Desalt the peptide mixture using Zip Tips C18 according to the manufacturer’s procedure.
Elute peptides with 10 ml of acetonitrile:trifluoroacetic acid (80:0.1%) and concentrate by evaporation to 2 ml.
Mix 0.5 ml of analyte solution with the same volume of matrix (10 mg α-cyano-4-hydroxy-trans-cinnamic acid in 1 ml of acetonitrile:trifluoroacetic acid, 50:0.1%).
Deposit 0.3 ml of this mixture on a stainless steel target.
Evaporate the solvent.
Introduce the target in the mass spectrometer.
MALDI-TOF mass spectra interpretation and database interrogation
Calibrate the mass spectra using the auto-proteolysis products of trypsin (m/z 842.51, 1045.56, 2211.10) as internal calibrates using an appropriate software (e.g. the XTOF software, Bruker, etc.).
Select mono isotopic peptides in the mass range of 800-3500 Da.
Remove contaminating peptides from the mass list (peptides found in the control digestion performed on a blank region of the gel).
Search database with the resulting peptide mass list using an appropriate software (such as Mascot or Peptident).
Micro purification of peptides for tandem mass spectrometry
Suspend 2 ml of POROS R2 sorbent (PerSeptive Biosystems) in 50% methanol.
Load suspension in a capillary (Protana Inc.) using Gel loader tips.
Centrifuge the capillary using the purification device.
Wash the micro column with 5 ml methanol:formic acid (50:5%) and store in 10 ml formic acid (5%). A new capillary with new POROS must be used for each analysis.
Dissolve peptide mixture in 5 ml formic acid (5%) and load onto the micro column.
Centrifuge the capillary using the purification device.
Wash the peptides on the column with 5 ml formic acid (5%).
Align a nano Electrospray (nanoES) capillary (ES380-ES382, Protana Inc) in continuation of the micro column.
Elute the peptides into the nanoES capillary twice with 0.5 ml methanol:formic acid (50:5%).
Place the nanoES capillary in the nanoES source head.
Tandem mass spectra interpretation and database searching
Select double charged ions in the MS spectrum and fragment them by MS/MS.
Perform database search with the mass lists from MS/MS spectra of multiple peptides from the same protein using the Mascot software (http://www.matrixscience.com).
Combine the mass of the precursor peptide, the mass of the first peak of an identified sequence ladder, the sequence ladder and the final peak of the sequence ladder to obtain a peptide sequence tag (21). The ladder is obtained manually by starting from the highest m/z of a Y ion series of the MS/MS spectrum using an appropriate software (e.g. BioAnalyst, Applied Biosystems).
Perform database search with this tag using the PeptideSearch software (http://www.narrador.embl-heidelberg.de).
Compare results in 2 and 4.
References
- Husi H, Ward MA, Choudhary JS, Blackstock WP, Grant SG. Proteomic analysis of NMDA receptor-adhesion protein signaling complexes. Nat Neurosci. 2000;3:661–669. doi: 10.1038/76615. [DOI] [PubMed] [Google Scholar]
- Kim M, Jiang LH, Wilson HL, North RA, Suprenant A. Proteomic and functional evidence for a P2X(7) receptor signalling complex. Embo J. 2001;20:6347–6358. doi: 10.1093/emboj/20.22.6347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brzostowski JA, Kimmel AR. Signaling at zero G: G-protein-independent functions for 7-TM receptors. Trends Biochem Sci. 2001;26:291–297. doi: 10.1016/S0968-0004(01)01804-7. [DOI] [PubMed] [Google Scholar]
- Milligan G, White J. Protein-protein interactions at G-protein-coupled receptors. Trends Pharmacol Sci. 2001;22:513–518. doi: 10.1016/s0165-6147(00)01801-0. [DOI] [PubMed] [Google Scholar]
- Brady AE, Limbird LE. G protein-coupled receptor interacting proteins: emerging roles in localization and signal transduction. Cell Signal. 2002;14:297–309. doi: 10.1016/s0898-6568(01)00239-x. [DOI] [PubMed] [Google Scholar]
- Bockaert J, Claeysen S, Bécamel C, Pinloche S, Dumuis A. G protein-coupled receptors: dominant players in cell-cell communication. Int Rev Cytol. 2002;212:63–132. doi: 10.1016/s0074-7696(01)12004-8. [DOI] [PubMed] [Google Scholar]
- Sheng M, Sala C. PDZ domains and the organization of supramolecular complexes. Annu Rev Neurosci. 2001;24:1–29. doi: 10.1146/annurev.neuro.24.1.1. [DOI] [PubMed] [Google Scholar]
- Hall RA, Premont RT, Chow CW, Blitzer JT, Pitcher JA, Claing A, Stoffel RH, Barak LS, Shenolikar S, Weinman EJ, Grinstein S, Lefkowitz RJ. The beta2- adrenergic receptor interacts with the Na+/H+-exchanger regulatory factor to control Na+/H+ exchange. Nature. 1998;392:626–630. doi: 10.1038/33458. [DOI] [PubMed] [Google Scholar]
- Ullmer C, Schmuck K, Figge A, Lubbert H. Cloning and characterization of MUPP1, a novel PDZ domain protein. FEBS Lett. 1998;424:63–68. doi: 10.1016/S0014-5793(98)00141-0. [DOI] [PubMed] [Google Scholar]
- Zitzer H, Honck HH, Bachner D, Richter D, Kreienkamp HJ. Somatostatin receptor interacting protein defines a novel family of multidomain proteins present in human and rodent brain. J Biol Chem. 1999b;274:32997–33001. doi: 10.1074/jbc.274.46.32997. [DOI] [PubMed] [Google Scholar]
- Raymond JR, Mukhin YV, Gelasco A, Turner J, Collinsworth G, Gettys TW, Grewal JS, Garnovskaya MN. Multiplicity of mechanisms of serotonin receptor signal transduction. Pharmacol Ther. 2001;92:179–212. doi: 10.1016/s0163-7258(01)00169-3. [DOI] [PubMed] [Google Scholar]
- Bécamel C, Alonso G, Galeotti N, Demey E, Jouin P, Ullmer C, Dumuis A, Bockaert J, Marin P. Synaptic multiprotein complexes associated with 5-HT(2C) receptors: a proteomic approach. Embo J. 2002;21:2332–2342. doi: 10.1093/emboj/21.10.2332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Misawa H, Kawasaki Y, Mellor J, Sweeney N, Jo K, Nicoll RA, Bredt DS. Contrasting localizations of MALS/LIN-7 PDZ proteins in brain and molecular compensation in knockout mice. J Biol Chem. 2001;276:9264–9272. doi: 10.1074/jbc.M009334200. [DOI] [PubMed] [Google Scholar]
- Bécamel C, Figge A, Poliak S, Dumuis A, Peles E, Bockaert J, Lubbert H, Ullmer C. Interaction of serotonin 5-hydroxytryptamine type 2C receptors with PDZ10 of the multi-PDZ domain protein MUPP1. J Biol Chem. 2001;276:12974–12982. doi: 10.1074/jbc.M008089200. [DOI] [PubMed] [Google Scholar]
- Laoudj-Chenivesse D, Marin P, Bennes R, Tronel-Peyroz E, Leterrier F. High performance two-dimensional gel electrophoresis using a wetting agent Tergitol(R) NP7. Proteomics. 2002;2:481–485. doi: 10.1002/1615-9861(200205)2:5<481::AID-PROT481>3.0.CO;2-G. [DOI] [PubMed] [Google Scholar]
- Shevchenko A, Wilm M, Vorm O, Mann M. Mass spectrometric sequencing of proteins silver-stained polyacrylamide gels. Anal Chem. 1996;68:850–858. doi: 10.1021/ac950914h. [DOI] [PubMed] [Google Scholar]
- Lee K, Bae D, Lim D. Evaluation of parameters in peptide mass fingerprinting for protein identification by MALDI-TOF mass spectrometry. Mol Cells. 2002;13:175–184. [PubMed] [Google Scholar]
- Karas M, Hillenkamp F. Laser desorption ionization of proteins with molecular masses exceeding 10,000 daltons. Anal Chem. 1988;60:2299–2301. doi: 10.1021/ac00171a028. [DOI] [PubMed] [Google Scholar]
- Garin J, Diez R, Kieffer S, Dermine JF, Duclos S, Gagnon E, Sadoul R, Rondeau C, Desjardins M. The phagosome proteome: insight into phagosome functions. J Cell Biol. 2001;152:165–180. doi: 10.1083/jcb.152.1.165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mann M, Wilm M. Error-tolerant identification of peptides in sequence databases by peptide sequence tags. Anal Chem. 1994;66:4390–4399. doi: 10.1021/ac00096a002. [DOI] [PubMed] [Google Scholar]
- Mann M, Hendrickson RC, Pandey A. Analysis of proteins and proteomes by mass spectrometry. Annu Rev Biochem. 2001;70:437–473. doi: 10.1146/annurev.biochem.70.1.437. [DOI] [PubMed] [Google Scholar]
- Ficarro SB, McCleland ML, Stukenberg PT, Burke DJ, Ross MM, Shabanowitz J, Hunt DF, White FM. Phosphoproteome analysis by mass spectrometry and its application to Saccharomyces cerevisiae. Nat Biotechnol. 2002;20:301–305. doi: 10.1038/nbt0302-301. [DOI] [PubMed] [Google Scholar]
- Jo K, Derin R, Li M, Bredt DS. Characterization of MALS/Velis-1, -2, and -3: a family of mammalian LIN- 7 homologs enriched at brain synapses in association with the postsynaptic density-95/NMDA receptor postsynaptic complex. J Neurosci. 1999;19:4189–4199. doi: 10.1523/JNEUROSCI.19-11-04189.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Butz S, Okamoto M, Südhof TC. A tripartite protein complex with the potential to couple synaptic vesicle exocytosis to cell adhesion in brain. Cell. 1998;94:773–782. doi: 10.1016/s0092-8674(00)81736-5. [DOI] [PubMed] [Google Scholar]
- Borg JP, Straight SW, Kaech SM, de Taddeo-Borg M, Kroon DE, Karnak D, Turner RS, Kim SK, Margolis B. Identification of an evolutionarily conserved heterotrimeric protein complex involved in protein targeting. J Biol Chem. 1998;273:31633–31636. doi: 10.1074/jbc.273.48.31633. [DOI] [PubMed] [Google Scholar]