

Abstract
The cytoplasmic [PSI+] determinant of Saccharomyces cerevisiae is the prion form of the Sup35 protein. Oligopeptide repeats within the Sup35 N-terminal domain (PrD) presumably are required for the stable [PSI+] inheritance that in turn involves fragmentation of Sup35 polymers by the chaperone Hsp104. The nonsense suppressor [PSI+] phenotype can vary in efficiency probably due to different inheritable Sup35 polymer structures. Here we study the ability of Sup35 mutants with various deletions of the oligopeptide repeats to support [PSI+] propagation. We define the minimal region of the Sup35–PrD necessary to support [PSI+] as amino acids 1–64, which include the first two repeats, although a longer fragment, 1–83, is required to maintain weak [PSI+] variants. Replacement of wild-type Sup35 with deletion mutants decreases the strength of the [PSI+] phenotype. However, with one exception, reintroducing the wild-type Sup35 restores the original phenotype. Thus, the specific prion fold defining the [PSI+] variant can be preserved by the mutant Sup35 protein despite the change of phenotype. Coexpression of wild-type and mutant Sup35 containing three, two, one, or no oligopeptide repeats causes variant-specific [PSI+] elimination. These data suggest that [PSI+] variability is primarily defined by differential folding of the Sup35–PrD oligopeptide-repeat region.
PRIONS are infectious agents responsible for a group of diseases typified by sheep scrapie, bovine spongiform encephalopathy, and human Creutzfeldt-Jacob disease (for reviews, see Horwich and Weissman 1997; Prusiner 1998). In mammals, the only known protein with prion properties is PrP, which in its conformationally altered prion form (PrPSc), is able to convert a normal host-encoded protein (PrPC) into the prion form. Several proteins of the yeast Saccharomyces cerevisiae, similarly to mammalian prions, can undergo autocatalytic conformational rearrangement. This process can last stably for many cell generations, resulting in some cases in heritable phenotypes (Wickner et al. 2000). The best studied of the yeast prion proteins is the translation termination factor Sup35 (eRF3) whose prion state is characterized by its aggregation and partial inactivation, which causes the associated nonsense-suppressor phenotype (Patino et al. 1996; Paushkin et al. 1996). Sup35 aggregates have a complex structure composed of multiple small Sup35 amyloid-like prion polymers and associated proteins (Kryndushkin et al. 2003).
Sup35 consists of the nonessential N-terminal domain responsible for [PSI+] appearance and maintenance (Chernoff et al. 1993; Ter-Avanesyan et al. 1994), the charged middle (M) domain, which is also important for [PSI+] propagation (Liu et al. 2002; Bradley and Liebman 2004), and the C-terminal domain fulfilling the essential translation termination activity (Ter-Avanesyan et al. 1993). The N domain of Sup35, the so-called prion-forming domain (PrD), is enriched in glutamine and asparagine residues and can be subdivided into two regions with different functions. The extreme N-terminal region (residues 1–41), designated as the N domain Q (NQ)-rich region, is particularly rich in glutamine and asparagine and implicated both in prion conversion (DePace et al. 1998) and in providing a species specificity determinant (Santoso et al. 2000; Hara et al. 2003). The NQ region is followed by the N domain repeat (NR) region, which contains five complete copies (R1–R5) and one partial copy (R6) of the imperfect oligopeptide repeat with the consensus sequence PQGGYQQ-YN. While the NQ region mediates sequence-specific aggregation, the NR region is required for the stable inheritance of these aggregates, possibly by mediating their fragmentation by the chaperone Hsp104 (Osherovich et al. 2004).
A systematic deletion analysis of Sup35–PrD defined the minimum length required for stable [PSI+] propagation as amino acids 1–93, up to and including repeat R5 (Parham et al. 2001; Osherovich et al. 2004). It is important to stress that this analysis was performed for only one [PSI+] isolate. However, prions can exist in multiple variants, also called strains. In mammals, different prion variants are defined by specific incubation times, distribution of vacuolar lesions, and the pattern of PrPSc accumulation (for review, see Prusiner 1998). For the yeast [PSI+] prion, they can be revealed by differences in the nonsense suppressor efficiency and mitotic stability of independently isolated [PSI+] strains (Derkatch et al. 1996). [PSI+] variants with strong suppression ([PSI+] “strong”) are highly stable, and the variants with weak suppression ([PSI+] “weak”) show decreased mitotic stability (Derkatch et al. 1996; Kochneva-Pervukhova et al. 2001). At the molecular level, mammalian prion strain differences can be correlated with stable variations in the prion protein structure (Bessen et al. 1995; Caughey et al. 1998; Safar et al. 1998). However, although there are some differences in the rate of Sup35 aggregation as well as in the size of prion polymers constituting Sup35 aggregates in cells with different [PSI+] variants (Zhou et al. 1999; Kochneva-Pervukhova et al. 2001; Kryndushkin et al. 2003), the molecular basis of [PSI+] variability is still poorly understood.
Here we report the use of Sup35 deletion mutants with different numbers of Sup35–PrD oligopeptide repeats to investigate the role of these repeats in [PSI+] prion variability. This was performed by [PSI+] transmission from wild-type Sup35 to various repeat-deleted mutants and then back to wild-type Sup35. The results obtained define the minimal region of Sup35–PrD able to support [PSI+] as amino acids 1–64 and suggest that the [PSI+] variability depends on differences in folding of the NR region of Sup35–PrD.
MATERIALS AND METHODS
Media, strains, plasmids, and genetic methods:
Yeast strains were grown at 30° on either complete (YPD) or synthetic (SC) media containing 2% glucose. Selective media lacking uracil, adenine, or histidine are designated here as SC–Ura, SC–Ade, or SC–His, respectively. To select for cells that had lost URA3 plasmids, cells were grown on 5-fluoroorotic acid (5-FOA) medium (Sherman et al. 1986). To cure cells of the [PSI+] determinants, cells were grown from single cells to colonies on medium containing 3 mm guanidine hydrochloride (GuHCl) (Tuite et al. 1981). For assaying the [PSI+] suppressor phenotype, SClow Ade–His (supplemented with 0.07 mg/ml of adenine sulfate) or modified YPD (YPDred: 0.5% yeast extract, 2% peptone, 4% glucose) medium was used because these media promote accumulation of red pigment in the ade2 mutants. Bacteria were grown at 37° on 2× YT medium (Sambrook et al. 1989). All solid media contained 2% (w/v) agar. DNA transformation of yeast cells was performed using the lithium acetate method (Gietz and Woods 2002).
Centromeric plasmids carrying HIS3 and either wild-type SUP35 or SUP35 deletion alleles encoding mutant proteins Sup35ΔN, Sup35R0, Sup35R1, Sup35R1–2, Sup35R1–3, Sup35R1–4, and Sup35R1–5 were previously described by Parham et al. (2001). The multicopy LEU2 plasmid YEp181–SUP35ΔSal contains the XhoI–SalI fragment of the SUP35 gene encoding a C-terminally truncated Sup35. This plasmid was used to induce [PSI+] appearance de novo. The centromeric URA3 plasmid pRS316–SUP35 carrying wild-type SUP35 and multicopy LEU2 plasmid pTR30-1 with SUP35 from Pichia methanolica were previously described (Kushnirov et al. 1990).
The S. cerevisiae strain 22V-H63 (MATa ade2-1 SUQ5 kar1 lys1 his3 ura3 leu2 cyhR) was used to disrupt the SUP35 gene. For this, 22V-H63 was transformed with the MluI–BamHI fragment of the pSTR4-SUP35∷TRP1 plasmid (Ter-Avanesyan et al. 1993) together with the pTR30-1 plasmid. The obtained Leu+ clones were screened by Western blotting to select strains expressing the homolog of Sup35 from P. methanolica only (this protein differs by molecular mass from Sup35 of S. cerevisiae). pTR30-1 was then replaced with pRS316–SUP35. The multicopy plasmid YEp181–SUP35ΔSal was transiently introduced into the obtained disruptant to induce [PSI+] de novo. The [psi−] colonies of the ade2-1 SUQ5 strain 22V-H63 were distinguished by their red color and adenine requirement, because the weak serine-inserting tRNA suppressor SUQ5 cannot suppress the ade2-1 ochre mutation in the absence of the [PSI+] determinant (Cox 1965). This allowed the identification of [PSI+] in transformants of this strain by the appearance of colonies with white or pink color, depending on the [PSI+] variant. The [PSI+] state of the transformants was then confirmed by growth in 3 mm GuHCl. To quantify the mitotic stability of [PSI+], cells of three colonies for each [PSI+] isolate were suspended in water and plated onto YPDred medium. Plates were incubated for 3 days and the percentage of red colonies was determined. The [PSI+]-eliminating effect of plasmids encoding Sup35 with a truncated PrD was estimated in a similar way, but cell suspensions of transformants carrying plasmids with wild-type and mutant SUP35 were plated onto a medium selective for the plasmid with wild-type SUP35 (SClow Ade–Ura). To calculate the efficiency of [PSI+] rescue upon the replacement of the plasmid bearing wild-type SUP35, with the plasmids carrying different SUP35 deletion alleles, three independent transformants for each combination were grown on 5-FOA medium to select for cells that had lost the URA3 SUP35 plasmid pRS316–SUP35. The resulting Ura− cells were grown overnight in liquid SC–His medium and plated onto SClow Ade–His medium. After 4 days of growth on this medium, colonies were replica plated onto SC–Ade medium. The [PSI+] status of all Ade+ colonies was then confirmed by growth in 3 mm GuHCl. To generate cells that had lost the HIS3 plasmids with the mutant SUP35, transformants carrying this plasmid along with the wild-type SUP35 plasmid were passaged five times on SC–Ura medium (∼70% of cells of each transformant had lost the HIS3 plasmids after this procedure) and replicated to SC–Ade medium to assess the suppressor phenotype. Then transformants were streaked to single cells on YPDred plates, and the [PSI+] status of clones that had lost the HIS3 plasmids was confirmed by the GuHCl test.
Preparation of yeast cell lysates:
Yeast cultures were grown in liquid media to an OD600 = 1. Cells were harvested, washed in water, and lysed by vortexing with glass beads in buffer L: 30 mm Tris–HCl, pH 7.4, 150 mm NaCl, and 10 mm dithiothreitol. To prevent proteolytic degradation, 25 mm EDTA, 10 mm phenylmethylsulfonyl fluoride, and complete protease inhibitor cocktail (Roche Applied Science) were added. Cell debris were removed by centrifugation at 10,000 × g for 5 min.
Electrophoresis:
For separation of prion particles, horizontal 1.8% agarose gels in Tris–acetate–EDTA buffer with 0.1% SDS were used as described by Kryndushkin et al. (2003). To analyze Sup35 polymers and monomers in a single gel, the standard SDS–PAGE system (Laemmli 1970) was modified as follows. The concentrating and separating gels contained 1 m urea and 6% and 8% polyacrylamide, respectively. Yeast cell lysates were mixed with 4× sample buffer I (0.25 m Tris–HCl, pH 6.8, 4 m urea, 8% w/v SDS, 8% 2-mercaptoethanol, 20% glycerol, and 0.2% w/v bromophenol blue), incubated for 2 min at a room temperature, loaded on a gel, and run for 30 min. Sup35 monomers separated, while polymers stopped at the start of stacking gel. To dissolve and analyze the polymers, 4× sample buffer II (0.1 m Tris–glycine pH 8.3, 4 m urea, 8% w/v SDS, 8% 2-mercaptoethanol, 20% glycerol, 0.2% w/v bromophenol blue) was loaded into the wells, and the gel was sealed and boiled for 5 min. The separation was continued after boiling. After electrophoresis, proteins were transferred to a Hybond ECL nitrocellulose membrane and decorated with anti-Sup35 antibody. Bound antibody was detected using the Amersham Biosciences ECL system.
RESULTS
Coexpression of wild-type Sup35 with truncated PrD mutants of Sup35 causes variant-specific effects on [PSI+] propagation:
A modification of the plasmid shuffle assay, developed to test the significance of Sup35 oligopeptide repeats in [PSI+] prion propagation (Parham et al. 2001), was used to study their role in [PSI+] variability (Figure 1). In the [psi−] strain 22V-H63, the SUP35 gene was disrupted by insertion of the TRP1 gene and wild-type SUP35 on the URA3–CEN plasmid pRS316–SUP35 was introduced to support the viability. [PSI+] derivatives of this strain were then induced de novo using the multicopy SUP35 plasmid YEp181–SUP35ΔSal. Five strong and five weak independent [PSI+] isolates were taken for further analysis. Strong [PSI+] (s1–s5) were similar to each other as judged by their suppressor phenotypes, while weak [PSI+] could be divided into two classes: weak (w1 and w2) and very weak (w3–w5) (Figure 2.)
Figure 1.

Overall scheme of the experiments on [PSI+] transfer to truncated Sup35 performed in this work. Strain 22V-H63 contains chromosomal SUP35 disrupted by insertion of TRP1. pSUP35Δ denotes SUP35 deletion plasmids; pSUP35 is pRS316–SUP35.
Figure 2.

Suppressor phenotype of [PSI+] cells. (A) [PSI+] variants with wild-type Sup35 used for the experiments: s1–s5, strong; w1–w5, weak. (B) [PSI+] s1 expressing indicated truncated Sup35 instead of complete Sup35.
Centromeric plasmids encoding Sup35 with a truncated PrD (Parham et al. 2001) were introduced into the obtained [PSI+] variants. The phenotypic effects were similar when the second plasmid encoded either complete Sup35 or Sup35 with five PrD repeats (Sup35R1–5), confirming that the sixth half repeat is not essential for the Sup35 prion properties (Parham et al. 2001). Suppression in all weak [PSI+] variants was increased, as determined by faster growth on adenine omission medium and whiter colony color. The suppressor phenotype of the strong [PSI+] variants was not changed, but without selection for suppression such cells grew slower than in the presence of a single-copy SUP35 plasmid (data not shown). These effects are likely to reflect the increase in the level of Sup35, which should accelerate prion conversion, thus strengthening suppression in weak [PSI+] and inhibiting growth of strong [PSI+] cells (Dagkesamanskaya and Ter-Avanesyan 1991). The plasmid encoding Sup35 with four PrD repeats (Sup35R1–4) weakened suppression in strong and weak [PSI+] variants, but strengthened it in very weak [PSI+] cells. This resembles the oppositely directed effects of the mutant PNM2 allele of SUP35 on strong and weak [PSI+] (Derkatch et al. 1999). The plasmids encoding Sup35 with three (Sup35R1–3), two (Sup35R1–2), one (Sup35R1), or no PrD repeats (Sup35R0) weakened nonsense suppression in all [PSI+] variants examined (data not shown).
Expression of Sup35R0 and Sup35R1 destabilized strong [PSI+] variants and caused their loss at the rate of 1–3% (Table 1). Expression of Sup35R0, Sup35R1, Sup35R1–2, and Sup35R1–3 caused an approximately twofold increase in the loss of very weak [PSI+]. Efficient elimination of [PSI+] w1 was observed in the presence of Sup35R0 and Sup35R1 while [PSI+] w2 was very efficiently eliminated by Sup35R1, Sup35R1–2, and Sup35R1–3 (Table 1. Therefore, Sup35 with a truncated PrD could cause efficient [PSI+] curing in a dominant manner with the curing efficiency depending greatly on both the [PSI+] variant used and the Sup35 deletion construct.
TABLE 1.
Mitotic stability of [PSI+] in transformants carrying plasmid pairs with the SUP35 wild-type and deletion alleles
| [PSI+] loss (%) in transformants with plasmids encoding
|
||||||
|---|---|---|---|---|---|---|
| Type of [PSI+] | [PSI+] isolate | Sup35 | Sup35 + Sup35R0 | Sup35 + Sup35R1 | Sup35 + Sup35R1–2 | Sup35 + Sup35R1–3 |
| Strong | s1 | < 0.3 | 0.8 ± 0.4 | 1.1 ± 0.2 | < 0.3 | < 0.3 |
| s2 | < 0.2 | 1.1 ± 0.3 | 2.3 ± 1.0 | < 0.3 | < 0.2 | |
| s3 | < 0.4 | 2.4 ± 0.9 | 1.9 ± 0.4 | < 0.2 | < 0.3 | |
| s4 | < 0.3 | 2.3 ± 0.5 | 3.1 ± 0.1 | < 0.3 | < 0.3 | |
| s5 | < 0.3 | 1.5 ± 0.1 | 2.6 ± 0.6 | < 0.4 | < 0.3 | |
| Weak | w1 | 0.3 ± 0.2 | 2.3 ± 1.3 | 4.8 ± 0.5 | 0.2 ± 0.1 | 0.2 ± 0.0 |
| w2 | 0.3 ± 0.1 | 2.6 ± 1.4 | 50.4 ± 5.4 | 43.3 ± 15.0 | 47.6 ± 3.9 | |
| w3 | 21.7 ± 3.3 | 43.6 ± 12.9 | 38.9 ± 1.1 | 24.6 ± 7.4 | 46.5 ± 5.4 | |
| w4 | 21.8 ± 5.9 | 38.9 ± 9.3 | 41.2 ± 7.1 | 35.2 ± 5.4 | 49.6 ± 5.4 | |
| w5 | 14.6 ± 4.2 | 49.7 ± 6.0 | 24.4 ± 3.0 | 29.6 ± 4.3 | 23.5 ± 1.7 | |
The transformants carried plasmids encoding Sup35 proteins as indicated. Transformants producing Sup35 only had the empty vector instead of the plasmid encoding mutant Sup35. [PSI+] maintenance in three independent transformants with each plasmid combination was analyzed and the standard error is indicated.
To monitor the incorporation of Sup35 into prion polymers, a novel method, based on the insolubility of Sup35 polymers in SDS at room temperature, was developed. Lysates of cells expressing both complete and truncated Sup35 were mixed with SDS sample buffer and loaded on a gel without boiling. The Sup35 monomers separated, while polymers were trapped at the origin of the gel. After about a quarter of the run time, the gel was taken out and heated to 100° to disassemble polymers, and then the run was continued (for details, see materials and methods). The amount of unpolymerized Sup35 correlated inversely with the suppressor strength (Figure 3) with the monomer fraction consisting mostly of truncated Sup35. Efficient incorporation of deleted Sup35 into polymers was achieved with two repeats (Sup35R1–2) for strong [PSI+], but required a longer construct (Sup35R1–4) for weak [PSI+]. Shorter Sup35 mutants incorporated less efficiently in correlation with the number of Sup35–PrD repeats. Strikingly, [PSI+] destabilization and curing always correlated with incorporation of only a small portion of truncated Sup35 into the prion polymers.
Figure 3.

Distribution of wild-type and mutant Sup35 (Sup35Δ) proteins between the monomer and polymer fractions in lysates of [PSI+] transformants coexpressing them. (A) Strong [PSI+] variant s5. (B) Weak [PSI+] variant w3.
[PSI+] destabilization was also accompanied by a slight but reproducible increase in the size of SDS-resistant prion polymers (Figure 4). The size of the Sup35 prion polymers depends on the rate of their elongation and accessibility to fragmentation by the Hsp104 chaperone (Kryndushkin et al. 2003). Truncated Sup35 variants showed reduced ability to copolymerize with complete Sup35, and thus they could not accelerate prion polymerization. Therefore, elimination of [PSI+] was primarily due to a reduced efficiency of fragmentation of such polymers. Increased size of Sup35 polymers should lead to a decreased number of prion seeds and lower [PSI+] stability (Kryndushkin et al. 2003). The increase in polymer size was not large. However, it is likely that the [PSI+] loss occurred in a subpopulation of cells in which the increase was more pronounced. These could be cells in which the proportion of truncated Sup35 to wild-type Sup35 was increased, possibly due to fluctuations in the copy number of the centromeric plasmid used.
Figure 4.

Coexpression of mutant Sup35 proteins with wild-type Sup35 increases the size of Sup35 prion polymers. Sup35 mutants coexpressed with wild-type Sup35 are indicated. Control: [PSI+] strains expressing wild-type Sup35 only. The transformants carried either strong [PSI+] variant s5 or weak [PSI+] w3.
Reduction in the number of Sup35–PrD repeats to two does not abolish [PSI+] maintenance but does weaken its suppressor phenotype:
Using the double transformants described above, cells lacking the URA3 plasmid with wild-type SUP35 were selected by growth of the transformants on 5-FOA-containing medium. The cells, which expressed only truncated Sup35, were tested for the suppressor phenotype. The fewest number of repeats able to support [PSI+] was two (Sup35R1–2) for strong [PSI+] and four (Sup35R1–4) for weak [PSI+]. A significant proportion of cells lost [PSI+] concomitantly with the loss of complete Sup35 (Table 2) with the proportion showing an inverse correlation with the number of repeats in the remaining Sup35 mutant. However, Sup35R1–4 was exceptional, being poorly compatible with the strong [PSI+] variants, rescuing strong [PSI+] variants less efficiently than Sup35R1–3 and Sup35R1–2, and rescuing weak [PSI+] much better than strong [PSI+] variants (Table 2).
TABLE 2.
The efficiency of [PSI+] rescue upon the replacement of SUP35 with SUP35 deletion alleles
| [PSI+] rescue (%)
|
||||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| Replacing SUP35 allele
|
Strong [PSI+] variants
|
Weak [PSI+] variants
|
||||||||
| s1 | s2 | s3 | s4 | s5 | w1 | w2 | w3 | w4 | w5 | |
| SUP35 | 100 ± 0.0 | 100 ± 0.0 | 100 ± 0.0 | 100 ± 0.0 | 100 ± 0.0 | 99.5 ± 0.1 | 98.6 ± 0.4 | 49.9 ± 0.7 | 44.0 ± 4.2 | 39.4 ± 3.6 |
| SUP35R1–5 | 82 ± 6.7 | 98.8 ± 0.2 | 98.3 ± 1.4 | 99.4 ± 0.4 | 98.7 ± 0.7 | 88.6 ± 1.0 | 59.6 ± 18.8 | 27.2 ± 3.1 | 33.2 ± 3.0 | 27.8 ± 0.5 |
| SUP35R1–4 | 3.1 ± 0.5 | 2.7 ± 1.3 | 3.1 ± 1.2 | 5.0 ± 0.9 | 3.8 ± 1.5 | 57.7 ± 2.5 | 27.9 ± 3.8 | 25.6 ± 2.2 | 23.9 ± 1.3 | 21.7 ± 3.1 |
| SUP35R1–3 | 7.7 ± 1.5 | 13.8 ± 1.7 | 14.8 ± 0.7 | 12.5 ± 0.9 | 11.6 ± 1.4 | 0.0 | 0.0 | 0.0 | 0.0 | 0.0 |
| SUP35R1–2 | 8.8 ± 0.6 | 13.2 ± 1.3 | 14.9 ± 1.1 | 13.6 ± 2.1 | 10.8 ± 1.1 | 0.0 | 0.0 | 0.0 | 0.0 | 0.0 |
| SUP35R1 | 0.0 | 0.0 | 0.0 | 0.0 | 0.0 | 0.0 | 0.0 | 0.0 | 0.0 | 0.0 |
| SUP35ΔN | 0.0 | 0.0 | 0.0 | 0.0 | 0.0 | 0.0 | 0.0 | 0.0 | 0.0 | 0.0 |
In the indicated [PSI+] variants, the plasmid with wild-type SUP35 was replaced with the indicated SUP35 alleles. Following loss of the wild-type SUP35 plasmid, clones that retained [PSI+] were counted. Three transformants were analyzed in each case and the standard error is indicated.
The replacement of Sup35 with its deletion variants reduced the strength of both the [PSI+] suppressor phenotype and its mitotic stability (Figure 2 and Table 3). The size of the Sup35 prion polymers increased significantly, being inversely related to the number of repeats (Figure 5). Such an increase most likely reflects a reduced susceptibility of these polymers to fragmentation. The increase of polymer size correlated well with the weakening of [PSI+] suppression and stability and may be considered as the cause of the weakening. In contrast to the overall trend, the [PSI+] stability was lower with Sup35R1–3 than with Sup35R1–2, while the polymer size was roughly similar.
TABLE 3.
Mitotic stability of [PSI+] in transformants carrying different SUP35 deletion alleles
| SUP35 allele | No. of clones | No. of [psi−] clones | % of [psi−] clones |
|---|---|---|---|
| SUP35R1–5 | 226 | 0 | < 0.5 |
| 205 | 0 | ||
| 190 | 0 | ||
| SUP35R1–4 | 171 | 29 | 20.6 ± 1.9 |
| 171 | 37 | ||
| 185 | 43 | ||
| SUP35R1–3 | 726 | 291 | 43.3 ± 1.7 |
| 494 | 217 | ||
| 433 | 199 | ||
| SUP35R1–2 | 612 | 123 | 27.7 ± 5.4 |
| 512 | 128 | ||
| 517 | 197 |
The transformants were obtained by replacement of wild-type SUP35 with mutant alleles in the strain carrying [PSI+] s1 variant. Data represent averages from three independent subclones of each [PSI+] transformant. The standard error is indicated.
Figure 5.
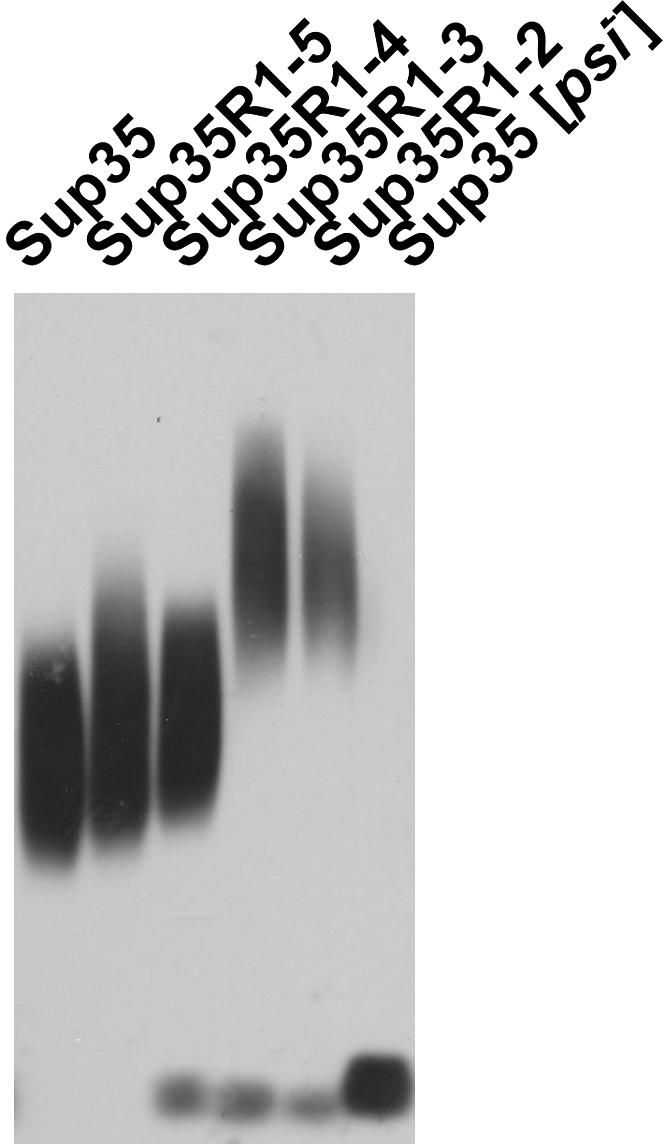
Reduction of the number of Sup35 PrD repeats increases the size of Sup35 prion polymers. The truncated Sup35 proteins propagating [PSI+] are indicated.
Strong [PSI+] is preserved by Sup35 deletants, with the exception of Sup35R1–4:
To find whether the strong [PSI+] fold was preserved by the mutant Sup35 molecules with truncated PrD's, the plasmid with wild-type SUP35 was reintroduced into [PSI+] cells expressing the different truncated Sup35, which derived from the strong [PSI+] variant s1. In each case, two types of transformants were observed (Table 4). Transformants of the first, major, type maintained [PSI+] stably and did not vary in suppression. Coexpression of full-length Sup35 strengthened the [PSI+] suppressor phenotype compared to the cells with truncated Sup35 proteins only. This suggests that in most cases the prion fold of the truncated Sup35 proteins was readily transferred to wild-type Sup35. Loss of the plasmids encoding Sup35R1–2, Sup35R1–3, or Sup35R1–5 always resulted in colonies showing the strong suppressor phenotype similar to that of the original [PSI+] s1 isolate. In contrast, the loss of the plasmid encoding Sup35R1–4 resulted in cells with the weak [PSI+] phenotype. Thus, the strong [PSI+] fold was preserved by Sup35R1–2, Sup35R1–3, and Sup35R1–5, but not by Sup35R1–4.
TABLE 4.
The [PSI+] phenotypes revealed after replacement of SUP35 deletion alleles with wild-type SUP35
| Transformants with stable [PSI+]
|
||||
|---|---|---|---|---|
| SUP35 deletion allele | Total no. of transformants with wild-type and deleted SUP35 | Producing cells with strong [PSI+] | Producing cells with weak [PSI+] | Transformants with unstable [PSI+] |
| SUP35R1–2 | 119 | 86 (72.3) | 0 | 33 (27.7) |
| SUP35R1–3 | 120 | 93 (77.5) | 0 | 27 (22.5) |
| SUP35R1–4 | 120 | 0 | 84 (70.0) | 36 (30.0) |
| SUP35R1–5 | 119 | 113 (95.0) | 0 | 6 (5.0) |
Transformants of the [PSI+] s1 variant carrying plasmid pairs with the SUP35 wild-type and deletion alleles had lost plasmids with the indicated SUP35 deletion allele. The [PSI+] status of the resulting strains was determined as described in materials and methods. Numbers in parentheses are percentages.
Transformants of the second, minor, type were unstable, losing [PSI+] with high frequency. After the loss of plasmids encoding truncated Sup35, these transformants always produced cells with weak and strong [PSI+], as well as [psi−] cells. Apparently, in these transformants copolymerization of truncated and complete Sup35 caused a change of the prion fold to an “unstable” type resembling undifferentiated [PSI+], described by Bradley and Liebman (2004).
DISCUSSION
In this study we have analyzed the ability of Sup35 proteins with reduced numbers of oligopeptide repeats in the PrD to maintain different [PSI+] variants. The minimal region of Sup35–PrD required for [PSI+] maintenance was established as amino acids 1–64, which includes the NQ region and the two first repeats. This agrees with observations that the region 1–57 was sufficient for Sup35 aggregation and [PSI+] induction (Osherovich et al. 2004) and that the polymers of fragment 1–61 obtained in vitro were able to transform [psi−] yeast cells to the [PSI+] state (King and Diaz-Avalos 2004). However, earlier tests for [PSI+] maintenance (Parham et al. 2001), performed with the same set of SUP35 constructs, showed the requirement for a significantly larger region, 1–93. The reason for this discrepancy may be the use in our work of different [PSI+] variants and a different genetic background. Very recently, the in vitro studies of Krishnan and Lindquist (2005) have shown that the core of the Sup35 amyloid fibrils comprises residues 21–121 of Sup35–PrD in a “weak” conformation and residues 31–86 in a “strong” conformation. Consequently, our data may suggest that a significant C-terminal fragment of this core is inessential for the [PSI+] maintenance. Furthermore, the Sup35–PrD region inessential for [PSI+] comprised the “tail” area, which represents an interface for interaction of neighboring Sup35 monomers in a fibril (Krishnan and Lindquist 2005). This suggests that the “tail” interface is not linked to a certain amino acid sequence. Rather, any residues found at the C terminus of the Sup35 amyloidogenic region would form such an interface.
The transfer of the strong [PSI+] character from full-length to deleted Sup35 always weakened the suppressor phenotype and the mitotic stability of [PSI+]. A significant increase in the size of Sup35 polymers was also observed, which could be the cause of these effects, since the number of prion polymers and the Sup35 polymerization rate are inversely proportional to the size of polymers. The number of higher-order prion aggregates, which are likely to represent heritable prion units (i.e., propagons), may decrease in even higher proportion, since bigger polymers have a higher propensity to aggregate (Kryndushkin et al. 2003). The cause for the increase in the size of polymers could be either their decreased fragmentation or increased polymerization speed. The second possibility appears unlikely, since in the [PSI+] cells coexpressing complete and truncated Sup35, the latter showed a reduced ability to polymerize.
Prion polymer fragmentation is performed by the Hsp104 chaperone, possibly with the aid of other chaperones (Ferreira et al. 2001; Kryndushkin et al. 2003), and the frequency of such fragmentation would be defined by the ability of Hsp104 to recognize prion polymers as abnormal structures. This frequency can vary greatly depending on the [PSI+] variant (Kryndushkin et al. 2003) and some Sup35 polymers are recognized poorly, if at all (Salnikova et al. 2005). Here we show that the fragmentation decreased with a reduction in the number of Sup35–PrD repeats, suggesting that the NR region of Sup35–PrD provides the key determinants for Hsp104 recognition. A similar suggestion was made by Osherovich et al. (2004), but without experimental evidence to support the proposal. The importance of the NR region for fragmentation of Sup35 polymers was inferred from the properties of the NQ region, which was able to polymerize, but unable to support [PSI+]. This would imply that the polymers of the Sup35R0 mutant do not experience fragmentation and thus that the NR region is required to allow fragmentation of Sup35 polymers by Hsp104. Here we have shown the importance of the NR region for fragmentation of the Sup35 polymers: the size of Sup35 prion polymers gradually increased with a reduction in the number of Sup35–PrD oligopeptide repeats. In the absence of repeats, the fragmentation efficiency was insufficient for [PSI+] maintenance. Thus, the NR region may serve as a target for recognition by Hsp104 for subsequent fibril fragmentation. These data may also be explained in a different way. The decreased recognition by Hsp104 may be due to shielding of the amyloid core by the Sup35 MC domains. If we assume the “nanotube” structure (Perutz et al. 2002) for the Sup35 prion domain, the length of amyloid fibril per Sup35 monomer is proportional to the length of the amyloidogenic sequence. The N-terminal deletions made in Sup35 would decrease the space between the Sup35 MC domains and thus reduce the Hsp104 access to the amyloid core. This scenario appears less likely since there is no evidence that the MC domains of Sup35 can efficiently shield the amyloid core. The phenotypic difference of [PSI+] variants is usually due to difference in fragmentation of Sup35 polymers, and in only one case was the rate of polymerization significantly altered (Kryndushkin et al. 2003; our unpublished data). Therefore, differential folding of the NR region should define the [PSI+] variability. Our coexpression experiments also confirmed the conclusion of Osherovich et al. (2004) that NQ is sufficient for Sup35 polymerization. However, an important reservation should be made: the NR region, or at least a part of it, was required for efficient Sup35 polymerization. Sup35 lacking the NR sequence polymerized ∼10-fold less efficiently than full-length Sup35 when they were coexpressed (Figure 3).
For weak [PSI+] variants, a minimum of four Sup35–PrD oligopeptide repeats was required, in contrast with the two repeats needed for strong [PSI+] variants. Two explanations for this are possible. First, a “qualitative” explanation, namely that Sup35–PrD with less than four repeats cannot form a stable polymer of the weak type. The second explanation would be a “quantitative” one; i.e., since weak folds are relatively poorly recognized by chaperones, a minimum of four repeats is required for fragmentation of polymers.
The strong [PSI+] phenotype weakened upon transfer from complete Sup35 to Sup35R1–2 (and to Sup35R1–3 and Sup35R1–5). This suggests that the missing repeats are involved in prion structure. When [PSI+] was returned to the full-length Sup35, the original phenotype was restored. Thus, the information about the specific fold of the missing repeats was preserved in their absence. This striking result may be explained if we assume that the NR region forms a β-helical nanotube (Perutz et al. 2002; Krishnan and Lindquist 2005) with two repeats (19 residues) per turn. It should be noted that the repeat length is not uniform, but regularly alters from 9 to 10 residues. This suggests that the actual repeating unit consists of two repeats, which also represents a minimal length of a turn in the nanotube model. In such tubes, other repeats would fold in the same way as the first two. Thus, the first two repeats would be sufficient to preserve the folding of the repeat region. Interestingly, the nanotube model can also explain the inability of Sup35R1–2 and Sup35R1–3 to maintain weak [PSI+] if one assumes that, in weak folds, four repeats are used per one helical turn. Then at least four repeats would be required for a stable “weak” structure.
Propagation of prion conformation represents a templated process, which suggests that the prion fold cannot alter during propagation; it can only appear and disappear. However, we have previously reported that some weak [PSI+] variants can become strong variants with a significant frequency (Kochneva-Pervukhova et al. 2001). To explain this observation, we propose that certain parts of Sup35–PrD (for example, NR and NQ) can fold into a prion conformation independently and that the loss or acquisition of the prion fold may be restricted to only a part of PrD. This would allow alteration of the [PSI+] variant during polymerization.
The repeat-truncated mutants of Sup35 could be a useful tool for changing conformation of only the NR region. One example of such change relates to Sup35R1–4. Contrary to general trends, strong [PSI+] was transferred to Sup35R1–4 significantly less efficiently than to Sup35R1–2 and Sup35R1–3 and than weak [PSI+] was transferred to it. Furthermore, the [PSI+] transfer back to full-length Sup35 revealed that Sup35R1–4 changed the strong prion fold to the weak type. Thus, Sup35R1–4 did not support the strong prion fold. It is unclear why only this construct, and not the shorter Sup35R1–2 and Sup35R1–3, showed such incompatibility. Nevertheless, one can assume that in this case we achieved a change of the [PSI+] variant by altering conformation of only the NR region. The [PSI+] change caused by the truncated Sup35 may be presumed to be this in the other cases. For example, a significant proportion of unstable [PSI+] appearing during [PSI+] transfer to full-length Sup35 may be regarded as a consequence of a structural alteration in the NR region caused by [PSI+] propagation by repeat-truncated Sup35. Finally, the [PSI+] curing upon coexpression of full-length Sup35 and its truncated variants may also be explained by a localized loss of conformation. Sup35R1 on its own cannot support [PSI+], while Sup35R1–2 and Sup35R1–3 cannot maintain the weak prion folds. This suggests that when Sup35R0 or Sup35R1 incorporate into a growing Sup35 “strong” prion polymer, and Sup35R1–2 or Sup35R1–3 are incorporated into a “weak” one, they transfer the conformational information for the NQ, but not for the NR region (Figure 6). The NR region will then lose its original prion fold and acquire a new one, which would be poorly recognized by Hsp104. According to our recent observations (Salnikova et al. 2005), most of the Sup35 amyloid-like polymers appearing de novo represent variants that are poorly fragmented by Hsp104. In an alternative model for [PSI+] curing, the truncated Sup35 could act as a “terminator” of polymerization. This should interfere with polymerization and decrease the size of Sup35 polymers, which contradicts the observation that the size of polymers increased (Figure 4). Thus, truncated Sup35 could cure [PSI+] by spoiling fragmentation, rather than by polymerization.
Figure 6.

Alteration of a Sup35 prion fold by insertion in a polymer of Sup35 with truncated PrD (Sup35Δ) (see discussion).
After this work was submitted, Ross et al. (2005) showed that the amino acid sequence of the Sup35 prion domain, including the repeat region, can be shuffled without apparently blocking its ability to form prion and different prion variants. This would suggest that the repeats per se are not required for prion formation and variability, which is consistent with the lack of oligopeptide repeats in the Ure2 and Rnq1 prion proteins. However, the observations made by Ross et al. (2005) are made with artificial Sup35 and do not conflict with our data, since we have considered the role of the Sup35 repeats specifically for the natural protein and its preexisting [PSI+] variants.
Acknowledgments
We thank Ilya Alexandrov for helpful ideas in developing modification of electrophoretic technique for Figure 3. Research in the Ter-Avanesyan laboratory was supported by grants from the Howard Hughes Medical Institute, the Wellcome Trust, the Russian Foundation for Basic Research, and the U.S. Army European Research Office. Research in the Tuite laboratory was supported by a project grant from the Biotechnology and Biological Sciences Research Council.
References
- Bessen, R. A., D. A. Kocisko, G. J. Raymond, S. Nandan, P. T. Lansbury et al., 1995. Non-genetic propagation of strain-specific properties of scrapie prion protein. Nature 375: 698–700. [DOI] [PubMed] [Google Scholar]
- Bradley, M. E., and S. W. Liebman, 2004. The Sup35 domains required for maintenance of weak, strong or undifferentiated yeast [PSI+] prions. Mol. Microbiol. 51: 1649–1659. [DOI] [PubMed] [Google Scholar]
- Caughey, B., G. J. Raymond and R. A. Bessen, 1998. Strain-dependent differences in β-sheet conformations of abnormal prion protein. J. Biol. Chem. 273: 32230–32235. [DOI] [PubMed] [Google Scholar]
- Chernoff, Y. O., I. L. Derkach and S. G. Inge-Vechtomov, 1993. Multicopy SUP35 gene induces de-novo appearance of psi-like factors in the yeast Saccharomyces cerevisiae. Curr. Genet. 24: 268–270. [DOI] [PubMed] [Google Scholar]
- Cox, B. S., 1965. [PSI+], a cytoplasmic suppressor of super-suppressor in yeast. Heredity 20: 505–521. [Google Scholar]
- Dagkesamanskaya, A. R., and M. D. Ter-Avanesyan, 1991. Interaction of the yeast omnipotent suppressors SUP1(SUP45) and SUP2(SUP35) with non-Mendelian factors. Genetics 128: 513–520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DePace, A. H., A. Santoso, P. Hillner and J. S. Weissman, 1998. A critical role for amino-terminal glutamine/asparagine repeats in the formation and propagation of a yeast prion. Cell 93: 1241–1252. [DOI] [PubMed] [Google Scholar]
- Derkatch, I. L., Y. O. Chernoff, V. V. Kushnirov, S. G. Inge-Vechtomov and S. W. Liebman, 1996. Genesis and variability of [PSI+] prion factors in Saccharomyces cerevisiae. Genetics 144: 1375–1386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Derkatch, I. L., M. E. Bradley, P. Zhou and S. W. Liebman, 1999. The PNM2 mutation in the prion protein domain of SUP35 has distinct effects on different variants of the [PSI+] prion in yeast. Curr. Genet. 35: 59–67. [DOI] [PubMed] [Google Scholar]
- Ferreira, P. C., F. Ness, S. R. Edwards, B. S. Cox and M. F. Tuite, 2001. The elimination of the yeast [PSI+] prion by guanidine hydrochloride is the result of Hsp104 inactivation. Mol. Microbiol. 40: 1357–1369. [DOI] [PubMed] [Google Scholar]
- Gietz, R. D., and R. A. Woods, 2002. Transformation of yeast by lithium acetate/single-stranded carrier DNA/polyethylene glycol method. Methods Enzymol. 350: 87–96. [DOI] [PubMed] [Google Scholar]
- Hara, H., T. Nakayashiki, C. G. Crist and Y. Nakamura, 2003. Prion domain interaction responsible for species discrimination in yeast [PSI+] transmission. Genes Cells 8: 925–939. [DOI] [PubMed] [Google Scholar]
- Horwich, A. L., and J. S. Weissman, 1997. Deadly conformations—protein misfolding in prion disease. Cell 89: 499–510. [DOI] [PubMed] [Google Scholar]
- King, C. Y., and R. Diaz-Avalos, 2004. Protein-only transmission of three yeast prion strains. Nature 428: 319–323. [DOI] [PubMed] [Google Scholar]
- Kochneva-Pervukhova, N. V., M. B. Chechenova, I. A. Valouev, V. V. Kushnirov, V. N. Smirnov et al., 2001. [PSI+] prion generation in yeast: characterization of the “strain” difference. Yeast 18: 489–497. [DOI] [PubMed] [Google Scholar]
- Krishnan, R., and S. L. Lindquist, 2005. Structural insights into a yeast prion illuminate nucleation and strain diversity. Nature 435: 765–772. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kryndushkin, D. S., I. M. Alexandrov, M. D. Ter-Avanesyan and V. V. Kushnirov, 2003. Yeast [PSI+] prion aggregates are formed by small Sup35 polymers fragmented by Hsp104. J. Biol. Chem. 278: 49636–49643. [DOI] [PubMed] [Google Scholar]
- Kushnirov, V. V., M. D. Ter-Avanesyan, S. A. Didichenko, V. N. Smirnov, Y. O. Chernoff et al., 1990. Divergence and conservation of SUP2 (SUP35) gene of yeast Pichia pinus and Saccharomyces cerevisiae. Yeast 6: 461–472. [DOI] [PubMed] [Google Scholar]
- Laemmli, U. K., 1970. Cleavage of structural proteins during the assembly of the head of bacteriophage T4. Nature 227: 680–685. [DOI] [PubMed] [Google Scholar]
- Liu, J. J., N. Sondheimer and S. L. Lindquist, 2002. Changes in the middle region of Sup35 profoundly alter the nature of epigenetic inheritance for the yeast prion [PSI+] Proc. Natl. Acad. Sci. USA 99(Suppl. 4): 16446–16453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Osherovich, L. Z., B. S. Cox, M. F. Tuite and J. S. Weissman, 2004. Dissection and design of yeast prions. PLoS Biol. 2: E86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parham, S. N., C. G. Resende and M. F. Tuite, 2001. Oligopeptide repeats in the yeast protein Sup35p stabilize intermolecular prion interactions. EMBO J. 20: 2111–2119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Patino, M. M., J. J. Liu, J. R. Glover and S. Lindquist, 1996. Support for the prion hypothesis for inheritance of a phenotypic trait in yeast. Science 273: 622–626. [DOI] [PubMed] [Google Scholar]
- Paushkin, S. V., V. V. Kushnirov, V. N. Smirnov and M. D. Ter-Avanesyan, 1996. Propagation of the yeast prion-like [PSI+] determinant is mediated by oligomerization of the SUP35-encoded polypeptide chain release factor. EMBO J. 15: 3127–3134. [PMC free article] [PubMed] [Google Scholar]
- Perutz, M. F., J. T. Finch, J. Berriman and A. Lesk, 2002. Amyloid fibers are water-filled nanotubes. Proc. Natl. Acad. Sci. USA 99: 5591–5595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prusiner, S. B., 1998. Prions. Proc. Natl. Acad. Sci. USA 95: 13363–13383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ross, E. D., H. K. Edskes, M. J. Terry and R. B. Wickner, 2005. Primary sequence independence for prion formation. Proc. Natl. Acad. Sci. USA 102: 12825–12830. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Safar, J., H. Wille, V. Itri, D. Groth, H. Serban et al., 1998. Eight prion strains have PrPSc molecules with different conformations. Nat. Med. 4: 1157–1165. [DOI] [PubMed] [Google Scholar]
- Salnikova, A. B., D. S. Kryndushkin, V. N. Smirnov, V. V. Kushnirov and M. D. Ter-Avanesyan, 2005. Nonsense suppression in yeast cells overproducing Sup35 (eRF3) is caused by its non-heritable amyloids. J. Biol. Chem. 280: 8808–8812. [DOI] [PubMed] [Google Scholar]
- Sambrook, J., E. E. Fritsch and T. Maniatis, 1989. Molecular Cloning: A Laboratory Manual, Ed 2. Cold Spring Harbor Laboratory Press, Cold Spring Harbor, NY.
- Santoso, A., P. Chien, L. Z. Osherovich and J. S. Weissman, 2000. Molecular basis of a yeast prion species barrier. Cell 100: 277–288. [DOI] [PubMed] [Google Scholar]
- Sherman, F., G. R. Fink and J. B. Hicks, 1986. Methods in Yeast Genetics. Cold Spring Harbor Laboratory Press, Cold Spring Harbor, NY.
- Ter-Avanesyan, M. D., V. V. Kushnirov, A. R. Dagkesamanskaya, S. A. Didichenko, Y. O. Chernoff et al., 1993. Deletion analysis of the SUP35 gene of the yeast Saccharomyces cerevisiae reveals two non-overlapping functional regions in the encoded protein. Mol. Microbiol. 7: 683–692. [DOI] [PubMed] [Google Scholar]
- Ter-Avanesyan, M. D., A. R. Dagkesamanskaya, V. V. Kushnirov and V. N. Smirnov, 1994. The SUP35 omnipotent suppressor gene is involved in the maintenance of the non-Mendelian determinant [psi+] in the yeast Saccharomyces cerevisiae. Genetics 137: 671–676. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tuite, M. F., C. R. Mundy and B. S. Cox, 1981. Agents that cause a high frequency of genetic change from [PSI+] to [psi−] in Saccharomyces cerevisiae. Genetics 98: 691–711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wickner, R. B., K. L. Taylor, H. K. Edskes, M. L. Maddelein, H. Moriyama et al., 2000. Prions of yeast as heritable amyloidoses. J. Struct. Biol. 130: 310–322. [DOI] [PubMed] [Google Scholar]
- Zhou, P., I. L. Derkatch, S. M. Uptain, M. M. Patino, S. Lindquist et al., 1999. The yeast non-Mendelian factor [ETA+] is a variant of [PSI+], a prion-like form of release factor eRF3. EMBO J. 18: 1182–1191. [DOI] [PMC free article] [PubMed] [Google Scholar]