

Abstract
Development of cancer requires the acquisition of multiple oncogenic mutations and selection of the malignant clone. Cancer evolves within a finite host lifetime and mechanisms of carcinogenesis that accelerate this process may be more likely to contribute to the development of clinical cancers. Mutator mutations are mutations that affect genome stability and accelerate the acquisition of oncogenic mutations. However, mutator mutations will also accelerate the accumulation of mutations that decrease cell proliferation, increase apoptosis, or affect other key fitness parameters. These “reduced-fitness” mutations may mediate “negative clonal selection,” i.e., selective elimination of premalignant mutator clones. Target reduced-fitness loci may be “recessive” (both copies must be mutated to reduce fitness) or “dominant” (single-copy mutation reduces fitness). A direct mathematical analysis is applied to negative clonal selection, leading to the conclusion that negative clonal selection against mutator clones is unlikely to be a significant effect under realistic conditions. In addition, the relative importance of dominant and recessive reduced-fitness mutations is quantitatively defined. The relative predominance of mutator mutations in clinical cancers will depend on several variables, including the tolerance of the genome for reduced-fitness mutations, particularly the number and potency of dominant reduced-fitness loci.
DEVELOPMENT of cancer is characterized by the accumulation and selection of multiple genetic changes in key genes altering at least six cancer-associated phenotypes (Hanahan and Weinberg 2000). Mutator mutations, defined as mutations that affect genomic stability, may accelerate this process. A mutator mutation in DNA polymerase proofreading activity leads to increased incidence of lymphomas and epithelial tumors in mice (Goldsby et al. 2001), and mutations that inactivate a variety of DNA repair enzymes have been shown to result in increased mutation frequency in mice and humans (Wood et al. 2001). Multiple mutator mutations can cooperate to further destabilize the genome, as has been demonstrated in yeast (Morrison et al. 1993; Datta et al. 2000).
Tumors evolve within a finite human lifetime. The timescale of appearance of most adult tumors is constant to within less than an order of magnitude, ranging from ∼5 years (secondary leukemias after chemotherapy), to 20 years (solid tumors after chemotherapy, occupational carcinogen exposure, or radiation exposure), to a maximum of 50–100 years based on the human life span. Within this fixed time frame, rather than reaching equilibrium, there may be kinetic competition between different mechanisms of carcinogenesis. Mechanisms that accelerate this process may have a greater chance of contributing to carcinogenesis.
The mutator phenotype hypothesis states that mutator mutations contribute to carcinogenesis by accelerating the accumulation of oncogenic mutations (see Figure 1)(Loeb et al. 1974, 2003; Loeb 1991, 1998). Mutator mutations and genetic instability are generalized concepts, referring not just to mutations leading to enhanced base substitution, but also to microsatellite instability (MIN) (Fishel et al. 1993; Ionov et al. 1993), chromosomal instability (CIN) (Lengauer et al. 1998), and alterations in checkpoint control (Sherr and McCormick 2002). Both MIN and CIN contribute to the development of colon cancer (Loeb et al. 2003). Tumors appear to contain thousands of mutations, and some of these are in codons and repetitive sequences in which one would not expect mutations to be directly selected (Levine 1997; Futreal et al. 2004).
Figure 1.


Fates of cell lineages without (A) and with (B) initial mutator mutations. The major pathway for normal stem cells (A) is to differentiate into somatic cells, but development of oncogenic mutations leading to cancer or of mutations leading to reduced fitness and clonal extinction also occurs at a low frequency. The introduction of a mutator mutation (B) results in increased frequency of oncogenic mutations and increased frequency of mutations leading to reduced fitness.
When mutator clones acquire increased fitness due to a mutation, they can undergo selection and clonal expansion, further accelerating subseqent steps in carcinogenesis (Nowell 1976). We term this phenomenon “positive clonal selection” of mutator clones.
One argument against the mutator phenotype hypothesis can be termed “negative clonal selection,” defined as selection against mutator clones due to their more rapid acquisition of “reduced-fitness” (RF) mutations. An RF mutation is defined as any mutation that reduces the fitness of the clone harboring it, including mutations that reduce proliferation, enhance apoptosis, or enhance vulnerability to environmental factors. More rapid accumulation of RF mutations could lead to negative clonal selection against mutator clones, offsetting the more rapid development of oncogenic and other increased fitness mutations in mutator clones compared to wild type (see Figure 1).
A direct, deterministic mathematical model is applied to evaluate the importance of negative clonal selection in tumor evolution, as a function of key parameters. These parameters include the number of loci that lead to reduced fitness when mutated, the fraction of these loci that require mutation in only one copy for an effect on fitness, the mutation rates for wild-type and mutator clones, and the number of cell generations before a cancer is seen. Both dominant and recessive RF loci are considered. These are defined as loci where mutation of one copy leads to reduced fitness (dominant) or where both copies must be mutated to reduce fitness (recessive). These require the use of one-hit and two-hit models, respectively. Several authors have discussed multihit models in the acquisition of a malignant phenotype (Knudson 1971; Moolgavkar and Knudson 1981; Nowak et al. 2002; Komarova et al. 2003; Iwasa et al. 2004); however, in this work we evaluate the effect of one- and two-hit mechanisms on negative clonal selection. Dominant RF mutations may also be codominant, as well as dominant negative. Although one would expect dominant RF mutations to be more important given that only one hit is required, it is likely that fewer loci are affected by this mechanism, so the relative importance of dominant and recessive RF mutations cannot be stated a priori. The mathematical model can evaluate the relative importance of dominant and recessive RF mutations as a function of key parameters.
Negative clonal selection is considered in isolation, in the absence of oncogenic or other increased fitness mutations that could lead to positive clonal selection of mutator clones. Results obtained are therefore a limiting case or upper bound to the potential importance of negative clonal selection in tumor evolution. The study of limiting cases can be useful for clarifying issues and obtaining firm qualitative conclusions. The results indicate that the importance of negative clonal selection, and therefore the potential for negative clonal selection to limit the role of mutator mutations in carcinogenesis, depends on a variety of factors, including the overall tolerance of the genome for mutation.
THE MODEL
Underlying assumptions:
The model we present does not consider mutations leading to increased fitness and positive selection of clones harboring them, nor are neutral mutations considered. Thus the model focuses only on the isolated effect of negative clonal selection during the period in which carcinogenesis takes place. It is implicitly assumed that once a full complement of oncogenic mutations has been obtained, the resulting tumor is strongly selected.
The model is therefore a limiting case designed to determine an upper bound for the contribution of negative clonal selection. A variety of other simplifying assumptions are applied.
Two types of RF loci are considered: recessive and dominant. The model is capable of handling the case where multiple recessive RF loci are inactivated in one copy only. In the case of recessive RF mutations, the model assumes the same mutation rates for both hits. However, the model can be adapted to the case of different mutation rates for the two steps, such as would occur in CIN mutant cells in which mutation of one copy would occur at a much slower rate than that of the second step, loss of heterozygosity (LOH) at the relevant locus. The case of CIN mutants is closely approximated by all RF loci being dominant, since the second hit is not rate limiting. Dominant RF mutations can be dominant negative, or they can be in codominant genes in which a quantitative reduction in function alters cellular fitness.
The model assumes that within a given lineage, multiple mutations are independent in their effect on fitness. For example, mutations that reduce the tendency to apoptosis in response to DNA damage are not considered. Such mutations would violate the assumption of independence in that they would alter the effect of subsequent mutations on fitness. This type of mutation could actually further reduce the role of negative clonal selection (Komarova and Wodarz 2003).
The model assumes a constant mutation rate per locus, a constant mutation rate over time (except as affected by mutator mutations), and a fixed number of cell generations to cancer. Reversions are also ignored, an assumption that is valid at low mutation densities (i.e., ≪1).
We assume that any clone acquiring reduced fitness becomes extinct immediately. If the clone is within a large population, or has a significant reduction in fitness, this assumption is very nearly true. The assumption may break down for small cell populations or for minor reductions in fitness. An exact expression for the probability of extinction, allowing one to calculate how accurate this assumption is under different conditions, is given in the appendix.
Key parameters and values:
The model utilizes a small number of key parameters. The central parameter is NRFLN, the net number of RF loci in the genome (reduced-fitness loci net, RFLN) on a per nucleotide basis. Assume RF loci can be divided into a variety of different groups (i = 1, 2, 3, …) on the basis of how they affect fitness. Then NRFLN is a sum over all groups i of the products of numbers of RF loci of group i (NRF loci,i) multiplied by the probability (Pfitness disadvantage,i) that mutation of loci of group i will actually affect the corresponding gene product and ultimately lead to reduced fitness:
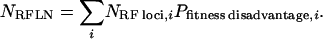 |
(1) |
This expresses the different types of RF mutations, each of which will lead to reduced fitness only in particular environmental or genetic contexts, and therefore Pfitness disadvantage,i ≤ 1.
One way to estimate an upper bound for Pfitness disadvantage,i is based on the probability of enzyme inactivation upon mutation of a coding locus. Guo et al. (2004) have estimated this parameter by constructing three random mutagenic polymerase chain reaction libraries of the human DNA repair enzyme 3-methyladenine DNA glycosylase, each with different average mutation burdens. These libraries were used to transfect a repair-deficient strain of Escherichia coli and protect it against methylmethane-sulfonate toxicity. The percentage of surviving colonies was measured as a function of the number of mutations in each clone. In this way, Pfitness disadvantage,i was estimated at 0.26 (on a per nucleotide basis), in agreement with estimates based on studies of lac repressor mutants (Markiewicz et al. 1994) and homologous human and chimpanzee protein pairs (Eyre-Walker and Keightley 1999). Estimating Pfitness disadvantage,i on the basis of inactivation of an enzyme is likely a maximal or worst-case scenario for clonal fitness reduction. Thus it is possible that inactivation of a particular enzyme may not actually reduce clonal fitness if there are redundant pathways or if the particular enzyme is not important in the given genetic or environmental context.
We obtain a worst-case value for NRFLN by multiplying an estimate of the total number of genes, 2.5 × 104 (International Human Genome Sequencing Consortium 2004), by an average number of coding bases per gene of 1.5 × 103 (International Human Genome Sequencing Consortium 2004) and by Pfitness disadvantage,i, estimated at 0.26, to obtain an estimate of NRFLN = 9.8 × 106.
We define NRFLN-D as the net number of dominant reduced-fitness loci. In the cases discussed below, we explore two possible values of this parameter representing 1 and 10% of NRFLN.
NRFLN-R is the net number of recessive reduced-fitness loci. Since any locus will be affected if both copies are genetically altered, NRFLN-R = NRFLN.
kmut is the wild-type mutation rate per nucleotide locus per cell generation. For stem cells, which likely are the populations from which cancers arise, this parameter may be as low as 10−11 (Cervantes et al. 2002).
α is the fold increase in kmut due to a mutator mutation. This can vary considerably depending on the type of mutator mutation. For purposes of this simulation, we utilize α = 102. This is approximately the order of the expected effect due to a mutation that abolishes proofreading (Beckman and Loeb 1993). If there is no mutator mutation, α = 1.
T is the number of cell generations to cancer. For cells lining colonic crypts, this is estimated at 5 × 103 (Tomlinson et al. 2002), but for other tumor types the number of cell generations may be significantly lower (Loeb et al. 2003). One way to estimate this parameter is to consider cell generation times for cycling cells and growth fractions for cells with reproductive potential, in the context of the available time to develop cancer (a human lifetime). Generation times for cycling cells are on the order of 36 hr for fibroblasts (Baca et al. 1985) and 48 hr in general (MITOPENCOURSEWARE 2005). A 36-hr generation time allows for ∼17,000 cell generations in a 70-year human lifetime. However, normal cells of reproductive potential are generally quiescent, cycling <1% of the time as measured by incorporation of bromodeoxyuridine (BrdUrd) with a 24-hr pulse as well as observation of cell growth kinetics for a 2-week period (Baker et al. 1995). Thus, as few as 170 cell generations may be available in some tissues for generation of a cancer.
Finally, we utilize the parameter Ngenes, the total number of genes in a genome, at 2.5 × 104 (International Human Genome Sequencing Consortium 2004).
Model for dominant reduced-fitness mutations:
The rate of increase in the fraction of clones with reduced fitness due to dominant reduced-fitness mutations is proportional to the mutation rate constant (and its increase due to a mutator mutation if applicable), the net number of dominant reduced-fitness loci, the fraction of clones still remaining with normal fitness, and the number 2 since there are two copies of the gene that may be inactivated. Reversions are ignored.
Thus, the following differential equation for PHI-D, the fraction of clones with haploid inactivation (HI) of at least one dominant (D) reduced-fitness locus, defines the model:
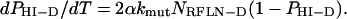 |
(2) |
Solving Equation 2 subject to the initial condition of no dominant reduced-fitness mutations initially (PHI-D = 0 at T = 0), we obtain
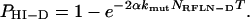 |
(3) |
Thus the fraction of clones with reduced fitness due to a dominant reduced fitness mutation increases as a saturating function over the number of cell generations T, approaching the limiting value of 1 in a simple exponential fashion, and the exponential rate constant is the product of the number 2, the mutation rate constant (and its increase due to a mutator mutation if applicable) and the number of dominant reduced fitness loci.
Model for recessive reduced-fitness mutations:
For the case where diploid inactivation of recessive reduced-fitness genes is the mechanism of cell death, define the fraction of fully fit cells at any moment as FF. Define the fraction of genes that have one copy inactivated at any moment as FHI, where HI stands for “haploid inactivated.” Multiple recessive RF genes can be haploid inactivated at once without affecting cell fitness, and this is considered in the model.
The rate of increase in the fraction of haploid-inactivated genes (i.e., the rate governing the “first hit”) is proportional to those not yet haploid inactivated (1 − FHI), the mutation rate constant (and its increase due to a mutator mutation if applicable), the number of reduced-fitness loci (NRFLN), and 2 since there are two copies of the gene that may be inactivated, all divided by the number of genes, since this quantity is on a per gene basis (Ngenes).
Thus, the differential equation for the first hit—i.e., inactivation of one copy of any given gene—is
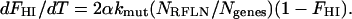 |
(4) |
We apply the boundary condition that the fraction of haploid inactivated genes is zero initially (FHI = 0 at T = 0) and derive the following equation for the first hit:
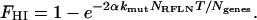 |
(5) |
For the second hit, the rate of decrease in the fraction of clones with full fitness is proportional to the product of the fraction of clones with full fitness remaining, the mutation constant (and its increase due to a mutator mutation if applicable), the total number of reduced fitness loci NRFLN, and the fraction of NRFLN involving genes that have already suffered the first hit, FHI.
Thus, the second differential equation describes the decrease in fully fit fraction due to accumulation of the second hit in genes that have already suffered the first:
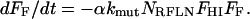 |
(6) |
This equation now quantifies the effect of reduced-fitness mutations on a full-genome basis, like Equation 2, rather than on a per gene basis, as in Equation 4.
We apply the boundary conditions that the fraction of clones suffering the first hit is zero and the fraction of fully fit clones is 1 initially (FHI = 0 and FF = 1 at T = 0). Substituting the value of FHI from Equation 5 into Equation 6, and separating the variables FF and T in the resulting equation, leads to an expression for FF, the fraction of fully fit clones as a function of cell generation number T under the influence of recessive reduced-fitness mutations,
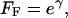 |
(7) |
where
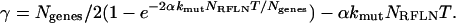 |
(8) |
Thus the fraction of fully fit clones is decreasing exponentially with a rate constant the absolute value of which is exponentially decreasing to a limiting value. The exponential rate constant and its rate of change are increasing functions of the mutation constant (and its increase due to a mutator mutation if applicable) and the total number of reduced fitness loci, NRFLN.
At low mutation densities, the time dependence of loss of fitness due to recessive reduced-fitness mutations is simpler, occurring as an exponential of the square of the number of cell generations. Thus, Equations 7 and 8 can be simplified, when 2αkmutNRFLNT/Ngenes ≪ 1, by expanding the exponential in Equation 8 in a Taylor series (which expresses the exponential as a sum of terms of increasing powers of 2αkmut NRFLN T / Ngenes) and truncating after terms of order 2 (as higher order terms are very small when 2αkmut NRFLN T / Ngenes ≪ 1), leading to
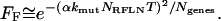 |
(9) |
The fraction of clones with reduced fitness, FRF, is equal to 1 − FF. Expanding FF in a Taylor series and truncating after the second term, we obtain
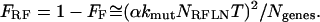 |
(10) |
Equation 10 is useful for estimating FRF when it is very small, where rounding errors in the calculation of the exponentials can lead to errors.
RESULTS
Four brief illustrative cases are presented, with the intent of assessing the percentage of fully fit clones, and therefore the influence of negative clonal selection, under a variety of conditions. The importance of dominant and recessive reduced-fitness mutations is assessed separately for each case. Parameters varied include the percentage of reduced-fitness loci that are dominant, the number of cell generations, and the presence/absence of a mutator mutation.
Case 1:
We assume that the wild-type mutation rate in human stem cells that give rise to tumors is equal to that in mouse embryonic stem cells (Cervantes et al. 2002) (α = 1, αkmut = 10−11), an average number of cell generations (T = 170), and that a high percentage of reduced-fitness loci yield a dominant phenotype (10%, NRFLN-D = 9.8 × 105). Using Equation 3, we determine that 0.3% of clones (1 in 300) has reduced fitness due to dominant reduced-fitness mutations, and if this is the only mechanism of negative clonal selection, 99.7% of clones will have normal fitness. Using Equation 10 we determine that ∼0.00000001% of clones (1 in 10 billion) has reduced fitness due to recessive reduced fitness mutations, and if this is the only mechanism of negative clonal selection 99.99999999% of clones will have normal fitness. Thus, with a wild-type mutation rate as reported in mouse embryonic stem cells and an average number of cell generations, negative clonal selection has only a negligible effect, almost entirely due to dominant reduced-fitness mutations.
Case 2:
We assume a mutator mutation imparting a 100-fold enhancement over the stem cell mutation rate (α = 100, αkmut = 10−9) and a high number of cell generations as may be the case for colon cancer(T = 5000), as well as a high percentage of reduced-fitness loci dominant (10%, NRFLN-D = 9.8 × 105). A mutation rate of 10−9 is also a reasonable approximation to that in differentiated cells in human tissues, as is seen at the hgprt locus in a variety of studies using human somatic cells in culture (Albertini et al. 1990). Using Equation 3, we determine that 99.994% of clones have reduced fitness due to dominant reduced-fitness mutations. Only 0.006% of clones (1 in 18,000) has normal fitness. Using Equations 7 and 8, we determine that ∼9% of clones (1 in 11) have reduced fitness due to recessive reduced fitness mutations, and if this is the only mechanism of negative clonal selection, 91% of the clones will have normal fitness. This case is the maximal case for negative clonal selection within the parameter ranges considered. Under these conditions, recessive reduced-fitness mutations have a measurable effect, but it involves <10% of the clones. Thus, only a 10% acceleration of mutator clones due to more rapid accumulation of oncogenic mutations in mutator clones compared to wild-type clones would be sufficient to offset this effect. Dominant reduced-fitness mutations produce a very large effect of negative clonal selection under these conditions, however, and an 18,000-fold effect of more rapid acquisition of oncogenic mutations by mutator clones compared to wild-type clones would be required to offset it.
Case 3:
We assume a mutator mutation imparting a 100-fold enhancement over the stem cell mutation rate (α = 100, αkmut = 10−9), a high number of cell generations as may be the case for colon cancer (T = 5000), and a medium percentage of reduced-fitness loci dominant (1%, NRFLN-D = 9.8 × 104). Using Equation 3, we determine that ∼62% of clones (5 of 8) have reduced fitness due to dominant reduced-fitness mutations, leaving 38% of clones (3 of 8) with normal fitness if only this mechanism is considered. Using Equations 7 and 8, we determine that ∼0.001% of clones (or 1 in 100,000) have reduced fitness due to recessive reduced-fitness mutations, leaving 99.999% of clones with normal fitness if only this mechanism is considered. Negative clonal selection has a slightly >2-fold effect, and thus a 2-fold more rapid acquisition of oncogenic mutations by mutator clones compared to wild-type clones would be required to offset it.
Case 4:
We again assume a mutator mutation imparting a 100-fold enhancement over the stem cell mutation rate (α = 100, αkmut = 10−9), but an average number of cell generations (T = 170), and a high percentage of reduced-fitness loci dominant (10%, NRFLN-D = 9.8 × 105). Using Equation 3, we determine that ∼28% of clones (or ∼2 in 7) have reduced fitness due to dominant reduced-fitness mutations, and 72% (5 in 7) retain normal fitness if this is the only mechanism of negative clonal selection (Figure 2). Using Equations 7 and 8 we determine that only 0.0001% of clones (1 in 1 million) has reduced fitness due to recessive reduced-fitness mutations, with 99.9999% of clones retaining normal fitness if this is the only mechanism of negative clonal selection (Figure 2). Under these conditions, negative clonal selection has a less than twofold effect, and again only a less than twofold effect of the more rapid acquisition of oncogenic mutations in mutator clones compared to wild-type clones would be required to offset it. The effect of negative clonal selection is nearly entirely due to dominant reduced-fitness mutations.
Figure 2.

Percentage of clones with reduced fitness, as a function of the number of cellular generations T for dominant (squares) and recessive (triangles) reduced-fitness mutational mechanisms, calculated using Equations 3 and 10, respectively, and converting to percentages, for case 4 (see results). Assumed parameter values are αkmut = 10−9 and NRFLN-D = 9.8 × 105.
DISCUSSION
This article considers a limiting-case or worst-case scenario in an effort to determine if negative clonal selection of mutator clones can serve as an argument against the mutator hypothesis of carcinogenesis. Positive clonal selection, which could in principle favor mutator clones, is not considered. Extinction is presumed certain with any slight fitness reduction, and the possibility of mutations that would diminish the probability of apoptosis in response to genetic alterations is ignored. Even with these extreme assumptions a severe effect due to negative clonal selection is not seen in our model unless all loci are reduced-fitness loci with an average probability of reduced fitness of 26%, 10% of these loci are dominant reduced-fitness loci, and 5000 cell generations occur prior to cancer development (case 2). Thus, it would seem to be unlikely that negative clonal selection is a realistic argument against the mutator hypothesis in most cases. A complete model that incorporates negative clonal selection together with the more rapid accumulation of oncogenic mutations in mutator clones could address that question from a theoretical perspective.
However, the possibility that some real cases may mimic our case 2 remains, and in these cases negative clonal selection may limit the importance of mutator mutations in carcinogenesis. It seems reasonable that tumors that arise in tissues that undergo rapid cell proliferation and that shed dividing cells such as colonic epithelium and skin could undergo large numbers of cell generations as in case 2. In contrast, tissues that do not divide rapidly in the adult, such as liver and breast, are likely to have fewer cell generations prior to tumor formation, as in cases 1 and 4.
The results of case 2 do indicate the existence of an upper limit to the mutation rate, beyond which negative clonal selection will begin to limit the survival of mutator clones. In fact, the critical mutation rate is approximately the reciprocal of the product 2NRFLN-DT, i.e., 2 times the net number of dominant reduced-fitness loci times the number of cell generations. At the critical mutation rate, 63% of the mutator clones will be eliminated due to negative clonal selection, a degree of negative clonal selection that could potentially be offset by the more rapid acquisition of oncogenic mutations by mutator clones compared to wild-type clones if that effect provides a threefold advantage. However, at 3 times this mutation rate, 95% of the mutator clones will be eliminated due to negative clonal selection. At 10 times the critical mutation rate, 99.995% of the mutator clones will be eliminated by negative clonal selection, with ∼1 in 20,000 mutator clones surviving. Over the range of assumptions for NRFLN-D and T considered herein, the critical mutation rate ranges from 1.1 × 10−10 to 3 × 10−8. However, these values presume the very high values of NRFLN-D that were assumed for this model. Lower values of NRFLN-D will lead to higher values of the critical mutation rate. Work in progress will more precisely define if and when negative clonal selection of this magnitude can outweigh the advantage of more rapid accumulation of oncogenic mutations, as a function of key parameters. Clonal expansion following favorable mutations can effectively limit the number of rate-limiting oncogenic mutation steps, also affecting the trade-off between negative clonal selection and oncogenic mutation in mutator clones.
Under the conditions explored, dominant reduced-fitness mutations are more important than recessive reduced-fitness mutations. However, the proportion of reduced-fitness loci assumed dominant has been varied from 1 to 10% in the above cases. It is possible that the proportion of reduced-fitness loci that are dominant is much less.
CIN cells have a high probability of chromosomal loss, up to 10−2 per chromosome pair per cell generation (Lengauer et al. 1998). In this case, recessive reduced-fitness loci may functionally be “partially” dominant, in that any haploid mutation has a very high probability of LOH, resulting in a loss of the second wild-type gene copy in half of the cases. However, since half of the clones will lose the mutated copy rather than the wild-type copy, and therefore have normal fitness, negative clonal selection can be no more than twofold by this mechanism at low mutation densities. Thus at low mutation densities, CIN mutations may accelerate the development of positively selected oncogenic mutations while having a minimal effect on negative clonal selection.
At higher mutation densities, each copy of a chromosome pair will potentially have a recessive reduced-fitness mutation, albeit in different loci. Under those conditions, loss of either copy of the chromosome would lead to fitness reduction. Thus, the survival of CIN cells as a function of mutation density is of interest. If CIN cells continue to survive at high mutation density, it can mean that only a very small minority of gene loci affect fitness. It is conceivable, for example, that redundancy in biochemical pathways requires multiple redundant genes to be inactivated before a reduction in fitness is seen. The requirement for mutation of two dominant RF loci to reduce fitness is mathematically similar to the need to mutate two copies of a single recessive RF locus. This would in turn imply a much lesser effect of negative clonal selection than the upper bound modeled in this article.
Other investigators (Iwasa et al. 2004) have considered the role of a deleterious mutation, followed by a favorable one, in carcinogenesis, using a stochastic model. In contrast, we consider up to two sequential deleterious mutations in the context of negative clonal selection, using a deterministic model. Deterministic models provide exact equations for the average behavior of the population as a whole, without considering the chance variation of individual members of the population, which is delineated by stochastic models. Deterministic models can yield only average expectation values, whereas stochastic models yield an entire probability distribution. On the other hand, deterministic models such as ours explicitly consider the case where the first hit may occur in more than one gene prior to the second hit occurring in any gene. This is difficult to achieve with stochastic models. In agreement with Iwasa et al. (2004), we find that for low cell numbers and low mutation density, the accumulation of two mutations is approximately proportional to the square of the number of cell generations. Iwasa et al. (2004) also consider different mutation rates for each of the two steps. While we have not considered this case, the mathematics presented herein are easily adaptable to it.
During actual cancer progression, a mutation reducing the probability of apoptosis as a function of further mutation is possible. This is part of the general concept of a mutator phenotype. Komarova and Wodarz (2003) consider which would occur first, a conventional mutator mutation or an anti-apoptotic mutator mutation. In our model, an anti-apoptosis mutation would result in a decrease in NRFLN. The model as currently constructed allows one to consider the effect of preexisting mutations of this nature, but as NRFLN and NRFLN-D are then assumed constant, the model would require modification to account for a change in these parameters. This could be accommodated by dividing the cell generations into those before and after the event, resulting in convolution integrals. For small α (≤10- to 30-fold, as expected for proofreading mutations in polymerases), our results appear to indicate that an anti-apoptotic mutation early would not be critical. However, for large α (>100-fold enhancement in error rate) and for CIN mutations, an early anti-apoptotic mutation could be crucial in mitigating extreme effects of negative clonal selection, as discussed above.
Given that our model is a limiting case designed to maximize the impact of negative clonal selection, we anticipate that clonal fitness as a function of increased mutation rate in any experimental system would exceed that predicted by this model, due to effects such as anti-apoptosis mutations and other mutations that would enhance clonal fitness. Our analysis indicates that dominant reduced-fitness mutations are more likely to be significant than recessive ones.
Verification of our model could be achieved by increasing the mutation rates in cultured tumor cells and measuring the frequency of reduced-fitness clones as a function of the number of cell generations. The most direct approach to increasing point mutation rates would be to culture cells in the presence of mutagenic nucleotide analogs. Fitness could be determined by growing aliquots of the culture after increasing numbers of generations and measuring cloning efficiencies. In the case of dominant mutations being the major mechanism of fitness reduction the natural logarithm of the fraction of fully fit clones should decrease with the number of cell generations in a linear fashion, with slope −2αkmutNRFLN-D. Given the ability to also measure αkmut by sequencing of various nonselected cellular loci in successive generations (Bielas and Loeb 2005), one could also estimate NRFLN-D.
We have examined the effect of negative clonal selection in possibly mitigating against the importance of a mutator phenotype in carcinogenesis. In general, it seems unlikely that negative clonal selection against mutator clones is a significant effect. However, the result depends equally on a variety of factors singly and in combination, including the mutation rate within the mutator clone, the number of cell generations, and, perhaps of greatest interest, the overall tolerance of the genome for mutation. Greater or lesser plasticity of the genome will tend to increase or decrease, respectively, the likely importance of mutator mutations in carcinogenesis.
Acknowledgments
We are indebted to Ern Loh, Ali Ozgenc, and Mike Schmitt for critical review of the manuscript. This work was supported in part by grants to L.A.L. from the National Cancer Institute (CA 78885) and National Institutes of Health (CA 102029).
APPENDIX: PROBABILITY OF EXTINCTION OF A CLONE WITH REDUCED FITNESS
An exact expression for the probability of extinction of a new cell clone with fitness r < 1 in a population of N cells is 1 minus the fixation probability (i.e., the probability that the cells take over the population), the latter given in Iwasa et al. (2004):
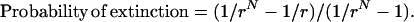 |
(A1) |
Let N equal the total number of cells in the population and n equal the number of cells corresponding to the new clone. Consider a random walk where in each step n either decreases or increases by 1. The random walk has “absorbing barriers”; that is, both extinction (n = 0) and complete takeover of the population (n = N) are irreversible (Feller 1957). Let Qn be the probability of extinction starting from n cells derived from the new clone. If the relative fitness of the new clone is r < 1, and the relative fitness of the remaining cells is 1, the single-step probability p of increasing the number of cells in the new clone is
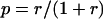 |
(A2) |
and the single-step probability q of decreasing the number of cells in the new clone is
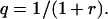 |
(A3) |
Starting from n cells in the new clone there are two possible next states, n + 1 cells or n − 1 cells in the new clone, and therefore
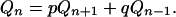 |
(A4) |
We can verify by substitution that two particular solutions of the difference equation (A4) are possible, Qn = 1 for all n and Qn = (q/p)n, and therefore the linear combination of these two particular solutions is also a solution,
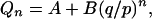 |
(A5) |
where A and B are constants. Applying the boundary conditions Q0 = 1 (extinction is irreversible) and QN = 0 (full takeover of the population is irreversible), we obtain the values of A and B, leading to
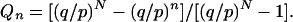 |
(A6) |
Substituting the values of p and q from (A2) and (A3) into (A6), and setting n = 1 for the case when the first new cell from a new clone is formed, we obtain Equation A1.
Inspection of Equation A1 shows that the probability of extinction approaches 1 as r approaches zero (large reduction in fitness) or N approaches infinity (large cell populations), and therefore under these conditions the approximation that a reduced fitness clone will always become extinct is nearly true. Using Equation A1, one can calculate how good that approximation is in real cases.
References
- Albertini, R. J., J. A. Nicklas, J. P. O'Neill and S. H. Robison, 1990. In vivo somatic mutations in humans: measurement and analysis. Annu. Rev. Genet. 24: 305–326. [DOI] [PubMed] [Google Scholar]
- Baca, O. G., O. S. Tacheeni, E. T. Akporiaye, R. Deblasie and H. A. Crissman, 1985. Cell cycle distribution patterns and generation times of L929 fibroblast cells persistently infected with Coxiella burnetti. Infect. Immun. 47: 366–369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baker, F. L., L. J. Sanger, R. W. Rodgers, K. Jabboury and O. R. Mangini, 1995. Cell proliferation kinetics of normal and tumour tissue in vitro: quiescent reproductive cells and the cycling reproductive fraction. Cell Proliferation 28: 1–15. [DOI] [PubMed] [Google Scholar]
- Beckman, R. A., and L. A. Loeb, 1993. Multistage proofreading in DNA replication. Q. Rev. Biophys. 26: 225–331. [DOI] [PubMed] [Google Scholar]
- Bielas, J. H., and L. A. Loeb, 2005. Quantitation of random genetic mutations. Nat. Methods 2: 285–290. [DOI] [PubMed] [Google Scholar]
- Cervantes, R. B., J. R. Stringer, C. Shao, J. A. Tishcfield and P. J. Stambrook, 2002. Embryonic stem cells and somatic cells differ in mutation frequency and type. Proc. Natl. Acad. Sci. USA 99: 3586–3590. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Datta, A., J. L. Schmeits, N. S. Amin, P. J. Lau, K. Myung et al., 2000. Checkpoint-dependent activation of mutagenic repair in Saccharomyces cerevisiae pol2–01 mutants. Mol. Cell 6: 593–603. [DOI] [PubMed] [Google Scholar]
- Eyre-Walker, A., and P. D. Keightley, 1999. High genomic deleterious mutation rates in hominids. Nature 397: 344–347. [DOI] [PubMed] [Google Scholar]
- Feller, W., 1957. An Introduction to Probability Theory and Its Applications, pp. 313–314. John Wiley & Sons, New York.
- Fishel, R., M. K. Lescoe, M. R. S. Rao, N. G. Copeland, N. G. Jenkins et al., 1993. The human mutator gene homolog MSH2 and its association with hereditary nonpolyposis colon cancer. Cell 75: 1027–1038. [DOI] [PubMed] [Google Scholar]
- Futreal, P. A., L. Coin, M. Marshall, T. Down, T. Hubbard et al., 2004. A census of human cancer genes. Nat. Rev. Cancer 4: 117–183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goldsby, R. E., N. A. Lawrence, L. E. Hays, E. A. Olmsted, X. Chen et al., 2001. Defective DNA polymerase-δ proofreading causes cancer susceptibility in mice. Nat. Med. 7: 638–639. [DOI] [PubMed] [Google Scholar]
- Guo, H. H., J. Choe and L. A. Loeb, 2004. Protein tolerance to random amino acid substitution. Proc. Natl. Acad. Sci. USA 101: 9205–9210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hanahan, D., and R. A. Weinberg, 2000. The hallmarks of cancer. Cell 100: 57–70. [DOI] [PubMed] [Google Scholar]
- International Human Genome Sequencing Consortium, 2004. Finishing the euchromatic sequence of the human genome. Nature 431: 931–945. [DOI] [PubMed] [Google Scholar]
- Ionov, Y., M. A. Peinado, S. Malkhosyan, S. Shibata and M. Perucho, 1993. Ubiquitous somatic mutations in simple repeated sequences reveal a new mechanism for colonic carcinogenesis. Nature 363: 558–561. [DOI] [PubMed] [Google Scholar]
- Iwasa, Y., F. Michor and M. A. Nowak, 2004. Stochastic tunnels in evolutionary dynamics. Genetics 166: 1571–1579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Knudson, A. G., 1971. Mutation and cancer: statistical study of retinoblastoma. Proc. Natl. Acad. Sci. USA 68: 820–823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Komarova, N. L., and D. Wodarz, 2003. Evolutionary dynamics of mutator phenotypes in cancer: implications for chemotherapy. Cancer Res. 63: 6335–6342. [PubMed] [Google Scholar]
- Komarova, N. L., A. Sengupta and M. A. Nowak, 2003. Mutation-selection networks of cancer initiation: tumor suppressor genes and chromosomal instability. J. Theor. Biol. 223: 433–450. [DOI] [PubMed] [Google Scholar]
- Lengauer, C., K. W. Kinzler and B. Vogelstein, 1998. Genetic instabilities in human cancers. Nature 396: 643–649. [DOI] [PubMed] [Google Scholar]
- Levine, A. J., 1997. p53, the cellular gatekeeper for growth and division. Cell 88: 323–331. [DOI] [PubMed] [Google Scholar]
- Loeb, L. A., 1991. Mutator phenotype may be required for multistage carcinogenesis. Cancer Res. 51: 3075–3079. [PubMed] [Google Scholar]
- Loeb, L. A., 1998. Cancer cells exhibit a mutator phenotype. Adv. Cancer Res. 72: 25–56. [DOI] [PubMed] [Google Scholar]
- Loeb, L. A., C. F. Springgate and N. Battula, 1974. Errors in DNA replication as a basis of malignant changes. Cancer Res. 34: 2311–2321. [PubMed] [Google Scholar]
- Loeb, L. A., K. R. Loeb and J. P. Anderson, 2003. Multiple mutations in cancer. Proc. Natl. Acad. Sci. USA 100: 776–781. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Markiewicz, P., L. G. Kleina, C. Cruz, S. Ehret and J. H. Miller, 1994. Genetic studies of the lac repressor. XIV. Analysis of 400 altered Escherichia coli lac repressors reveals essential and non-essential residues, as well as “spacers” which do not require a specific sequence. J. Mol. Biol. 240: 421–433. [DOI] [PubMed] [Google Scholar]
- MITOPENCOURSEWARE, 2005. Cell, tissue and tumor kinetics (http://ocw.mit.edu).
- Moolgavkar, S. H., and A. G. Knudson, 1981. Mutation and cancer: a model for human carcinogenesis. J. Natl. Cancer Inst. 66: 1037–1052. [DOI] [PubMed] [Google Scholar]
- Morrison, A., A. L. Johnson, L. H. Johnston and A. Sugino, 1993. Pathway correcting DNA replication errors in Saccharomyces cerevisiae. EMBO J. 12: 1467–1473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nowak, M. A., N. L. Komarova, A. Sengupta, P. V. Jallepalli, I. M. Shin et al., 2002. The role of chromosomal instability in tumor initiation. Proc. Natl. Acad. Sci. USA 99: 16226–16231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nowell, P. C., 1976. The clonal evolution of tumor cell populations. Science 194: 23–28. [DOI] [PubMed] [Google Scholar]
- Sherr, C. J., and F. McCormick, 2002. The RB and p53 pathways in cancer. Cancer Cell 2: 103–112. [DOI] [PubMed] [Google Scholar]
- Tomlinson, I., P. Sasieni and W. Bodmer, 2002. How many mutations in a cancer? Am. J. Pathol. 160: 755–758. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wood, R. D., M. Mitchell, J. Sgouros and T. Lindahl, 2001. Human DNA repair genes. Science 29: 1284–1289. [DOI] [PubMed] [Google Scholar]