

Abstract
The prune gene of Drosophila melanogaster is predicted to encode a phosphodiesterase. Null alleles of prune are viable but cause an eye-color phenotype. The abnormal wing discs gene encodes a nucleoside diphosphate kinase. Killer of prune is a missense mutation in the abnormal wing discs gene. Although it has no phenotype by itself even when homozygous, Killer of prune when heterozygous causes lethality in the absence of prune gene function. A screen for suppressors of transgenic Killer of prune led to the recovery of three mutations, all of which are in the same gene. As heterozygotes these mutations are dominant suppressors of the prune-Killer of prune lethal interaction; as homozygotes these mutations cause early larval lethality and the absence of imaginal discs. These alleles are loss-of-function mutations in CG10065, a gene that is predicted to encode a protein with several zinc finger domains and glutathione S-transferase activity.
THE prune (pn) gene was identified by viable mutations that cause a brownish purple eye-color phenotype (Beadle and Ephrussi 1936). The pn gene encodes a single 1.8-kb transcript that is predicted to be translated into a 44.5-kDa protein (Frolov et al. 1994). Nucleotide sequence analysis of pn mutants suggested that many of them are null alleles (Timmons and Shearn 1996). Immunoblotting of these same alleles was consistent with this interpretation; no PN protein was detected (Timmons and Shearn 1996). On the basis of sequence similarities PN was predicted to have pyrophosphatase activity (Aravind and Koonin 1998). A recent study of the human ortholog of PN revealed that it has phosphodiesterase activity, specifically cyclic AMP phosphodiesterase activity (D'Angelo et al. 2004). Moreover, there appears to be a correlation between highly metastatic tumors and high levels of PN protein (Forus et al. 2001).
The abnormal wing discs (awd) gene was identified in a hybrid dysgenic screen for lethal mutations that cause imaginal disc abnormalities (Dearolf et al. 1988a). The awd gene encodes a single 0.8-kb transcript (Dearolf et al. 1988b) that is translated into a 17-kDa subunit of a 100-kDa homo-hexameric protein that accounts for 98% of the nucleoside diphosphate kinase activity of larval extracts (Biggs et al. 1990; Timmons et al. 1995). Nucleoside diphosphate kinases can transfer the terminal phosphate from any nucleoside triphosphate donor to any nucleoside diphosphate acceptor via a ping-pong mechanism. Drosophila AWD is 78% identical to the human NM23A and NM23B proteins that also have nucleoside diphosphate kinase activity (Rosengard et al. 1989). The nm23 gene was identified in a screen for genes whose expression is downregulated in metastatic tumor cell lines (Steeg et al. 1988). When expressed in Drosophila, either NM23A or NM23B produces nucleoside diphosphate kinase activity and can, to varying extents, rescue the awd mutant phenotype (Xu et al. 1996).
Killer of prune (Kpn) was identified by Sturtevant (1956) as a nonlethal mutation that causes lethality in flies that lack PN function. Lifschytz and Falk (1969a) recovered X-ray-induced revertants of Kpn that were homozygous lethal. They interpreted these revertants as deletions of one or more vital genes. Lifschytz and Falk (1969b) also recovered EMS-induced revertants of Kpn that were homozygous lethal and allelic to each other. They interpreted these revertants as mutations of a single vital gene. We discovered that this vital gene is awd (Biggs et al. 1988). Kpn is actually a missense mutation that causes a substitution of Ser for Pro at position 97 of the AWD protein (Lascu et al. 1992; Timmons et al. 1995). We renamed Kpn as awdKpn.
In hopes of discovering the molecular mechanism of the pn-awdKpn lethal interaction we set out to generate mutations that would suppress this interaction and to identify the function of the relevant genes.
MATERIALS AND METHODS
Stocks:
Flies were reared on a cornmeal-molasses-agar-yeast extract medium seeded with live yeast at 20°. The y1 w67c and awdKpn stocks, all of the deficiencies listed in Table 2, most of the lethal mutations listed in Table 4, all of the stocks with nonlethal transposon insertions, and the TM3 balancer chromosome marked with Kr-GFP were obtained from the Bloomington Stock Center. The lethal mutations listed in Table 4 that have an “s” in their symbols were obtained from the Szeged Stock Center. The prune alleles used (pn18a and pn12c) are null alleles generated in this laboratory on a y1 w67c chromosome (Timmons and Shearn 1996). The TM3 balancer chromosome marked with y+ was also generated in this laboratory.
TABLE 2.
Cytogenetic localization of Su(Kpn)
| Deficiency (3) | Deleted region | Complementation |
|---|---|---|
| th102 | 72A21; 72D10 | + |
| W10 | 75A6; 75C2 | + |
| W4 | 75B10; 75C5 | + |
| H99 | 75C1; 75C2 | + |
| Cat | 75C1; 75F1 | + |
| VW3 | 76A3; 76B2 | + |
| in61 | 76F1–3; 77D1–2 | + |
| rdgC-co2 | 77A1; 77D1 | + |
| ri79c | 77B7; 77F5 | + |
| ME107 | 77F3; 78C8–9 | + |
| 31A | 78A; 78E | + |
| Pc-2q | 78C5–6; 79A1 | + |
| Delta1AK | 79F; 80A | + |
| 2-2 | 81F4; 83A | + |
| 6-7 | 82D5; 82F6 | + |
| Tp110 | 83C1–2; 84B1–2 | + |
| Scr | 84A1–2; 84B1–2 | + |
| Antp17 | 84A5; 84D11–14 | − |
| p712 | 84D4–6; 85B6 | + |
| XT103 | 85A2; 85C1–2 | + |
TABLE 4.
Complementation tests with lethal mutations in cytogenetic region 84
| Lethal mutation | Gene symbol | Cytogenetic region | Complementation |
|---|---|---|---|
| l(3)s093501 | 84A3–6 | + | |
| l(3)84Bb | 84B | + | |
| l(3)84Bc | 84B | + | |
| l(3j7A6 | 84B1–2 | + | |
| l(3)L2100 | 84B2–3 | + | |
| l(3)s147406 | 84B3–6 | + | |
| l(3)84Cb | 84C | + | |
| l(3)84Ce | 84C | + | |
| l(3)s057809 | 84C | + | |
| l(3)s006313 | 84C1–2 | + | |
| l(3j8C8 | 84C1–2 | + | |
| l(3)02267 | Aly = CG1101 | 84C1–2 | + |
| l(3)S2214 | = CG10061 | 84C4–6 | + |
| l(3)84Cc | gfzf = CG10065 | 84C6 | − |
| l(3)s001813 | 84D | + | |
| l(3)s007403 | 84D | + | |
| l(3)s023931 | 84D | + | |
| l(3)s097301 | 84D | + | |
| l(3)s127405 | 84D | + | |
| l(3)84Da | 84D | + | |
| l(3)84Db | 84D | + | |
| l(3)84Dc | 84D | + | |
| l(3)84Dd | 84D | + | |
| l(3)neo34 | lap = CG2520 | 84D1–14 | + |
| l(3)s127416 | 84D1–4 | + | |
| l(3)s145110 | 84D1–7 | + | |
| l(3)s050115 | 84D4–8 | + | |
| l(3)s002237 | 84A5; 84D11–14 | + | |
| l(3)s015110 | 84A5; 84D11–14 | + | |
| l(3)s091911 | 84A5; 84D11–14 | + | |
| l(3)s026909 | 84A5; 84D11–14 | + | |
| l(3)s026915 | 84A5; 84D11–14 | + | |
| l(3)s036809 | 84A5; 84D11–14 | + | |
| l(3)s042302 | 84A5; 84D11–14 | + | |
| l(3)s044324 | 84A5; 84D11–14 | + | |
| l(3)s051513 | 84A5; 84D11–14 | + | |
| l(3)s113302 | 84A5; 84D11–14 | + | |
| l(3)s118306 | 84A5; 84D11–14 | + | |
| l(3)s139908 | 84A5; 84D11–14 | + | |
| l(3)s140410 | 84A5; 84D11–14 | + |
Screen for suppression of endogenous awdKpn:
Young male flies (0–24 hr posteclosion) that were hemizygous for pn18a on the X chromosome and homozygous for ebony (e) on the third chromosome were fed EMS for 24 hr as described by the Shearn and Garen (1974) modification of the procedure initially described by Lewis and Bacher (1968). After this mutagenesis, the males were mated to females that were homozygous for yellow (y) on attached X chromosomes and were homozygous for awdKpn. All of the male progeny of this cross were expected to die because they were hemizygous for pn and heterozygous for awdKpn (Table 1) unless a suppressor mutation had been induced.
TABLE 1.
Suppressor activity on endogenous and transgenic awdKpn
| ♀ genotype | No. | ♂ genotype | No. |
|---|---|---|---|
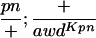 |
1804 | 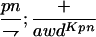 |
0 |
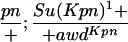 |
592 | 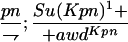 |
0 |
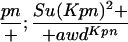 |
305 | 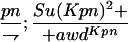 |
0 |
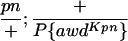 |
687 | 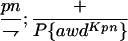 |
0 |
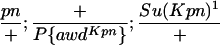 |
456 | 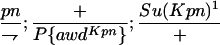 |
297** |
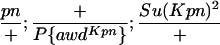 |
398 | 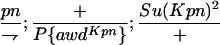 |
386 |
P{awdKpn}, second chromosome insert of transgene. **Number of males highly significantly different (P < 0.01) than number of females according to the G-test.
Assay for suppressor activity on endogenous and transgenic awdKpn:
As controls, females that were homozygous for pn on the X chromosomes were mated to males that were homozygous for endogenous awdKpn on the third chromosomes or for transgenic awdKpn on the second chromosomes. The number of male progeny hemizygous for pn and heterozygous for endogenous or transgenic awdKpn was compared to the number of female progeny heterozygous for pn and heterozygous for endogenous or transgenic awdKpn. As experimentals, females that were homozygous for pn on the X chromosomes and heterozygous for a suppressor on the third chromosome were mated to males that were homozygous for endogenous awdKpn on the third chromosome or for transgenic awdKpn on the second chromosome. The number of male progeny hemizygous for pn, heterozygous for endogenous or transgenic awdKpn, and heterozygous for a suppressor was compared to the number of female progeny heterozygous for pn, heterozygous for endogenous or transgenic awdKpn, and heterozygous for a suppressor.
Screen for suppression of transgenic awdKpn:
Young male flies (0–24 hr posteclosion) that were hemizygous for y, pn, and white (w) on the X chromosome were fed EMS for 24 hr as described above. After this mutagenesis, the males were mated to females that were homozygous for y and w on attached X chromosomes, were homozygous for transgenic awdKpn (marked with w+) on the second chromosome, and were homozygous for e and a null allele of awd on the third chromosome. The NDP kinase activity supplied by transgenic awdKpn is sufficient to rescue the lethality caused by the null awd allele. All of the male progeny of this cross were expected to die because they were hemizygous for pn and heterozygous for transgenic awdKpn (Table 1) unless a suppressor mutation had been induced.
Deletion mapping of Su(Kpn):
Female flies heterozygous for Su(Kpn)1 were mated to males heterozygous for third chromosome deficiencies. A recovery of <1% of adult flies trans-heterozygous for Su(Kpn)1 and a deficiency relative to their heterozygous sibs were considered a failure to complement.
Screen for mutations that fail to complement the lethality of Su(Kpn):
Young male flies (0–24 hr posteclosion) that were hemizygous for y and w on the X chromosome and homozygous for P[neoFRT82B] and P[NM88Cw+] on the third chromosome were fed EMS for 24 hr as described above. After this mutagenesis, the males were mated at 27° to females that were homozygous for y and w on the X chromosome and heterozygous for Glued (Gl) and a dominant temperature-sensitive mutation (DTS) and a TM3 balancer chromosome marked with Serrate (Ser) and a y+ transgene. The y+ transgene was derived (A. Shearn, unpublished data) from the y+ transgene on a second chromosome balancer (Timmons et al. 1993). Male progeny were mated individually to two to three females that were homozygous for y and w on the X chromosome and heterozygous for Su(Kpn)1. If no progeny trans-heterozygous for a mutagenized third chromosome and Su(Kpn)1 were recovered from an individual cross, then males heterozygous for the mutagenized chromosome, recognizable by the w+ marker, were retested by mating them to females that were homozygous for y and w on the X chromosome and heterozygous for Su(Kpn)2. If no progeny trans-heterozygous for a mutagenized third chromosome and Su(Kpn)2 were recovered from an individual cross, then males and females heterozygous for the mutagenized chromosome were mated to each other to produce a stock.
Test for suppression of pn-awdKpn lethal phenotype:
Females homozygous for y pn18a w− on the X chromosome, heterozygous for transgenic <w+ awdKpn> over the y+ Cyo balancer on the second chromosome, and heterozygous for Su(Kpn)1 over the y+ TM3 e Ser balancer were mated to y w− males heterozygous for the putative suppressor alleles, Su(Kpn)X w+ FRT balanced over w+ TM3 e Sb KrGFP. Males that were pn P{awdKpn} in combination with either Su(Kpn)1 or Su(Kpn)X alleles were identified on the basis of the presence of the balancer chromosomes. The relative suppression was calculated as the ratio of [pn-Su(Kpn)1/pn P{awdKpn}-Su(Kpn)1]/[pn-Su(Kpn)X/pn P{awdKpn}-Su(Kpn)X]. A G-test for independence was used to test the significance of the difference in the ability to suppress the pn-awdKpn lethality between Su(Kpn)1 and Su(Kpn)X. Nonsignificant P-values indicate the ratios of Su(Kpn)1 to Su(Kpn)X are equivalent. Ratios <1 with significant P-values indicate Su(Kpn)X is weaker than Su(Kpn)1. Ratios >1 with significant P-values indicate Su(Kpn)X is stronger than Su(Kpn)1. An average of 298 male flies were scored from individual crosses. All crosses were set up twice independently. At least 45 suppressor males were counted in each cross. All crosses were carried out at 25°.
Lethal excisions of an insertion near Su(Kpn):
Males that were hemizygous for y and w on the X chromosome and homozygous for an insertion in 84C6–8 marked with y+ and w+ were mated to females that were homozygous for y and w on the X chromosome and heterozygous for Dropped (Dr) and a balancer with a transgene that encodes transposase activity (cross I). Male progeny heterozygous for the insertion and the balancer were mated to females homozygous for y and w on the X chromosome and heterozygous for a deficiency that uncovers Su(Kpn) and a TM3 balancer mutation marked with Ser and a y+ transgene (cross II). Male progeny lacking the w+ marker of the insertion and the marker of the balancer with transposase activity were considered putative excisions of the insertions. These males were mated to females of the same genotype as their mothers (cross III). If no progeny were recovered that were trans-heterozygous for the putative excision and the deficiency, then the excision was deemed to have caused a lethal mutation somewhere within the limits of the deficiency. Male progeny heterozygous for this lethal excision were mated to females homozygous for y and w on the X chromosome and heterozygous for Su(Kpn)2 (cross IV). If no progeny were recovered that were trans-heterozygous for the lethal excision and for Su(Kpn)2, then the excision was deemed to have caused a lethal mutation in Su(Kpn).
Immunoblot analysis of endogenous and transgenic AWDKpn:
To compare the protein levels of endogenous AWDKpn to transgenic AWDKpn, an immunoblot of third instar larvae homozygous for awdKpn or <awdKpn> on an awd null background was done. A 15% SDS-PAGE gel was loaded with the equivalent of three larvae per well in 2× SDS sample buffer. Purified AWD polyclonal antibody previously generated in the laboratory (Timmons et al. 1995) was used at 1:750 to detect the 15-kDa monomers of AWDKpn. The blot was normalized to β-tubulin (E7: Developmental Studies Hybridoma Bank) at 1:500. HRP-conjugated secondary antibodies were used at 1:5000. The blot was quantitated using NIH Image Software.
Determination of DNA sequence alterations in Su(Kpn) mutants:
Homozygous larvae were identified as viable, nonfluorescent larvae from the stock y w; Su(Kpn)X w+FRT/w+TM3 e Sb Kr-GFP. Larvae were collected between the first and third instar. Heterozygous larvae were used for samples lethal before the first instar. Twenty to 40 larvae were dounced in buffer A (10 mm Tris-HCl pH 7.5, 60 mm NaCl, 10 mm EDTA pH 8.0, 150 nm spermine, 150 nm spermidine, 5% sucrose). An equal volume of buffer B (1.25% SDS, 300 mm Tris-HCl pH 9.0, 100 mm EDTA pH 8.0, 5% sucrose, 100 ug/ml Proteinase K) was added and the lysate was incubated at 37° for 1–2 hr for proteinase K digestion. Following digestion, standard phenol/chloroform/isoamylalcohol extraction and ethanol/NaOAc precipitation recovered genomic DNA. Genomic DNA was PCR amplified using standard conditions with Amplitaq Gold Hotstart Taq (Applied Biosciences, Foster City, CA). Primers covered the four exons of CG10065 in six reactions covering the full gene, including intron/exon splice sites: exon 1 primers, (forward) for 5′-TAA ATC GCC GGC GTT TGC, (reverse) rev 5′-GGC GGG AAA CCA TCT CAT TA; exon 2 set A, for 5′-TAC TGA AGG GAA TAT CCA AGG G, rev 5′-GTC GGG CAG TAT CTC GAT CTT; exon 2 set B, for AGC CAC AAT CCA CTC ACG G, rev 5′-GCT CTT GCA ATT GAT GTC CGG AT; exon 2 set C, for 5′-ATA CCA TGC CAA GAA CCG TG, rev 5′-GGT TCG CAA ATA CTC TGC CT; exon 3 primers, for 5′-AAG CTA CAA GCA AAC CGG TC, rev 5′-CCG CTG ATT GAT TAC TGC TC; and exon 4 primers, for 5′-TAC CTC TGC GAC AAG TAC G, rev 5′-CAA TTT GGA AGG CAA AAC AAC CTA. Post-PCR, the product was purified from TAE 1% agarose gel by a QIAGEN (Valencia, CA) gel extraction kit and TA cloned (Invitrogen, San Diego) using standard manufacturer protocol. DNA was purified from three to six colonies by standard miniprep protocol (QIAGEN). Homozygous samples were sent in triplicate and heterozygous samples were sent in quadruplicate for direct sequencing (Johns Hopkins University Genetics Core). Samples were sequenced in both directions using primers included in the TA cloning kit, M13 forward and M13 reverse. Sequence data were aligned using Vector NTI software (Invitrogen).
Stage of lethality of trans-heterozygotes:
Female flies heterozygous for Su(Kpn)1, Su(Kpn)2, or Su(Kpn)3 balanced over w+ TM3 e Sb KrGFP were crossed to males of each of the Su(Kpn)X alleles balanced over w+ TM3 e Sb KrGFP. Larvae that were trans-heterozygous were identified on a fluorescent dissecting microscope as lacking GFP expression. These larvae were moved to a fresh vial and reared in the absence of heterozygous sibling competition at 20°. Those that eclosed as adults were scored for non-Sb bristles to confirm that true trans-heterozygotes had been selected.
RESULTS
Screen for suppression of endogenous awdKpn:
Repeating the work of Lifschytz and Falk (1969b), we had previously mutagenized awdKpn males with EMS and recovered revertants of pn-awdKpn lethality. All of the revertants we recovered were either second-site point mutations in the awd gene or deletions of the awd gene (Timmons et al. 1995). To try to identify suppressors of pn-awdKpn lethality, we used EMS to mutagenize pn males and mated them to awdKpn homozygous females with attached X chromosomes. All of the male progeny of such crosses would be hemizygous for pn and heterozygous for awdKpn. We expected that such males would survive only if a suppressor mutation were present. We recovered >240,000 female progeny from this screen and only two males (Figure 1). These males represented putative suppressors. However, even though these putative suppressors were located on the third chromosome they were not on third chromosomes that contained the marker ebony (e) that was present on mutagenized third chromosomes. Further analysis revealed that these males resulted from second-site mutations of the awdKpn gene in the unmutagenized female parents (data not shown). This result demonstrated the extreme selectivity of this screen but indicated that it was unlikely that this protocol would lead to the recovery of suppressor mutations.
Figure 1.

Screen for suppressors of endogenous awdKpn. Males were mutagenized with EMS. No suppressors were recovered. The two males recovered were derived by reversion of awdKpn in the unmutagenized female parent as described in the text.
Transgenic awdKpn is viable in the absence of awd but still kills prune:
The AWD protein is normally present at higher levels than are required for viability. Transgenes containing 750 bp of upstream sequence fused to an awd cDNA lead to the accumulation of lower levels of AWD than does the endogenous gene, but can nevertheless rescue the lethal phenotype caused by an awd null mutation. As examples, two different insertions of such a transgene lead to 42 and 87% viability, respectively, but have 4 and 14%, respectively, as much nucleoside diphosphate kinase activity as wild type (Xu et al. 1996). This finding led to the idea of modifying our suppressor screen using transgenic rather than endogenous awdKpn. We hypothesized that with a lower level of awdKpn mutant enzyme, a mutation might be able to suppress the lethal interaction. We constructed an awdKpn transgene by mutating the wild-type awd transgene described above (Timmons and Shearn 1997). A second chromosome insertion of this awdKpn transgene can rescue the lethality caused by an awd null mutation (data not shown) but still causes lethality in the absence of pn function (Table 1). To compare levels of transgenic and endogenous AWDKpn protein, we immunoblotted larval extract using an AWD polyclonal antibody. We found, as expected, endogenous AWDKpn was present at twice the level of transgenic <AWDKpn> (Figure 2).
Figure 2.

Endogenous AWDKpn protein is present at higher levels than transgenic AWDKpn protein. Lysate from third instar larvae homozygous for endogenous awdKpn or homozygous for the transgenic P{awdKpn} on an awd null background were blotted with AWD antibody. A single band at 15 kDa was detected for both samples, representing AWD monomer. The endogenous AWDKpn protein is present at higher levels than the AWDKpn protein. The blot was normalized with β-tubulin to show equal loading of protein.
Screen for suppression of transgenic awdKpn:
To identify suppressors of pn-awdKpn lethality, we used EMS to mutagenize pn males and mated them to females with attached X chromosomes that were homozygous for transgenic awdKpn on the second chromosome and homozygous for an awd null mutation on the third chromosome. All of the male progeny of such a cross would be hemizygous for pn, heterozygous for transgenic awdKpn, and heterozygous for an awd null mutation. We expected that such males would survive only if a suppressor mutation were present. We recovered >78,000 female progeny from this screen and four males (Figure 3). One of the males had a second-site mutation in the pn gene that reverted the pn mutant phenotype (data not shown). The other three males had suppressor mutations on the third chromosome that are allelic. We named the gene that had suffered these mutations Suppressor of Killer of prune, Su(Kpn).
Figure 3.

Screen for suppressors of transgenic awdKpn. Males were mutagenized with EMS. The four males recovered included three suppressors as well as one pn revertant.
Su(Kpn) mutations cannot suppress endogenous awdKpn but can suppress transgenic awdKpn:
The rationale for the modified screen was that putative suppressors might be able to suppress transgenic awdKpn because it would produce a lower level of AWDKPN protein but not endogenous awdKpn because it would produce a higher level of AWDKPN protein. We directly tested this with the first two of the suppressor alleles recovered (Table 1). We found that neither of these alleles could suppress the lethal interaction between pn and endogenous awdKpn. Females that were homozygous for pn and heterozygous for a suppressor allele were crossed to males that were homozygous for awdKpn. Hundreds of female progeny that were heterozygous for pn, heterozygous for a suppressor allele, and heterozygous for awdKpn were recovered but no males that were hemizygous for pn, heterozygous for a suppressor allele, and heterozygous for awdKpn were recovered. By contrast, we found that both of these alleles could suppress the lethal interaction between pn and transgenic awdKpn to varying extents. Females that were homozygous for pn and heterozygous for a suppressor allele were crossed to males that were homozygous for transgenic awdKpn. Hundreds of female progeny that were heterozygous for pn, heterozygous for a suppressor allele, and heterozygous for transgenic awdKpn were recovered and hundreds of males that were hemizygous for pn, heterozygous for a suppressor allele, and heterozygous for transgenic awdKpn were also recovered. The ratio of males to females was 0.65 for Su(Kpn)1 and 0.97 for Su(Kpn)2.
Su(Kpn) cannot suppress pn:
The suppressor alleles were recovered in flies that were not only mutant for pn but also mutant for white. This was done to follow the awdKpn transgene that is marked with w+. One possibility was that the suppressor acted by suppressing pn rather than by suppressing the lethal interaction between pn and awdKpn. To examine this possibility we generated females trans-heterozygous for pn18a and pn12c, heterozygous for white, and heterozygous for each of the first three suppressor alleles. None of the alleles suppressed the prune eye-color phenotype (data not shown). Moreover, as heterozygotes Su(Kpn) alleles are not lethal with either endogenous or transgenic awdKpn.
The Su(Kpn) gene is in 84C:
All three of the suppressor alleles recovered are homozygous lethal and lethal in all three trans-heterozygous combinations. To discover the approximate location of this gene we mapped the lethality by recombination mapping using a previously described protocol (Shearn and Garen 1974). The lethality mapped to the middle of the third chromosome between Gl and Sb (data not shown). To determine the cytogenetic location of Su(Kpn), deficiencies known to remove regions of the chromosome between the Gl gene at 70C and the Sb gene at 89B were crossed to suppressor alleles and assayed for lack of complementation for the lethal phenotype (Table 2). The failure of Df(3R)Antp17 to complement indicated that the Su(Kpn) gene was located in the region between 84A5 and 84D14. The location was further refined to 84C6–8 by crossing to a second set of deficiencies (Figure 4). The lethality of Su(Kpn) is complemented by Df(3R)Antp6 that removes 84B1–C6 and also by Df(3R)dsx29 that removes 84C8–F6. These data suggest that the cytogenetic location of Su(Kpn) is within 84C6–8.
Figure 4.

Cytogenetic localization of Su(Kpn). Map of section of right arm of polytene chromosome 3 illustrates the 84 region. Deficiencies lacking specific parts of this region, indicated by gaps in solid bars, were combined with a mutation in Su(Kpn). If this combination was viable it was indicated as “+” in the complement column; if this combination was lethal it was indicated as “−” in the complement column.
Su(Kpn) alleles are loss-of-function mutations:
Our initial expectation was that the suppressor alleles would be gain-of-function alleles. Finding deficiencies that failed to complement the lethality caused by Su(Kpn) alleles allowed us the opportunity to test this expectation. Females that were homozygous for pn and heterozygous for Df(3R)Antp17 were crossed to males that were homozygous for transgenic awdKpn. Hundreds of female progeny that were heterozygous for pn, heterozygous for the deficiency, and heterozygous for transgenic awdKpn were recovered and hundreds of males that were hemizygous for pn, heterozygous for the deficiency, and heterozygous for transgenic awdKpn were also recovered (data not shown). The fact that the loss of one dose of Su(Kpn)+ can provide suppression implies that the suppressor alleles we had recovered were loss-of-function mutations.
Trans-heterozygous Su(Kpn) mutations cause early larval lethality and an autonomous small disc phenotype:
Homozygous Su(Kpn)1 or Su(Kpn)2 mutants or trans-heterozygous Su(Kpn)1/Su(Kpn)2 mutants are lethal at the embryo/first larval instar interphase. To examine the imaginal disc phenotype caused by these mutations, anterior halves of trans-heterozygous mutant and nonmutant control larvae were cultured in the abdomens of female hosts using a procedure described previously (Simcox et al. 1989). The anterior halves of larvae continue to develop in host females. When recovered, 10/10 of the nonmutant larvae had normal-appearing imaginal discs (Figure 5A). The anterior halves of mutant larvae also continue to develop in host females. However, when recovered, 0/15 of these mutant larvae had detectable imaginal discs (Figure 5B).
Figure 5.

Su(Kpn) function is required for growth of imaginal discs. The anterior halves of control and mutant larvae were cultured in the abdomens of host adult females for 3 days. (A) Control halves developed imaginal discs; (B) mutant halves did not develop imaginal discs. Disc, imaginal discs; fb, fat body; sg, salivary gland; cut, larval cuticle; bl, brain lobe; vg, ventral ganglion; mp, mouthparts.
Additional alleles were recovered in a noncomplementation screen:
The fact that a deficiency can act as a suppressor made it seem possible that our screen for suppressors selected for extreme alleles. To recover a wider range of mutations, we performed a screen for mutations that fail to complement the lethality caused by the original suppressor mutation. The screen was similar in design to ones we have previously reported (Shearn et al. 1978, 1987; Simcox et al. 1987; Mansfield et al. 1994). We mutagenized males that had a lethal free isogenic third chromosome marked with a w+ transgene and mated them to females that were homozygous for y and w on the X chromosome and were heterozygous for a dominant mutation and a balancer on the third chromosome. Male progeny heterozygous for a mutagenized third chromosome and a balancer chromosome were mated individually to females heterozygous for Su(Kpn)1. Crosses in which only balancer progeny were recovered represented ones in which the male parent had a putative Su(Kpn) mutation. Male progeny were retested by crossing to Su(Kpn)2 heterozygous females (Figure 6). Out of 24,616 single-male crosses, we recovered 16 new alleles.
Figure 6.

Screen for mutations that fail to complement Su(Kpn) mutations. Males were mutagenized with EMS and crossed to balancer females (cross I). Male progeny heterozygous for a mutagenized third chromosome were mated to females heterozygous for Su(Kpn)1 (cross II). If no progeny trans-heterozygous for the mutagenized chromosome and Su(Kpn)1 survive, then a putative new allele has been recovered. Male progeny heterozygous for the putative new allele were mated to females heterozygous for Su(Kpn)2 (cross III). If no progeny trans-heterozygous for the putative new allele and Su(Kpn)2 survive, then a new allele has been confirmed. Su(Kpn)i indicates new allele.
Because the recovery of alleles from the noncomplementation screen was at a higher frequency than the recovery of suppressors of the pn-awdKpn lethal phenotype, we believed the new alleles were of equivalent strength or weaker than the original Su(Kpn) alleles. Further, because these alleles were identified by noncomplementation, their ability to suppress the pn-awdKpn lethal phenotype was unknown. To test their ability to also suppress the pn-P{awdKpn} lethal interaction, males heterozygous for the putative Su(Kpn)X were crossed to females homozygous for pn18a, heterozygous for P{awdKpn}, and heterozygous for Su(Kpn)1. Male progeny that were pn P{awdKpn} and either Su(Kpn)1 or the putative Su(Kpn)X from this cross were counted. The strength of the candidate suppressors was compared to that of Su(Kpn)1 and tested for statistical significance using the G-test. Our results show that at 25°, 15/16 Su(Kpn) alleles suppress the pn-awdKpn lethality (Table 3). AB924 did not suppress the lethality at all. Ten of the alleles were equivalent to Su(Kpn)1 in their ability to suppress, their ratio being equivalent to 1. Six of the alleles were weaker than Su(Kpn)1. The ratios of the weaker alleles were <1 and statistically significant with a P-value <0.005. As previously observed (Table 1), Su(Kpn)2 was stronger than Su(Kpn)1, having a statistically significant ratio >1.
TABLE 3.
Suppression of pn−-awdKpn lethality by suppressor alleles
| Su(Kpn)X | Na | Relative suppressionb | Significancec |
|---|---|---|---|
| AB621 | 56 | 0.73 | NS |
| AB924 | 57 | 0 | P < 0.005 |
| AP320 | 78 | 1.25 | NS |
| AP434 | 141 | 0.91 | NS |
| AV739 | 59 | 0.01 | P < 0.005 |
| BS304 | 73 | 0.24 | P < 0.005 |
| BU640 | 46 | 1.4 | NS |
| BW914 | 114 | 0.73 | NS |
| BX123 | 73 | 0.76 | NS |
| CK438 | 117 | 1.12 | NS |
| CL1027 | 76 | 0.11 | P < 0.005 |
| CU338 | 102 | 0.21 | P < 0.005 |
| CX322 | 63 | 0.83 | NS |
| CZ811 | 63 | 1.07 | NS |
| DC727 | 56 | 1.39 | NS |
| DC806 | 57 | 0.11 | P < 0.005 |
| Su(Kpn)2 | 98 | 2.15 | P < 0.005 |
| Su(Kpn)3 | 52 | 0.27 | P < 0.005 |
The number of male pn-awdKpn-Su(Kpn)1 progeny recovered. An average of 298 total males were scored per cross.
The ratio of [pn-Su(Kpn)1/pn-<awdKpn>-Su(Kpn)1] [pn-Su(Kpn)X/pn-<awdKpn>-Su(Kpn)X].
The P-value obtained from the G-test for independence.
NS denotes nonsignificance, that is, the ratio of Su(Kpn)1:Su(Kpn)X is not statistically different from 1:1. A ratio <1 reflects that Su(Kpn)X is a weaker suppressor of the pn-awdKpn lethality than Su(Kpn)1. A ratio >1 reflects that Su(Kpn)X is a stronger suppressor of the pn-awdKpn lethality than Su(Kpn)1.
Su(Kpn) fails to complement l(3)84Cc but complements all lethal insertion mutations in 84C:
We hoped to use transposon tagging to help identify the DNA sequence that corresponds to the Su(Kpn) gene. Toward that end we tested available lethal mutations in 84C, especially lethal transposon insertion mutations for failure to complement Su(Kpn) alleles. We found that Su(Kpn) alleles fail to complement l(3)84Cc that was discovered in a screen for EMS-induced mutations that fail to complement a deletion of the 84B–D region (Lewis et al. 1980; Cavener et al. 1986) but complement every other lethal mutation in the 84B–D region that we tested (Table 4), including all lethal insertion mutations. So, we needed an alternative approach to locate the Su(Kpn) DNA sequence.
Some lethal excisions of P{EPgy2}EY00044 fail to complement Su(Kpn):
Several nonlethal transposon insertion mutations are available that are located in 84C. We hoped to generate lethal excisions of one of these to help identify the DNA sequence that corresponds to the Su(Kpn) gene. To decide which one to excise, we used genetic recombination to find the insertion that maps closest to Su(Kpn). We generated females that were trans-heterozygous for Su(Kpn) and insertions marked with w+ and crossed them to males heterozygous for a deficiency of 84C, Df(3R)Antp7. One class of recombinant between the insertion and Su(Kpn) would generate viable individuals that were w−. We reasoned that the insertion that gave the lowest frequency of such individuals would be the one closest to Su(Kpn). This process led us to focus our attention on EY00044, a nonlethal EP insertion into 84C6 that is marked with both y+ and w+ (data not shown). We generated excisions of EY00044 that failed to complement Df(3R)Antp7 for lethality and then tested them for failure to complement Su(Kpn)1 (Figure 7). We recovered 88 males that had lethal excisions of EY00044 from a sample of 2769 males (3.2%). Fifty-eight of these excision males were fertile and 12 of them failed to complement Su(Kpn). From this 21% frequency of lethal excisions that fail to complement Su(Kpn), we concluded that Su(Kpn) is near to the site of the EY00044 insertion. The EY00044 insertion is within the interval of 2976K and 2977K on the genomic map between CG10061 on the distal side and CG2656 and CG10065 on the proximal side (FlyBase). Lethal mutations in CG10061 complement Su(Kpn) (Table 4), so we focused our attention on CG2656 and CG10065. CG10065 is within a large intron of CG2656.
Figure 7.

Screen for excision of insertion that maps near to Su(Kpn). Males homozygous for nonlethal insertion were mated to females carrying the source of transposase {Δ2-3} (cross I). Males doubly heterozygous for insertion and transposase were mated to females carrying a balancer chromosome (cross II). Male progeny with white eyes represent putative excisions; they were mated individually to deficiency females to assay for the presence of lethal mutation (cross III). Male progeny carrying lethal mutation were mated to females heterozygous to Su(Kpn)2 to test for allelism. Su(Kpn)i indicates homozygous lethal excision that fails to complement Su(Kpn) mutations.
Detection of a broad range of mutations in CG10065 identifies it as the Su(Kpn):
To determine if either CG2656 or CG10065 is the gene identified by Su(Kpn) mutations, we used PCR to amplify both genes from mutant larval DNA and determined their nucleotide sequences. Ten Su(Kpn) mutants are homozygous lethal in larval stages. These lines were collected as homozygous larvae and analyzed for nucleotide changes in both CG2656 and CG10065. The remaining nine mutant lines are embryonic lethal and were collected as heterozygotes for sequence analysis. PCR and sequencing of homozygous mutant larvae revealed only wild-type sequence in all three exons of CG2656 from eight different alleles (Table 5). However, sequence analysis of the four exons of CG10065 detected mutations in eight homozygous and eight heterozygous larval samples. The nucleotide sequence alterations detected include nine missense mutations [AB621, AB924, AP434, BX123, CL1027, CU338, DC727, DC806, and Su(Kpn)1], one small deletion that remains in frame (BU640), four premature stop codons [BW914, CK438, CX322, and Su(Kpn)2], and two deletions that cause a frameshift and premature stop codon [CZ811 and Su(Kpn)3]. The presence of nucleotide sequence alterations in CG10065 from nearly every allele examined identifies it as Su(Kpn).
TABLE 5.
Nucleotide alterations in either of two candidate Su(Kpn) genes
| CG2656a | CG10065b | |
|---|---|---|
| Control | WT | WT |
| AB621 | NA | P134S |
| AB924 | WT | P46 |
| AP434 | NA | R169H |
| BU640 | WT | ΔV160–R166 |
| BW914 | WT | Q896* |
| BX123 | WT | Y882N |
| CK438 | WT | W995* |
| CL1027 | NA | P821L |
| CU338 | NA | L27F |
| CX322 | NA | Q117* |
| CZ811 | WT | ΔfsF469–476* |
| DC727 | WT | G40R |
| DC806 | WT | D30G |
| Su(Kpn)1 | NA | R169C |
| Su(Kpn)2 | NA | W47* |
| Su(Kpn)3 | NA | ΔfsA405–408* |
NA, sequence not determined. WT, wild-type sequence.
*, stop codon. Δ, a deletion. fs, frameshift.
Recently, another group serendipitously studied CG10065 independent of its role in the pn-awdKpn lethal interaction (Dai et al. 2004). Dai et al. reported that CG10065 has a single 3.4-kb transcript that encodes a 140-kDa protein. This protein is a novel glutathione S-transferase (GST) containing FLYWCH zinc-finger protein symbolized by the gene name as gfzf. Structurally, the protein contains four zinc fingers, an acidic domain, and a GST domain. The mutations we have identified in this gene that we named Su(Kpn) occur in both the zinc-finger and GST domains (Figure 8). This suggests that both domains are functionally important in the Su(Kpn)-mediated toxicity of the pn-awdKpn lethal interaction.
Figure 8.

Diagram of the SU(KPN) protein and amino acid changes in the EMS-generated mutants of Su(Kpn). The SU(KPN) contains four zinc-finger domains (ZF1–4), an acidic domain, and a glutathione S-transferase (GST) domain. A broad range of mutations have been identified in both the zinc-finger and the GST domains, including missense mutations, nonsense mutations (*), deletions (Δ), and frameshifts (fs) that introduce nonsense mutations. The stage of lethality of the alleles was evaluated in trans—with Su(Kpn)1, Su(Kpn)2, and Su(Kpn)3 at 20°. Trans-heterozygotes lethal at larval stages are above the diagram, while trans-heterozygotes that are viable as adults are below it.
To determine the strength of each mutation with regard to larval viability, we staged larvae trans-heterozygous for Su(Kpn)1, Su(Kpn)2, or Su(Kpn)3 and for each of the Su(Kpn)X alleles. We used trans-heterozygotes because mutants generated by EMS may give misleading staging information as homozygotes since other mutations may have been simultaneously generated in the genome. Creating trans-heterozygous individuals dilutes the second-site mutation effects and allows evaluation of viability, as a result of the Su(Kpn) mutations. We conducted the staging experiments at 20° and crossed our original alleles Su(Kpn)1, Su(Kpn)2, or Su(Kpn)3 to each of the Su(Kpn)X alleles. Our results reveal that the trans-heterozygotes lethal at larval stages include missense mutations and all premature stop codons and deletions. Alleles that were viable as trans-heterozygous adults were all missense mutations (Figure 8). This suggests that some of the missense mutations are weak mutants that are able to compensate for the loss of function of the extreme alleles and produce viable adult flies.
DISCUSSION
Suppressor recovery:
Our screen to recover suppressors of endogenous awdKpn did not lead to the recovery of any verified suppressors (Figure 1). We did recover two males, both of which turned out to have revertant mutations in the awdKpn gene that was derived from unmutagenized female parents. These revertants provide an estimate of the spontaneous mutation frequency. We recovered 242,991 females. Assuming that a similar number of males would have been recovered but for the pn: awdKpn lethality, then that frequency can be estimated to be 2/242,991 or 8/1,000,000 chromosomes tested. The EMS-induced frequency of awdKpn revertants we have reported is 65/1,000,000 chromosomes tested (Timmons et al. 1995). So the induced frequency we recovered is 8.1 times higher than the spontaneous frequency. We had been surprised that 3 of the 16 revertants we recovered in the EMS-induced screen for revertants were deletions (Timmons et al. 1995). It is possible that one or more of those deletions were actually of spontaneous origin.
Our screen to recover suppressors of transgenic awdKpn did lead to the recovery of three verified suppressors. This frequency of recovery, 3/78,000, is significantly different from the lack of recovery of suppressors of endogenous awdKpn (0/242,991) at the 5% level according to the G-test. This recovery is consistent with the hypothesis that such mutations could not suppress endogenous levels of AWDKPN but could suppress transgenic levels of AWDKPN. The first two suppressors recovered were used to directly test this hypothesis. Indeed neither Su(Kpn)1 nor Su(Kpn)2 can suppress endogenous awdKpn but both can suppress transgenic awdKpn (Table 1). The Su(Kpn)2 allele suppresses to a greater extent than does Su(Kpn)1. We recovered as many males that were hemizygous for pn, heterozygous for transgenic awdKpn, and heterozygous for Su(Kpn)2 as females that were heterozygous for pn, heterozygous for transgenic awdKpn, and heterozygous for Su(Kpn)2. However, we recovered significantly fewer males hemizygous for pn, heterozygous for transgenic awdKpn, and heterozygous for Su(Kpn)1 than females heterozygous for pn, heterozygous for transgenic awdKpn, and heterozygous for Su(Kpn)1. This indicates that Su(Kpn)2 is a stronger suppressor than Su(Kpn)1. Subsequent sequencing revealed that Su(Kpn)2 causes a nonsense mutation at amino acid 47, whereas Su(Kpn)1 causes a missense mutation at amino acid 169.
Allele recovery:
The F2 screen for Su(Kpn) noncomplementers yielded, 6.5 alleles per 10,000 chromosomes tested. This is comparable to frequencies recovered in similarly designed noncomplementation screens. For example, in a screen to recover alleles of five different mutations on the third chromosome, we recovered on average 3.6 alleles per 10,000 chromosomes (Shearn et al. 1978). In a screen designed to recover alleles of a different mutation on the third chromosome, we recovered 6 alleles per 10,000 chromosomes (Simcox et al. 1987). In a screen designed to recover alleles of two other mutations on the third chromosome, we recovered on average 5.2 alleles per 10,000 chromosomes (Shearn et al. 1987). In yet another screen designed to recover alleles of a mutation on the third chromosome, we recovered on average 5.5 alleles per 10,000 chromosomes (Mansfield et al. 1994). By contrast, the screen for suppressors of transgenic awdKpn yielded only 0.4 alleles per 10,000 chromosomes tested. This much lower frequency suggests that only certain kinds of alleles were recovered in that screen. Since all three alleles recovered in the suppressor screen cause early larval lethality but some alleles recovered in the noncomplementation screen cause later larval lethality, we propose that the suppressor screen selected for extreme alleles. Our analysis of the nucleotide alterations among the various alleles supports this interpretation (Table 5).
Nucleotide alterations among alleles:
Sequence analysis of mutant Su(Kpn) larvae identified a variety of nucleotide alterations in CG10065, ranging from missense mutations to deletions resulting in frameshifts and premature stop codons (Table 5). As we hypothesized from the low recovery rate of mutants from our screen for suppressors of transgenic awdKpn (Figure 3), Su(Kpn)1, Su(Kpn)2, and Su(Kpn)3 are all severe mutant alleles. We found that both Su(Kpn)2 and Su(Kpn)3 result in premature stop codons. While we have not yet evaluated these mutants for protein accumulation, we believe that Su(Kpn)2 and Su(Kpn)3 will be null mutants; neither one will accumulate any SU(KPN) protein because of the N-terminal stop codon in the mRNA sequence. Su(Kpn)1 is a point mutation in arginine 169. We believe that this residue is essential for proper function of SU(KPN) because another missense mutation in this residue was recovered in a second Su(Kpn) allele, AP434. AP434 is equivalent to Su(Kpn)1 in its suppression of the pn-awdKpn lethal phenotype (Table 3). Further, AP434 and Su(Kpn)1 are larval lethal in trans-heterozygous combinations (Figure 8), making these residues essential for both suppression of the lethal pn-awdKpn interaction and viability. BW914 and CK438 have C-terminal stop codons in the region that encodes the GST domain. If these mutant proteins accumulate they would lack GST function. They could be extremely valuable for studying the role of GST function in the suppressor activity.
The missense mutations we have detected occur in a wide variety of residues. It is unclear at this time how these residues affect the function of SU(KPN). Because the residues affected in these mutants are found in both the zinc-finger and the GST domains of the protein, we believe that both domains are essential to the function of SU(KPN) in vivo. Future structure-function studies will elucidate the role of these residues in SU(KPN) function. Analysis of the Su(Kpn) alleles in trans with our extreme alleles tested their developmental viability. Interestingly, when these viability data (Figure 8) are compared to their ability to suppress the pn-awdKpn lethal interaction (Table 3), it is apparent that there is an uncoupling between survival and suppression. For example, as expected, AB924, which is not able to suppress the pn-awdKpn lethal interaction, is viable as adults in trans-heterozygous combinations with our extreme alleles. However, Su(Kpn)3, a null allele, is larval lethal at the second instar, but is a rather weak suppressor of the pn-awdKpn lethal interaction. It is unclear what the reason for this is at this time. It is possible, although a stop codon exists in the coding region, that there may be read-through and that some protein may be accumulating. Further analysis will be required to determine what protein levels and function of the SU(KPN) are required for the pn-awdKpn lethal interaction vs. for viability.
In addition to the uncoupling of suppression and viability that emerges from our analysis of the trans-heterozygotes, this analysis suggests that the SU(KPN) protein may function as a dimer or multimer. Some alleles, such as AB621 and DC727, that are viable as trans-heterozygotes with Su(Kpn)1 are nevertheless strong suppressors as heterozygotes. Because GSTs are known to require dimerization for their function and because zinc fingers can mediate protein-protein interactions (Hayes et al. 2005), we hypothesize that the SU(KPN) protein forms multimeric complexes in vivo to mediate its effects. Identifying these binding partners will be the subject of future studies.
Although we have not completed analysis of 3 of the 19 alleles recovered in our EMS screens, so far, all of the nucleotide alterations detected have been in the coding region of the SU(KPN). This suggests that protein function, rather than misregulation of gene expression, is responsible for the lethality of the SU(KPN) in the pn-awdKpn interaction. Interestingly, we have not yet recovered any mutations in the acidic domain of the protein. The role of the acidic domain is unclear on the basis of its protein sequence and lack of homology to other known domains. One possible reason we have not recovered mutants in this domain is that it is dispensable for SU(KPN) function.
Protein product of CG10065:
The Su(Kpn) gene encodes a novel GST-containing zinc-finger protein (Dai et al. 2004). The zinc finger is a very common protein motif, occurring in up to 2% of identified proteins. Zinc-finger proteins have been described as binding both DNA and RNA, as well as mediating protein:protein interactions (Rubin et al. 2000). GSTs are likewise abundant cellular proteins. Thirty-seven GST genes are in Drosophila. The majority of GSTs exist as independent proteins that require dimerization to mediate their glutathione-conjugating activity, and it is very rare to find GST domains endogenously fused to other protein domains (Dai et al. 2004; Udomsinprasert et al. 2004). SU(KPN) is a unique protein because it endogenously fuses a GST domain to multiple zinc-finger domains. Because of the redundancy of GSTs and their overlapping functions, GSTs are able to compensate for each other. We believe SU(KPN) must be unique in its cellular function to have been identified in our EMS screen for suppressors of the pn-awdKpn lethal interaction. If SU(KPN) were functioning as a “classic” GST, it would have likely not been detected in our screen due to compensation by other GSTs present in Drosophila. Because of the unique fusion of zinc-finger and GST domains in SU(KPN), and because we have identified multiple mutations in both domains, we believe that both the zinc-finger and GST domains are essential for proper function of the SU(KPN). We also know that the wild-type function of SU(KPN) is essential for the proliferation of imaginal discs, because the alleles Su(Kpn)1, Su(Kpn)2, and Su(Kpn)3 recovered in our first screen lack imaginal discs (Figure 5). Future studies will determine the role of zinc-finger and GST domains in the function of SU(KPN) and its role in imaginal disc proliferation.
GSTs are responsible for conjugating reduced glutathione (GSH) to a wide range of native and xenobiotic substrates in vivo. Historically, GSTs are considered to be detoxifying enzymes and play a role in the host defense response. However, examples in which GST addition of GSH to substrates creates a product more toxic than the parent substrate exist. An example of this phenomenon is conjugation of GSH with the solvent dichloromethane. This reaction results in the formation of the highly unstable S-chloromethylglutathione, which contains an electrophilic center capable of modifying DNA (Hayes et al. 2005). To account for both the pn-awdKpn lethal interaction and its suppression by reduced Su(Kpn) function we have proposed a hypothetical biochemical model. According to this model, the SU(KPN) conjugation of GSH to a substrate accumulated in pn-awdKpn larvae is a toxic event leading to lethality in pn-awdKpn flies. Mutations in the Su(Kpn) that disrupt the GSH-conjugating ability of SU(KPN) allow pn-awdKpn male larvae to survive (Figure 9). Future studies will be directed at identifying the substrate of SU(KPN) and its mechanism in the pn-awdKpn lethal interaction.
Figure 9.

Model of pn-awdKpn lethal interaction. XP is the normal phosphodiester substrate of PN; X and Pi are the normal products. NDP and ATP are the normal substrates of AWD nucleoside diphosphate kinase; NTP and ADP are the normal products. Y and GSH are the normal substrates of SU(KPN) glutathione S-transferase; Y-GSH is the normal product. Either Y-GSH is required for imaginal disc growth or Y prevents imaginal disc growth. In a pn mutant XP accumulates. By itself this is not harmful because XP is not a substrate for normal AWD enzyme. In an awdKpn mutant there is slightly less nucleoside diphosphate kinase activity, but this does not cause a phenotype. However, in a pn-awdKpn mutant XP accumulates and is phosphorylated by AWDKPN to XPP. GSH is added to XPP by SU(KPN) and is converted to XPP-GSH, which is toxic. Loss-of-function mutations in Su(Kpn) can suppress the pn-transgenic awdKpn lethal interaction because they limit the amount of XPP-GSH that can be produced from the already reduced amount of XPP substrate. Loss-of-function mutations in Su(Kpn) cannot suppress the pn-endogenous awdKpn lethal interaction because the amount of XPP-GSH that can be produced from the abundant amount of XPP substrate is toxic.
Acknowledgments
We thank Douglas Cavener, Peter Deak, the Bloomington Stock Center, and the Szeged Stock Center for sending fly stocks. We thank Allison Jenkins Milutinovich for working on this project as a rotation student, Isaac Shearn for working on the figures and tables, and members of the Shearn lab for helpful suggestions and stimulating discussions. This work was supported by the National Science Foundation under grant no. 9974403.
References
- Aravind, L., and E. V. Koonin, 1998. A novel family of predicted phosphoesterases includes Drosophila prune protein and bacterial RecJ exonuclease. Trends Biochem. Sci. 23: 17–19. [DOI] [PubMed] [Google Scholar]
- Beadle, G. W., and B. Ephrussi, 1936. The differentiation of eye pigments in Drosophila as studied by transplantation. Genetics 21: 225–247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Biggs, J., N. Tripoulas, E. Hersperger, C. Dearolf and A. Shearn, 1988. Analysis of the lethal interaction between the prune and Killer of prune mutations of Drosophila. Genes Dev. 2: 1333–1343. [DOI] [PubMed] [Google Scholar]
- Biggs, J., E. Hersperger, P. S. Steeg, L. A. Liotta and A. Shearn, 1990. A Drosophila gene, which is homologous to a mammalian gene associated with tumor metastasis, codes for a nucleoside diphosphate kinase. Cell 63: 933–940. [DOI] [PubMed] [Google Scholar]
- Cavener, D. R., D. C. Otteson and T. C. Kaufman, 1986. A rehabilitation of the genetic map of the 84B–D region in Drosophila melanogaster. Genetics 114: 111–123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dai, M. S., X.-X. Sun, J. Qin, S. M. Smolik and H. Lua, 2004. Identification and characterization of a novel Drosophila melanogaster glutathione-S-transferase-containing FLYWCH zinc finger protein. Gene 342: 49–56. [DOI] [PubMed] [Google Scholar]
- D'Angelo, A., L. Garzia, A. Andre, P. Carotenuto, V. Aglio et al., 2004. Prune cAMP phosphodiesterase binds nm23–H1 and promotes cancer metastasis. Cancer Cell 5: 137–149. [DOI] [PubMed] [Google Scholar]
- Dearolf, C. R., E. Hersperger and A. Shearn, 1988. a Developmental consequences of awdb3, a cell-autonomous lethal mutation of Drosophila induced by hybrid-dysgenesis. Dev. Biol. 129: 159–168. [DOI] [PubMed] [Google Scholar]
- Dearolf, C. R., N. Tripoulas, J. Biggs and A. Shearn, 1988. b Molecular consequences of awdb3 a cell-autonomous lethal mutation of Drosophila induced by hybrid-dysgenesis. Dev. Biol. 129: 169–178. [DOI] [PubMed] [Google Scholar]
- Forus, A., A. D'Angelo, J. Henriksen, G. Merla, G. M. Maelandsmo et al., 2001. Amplification and overexpression of PRUNE in human sarcomas and breast carcinomas—a possible mechanism for altering the nm23–H1 activity. Oncogene 20: 6881–6890. [DOI] [PubMed] [Google Scholar]
- Frolov, M. V., V. V. Zverlov and V. E. Alatortsev, 1994. The mRNA product of the Drosophila gene prune is spliced and encodes a protein containing a putative transmembrane domain. Mol. Gen. Genet. 242: 478–483. [DOI] [PubMed] [Google Scholar]
- Hayes, J. D., J. U. Flanagan and I. R. Jowsey, 2005. Glutathione transferases. Annu. Rev. Pharmacol. Toxicol. 45: 51–88. [DOI] [PubMed] [Google Scholar]
- Lascu, I., A. Chaffotte, B. Limbourg-Bouchon and M. Veron, 1992. A Pro/Ser substitution in nucleoside diphosphate kinase of Drosophila melanogaster (mutation killer of prune) affects stability but not catalytic efficiency of the enzyme. J. Biol. Chem. 267: 12775–12781. [PubMed] [Google Scholar]
- Lewis, E. B., and F. Bacher, 1968. Methods of feeding ethyl methane sulfonate (EMS) to Drosophila males. Dros. Inf. Serv. 43: 193. [Google Scholar]
- Lewis, R. A., T. C. Kaufman, R. E. Denell and P. Tallerico, 1980. Genetic analysis of the Antennapedia gene complex (ANT-C) and adjacent chromosomal regions of Drosophila melanogaster. Genetics 95: 367–381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lifschytz, E., and R. Falk, 1969. a A system for screening of rare events in genes of Drosophila melanogaster. Genetics 62: 343–352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lifschytz, E., and R. Falk, 1969. b A genetic analysis of the Killer-prune (K-pn) locus of Drosophila melanogaster. Genetics 62: 353–358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mansfield, E., E. Hersperger, J. Biggs and A. Shearn, 1994. Genetic and molecular analysis of hyperplastic discs, a gene whose product is required for regulation of cell proliferation of Drosophila melanogaster imaginal discs and germ cells. Dev. Biol. 165: 507–526. [DOI] [PubMed] [Google Scholar]
- Rosengard, A. M., H. C. Krutzch, A. Shearn, J. R. Biggs, E. Barker et al., 1989. Reduced Nm23/Awd protein in tumour metastasis and aberrant Drosophila development. Nature 342: 177–180. [DOI] [PubMed] [Google Scholar]
- Rubin, G. M., M. D. Yandell, J. R. Worman, G. L. Gabor Miklos, C. R. Nelson et al., 2000. Comparative genomics of the eukaryotes. Science 287: 2204–2215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shearn, A., and A. Garen, 1974. Genetic control of imaginal disc development in Drosophila. Proc. Natl. Acad. Sci. USA 71: 1393–1397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shearn, A., G. Hersperger, E. Hersperger, E. S. Pentz and P. Denker, 1978. Multiple allele approach to the study of genes in Drosophila melanogaster which are involved in imaginal disc development. Genetics 89: 355–370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shearn, A., E. Hersperger and G. Hersperger, 1987. Genetic studies of mutations at two loci of Drosophila melanogaster which cause a wide variety of homeotic transformations. Roux's Arch. Dev. Biol. 196: 231–242. [DOI] [PubMed] [Google Scholar]
- Simcox, A. A., G. Wurst, E. Hersperger and A. Shearn, 1987. The defective dorsal discs gene of Drosophila is required for the growth of specific imaginal discs. Dev. Biol. 122: 559–567. [DOI] [PubMed] [Google Scholar]
- Simcox, A. A., I. J. H. Roberts, E. Hersperger, M. C. Gribbin, A. Shearn et al., 1989. Imaginal discs can be recovered from cultured embryos mutant for the segment-polarity genes engrailed, naked and patched but not from wingless. Development 107: 715–722. [DOI] [PubMed] [Google Scholar]
- Steeg, P. S., G. Bevilacqua, R. Pozzatti, L. A. Liotta and M. E. Sobel, 1988. Altered expression of NM23, a gene associated with low tumor metastatic potential, during adenovirus 2 Ela inhibition of experimental metastasis. Cancer Res. 48: 6550–6554. [PubMed] [Google Scholar]
- Sturtevant, A. H., 1956. A highly specific complementary lethal system in Drosophila melanogaster. Genetics 41: 118–123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Timmons, L., and A. Shearn, 1996. Germline transformation using a prune cDNA rescues prune/Killer of prune lethality and the prune eye color phenotype in Drosophila. Genetics 144: 1589–1600. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Timmons, L., and A. Shearn, 1997. prune/Killer of prune: a conditional dominant lethal interaction in Drosophila. Adv. Genet. 35: 207–252. [DOI] [PubMed] [Google Scholar]
- Timmons, L., E. Hersperger, E. Woodhouse, J. Xu, L.-Z. Liu et al., 1993. The expression of the Drosophila awd gene during normal development and in neoplastic brain tumors caused by lgl mutations. Dev. Biol. 158: 364–379. [DOI] [PubMed] [Google Scholar]
- Timmons, L., J. Xu, G. Hersperger, X.-F. Deng, M. Tharakan et al., 1995. Point mutations in awdKpn which revert the prune/Killer of prune lethal interaction affect conserved residues that are involved in nucleotide diphosphate kinase substrate binding and catalysis. J. Biol. Chem. 270: 23021–23030. [DOI] [PubMed] [Google Scholar]
- Udomsinprasert, R., M. A. Bogoyevitch and A. J. Ketterman, 2004. Reciprocal regulation of glutathione S-transferase spliceforms and the Drosophila c-Jun N-terminal kinase pathway components. Biochem. J. 383: 483–490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu, J., L.-Z. Liu, X.-F. Deng, L. Timmons, E. Hersperger et al., 1996. The enzymatic activity of AWD/NDP kinase is necessary but not sufficient for its biological function. Dev. Biol. 177: 544–557. [PubMed] [Google Scholar]