

Abstract
Whether speciation results more frequently from the genetic consequences of founder events or from gradual genetic divergence of large populations is a matter of debate. In this study, multiple analyses were applied to data from three loci (cytochrome b, α-enolase intron VIII, and MHC class II B) to test for founder effects associated with speciation in Aethia (Aves: Alcidae), a genus of seabirds thought to have undergone a rapid founder-induced radiation. Effective population sizes (Ne) were derived from estimators of θ based on allelic diversity and the coalescent and from data on trans-species polymorphism. Results indicated that Ne has been on the order of 105–106 individuals throughout the evolutionary histories of least and crested auklets (A. pusilla and A. cristatella, respectively) and that Ne of the ancestral species was at least 16,000 individuals. Computer simulations of MHC evolution indicated that a single-generation bottleneck at speciation could not have involved <85 individuals for each species. More moderate simulation scenarios indicated that population size could not have dropped below 2000 individuals at the time of species founding. Demographic history appears to have been stable for the auklets throughout the past several million years, and a founder effect associated with their speciation is unlikely.
FOUNDER-INDUCED speciation is arguably one of the most controversial of all potential mechanisms of speciation (Mayr 1942; Barton and Charlesworth 1984; Carson and Templeton 1984; Templeton 1986; Provine 1989; Gavrilets and Hastings 1996; Hollocher 1996; Clegg et al. 2002; Rundle 2003; Coyne and Orr 2004). As a result of a founder event (i.e., population founding by a small number of individuals), gene frequencies may change rapidly and particular alleles may be lost or fixed due to sampling error alone. Founder events also may induce linkage disequilibrium, and new or rare mutations and/or gene combinations (including those that are slightly deleterious) have a higher probability of drifting to fixation in small populations (Wright 1969; Gavrilets and Boake 1998). Such founder effects, in concert with environmental differences experienced by founder populations, have the potential to precipitate speciation (but see Rundle 2003). For example, by accelerating divergence in small populations, founder effects may facilitate rapid and/or adaptive radiations of taxa. Founder effect speciation is therefore among the speciational hypotheses considered most commonly for taxa whose histories appear to have contained radiations (Carson 1983; D. Klein et al. 1993; Hollocher 1996; Vincek et al. 1997; Clegg et al. 2002). Founder effect speciation is consistent with both vicariant and dispersal scenarios for population isolation.
Although the incidence of a founder event alone is not sufficient proof of the occurrence of speciation by founder effect, a first assessment of the relevance of a founder effect model to a given case of speciation involves evaluation of whether or not a founder event (such as a bottleneck in population size) plausibly occurred in the history of the examined taxa. To date, however, no empirical data have demonstrated a convincing association between bottlenecks in population size and speciation in vertebrates. Among animals, documented examples of what may be founder effect speciation exist, controversially, for Drosophila species (Carson 1983; Hollocher 1996; Templeton 1996), and cases of founder-induced speciation may have occurred in seabirds such as the shy albatross (Thalassarche cauta) (Abbott and Double 2003) and Armenian gull (Larus armenicus) (Liebers et al. 2001). The genus Drosophila, for example, has radiated throughout the Hawaiian islands to comprise almost 1000 species in the few million years since formation of the archipelago, and a founder-flush mechanism of speciation has been proposed for these flies (Carson 1983; Hollocher 1996).
Founder effect has been viewed as a theoretically attractive mechanism for vertebrate speciation (Templeton 1986; Gavrilets and Hastings 1996; Gavrilets and Boake 1998; Rundle et al. 1998; Turelli et al. 2001), but its occurrence may be rare (Gavrilets 2004) and it is not supported in the few cases in which it has been tested empirically. For example, hundreds of species of cichlid fishes originated in the African Rift Lakes in <1 million years (Meyer et al. 1990; Johnson et al. 1996), but extensive intraspecific major histocompatibility complex (MHC) variability suggests that their speciation was not due to a founder effect (D. Klein et al. 1993). Similarly, despite evidence for rapid radiation, no evidence indicates that speciation was associated with founder effects in either threespine sticklebacks (Gasterosteus aculeatus) (McKinnon and Rundle 2002) or island-colonizing birds in the silvereye species complex (Zosterops lateralis) (Clegg et al. 2002). On the basis of human leukocyte antigen (HLA) polymorphism and evidence from nuclear introns, several researchers concluded that founder effect was not an important component of human evolution (Takahata 1991, 1993; Takahata et al. 1995; Ayala and Escalante 1996). Likewise, founder effect has been discounted as the primary mode of speciation among Darwin's finches (Geospiza spp.) since the discovery of extensive MHC variability in these species (Vincek et al. 1997).
Methods have been developed for inferring both long-term and ancestral effective population sizes (Ne and Na, respectively) from DNA polymorphism in extant populations, as well as from variation shared by closely related species (e.g., Kimura 1969; Watterson 1975; Kingman 1982a,b; Tajima 1983; Rogers and Harpending 1992; Takahata et al. 1995; Yang 1997, 2002; Rannala and Yang 2003; Wall 2003). These methods allow tests of explicit hypotheses on the origin and radiation of taxa and have been particularly informative in the controversy surrounding the origin of modern humans (Takahata 1993; Rogers 1995; Takahata et al. 1995; Ayala and Escalante 1996). In this study, we applied several of these methods to test the hypothesis that founder effects were involved in speciation in Aethia auklets, a group of North Pacific seabirds.
Aethia (Charadriiformes: Alcidae) comprises four species (Aethia cristatella, A. psittacula, A. pusilla, and A. pygmaea) endemic to the North Pacific, Bering Sea, and Sea of Okhotsk. As wing-propelled divers, auklets spend most of their lives at sea, returning to islands and continental coasts to nest in sympatry in the spring. All four species are sexually monomorphic and display facial plumes and bill ornamentation during the breeding season; they appear to be monogamous and can produce only a single chick per breeding pair each breeding season (Gaston and Jones 1998; Jones 1999). Phylogenetic relationships within the genus are unresolved, and power analysis suggested that internodes between divergence events in this group were extremely short (relative to population sizes) (Walsh et al. 1999). The above results have led to the hypothesis that speciation in the genus was facilitated by population bottlenecking and/or founder events, precipitated by one or more vicariant events (Jones 1999; Walsh et al. 1999). Contemporary census population sizes are on the orders of millions of individuals for both least and crested auklets (A. pusilla and A. cristatella, respectively) (Stephensen and Irons 2003) and, on the basis of life history tables, generation times are 6.7 and 8.3 years, respectively (Walsh 1999; Walsh and Friesen 2003). DNA sequences of the cytochrome b gene indicate that lineages leading to least and crested auklets diverged ∼2.8 MYA (Friesen et al. 1996; Walsh and Friesen 2003). Given this time frame, divergence of the auklets may have followed isolation within glacial refugia of the Plio-Pleistocene, consistent with the glacier-induced speciation models proposed for many groups of species native to the Americas (e.g., O'Reilly et al. 1993; Hewitt 1996; Avise and Walker 1998; Holder et al. 1999; but see Klicka and Zink 1997, 1999). Auklets exhibit both strong mutual sexual selection and behavioral isolation among species (Jones and Hunter 1993; Gaston and Jones 1998), both of which are among the proposed consequences of founder effect speciation (Powell 1989).
Recent methods for estimating ancestral effective population sizes (Na) rely on identifying conflicts between gene trees and the presumed known species tree (Yang 2002; Rannala and Yang 2003) or on maximizing the likelihood of a given number of inferred mutations along branches in a known phylogeny (Wall 2003). For auklets, however, the species tree is unknown despite analyses of a large amount of sequence data (Moum et al. 1994; Friesen et al. 1996; Walsh et al. 1999). Without a known species tree, some methods for estimating Na cannot be applied; however, data from loci that exhibit trans-species polymorphism can be used to evaluate Na for species with unknown species trees. Using data on genetic diversity and allelic genealogy at three loci, we estimated Ne at several depths in the evolutionary histories of least and crested auklets to test the hypothesis that their speciation was associated with a founder effect.
THEORY AND METHODS
Because genetic diversity is a function of long-term Ne, estimates of Ne can be derived from the neutral mutation parameter, θ (= 4Neμ for nuclear DNA and 2Nfμ for mitochondrial DNA, where μ is the neutral mutation rate per sequence per generation, and Nf is female effective population size). (In what follows, we adopt the convention of Felsenstein to use Θ to denote 4Neμ per site and θ to denote 4Neμ per locus.)
Several methods have been advanced to estimate θ from DNA sequences (Kimura 1969; Watterson 1975; Nei 1987; Felsenstein 1992; Griffiths and Tavaré 1993; Fu 1994a,b; Kuhner et al. 1995; Takahata et al. 1995; Tajima 1996), and it can be scaled for balancing selection by applying Takahata's (1990) correction factor. In this study, θ was estimated using data on nucleotide polymorphism previously collected for least and crested auklets (Walsh and Friesen 2003). These data include 306 bp of sequence from the 5′ end of the mitochondrial cytochrome b gene, intron VIII (248 bp) from the nuclear gene for α-enolase, and a 154-bp fragment of the second exon of an MHC class II B gene. Analyses of 89 least auklets and 81 crested auklets sampled from throughout their breeding ranges revealed 5 and 8 alleles for cytochrome b, and 19 and 33 alleles for α-enolase, respectively. At least 40 alleles at each of two MHC II B loci were observed for each species (Walsh and Friesen 2003).
Substitution rates (μ):
Given the 2%/MY rate of cytochrome b divergence reported by Avise and Walker (1998), the substitution rate for cytochrome b is 2.1 × 10−5 and 2.5 × 10−5 substitutions per sequence per generation for least and crested auklets, respectively (given generation times of 6.7 and 8.3 years, respectively). The substitution rate for α-enolase intron VIII was estimated at 3.7 × 10−6 and 4.5 × 10−6 substitutions per sequence per generation for least and crested auklets, respectively, by comparison of auklet sequences with marbled murrelet sequences (Friesen et al. 1997) and assuming a divergence time of 12 MYA (Friesen et al. 1996). The substitution rate for the MHC II B fragment was estimated on the basis of the rate of 3.5 × 10−9 substitutions per site per year of Vincek et al. (1997), adjusted for generation times of the auklets to yield μ = 3.6 × 10−6 and 4.5 × 10−6 substitutions per sequence per generation, respectively.
Estimating Ne:
For cytochrome b and α-enolase, both Π (average pairwise differences) (Tajima 1983) and θW (Watterson's θ) (Watterson 1975) were calculated. Ne was derived from Π and θW, assuming substitution rates given above. We used the algorithm FLUCTUATE (Kuhner et al. 1995) to estimate contemporary Θ for least and crested auklets, using data on intraspecific allelic sequences for cytochrome b and α-enolase intron VIII.
We used the maximum-likelihood methods of Takahata et al. (1995) to estimate Ne for each species and their associated Na, given substitution rates for each locus. Ne for each species was estimated by evaluating Takahata et al.'s (1995) Equation 2 at its maximum (ΘML), given the data for cytochrome b and α-enolase. The maximum-likelihood value for Na (and associated 95% confidence interval) was estimated numerically from Takahata et al.'s (1995) Equation 3, with divergence time fixed at 2.8 MYA and a mean of four substitutions between two random sequences (Walsh and Friesen 2003).
Six polymorphisms are shared between least and crested auklets in intron VIII of the gene for α-enolase (i.e., SS = 6), and no fixed differences were observed between the two species for this locus. We thus used an abbreviated form of the method developed by Wakeley and Hey (1997) to estimate θA (= 4Naμ) from the data for α-enolase. We equated the number of polymorphisms shared between least and crested auklets with E(SS), the expectation for this quantity [given by Wakeley and Hey's (1997) Equation 12], substituting estimated θ-values from α-enolase intron VIII for θ1 and θ2. This equation was solved for θA and the corresponding value for Na was derived, assuming that the substitution rate for this locus in auklets is 4 × 10−6 substitutions per sequence per generation (the average for the two species, above).
Ne from trans-species polymorphism:
If trans-species polymorphism exists in a sample of nucleotide sequences (and is sufficiently common that it cannot reasonably be attributed to convergence), then coalescence of alleles within species predates speciation (Takahata 1991). Measured genetic distances between alleles can be superimposed on a genealogy to estimate k, the number of lineages extant today that also were present prior to a speciation event (Takahata 1991; J. Klein et al. 1993).
If time is measured in units of 2Ne generations [that is, 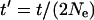 , where t is the known number of generations since a speciation event], the probability gnk(t′) that at t′ (in the past) there were k alleles in a population, given that the population now carries n alleles, is given by
, where t is the known number of generations since a speciation event], the probability gnk(t′) that at t′ (in the past) there were k alleles in a population, given that the population now carries n alleles, is given by
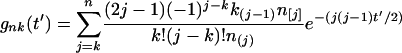 |
(1) |
when 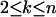 (Tavaré 1984), where
(Tavaré 1984), where 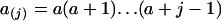 and
and 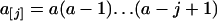 [a represents variables with subscripts in (1)]. k was determined as described below.
[a represents variables with subscripts in (1)]. k was determined as described below.
Relationships among least and crested auklet alleles for α-enolase intron VIII were estimated by the neighbor-joining method using Jukes-Cantor distances in MEGA v. 1.02 (Kumar et al. 1993). The expected Jukes-Cantor corrected distance (Jukes and Cantor 1969) between sequences that diverged 2.8 MYA (i.e., about the time of divergence of least and crested auklets) is 0.0125 substitutions per site. For each species, the minimum number of allelic lineages present prior to the speciation event, k, was estimated as the number of lineages whose coalescence on the tree occurs prior to 0.0125 distance units ago (J. Klein et al. 1993). For each species, the maximum of Equation 1 was estimated numerically and Ne at this point was calculated from its relation with t′.
The timescales over which our estimates of long-term Ne apply are given by an exponentially distributed time parameter with mean 4Ne[1 − (1/i)], where i is the number of sampled alleles (4Ne is replaced with 2Nf for mitochondrial DNA) (Kingman 1982a,b).
Ne by computer simulation:
Extant lineages whose coalescence predates speciation are lineages that persisted through any putative bottleneck at the time of species founding. Thus, if the number of extant allelic lineages whose coalescence occurred at or prior to species divergence is known, then the minimum size of the founding population can be inferred from results of computer simulations. Allelic lineages for which coalescence predates speciation generally are observed at loci under balancing selection, which acts to maintain variation through time. Trans-species polymorphism also may be evident at neutral loci when θ is large with respect to time since divergence, as noted by Takahata (1986, 1989) and observed in our study (see results, Figure 1). Vincek et al. (1997) used a computer simulation to determine the number of alleles retained in a population through bottlenecks of various sizes, given data on polymorphism for the second exon of an MHC II B gene. This locus is known to be under balancing selection of an intensity on the order of s = 0.01 in humans (Satta et al. 1994).
Figure 1.

Neighbor-joining tree of α-enolase alleles for least (L) and crested (C) auklets, based on Jukes-Cantor distances. Allelic lineages present prior to the speciation event (n = 7) are those that cross the vertical line indicated in boldface type (which marks the estimated divergence time of lineages leading to modern least and crested auklets). L12 and C13 are identical, as are C1 and L3.
In this study we used the simulation algorithm of Vincek et al. (1997) to determine the minimum population size of the immediate ancestor of least and crested auklets. We used available data on allelic polymorphism in a 154-bp fragment of the second exon of an MHC class II B gene for the auklets, at which a minimum of 40 alleles have been documented in each of least and crested auklets (Walsh and Friesen 2003). The number of allelic lineages older than the speciation event (2.8 MYA) was estimated by two methods. First, we estimated the average time to coalescence of two alleles under balancing selection, 2Nefs generations [where fs is given by Takahata's (1990) Equation 15], using a selection intensity of 0.01 (Satta et al. 1994). Ne was taken as 253,000 individuals for least auklets and 375,000 individuals for crested auklets (the average of four estimates of long-term Ne based on neutral markers, for each species; see results). Thus, the average time to coalescence of two alleles for exon 2 of the class II MHC B gene is 1.9 × 106 generations (12.6 × 106 years) for least auklets and 2.4 × 106 generations (20.3 × 106 years) for crested auklets. This result is consistent with the observation that some human allelic lineages at this locus are >30 million years old (J. Klein et al. 1993). Given this result, most extant auklet alleles at this locus predate the divergence of least and crested auklets.
To obtain a more conservative estimate of the number of alleles at this locus that predate the speciation event, we considered the observation that for some species that diverged within the last 5 MY [Darwin's finches (Vincek et al. 1997) and several extant primate species (J. Klein et al. 1993)], no less than 62% of the allelic lineages present today (for MHC II B exon 2) also were present prior to speciation. Given this generalization, we estimated that of the (minimum) 40 alleles currently present in each of least and crested auklets, 25 (∼62%) were present prior to speciation.
We ran the computer simulation to determine the minimum number of founder individuals that would allow persistence of 40 and 25 alleles for the second exon of an MHC II B locus. The ultimate parental population was assumed to have the same number and frequency of alleles as a known human (Caucasoid) population (as in Vincek et al. 1997). Simulations were run under conditions of immediate expansion following an initial bottleneck of size Nb and expansion following a bottleneck 10 generations in duration. As in Vincek et al. (1997), two growth rates were applied for each of the expansion scenarios (5 and 50% per generation), and an additional case, that of no expansion (growth = 0%), also was considered.
Historical demographic events inferred from mismatch distributions:
Rogers and Harpending (1992) argued that the distribution of pairwise nucleotide differences between individuals in a population sample (the mismatch distribution) carries information on the demographic history of the population (but see Di Rienzo and Wilson 1991; Lundstrom et al. 1992). We plotted the mismatch distributions for cytochrome b and α-enolase intron VIII for our sample of least and crested auklets to judge whether evidence exists for a population expansion in either species. Comparisons between all pairs of individuals were conducted using Arlequin ver. 2.000 (Schneider et al. 2000). Ninety-five percent confidence intervals on the raggedness statistics (r) (Harpending 1994) were generated in DnaSP v 4.0 (Rozas et al. 2003) by 5000 replicates in coalescent simulations based on the observed genetic diversities and samples sizes, under no recombination and under intermediate recombination (R = 10).
To address some of the assumptions of the above methods, tests of intraspecies population differentiation and of deviations from Hardy-Weinberg equilibrium were conducted using GENEPOP (v. 3.1d) (Raymond and Rousset 1995) and Arlequin (data given in Walsh and Friesen 2003).
RESULTS
Estimates of θ and Θ and corresponding values for Ne or Nf derived from variation in cytochrome b and the α-enolase intron indicated that population sizes for the auklets have been large throughout the course of their evolution (minimum 201,000 and 220,000 individuals for least and crested auklets, respectively; Tables 1 and 2). Timescales over which estimates of θ (or Θ) and N apply varied from 5.1 × 105 to 1.1 × 107 years (Tables 1 and 2). Fixing divergence time at 2.8 MYA gave a maximum-likelihood value of Na = 3.4 × 104 individuals (95% C.I. ∼0.0–1.9 × 106; maximum L = −1.69) by the method of Takahata et al. (1995).
TABLE 1.
Estimates of θ and Θ and corresponding values for Ne or Nf for least auklets, based on data for cytochrome b and α-enolase intron VIII
| Estimator |
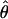 or or 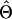
|
Nf | Generations (yra) before present throughout which estimates apply |
|---|---|---|---|
| Cytochrome b | |||
| Π (Tajima 1983) | 2.00 | ∼48,000 | ∼7.6 × 104 (5.1 × 105) |
| θW (Watterson 1975) | 2.40 | ∼57,000 | ∼9.1 × 104 (6.1 × 105) |
| ΘP (Kuhner et al. 1995) | 0.012 | ∼87,000 | Contemporary Nf |
| ΘML (Takahata et al. 1995) | 0.0065 | ∼47,000 | ∼7.6 × 104 (5.1 × 105) |
| Estimator |
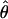 or or 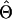
|
Ne | Generations (yra) before present throughout which estimates apply |
| α-Enolase | |||
| Π (Tajima 1983) | 3.08 | ∼208,000 | ∼7.9 × 105 (5.3 × 106) |
| θW (Watterson 1975) | 4.29 | ∼290,000 | ∼1.1 × 106 (7.4 × 106) |
| ΘP (Kuhner et al. 1995) | 0.043 | ∼720,000 | Contemporary Ne |
| ΘML (Takahata et al. 1995) | 0.012 | ∼201,000 | ∼7.6 × 105 (5.1 × 106) |
| Trans-species polymorphism | NAb | 313,000 | 4.2 × 105 (2.8 × 106) |
Given a generation time of 6.7 years for least auklets (Walsh and Friesen 2003).
Not applicable.
TABLE 2.
Estimates of θ and Θ and corresponding values for Ne or Nf for crested auklets, based on data for cytochrome b and α-enolase intron VIII
| Estimator |
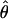 or or 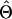
|
Nf | Generations (yra) before present throughout which estimates apply |
|---|---|---|---|
| Cytochrome b | |||
| Π (Tajima 1983) | 2.61 | ∼52,000 | ∼9.1 × 104 (7.6 × 105) |
| θW (Watterson 1975) | 3.47 | ∼69,000 | ∼1.2 × 105 (1.0 × 106) |
| ΘP (Kuhner et al. 1995) | 0.017 | ∼104,000 | Contemporary Nf |
| ΘML (Takahata et al. 1995) | 0.0098 | ∼60,000 | ∼1.0 × 105 (8.7 × 105) |
| Estimator |
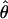 or or 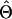
|
Ne | Generations (yra) before present throughout which estimates apply |
| α-Enolase | |||
| Π (Tajima 1983) | 3.96 | ∼220,000 | ∼8.5 × 105 (7.1 × 106) |
| θW (Watterson 1975) | 6.41 | ∼360,000 | ∼1.4 × 106 (1.1 × 107) |
| ΘP (Kuhner et al. 1995) | 0.092 | ∼1,300,000 | Contemporary Ne |
| ΘML (Takahata et al. 1995) | 0.016 | ∼220,000 | ∼8.6 × 105 (7.1 × 106) |
| Trans-species polymorphism | NAa | 699,000 | 3.4 × 105 (2.8 × 106) |
Given a generation time of 8.3 years for crested auklets (Walsh and Friesen 2003).
Not applicable.
Equating SS with Wakeley and Hey's (1997) Equation 12 and substituting estimated θW values for θ1 and θ2 gave a value of 0.256 for θA at the α-enolase locus. The corresponding value for the effective size of the ancestral population was 16,000 individuals. Substituting values obtained from other estimators of θ (Tables 1 and 2) for θ1 and θ2 produced higher values for Na (∼117,000 using Π and ∼123,000 using θML).
According to the phylogeny of least and crested auklet alleles for α-enolase intron VIII (Figure 1), seven allelic lineages predate the divergence of the two species, three of which have descendants in our sample of modern least auklets and all seven of which have descendants in our sample of modern crested auklets. The maximum of Equation 1 occurred at t′ = 0.673 for least auklets (k = 3, n = 19) and at t′ = 0.243 for crested auklets (k = 7, n = 33), giving Ne's of 3.13 × 105 and 6.99 × 105 individuals, respectively. These are long-term Ne's for the last 2.8 MY (i.e., since divergence of the lineages leading to the two species; Tables 1 and 2).
Results of the computer simulations (Figure 2) showed that the minimum population size required to maintain 25 MHC class II B allelic lineages through a founder event was 85 individuals (under simulation of immediate expansion from bottleneck size, with a population growth rate of 50% per generation). The largest estimate was 650 founder individuals (under simulation of expansion with a growth rate of 5% following a 10-generation bottleneck). For retention of all or almost all (90% of) 40 alleles through an initial founder event, 740 individuals were required under immediate expansion with growth rate 50% and >2000 individuals were required under expansion with growth rate 5% after 10 bottlenecked generations. Under a scenario of no expansion, 310 and >2000 individuals were required for retention of 25 and 40 alleles, respectively. In all cases, allelic loss was minimal once the size of the population was > ∼2000 individuals.
Figure 2.

The number of alleles retained in a population as a function of founder population size, after a 0- or 10-generation bottleneck followed by 0, 5, or 50% per generation population growth (from computer simulations). Bottleneck duration and subsequent population growth rate are indicated for each curve.
None of the frequency distributions of pairwise nucleotide differences for cytochrome b and α-enolase intron VIII in least and crested auklets (Figure 3) displayed a unimodal shape characteristic of a population expansion following an extended bottleneck, nor were many peaks observed at high numbers of differences, as expected for a brief bottleneck (Rogers and Harpending 1992). Raggedness statistics (r; Figure 3) all were within 95% confidence intervals for r under coalescent simulations given observed sample sizes and nucleotide diversities, under conditions of both no recombination and intermediate recombination.
Figure 3.

Frequency distributions of nucleotide differences among pairs of individuals in population samples of least and crested auklets. (A) Cytochrome b alleles among least auklets [r = 0.704; raggedness statistic (Harpending 1994)]; (B) cytochrome b alleles among crested auklets (r = 0.137); (C) α-enolase alleles among least auklets (r = 0.0926); (D) α-enolase alleles among crested auklets (r = 0.0472).
Exact tests of population differentiation showed no significant heterogeneity in haplotype or genotype distributions among populations for either cytochrome b or α-enolase (P > 0.10). Similarly, pairwise FST values detected no significant genetic structure among populations (all P > 0.05), and no significant deviations from Hardy-Weinberg equilibrium were observed (all P > 0.21).
DISCUSSION
Population sizes unequivocally have been large for least and crested auklets throughout the course of their evolution. This conclusion is consistent across loci and estimation methods (Tables 1 and 2). The absence of detectable population bottlenecks during the evolutionary history of the auklet species implies that founder effect could not have been a causal factor in their speciation. Large values for estimates of Ne at different depths in their history indicate that the observed genetic variation has been maintained over the long term and is not a product of a recent population expansion. Absence of detectable genetic structure among populations indicates that estimated Ne values are unlikely to have been inflated due to population subdivision.
Due to its haploid state and primarily maternal transmission, Ne for mtDNA (i.e., Nf) is expected to be approximately one-quarter that for nuclear DNA (Tajima 1983). Effective population sizes estimated from mtDNA in our study are consistent with this expectation (Tables 1 and 2).
Tajima (1996) showed that, given sufficient substitution rate variation across sites, estimates of θ based on the infinite-sites model will be substantial underestimates. Although site-specific substitution rate variation was not addressed in our work, such rate variation is observed frequently for samples of DNA sequences (e.g., Wakeley 1993; Voelker and Edwards 1998), and Tajima's (1996) conclusion makes our estimates of Ne more conservative.
The estimates of Ne in Tables 1 and 2 assume a single panmictic population in mutation-drift equilibrium. To address these assumptions, pairwise FST values and raggedness statistics on mismatch distributions were computed. FST values did not reveal any significant genetic structure among populations, suggesting panmixia, and r-values of all mismatch distributions fell within 95% confidence intervals of r estimated by coalescent simulations (Figure 3), suggesting equilibrium between mutation and genetic drift.
Our estimate of Na derived by the method of Takahata et al. (1995) may be biased in that it is based on data for a single locus, using a method that originally was developed to treat data for multiple loci (specifically, the variation of sequence divergences among loci). This may explain the width of the confidence interval on our estimate of Na by this method (Yang 1997). However, we do not expect among-locus variation in sequence divergences for these species to be very high because their recent divergence (with respect to effective population sizes, so much so that trans-species polymorphism is observed) predicts low divergence at all loci. Therefore, per locus sequence divergence for least and crested auklets may not be as biased by factors such as substitution rate variation among loci (Yang 1997), and single-locus estimates of ancestral parameters may be more robust for these species than they would be for species whose divergence times are older.
Bias is introduced into the estimation of θA when using empirically derived values for θ1 and θ2 in the evaluation of Wakeley and Hey's (1997) Equation 12. Watterson's θ, for example, assumes constant population size through time, a hypothesis that has not been rejected for these birds, but that remains an assumption by virtue of it being a null hypothesis. The use of different θ-estimators as variables in this equation suggests, however, that θW-values are underestimates (see results). Thus, our estimates of θA are conservatively low within the framework of testing the plausibility of founder effect as a mechanism for speciation of the auklets.
If fewer than seven allelic lineages for α-enolase intron VIII actually predate the speciation event (as may occur either if divergence of the two species is older than estimated or due to error associated with phylogeny estimation), the resulting Ne is reduced, although still sufficiently large to reject the hypothesis of founder effect speciation in the auklets. For example, if k = 3 for crested auklets, the maximum of Equation 1 occurs approximately at t′ = 0.8, and if k = 2 (the minimum value that k can take because trans-species polymorphism is observed), the maximum of (1) occurs approximately at t′ = 1.2. Given these values, estimates of Ne are ∼212,000 and ∼142,000 individuals, respectively.
The computer simulations in this study were based on conservative scenarios for the duration of population bottlenecks and the rate of expansion following the bottleneck phase and consider the minimum amount of variability at an MHC II B locus for least and crested auklets. Because single-generation bottlenecks allow retention of a greater number of alleles than do multigeneration bottlenecks, the estimates derived from computer simulation represent the minimum number of individuals required in a bottlenecked founder population. Given population bottlenecks of >10 generations, even larger population sizes would be required for retention of the observed number of ancient alleles.
The assumption of the computer model that the ancestral auklet population contained the same number and frequencies of alleles as in the current Caucasoid population is probably unrealistic, although less so than in the original simulations of Vincek et al. (1997) due to greater similarity of population size estimates for auklets and humans than for Darwin's finches and humans (Vincek et al. 1997). The model also assumes that mating is random and that rates of population growth are constant through time, and thus it ignores fluctuations due to sudden population explosions and/or catastrophes in addition to the original bottleneck; however, these assumptions are typical of similar models (e.g., Kuo and Janzen 2004) and are necessary to assure feasibility of the computer analysis. The intensity of selection at MHC II B loci in least and crested auklets is unknown and the value of s (0.01) used in the computer model may not be accurate. However, empirical data for humans (Satta et al. 1994) indicate that s for exon 2 of MHC II B loci is generally less than or equal to this value. Because balancing selection acts to maintain more variation than would be maintained by drift alone, a smaller value for s (i.e., reduced selection intensity) would necessitate larger population sizes through bottlenecks to allow persistence of the same amount of variation.
Our simulations suggest that throughout the evolutionary history of least and crested auklets and their immediate ancestor, no population size reductions to fewer than several thousand individuals occurred. This result is similar to that obtained for human populations over the past several million years (Ayala and Escalante 1996). In addition, mismatch distributions for the auklets do not fit models of population expansion subsequent to either long or brief bottlenecks. The above results suggest that the demographic history of the auklets has been relatively stable throughout the past several million years. For effective population sizes to be of the orders of magnitude reported in Tables 1 and 2, no dramatic bottlenecks, even for a single generation, could have occurred in the evolutionary histories of least and crested auklets. To have maintained observed levels of variability, even population reductions of moderate size lasting many generations are unlikely.
Methods for estimating ancestral population parameters are in an early stage of development. As such, estimates presented herein incorporate the biases of their respective models (e.g., model assumptions described above). Future development of more realistic models and their application to data for a large number of unlinked loci will allow more accurate estimation of ancestral population sizes (Rannala and Yang 2003). Despite uncertainties in the estimation methods applied herein, results across multiple estimation methods and three types of molecular markers provide unambiguous evidence that auklets did not speciate as a consequence of founder effect. Alternative explanations for both the relatively short internodes that separated divergence events among species in this group and their mode of speciation should focus on the demonstrated strong and mutual sexual selection exhibited by these species (Jones and Hunter 1993).
Though hypothesized to be a plausible and even widespread mechanism of speciation, to date available data do not support the hypothesis that speciation by founder effect has occurred commonly in vertebrates. Whether this is due to a deficit of relevant data or to lack of the effect itself is difficult to determine. A systematic evaluation across many vertebrate taxa, especially those expected to have experienced a history of reduced population size (e.g., island species, refugial populations, species in areas of wide ecological gradients), is required before generalizations can be established regarding the overall importance of this potential mechanism of speciation.
Acknowledgments
We are grateful to C. O'hUigin for providing the original source code for the simulation and to S. Uellenberg for assistance in compiling and running many versions of the algorithm. We thank J. Felsenstein, P. Beerli, and the members of the Friesen, Rohwer, and Swanson lab groups, who participated in helpful discussions and commented on the manuscript. We are indebted to Y. Artukhin, G. V. Byrd, L. Climo, S. Ebbert, J. Piatt, L. Scharf, D. Siegel-Causey, A. Sowls, S. Stephensen, J. Wachtel, J. Williams, and the crew of R/V Tiglax for their generous assistance with tissue collections and to F. Tajima and the anonymous reviewers who provided valuable comments on the manuscript. Financial and/or logistic support for this study was provided by the Natural Sciences and Engineering Research Council of Canada (NSERC discovery grant held by V.L.F. and postgraduate scholarships to H.E.W.); the Northern Sciences Training Program; the U.S. Fish and Wildlife Service, Alaska Maritime National Wildlife Refuge, Bering Sea and Aleutian Islands units; and the Leaders Five endowed fellowship of the University of Washington Burke Museum.
References
- Abbott, C. L., and M. C. Double, 2003. Genetic structure, conservation genetics and evidence of speciation by range expansion in shy and white-capped albatrosses. Mol. Ecol. 12: 2953–2962. [DOI] [PubMed] [Google Scholar]
- Avise, J. C., and D. Walker, 1998. Pleistocene phylogeographic effects on avian populations and the speciation process. Proc. R. Soc. Lond. Ser. B 265: 457–463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ayala, F. J., and A. A. Escalante, 1996. The evolution of human populations: a molecular perspective. Mol. Phylogenet. Evol. 5: 188–201. [DOI] [PubMed] [Google Scholar]
- Barton, N. H., and B. Charlesworth, 1984. Genetic revolutions, founder effects, and speciation. Annu. Rev. Ecol. Syst. 15: 133–164. [Google Scholar]
- Carson, H. L., 1983. Chromosomal sequences and interisland colonizations in Hawaiian Drosophila. Genetics 103: 465–482. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carson, H. L., and A. R. Templeton, 1984. Genetic revolutions in relation to speciation phenomena: the founding of new populations. Annu. Rev. Ecol. Syst. 15: 97–131. [Google Scholar]
- Clegg, S. M., S. M. Degnan, J. Kikkawa, C. Moritz, A. Estoup et al., 2002. Genetic consequences of sequential founder events by an island-colonizing bird. Proc. Natl. Acad. Sci. USA 99: 8127–8132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coyne, J. A., and H. A. Orr, 2004. Speciation. Sinauer Associates, Sunderland, MA.
- Di Rienzo, A., and A. C. Wilson, 1991. Branching pattern in the evolutionary tree for human mitochondrial DNA. Proc. Natl. Acad. Sci. USA 88: 1597–1601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Felsenstein, J., 1992. Estimating effective population size from samples of sequences: a bootstrap Monte Carlo integration method. Genet. Res. 60: 209–220. [DOI] [PubMed] [Google Scholar]
- Friesen, V. L., A. J. Baker and J. F. Piatt, 1996. Phylogenetic relationships within the Alcidae (Charadriiformes: Aves) inferred using total molecular evidence. Mol. Biol. Evol. 13: 359–367. [DOI] [PubMed] [Google Scholar]
- Friesen, V. L., B. C. Congdon, H. E. Walsh and T. P. Birt, 1997. Intron variation in marbled murrelets detected using analyses of single-stranded conformational polymorphisms. Mol. Ecol. 6: 1047–1058. [DOI] [PubMed] [Google Scholar]
- Fu, Y.-X., 1994. a A phylogenetic estimator of effective population size or mutation rate. Genetics 136: 685–692. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fu, Y.-X., 1994. b Estimating effective population size or mutation rate using the frequencies of mutations of various classes in a sample of DNA sequences. Genetics 138: 1375–1386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gaston, A. J., and I. L. Jones, 1998. The Auks: Alcidae. Oxford University Press, Oxford.
- Gavrilets, S., 2004. Fitness Landscapes and the Origin of Species. Princeton University Press, Princeton, NJ.
- Gavrilets, S., and C. R. B. Boake, 1998. On the evolution of premating isolation after a founder event. Am. Nat. 152: 706–716. [DOI] [PubMed] [Google Scholar]
- Gavrilets, S., and A. Hastings, 1996. Founder effect speciation: a theoretical reassessment. Am. Nat. 147: 466–491. [Google Scholar]
- Griffiths, R. C., and S. Tavaré, 1993. Sampling theory for neutral alleles in a varying environment. Philos. Trans. R. Soc. Lond. Ser. B 344: 403–410. [DOI] [PubMed] [Google Scholar]
- Harpending, H. C., 1994. Signature of ancient population growth in a low-resolution mitochondrial DNA mismatch distribution. Hum. Biol. 66: 591–600. [PubMed] [Google Scholar]
- Hewitt, G, 1996. Some genetic consequences of ice ages, and their role in divergence and speciation. Biol. J. Linn. Soc. 58: 247–276. [Google Scholar]
- Holder, K., R. Montgomerie and V. L. Friesen, 1999. A test of the glacial refugium hypothesis using patterns of mitochondrial and nuclear DNA sequence variation in rock ptarmigan (Lagopus mutus). Evolution 53: 1936–1950. [DOI] [PubMed] [Google Scholar]
- Hollocher, H., 1996. Island hopping in Drosophila: patterns and processes. Philos. Trans. R. Soc. Lond. B 351: 735–743. [DOI] [PubMed] [Google Scholar]
- Johnson, T. C., C. A. Scholz, M. R. Talbot, K. Kelts, R. D. Ricketts et al., 1996. Late Pleistocene desiccation of Lake Victoria and rapid evolution of cichlid fishes. Science 273: 1091–1093. [DOI] [PubMed] [Google Scholar]
- Jones, I. L., 1999. Assessing the role of sexual selection in adaptive radiation of the auklets (Alcidae, Aethiini). Proceedings of the 22nd International Ornithological Congress, edited by N. Adams and R. Slotow. University of Natal, Durban, South Africa, CD-ROM.
- Jones, I. L., and F. M. Hunter, 1993. Mutual sexual selection in a monogamous seabird. Nature 362: 238–239. [Google Scholar]
- Jukes, T. H., and C. R. Cantor, 1969. Evolution of protein molecules, pp. 21–132 in Mammalian Protein Metabolism, edited by H. N. Munro. Academic Press, New York.
- Kimura, M., 1969. The number of heterozygous nucleotide sites maintained in a finite population due to steady flux of mutations. Genetics 61: 893–903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kingman, J. F. C., 1982. a The coalescent. Stoch. Proc. Appl. 13: 235–248. [Google Scholar]
- Kingman, J. F. C., 1982. b On the genealogy of large populations. J. Appl. Probab. 19A: 27–43.
- Klein, D., H. Ono, C. O'hUigin, V. Vincek, T. Goldschmidt et al., 1993. Extensive Mhc variability in cichlid fishes of Lake Malawi. Nature 364: 330–334. [DOI] [PubMed] [Google Scholar]
- Klein, J., Y. Satta, N. Takahata and C. O'hUigin, 1993. Trans-specific Mhc polymorphism and the origin of species in primates. J. Med. Primatol. 22: 57–64. [PubMed] [Google Scholar]
- Klicka, J., and R. M. Zink, 1997. The importance of recent ice ages in speciation: a failed paradigm. Science 277: 1666–1669. [Google Scholar]
- Klicka, J., and R. M. Zink, 1999. Pleistocene effects on North American songbird evolution. Proc. R. Soc. Lond. Ser. B. 266: 695–700. [Google Scholar]
- Kuhner, M. K., J. Yamato and J. Felsenstein, 1995. Estimating effective population size and mutation rate from sequence data using Metropolis-Hastings sampling. Genetics 140: 1421–1430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kumar, S., K. Tamura and M. Nei, 1993. MEGA: Molecular Evolutionary Genetics Analysis (v. 1.0). Pennsylvania State University, University Park, PA.
- Kuo, C.-H., and F. J. Janzen, 2004. Genetic effects of a persistent bottleneck on a natural population of ornate box turtles (Terrapene ornata). Conserv. Genet. 5: 425–437. [Google Scholar]
- Liebers, D., A. J. Helbig and P. De Knijff, 2001. Genetic differentiation and phylogeography of gulls in the Larus cachinnans-fuscus group (Aves: Charadriiformes). Mol. Ecol. 10: 2447–2462. [DOI] [PubMed] [Google Scholar]
- Lundstrom, R., S. Tavaré and R. H. Ward, 1992. Modeling evolution of the human mitochondrial genome. Math. Biosci. 112: 319–335. [DOI] [PubMed] [Google Scholar]
- Mayr, E., 1942. Systematics and the Origin of Species. Columbia University Press, New York.
- McKinnon, J. S., and H. D. Rundle, 2002. Speciation in nature: the threespine stickleback model systems. Trends Ecol. Evol. 17: 480–488. [Google Scholar]
- Meyer, A., T. D. Kocher, P. Basasibwaki and A. C. Wilson, 1990. Monophyletic origin of Lake Victoria cichlid fishes suggested by mitochondrial DNA sequences. Nature 347: 550–553. [DOI] [PubMed] [Google Scholar]
- Moum, T., S. Johansen, K. E. Erikstad and J. F. Piatt, 1994. Phylogeny and evolution of the auks (subfamily Alcinae) based on mitochondrial DNA sequences. Proc. Natl. Acad. Sci. USA 91: 7912–7916. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nei, M., 1987. Molecular Evolutionary Genetics. Columbia University Press, New York.
- O'Reilly, P., T. E. Reimchen, R. Beech and C. Strobeck, 1993. Mitochondrial DNA in Gasterosteus and Pleistocene glacial refugium on the Queen Charlotte Islands, British Columbia. Evolution 47: 679–684. [DOI] [PubMed] [Google Scholar]
- Powell, J. R., 1989. The effects of founder-flush cycles on ethological isolation in laboratory populations of Drosophila, pp. 239–251 in Genetics, Speciation and the Founder Principle, edited by L. V. Giddings, K. Y. Kaneshiro and W. W. Anderson. Oxford University Press, New York.
- Provine, W. B., 1989. Founder effects and genetic revolutions in microevolution and speciation: an historical perspective, pp. 43–76 in Genetics, Speciation and the Founder Principle, edited by L. V. Giddings, K. Y. Kaneshiro and W. W. Anderson. Oxford University Press, New York.
- Rannala, B., and Z. H. Yang, 2003. Bayes estimation of species divergence times and ancestral population sizes using DNA sequences from multiple loci. Genetics 164: 1645–1656. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raymond, M., and F. Rousset, 1995. GENEPOP (version 1.2): population genetics software for exact tests and ecumenicism. J. Hered. 86: 248–249. [Google Scholar]
- Rogers, A. R., 1995. Genetic evidence for a Pleistocene population explosion. Evolution 49: 608–615. [DOI] [PubMed] [Google Scholar]
- Rogers, A. R., and H. C. Harpending, 1992. Population growth makes waves in the distribution of pairwise genetic differences. Mol. Biol. Evol. 9: 552–569. [DOI] [PubMed] [Google Scholar]
- Rozas, J., J. C. Sánchez-DelBarrio, X. Messeguer and R. Rozas, 2003. DnaSP, DNA polymorphism analyses by the coalescent and other methods. Bioinformatics 19: 2496–2497. [DOI] [PubMed] [Google Scholar]
- Rundle, H. D., 2003. Divergent environments and population bottlenecks fail to generate premating isolation in Drosophila pseudoobscura. Evolution 57: 2557–2565. [DOI] [PubMed] [Google Scholar]
- Rundle, H. D., A. O. Mooers and M. C. Whitlock, 1998. Single founder-flush events and the evolution of reproductive isolation. Evolution 52: 1850–1855. [DOI] [PubMed] [Google Scholar]
- Satta, Y., C. O'hUigin, N. Takahata and J. Klein, 1994. Intensity of natural selection at the major histocompatibility complex loci. Proc. Natl. Acad. Sci. USA 91: 7184–7188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schneider, S., D. Roessli and L. Excoffier, 2000. Arlequin: A Software for Population Genetic Data Analysis. Genetics and Biometry Laboratory, University of Geneva, Geneva.
- Stephensen, S. W., and D. B. Irons, 2003. Comparison of colonial breeding seabirds in the eastern Bering Sea and Gulf of Alaska. Mar. Ornithol. 31: 167–173. [Google Scholar]
- Tajima, F., 1983. Evolutionary relationship of DNA sequences in finite populations. Genetics 105: 437–460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tajima, F., 1996. The amount of DNA polymorphism maintained in a finite population when the neutral mutation rate varies among sites. Genetics 143: 1457–1465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takahata, N., 1986. An attempt to estimate the effective size of the ancestral species common to 2 extant species from which homologous genes are sequenced. Genet. Res. 48: 187–190. [DOI] [PubMed] [Google Scholar]
- Takahata, N., 1989. Gene genealogy in three related populations: consistency probability between gene and population trees. Genetics 122: 957–966. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takahata, N., 1990. A simple genealogical structure of strongly balanced allelic lines and trans-species evolution of polymorphism. Proc. Natl. Acad. Sci. USA 87: 2419–2423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takahata, N., 1991. Trans-species polymorphism of HLA molecules, founder principle, and human evolution, pp. 29–49 in Molecular Evolution of the Major Histocompatibility Complex, edited by J. Klein and D. Klein. Springer-Verlag, New York.
- Takahata, N., 1993. Allelic genealogy and human evolution. Mol. Biol. Evol. 10: 2–22. [DOI] [PubMed] [Google Scholar]
- Takahata, N., Y. Satta and J. Klein, 1995. Divergence time and population size in the lineage leading to modern humans. Theor. Popul. Biol. 48: 198–221. [DOI] [PubMed] [Google Scholar]
- Tavaré, S., 1984. Lines-of-descent and genealogical processes, and their applications in population genetics models. Theor. Popul. Biol. 26: 119–164. [DOI] [PubMed] [Google Scholar]
- Templeton, A. R., 1986. The relation between speciation mechanisms and macroevolutionary patterns, pp. 497–512 in Evolutionary Processes and Theory, edited by S. Karlin and E. Nevo. Academic Press, New York.
- Templeton, A. R., 1996. Experimental evidence for the genetic-transilience model of speciation. Evolution 50: 909–915. [DOI] [PubMed] [Google Scholar]
- Turelli, M., N. H. Barton and J. A. Coyne, 2001. Theory and speciation. Trends Ecol. Evol. 16: 330–343. [DOI] [PubMed] [Google Scholar]
- Vincek, V., C. O'hUigin, Y. Satta, N. Takahata, P. T. Boag, et al., 1997. How large was the founding population of Darwin's finches? Proc. R. Soc. Lond. Ser. B 264: 111–118. [Google Scholar]
- Voelker, G., and S. V. Edwards, 1998. Can weighting improve bushy trees? Models of cytochrome b evolution and the molecular systematics of pipits and wagtails (Aves: Motacillidae). Syst. Biol. 47: 589–603. [DOI] [PubMed] [Google Scholar]
- Wakeley, J., 1993. Substitution rate variation among sites in hypervariable region 1 of human mitochondrial DNA. J. Mol. Evol. 37: 613–623. [DOI] [PubMed] [Google Scholar]
- Wakeley, J., and J. Hey, 1997. Estimating ancestral population parameters. Genetics 145: 847–855. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wall, J. D., 2003. Estimating ancestral population sizes and divergence times. Genetics 163: 395–404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walsh, H. E., 1999. Polymorphism at neutral and non-neutral loci in least and crested auklets (Aethia spp.) and implications for speciation. M.Sc. Thesis, Queen's University, Kingston, ON, Canada.
- Walsh, H. E., and V. L. Friesen, 2003. A comparison of intraspecific patterns of DNA sequence variation in mitochondrial DNA, α-enolase, and MHC Class II B loci in auklets (Charadriiformes: Alcidae). J. Mol. Evol. 57: 681–693. [DOI] [PubMed] [Google Scholar]
- Walsh, H. E., M. G. Kidd, T. Moum and V. L. Friesen, 1999. Polytomies and the power of phylogenetic inference. Evolution 53: 932–937. [DOI] [PubMed] [Google Scholar]
- Watterson, G. A., 1975. On the number of segregating sites in genetical models without recombination. Theor. Popul. Biol. 7: 256–276. [DOI] [PubMed] [Google Scholar]
- Wright, S., 1969. Evolution and the Genetics of Populations, Vol. 2: The Theory of Gene Frequencies. University of Chicago Press, Chicago.
- Yang, Z., 1997. On the estimation of ancestral population sizes of modern humans. Genet. Res. 69: 111–116. [DOI] [PubMed] [Google Scholar]
- Yang, Z., 2002. Likelihood and Bayes estimation of ancestral population sizes in hominoids using data from multiple loci. Genetics 162: 1811–1823. [DOI] [PMC free article] [PubMed] [Google Scholar]