

Abstract
Cryptic genetic variation accumulates under weakened selection and has been proposed as a source of evolutionary innovations. Weakened selection may, however, also lead to the accumulation of strongly deleterious or lethal alleles, swamping the effect of any potentially adaptive alleles when they are revealed. Here I model variation that is partially shielded from selection, assuming that unconditionally deleterious variation is more strongly deleterious than variation that is potentially adaptive in a future environment. I find that cryptic genetic variation can be substantially enriched for potential adaptations under a broad range of realistic parameter values, including those applicable to alternative splices and readthrough products generated by the yeast prion [PSI+]. This enrichment is dramatically stronger when multiple simultaneous changes are required to generate a potentially adaptive phenotype. Cryptic genetic variation is likely to be an effective source of useful adaptations at a time of environmental change, relative to an equivalent source of variation that has not spent time in a hidden state.
THERE has recently been much interest in the ability of evolutionary capacitors to tap into cryptic or hidden genetic variation and reveal a range of phenotypes (Rutherford and Lindquist 1998; True and Lindquist 2000; Queitsch et al. 2002; Bergman and Siegal 2003; Gibson and Dworkin 2004). The study of such variation goes back to Waddington's classic experiments on genetic assimilation (Waddington 1942, 1953, 1956). It was speculated that revealing and assimilating cryptic genetic variation might be a powerful force in evolution (Schmalhausen 1949; Waddington 1957). In particular, it has been speculated that adaptive combinations of mutations may appear more readily when variation is subject to weakened selection, and that these combinations of mutations may be relevant to the evolution of really novel adaptations (Koch 1972; True and Lindquist 2000; Harrison and Gerstein 2002). In contrast, Williams argued that genetic assimilation and related phenomena were unlikely to be of importance in providing for the rapid development of really novel adaptation, stating that “the arguments against hopeful monsters are equally valid whether the monstrosities are genetic or epigenetic in origin” (Williams 1966, p. 78).
Here I propose a scenario to refute Williams' claim. Consider a system in which mutations come in two distinct kinds. “Hopeless monster” mutations are unconditionally and strongly deleterious in all environments. “Hopeful monster” mutations, on the other hand, are also deleterious in most environments, but only weakly so, and may potentially be adaptive under some rare circumstances. Hopeful monster mutations, as defined here, are the raw material of evolutionary innovation in response to environmental change.
Note that the definitions of hopeful and hopeless monsters used here are not standard: they refer to the probability that a mutation will be adaptive, rather than to the size of the phenotypic effect of a mutation. When cryptic genetic variation is revealed, multiple mutations are revealed in various combinations. Even if the phenotypic effects of the individual mutations are small, in combination they may have a large cumulative phenotypic effect, producing individuals that are phenotypically monsters in the conventional sense (Goldschmidt 1940).
When hidden, selection on both kinds of mutant allele is muted by a constant factor, allowing both to accumulate. The number of hopeful monster alleles increases, but since many or all individuals also carry hopeless monster alleles, any potential adaptations in revealed variation may be swamped by strongly deleterious effects. I calculate the number of hopeful monster genotypes that appear in individuals lacking hopeless monster alleles as a function of the population size, the environmental change rate, the deleterious mutation rate, the strength of selection, and the number of hopeful monster alleles required in combination to generate an adaptation. This is compared to the number that would be present if variation were not cryptic, to calculate the extent to which cryptic genetic variation is enriched for viable hopeful monster genotypes.
OUTLINE OF MODEL
Consider, for mathematical simplicity, a haploid population of constant size N. Consider a class of hopeful monster mutations appearing with probability m per replication that may be adaptive in some environments, but are deleterious in all others with fitness 1 − s1. Assume an infinite number of sites for these mutations. When a mutant fixes, it is reclassified as wild type, and so fixation events are not tracked. The mean number of potentially adaptive alleles segregating in the population is then given by mNτ(N, s1), where τ(N, s) is the sojourn time as a function of N and s. τ(N, s) can be calculated for the Moran model according to Equation A2 in the appendix and is expressed in terms of number of generations.
Now consider the case when these alleles are partially shielded from selection, such that their fitness is given by 1 − s1s2. The shielding parameter s2 represents the penetrance of cryptic alleles, i.e., the phenotype corresponding to a given allele is present with probability s2. When cryptic variation is controlled by a single switch such as the yeast prion [PSI+] (True and Lindquist 2000), at least one component of s2 will be constant, in this case equal to the frequency of [PSI+] individuals in a population in the absence of environmental change. Here we assume that s2 is generally constant across sites.
Once variation is shielded, a larger number of potentially adaptive alleles will segregate, in principle increasing the availability of suitable phenotypic variation revealed at a time of environmental change. Working against this is the possibility that selection might also shield a second class of hopeless monster alleles from selection. These appear at rate U and are deleterious in all environments. For simplicity, assume that hopeless monsters are lethal when revealed and, according to the assumption of a constant penetrance factor, have fitness 1 − s2 when hidden. Accumulated lethal mutations may thwart a role for revelation in adaptation.
The probability that a lethal allele will go on to become fixed is given by pfix(N, s2). This probability can be calculated for the Moran model by using Equation A1 in the appendix. Assume that environmental change events occur with probability θ in each generation. The probability that no unconditionally deleterious mutant headed for fixation has appeared by the next environmental change event is then given by
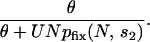 |
Note that this formula considers only the possibility that an unconditionally deleterious mutant fixed during the last environmental cycle. It neglects degradation that may occur over multiple cycles. Other work shows that this approximation has little effect, since fixation of unconditionally deleterious mutants is negligible unless U − s2 > θ (J. Masel and H. Maughan, unpublished results). This criterion is well captured even by the formula for a single cycle.
I make the conservative approximation that once a mutant headed for fixation has appeared, then fixation is essentially instant, such that there is no possibility for environmental change in the interim. Assume that when an environmental change event occurs, all individuals in the population reveal their cryptic genetic variation. We approximate environmental change as rare relative to the timescale of genetic drift and consider the case when no lethal has become fixed. There are now on average mNτ(N, s1s2) potentially adaptive alleles and UNτ*(N, s2) lethal alleles segregating in the population, where τ* indicates the sojourn time conditional on extinction rather than on fixation occurring. These formulas give a stochastic measure of mutation–selection balance. We approximate the probability distribution of the number of segregating deleterious and potentially adaptive alleles as Poisson.
Let the number of individuals free of all lethal alleles be x and the number of individuals carrying a particular potentially adaptive allele or combination of alleles be y. The probability that a particular potential adaptation appears in an individual free of unconditionally deleterious alleles is given by
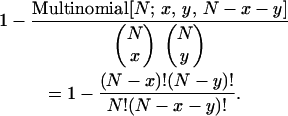 |
Assume that a particular set of j potentially adaptive alleles is segregating in the population and that in combination this set leads to an actual adaptation. Let 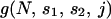 be the probability that this combination appears in at least one individual not carrying a lethal allele, given that no lethal mutant destined for fixation has appeared. We then have
be the probability that this combination appears in at least one individual not carrying a lethal allele, given that no lethal mutant destined for fixation has appeared. We then have
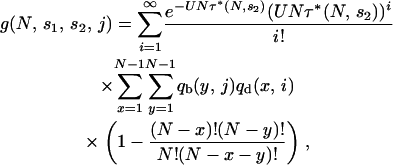 |
(1) |
where the first summation term corresponds to the Poisson probability distribution that i deleterious mutations segregate in the population, qd(x, i) is the probability that exactly x individuals will be free of all deleterious mutations, given i mutations segregating in the population, and qb(y, j) is the probability that exactly y individuals carry the adaptive combination of j alleles. The formula for τ* is given in the appendix by Equation A3, using results from the Moran model. Calculations for qd(x, i) and qb(y, j) are also described in the appendix.
The mean number of combinations of j alleles segregating in a population is given by
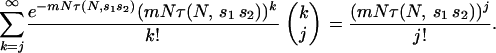 |
The mean number of potentially adaptive combinations of j alleles available for selection at a time of environmental change is then given by
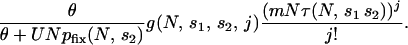 |
Normalizing this relative to the case when s2 = 1, we can see whether hiding and revealing variation increases or decreases the pool of variation available for adaptation. The normalized extent of enrichment is given by
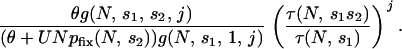 |
(2) |
The parameter m factors out during normalization, so we can determine which scenario is the case over the full range of parameters N, s1, s2, θ, and U.
RESULTS
In Figure 1A we see that the number of potential adaptations is highest for intermediate values of the hiding parameter s2. Figure 1B shows the same curve for combinations of two potentially adaptive alleles (j = 2) rather than for the appearance of a single allele. In this case, we see the same shaped curve, but dramatically larger values of enrichment. Enrichment is much greater again for j > 2 (not shown).
Figure 1.

The normalized extent to which cryptic genetic variation is enriched for potential adaptations, as calculated by Equation 2. N is the population size, U is the frequency per replication of unconditionally deleterious mutations, θ is the probability of environmental change per generation, s1 is the selection coefficient against potentially adaptive alleles in the ancestral environment, and s2 is the penetrance of alleles in the hidden state, representing the extent to which variation is shielded. (A) Enrichment is greatest for an intermediate level of shielding s2. Note that the curves for U = 10−2 and θ = 10−5 are almost superimposable. (B) A similar curve is seen when j = 2 alleles are required in combination for an adaptation, but in this case the magnitude of enrichment is greater. (C) For enrichment to occur at all, we need either θ > U (environmental change occurs more often than deleterious mutation in one individual) or s2N ≫ 1 (selection on strongly deleterious mutations is still appreciable in the hidden state). (D) The maximum extent of enrichment increases with s1N (the effectiveness of selection against nonhidden potential adaptative alleles), subject to an upper bound determined by the deleterious mutation rate U. (E) When j = 2 alleles are required in combination, very high levels of enrichment, such as 1000-fold, are easily possible. (F) Enrichment is typically greater for low levels of deleterious mutation. Note that this plot is arranged as a set of four curves for j = 1 plus a set of four curves for j = 2, and the inset refers to both sets.
Figure 1C shows the minimum value of s2 for which hiding variation increases the number of potential adaptations. For cryptic genetic variation to be enriched for potential adaptations, either U < θ or s2N ≫ 1 must be satisfied. The former condition is not appropriate, since we have assumed that θ is sufficiently rare such that variation has had time to accumulate between environmental change events. Note, however, that when j > 1 this condition is significantly relaxed and may become applicable. The latter condition corresponds to selection on hidden lethals being appreciable and is always a sufficient condition.
In Figure 1, D and E, we look at the extent to which hidden variation can be enriched for potential adaptations. This is assessed in a consistent way by taking the largest fold change in potential adaptations as s2 is allowed to vary. Actual enrichments will be lower and may vary across categories of sites if s2 is not constant. In most cases, however, enrichments at other values of s2 are strongly correlated to the optimal enrichment, as can be seen by the way curves shift in Figure 1, A and B.
We see in Figure 1D that for j = 1 the maximal extent of enrichment depends primarily on s1N, which corresponds to the effectiveness of selection on unhidden potentially adaptive alleles. This means that large populations generally have the greatest enrichment. This increase continues until s1N ≫ 1/U, at which point the extent of enrichment levels off. We see in Figure 1E that for j = 2 we get dramatically higher levels of enrichment, with leveling off occurring later.
In Figure 1F we see that enrichment is lower for higher rates of deleterious mutation U, but that this effect is smaller than that of s1N or j.
In summary, not only is cryptic genetic variation not significantly eroded by the accumulation of deleterious mutations, but instead it is positively enriched for potential adaptations. The minimum condition for enrichment is that selection on hidden lethals is appreciable, and the optimal condition is when selection on unhidden potentially adaptive alleles is highly effective. Enrichment is weak with respect to potential adaptations resulting from a single mutation, but is dramatic for potential adaptations based on a combination of mutations. This provides a powerful mechanism for achieving an adaptation involving multiple mutations when each mutation, taken by itself, is deleterious. This gives a rigorous basis to previous speculations that adaptive combinations of mutations may appear more readily when variation is subject to weakened selection (Koch 1972; True and Lindquist 2000; Harrison and Gerstein 2002). Recent models of the rate of obtaining an adaptive combination of mutations (Behe and Snoke 2004; Lynch 2005) are therefore substantial underestimates, since they do not take this enrichment into account.
DISCUSSION
Cryptic genetic variation is optimized by a “Goldilocks” level of selection in the hidden state: not too heavy and not too light. Is there empirical evidence both that cryptic genetic variation is under weakened selection and that some level of selection, however weak, remains? I look at two evolutionary capacitance systems that tap known pools of cryptic genetic variation, namely the yeast prion [PSI+] and alternative splicing.
The yeast prion [PSI+]:
The [PSI+] prion consists of aggregates of the Sup35 protein (Paushkin et al. 1996), a translation termination factor (Stansfield et al. 1995; Zhouravleva et al. 1995). The prion state is epigenetically inherited, with rare switching events between the prion and nonprion states (Wickner et al. 1995). When the prion appears, Sup35 is depleted due to its incorporation into [PSI+] aggregates, and so the extent of readthrough translation increases from a baseline of ∼5–10% to 30–40% (Bidou et al. 2000). In this way, the yeast prion [PSI+] reveals cryptic genetic variation beyond stop codons by impairing translation termination (True and Lindquist 2000; True et al. 2004; Wilson et al. 2005).
It is clear how stop codons shield cryptic genetic variation, but how much selection remains in the hidden state? Multiple stop codons occur in tandem more often than expected by chance (Liang et al. 2005), proving that selection on translation beyond stop codons is strong enough to affect evolution. More quantitatively, selection on hidden variation could occur either via a normal level of leakiness in translation termination or by occasional short-lived revelation events. The prion spontaneously appears with a frequency of ∼10−6 per replication (Lund and Cox 1981; Liu and Lindquist 1999; Nakayashiki et al. 2001). This means that all hidden variation is expressed with a probability of 10−6 and so gives a lower bound for s2. The effective population size of yeast is ∼107–108 (Lynch and Conery 2003; Wagner 2005). The condition s2N ≫ 1 seems to be minimally satisfied. In addition, baseline leakiness in translation termination and persistence of the [PSI+] state for more than one generation would increase s2N further. Since revelation potentially affects each of the ∼5000 genes in the genome, U and m are potentially quite high. Combined with high population size, it seems likely that U rather than s1N sets the limit on enrichment. High m increases the absolute number of potentially adaptive alleles segregating, although it has no impact on the extent to which they are enriched by shielding.
Alternative splicing:
Alternative splicing may affect as many as three-quarters of all human genes (Johnson et al. 2003; Kampa et al. 2004). Alternative splicing may sometimes represent a regulated mechanism of switching between states, and it may also introduce diversity via aberrant splicing or “noise” (Sorek et al. 2004). Alternative splicing can create novel isoforms during evolution by the insertion of new, functional protein sequences (Kondrashov and Koonin 2003) and is associated with a large increase in the frequency of recent exon creation and/or loss (Modrek and Lee 2003). It has been proposed that this is due to relaxed selection, leaving weakly expressed minor forms free to evolve rapidly in their cryptic state (Modrek and Lee 2003).
Alternatively spliced isoforms have a much higher frequency of premature termination codons than major transcript forms (Xing and Lee 2004), showing that alternatively spliced isoforms are subject to weakened selection. The extent to which selection is weakened is comparable to the effect of diploidy. Nevertheless, a detectable level of selection remains: alternative splices tend to insert or delete complete protein domains rather than disrupt structural modules (Kriventseva et al. 2003). Also, exons are biased toward exact multiples of three nucleotides, thus preserving the protein reading frame in both exon-inclusion and exon-skip splice forms (Resch et al. 2004). In addition, exons with low expression have Ka/Ks ratios closer to, but not equal to unity, relative to constitutively expressed exons (Xing and Lee 2005). Taken together, the evidence supports an intermediate level of selection on cryptic genetic variation.
Relaxing the assumption of two mutation types:
For mathematical simplicity, we have assumed that mutations fall into two categories, either unconditionally lethal under all circumstances or mildly deleterious under most circumstances but potentially adaptive on rare occasions. The key feature captured by this model is a correlation between the degree to which a mutation is deleterious in the ancestral environment and the probability that it will be adaptive in a new environment. Although for practical reasons it is not clear how to model a more realistic range of mutation types, it seems likely that any system that retains this key correlation will generate similar results to those described here. This correlation is critical, but seems highly plausible.
Preadaptation:
The enrichment of cryptic genetic variation for potential adaptations has implications for the concept of preadaptation. A preadaptation is a trait that evolved for one function but that is easily able to assume a new function when the need arises (Bock 1959). The problem with this concept of preadaptation is that the very word seems to imply that evolution somehow has foresight as to what will be adaptive in the future and directs evolution accordingly in advance. For this reason Gould and Vrba (1982) suggested the term exaptation instead. Exaptation describes how a trait originally adapted for one task can be coopted to perform another. By this scheme, “what we now incorrectly call ‘preadaptation’ is merely a category of exaptation considered before the fact” (Gould and Vrba 1982, p. 11). In certain cases, such as the uses of repetitive DNA, neutral or cryptic variation that Gould called a “nonaptation” may also be coopted through a process of exaptation.
Although we can never tell for certain what will be adaptive, in many cases we can make statements about the relative likelihood of being adaptive. Adaptation is always relative to the niche for which one is adapted. Nevertheless, some variation is self-evidently deleterious, irrespective of the environment. A stock of variation in which such self-evidently deleterious traits have been screened out can be regarded as preadapted. Even without the foresight of knowing exactly which adaptation will be needed in the future, we can still say that a population that is preadapted in this way is more likely to successfully adapt to a new environment than one that is not. When a trait previously adapted for one purpose is coopted for another, self-evidently deleterious variation is selected against throughout the evolutionary history of the two traits. In the case of a phenotypically invisible nonaptation, this screening process is less obvious and has been described here.
Eshel and Matessi argued that hidden variation is likely to be enriched in a direction that is adaptive in some spatial or temporal margin of the original environment (Eshel and Matessi 1998). Since environmental change events will frequently resemble some margin of the old environment, previously hidden variation is more likely to be adaptive than one would predict from an equivalent amount of variation generated de novo by mutation. They referred to this phenomenon as preadaptation, since selection in advance of the environmental change event at the population margin was increasing the likelihood of a successful adaptive response to environmental change. Here I have shown another scenario for the preadaptation of cryptic genetic variation, according to the same definition.
Standing genetic variation vs. new mutations:
Other work has assessed the circumstances under which adaptations are more likely to arise from standing genetic variation than from new mutations (Hermisson and Pennings 2005). The model presented here can be seen to modify this work, tipping the balance toward standing genetic variation. It has been shown here that cryptic sources can increase both the quantity and the quality of standing genetic variation. The model presented here can be seen as a new model of the variation present at mutation–selection balance.
Evolution of evolvability:
For cryptic genetic variation to be enriched, we need s2N ≫ 1, i.e., appreciable levels of preadapting selection. This sets the precondition for the evolution of evolutionary capacitance mechanisms that can tap into this variation (Masel and Bergman 2003). Indeed, once the preconditions are met, evolutionary capacitance can evolve quite readily, particularly in large populations, allowing the evolution of evolvability (Masel 2005). Note that this work does not imply that the level of shielding s2 evolves to optimize the number of potentially adaptive mutants. Selection on modifiers for evolvability is very weak and may not be relevant in a finite population. A modifier approach in a finite population, similar to that used for modifiers of revelation, could be used to assess whether selection on s2 modifiers could or has overcome random drift.
Acknowledgments
I acknowledge Lilach Hadany for helpful discussions, the editor Greg Gibson and two anonymous reviewers whose extensive comments helped improve the presentation of this work, and the BIO5 Institute for financial support.
APPENDIX
To calculate the sojourn times and the probability of an allele fixing by drift, I treat the dynamics of one locus and two alleles according to a Moran model. This means that at each time step, one individual is chosen at random to die and one to reproduce, and a number of important quantities are analytically accessible through the theory of branching Markov chain models (Ewens 2004). Specifically, consider a point in time in which i individuals have a mutant allele with fitness 1 − s. Neglecting recurrent and back mutation events since this is an infinite-sites model, the probability that the next individual chosen to reproduce has the mutant allele is given by
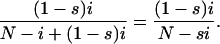 |
The probability that the next individual chosen to die has the mutant allele is given by i/N. The probability that the number of mutants increases from i by 1 is then given by the probability that a mutant individual is chosen to reproduce while a wild-type individual is chosen to die:
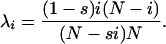 |
The probability that the number of mutants decreases from i by 1 is given by the probability that a wild-type individual is chosen to reproduce while a mutant individual is chosen to die:
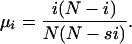 |
Following Ewens (2004), define
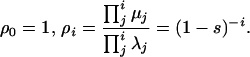 |
The probability of fixation by drift starting from i individuals is then
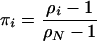 |
(Ewens 2004), and the probability of fixation by drift starting from a single mutant individual is
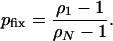 |
(A1) |
Then the sojourn time τi during which there are i descendants of a single original mutant is given by
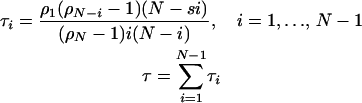 |
(A2) |
(Ewens 2004), where the unit of time is one generation or N rounds in the Moran model. The sojourn times conditional on elimination rather than on fixation by drift ultimately occurring are given by
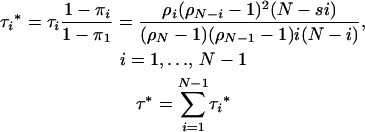 |
(A3) |
(Ewens 2004).
Note that calculation of g in Equation 1 scales with Ni+j+2, so in most cases g was calculated by randomly sampling probability distributions rather than by exhaustive enumeration of sums. First i was sampled, then x and y, and then the resulting final term was noted and averaged over 10,000–100,000 samples.
qd(x, i) is the probability that exactly x individuals are free of all deleterious mutations, given i mutations segregating in the population. To sample from this probability distribution, let the number of individuals with the nth mutation be xn. Next, x1 and x2 were randomly sampled according to 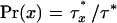 . Next, the number of individuals carrying neither mutation was randomly sampled. k1,2 was set with an initial value of N − x1 and decremented k1,2 by 1 with probability k1,2/N, k1,2/(N − 1), … , k1,2/(N − x2 + 1) for a total of x2 iterations. Then a value for x3 was randomly generated, and the process was repeated to sample k1,2,3 from k1,2 and x3 in the same way. This process was continued either until some value of k was decremented to zero or until a value for k1,2,…,i was achieved as the answer. The probability qb(y, j) that exactly y individuals carry all j potentially adaptive mutations is sampled in an analogous way.
. Next, the number of individuals carrying neither mutation was randomly sampled. k1,2 was set with an initial value of N − x1 and decremented k1,2 by 1 with probability k1,2/N, k1,2/(N − 1), … , k1,2/(N − x2 + 1) for a total of x2 iterations. Then a value for x3 was randomly generated, and the process was repeated to sample k1,2,3 from k1,2 and x3 in the same way. This process was continued either until some value of k was decremented to zero or until a value for k1,2,…,i was achieved as the answer. The probability qb(y, j) that exactly y individuals carry all j potentially adaptive mutations is sampled in an analogous way.
Note that even with this sampling scheme, calculations are prohibitively slow for large values of N. Fortunately, smooth curves with systematic trends were obtained with values of N up to 104, giving confidence in the extrapolation of those trends to larger values of N.
References
- Behe, M. J., and D. W. Snoke, 2004. Simulating evolution by gene duplication of protein features that require multiple amino acid residues. Protein Sci. 13: 2651–2664. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bergman, A., and M. L. Siegal, 2003. Evolutionary capacitance as a general feature of complex gene networks. Nature 424: 549–552. [DOI] [PubMed] [Google Scholar]
- Bidou, L., G. Stahl, I. Hatin, O. Namy, J. P. Rousset et al., 2000. Nonsense-mediated decay mutants do not affect programmed-1 frameshifting. RNA 6: 952–961. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bock, W. J., 1959. Preadaptation and multiple evolutionary pathways. Evolution 13: 194–211. [Google Scholar]
- Eshel, I., and C. Matessi, 1998. Canalization, genetic assimilation and preadaptation: a quantitative genetic model. Genetics 149: 2119–2133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ewens, W. J., 2004. Mathematical Population Genetics I. Theoretical Introduction. Springer-Verlag, New York.
- Gibson, G., and I. Dworkin, 2004. Uncovering cryptic genetic variation. Nat. Rev. Genet. 5: 681–690. [DOI] [PubMed] [Google Scholar]
- Goldschmidt, R., 1940. The Material Basis of Evolution. Yale University Press, New Haven, CT.
- Gould, S. J., and E. S. Vrba, 1982. Exaptation—a missing term in the science of form. Paleobiology 8: 4–15. [Google Scholar]
- Harrison, P. M., and M. Gerstein, 2002. Studying genomes through the aeons: protein families, pseudogenes and proteome evolution. J. Mol. Biol. 318: 1155–1174. [DOI] [PubMed] [Google Scholar]
- Hermisson, J., and P. S. Pennings, 2005. Soft sweeps: molecular population genetics of adaptation from standing genetic variation. Genetics 169: 2335–2352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnson, J. M., J. Castle, P. Garrett-Engele, Z. Y. Kan, P. M. Loerch et al., 2003. Genome-wide survey of human alternative pre-mRNA splicing with exon junction microarrays. Science 302: 2141–2144. [DOI] [PubMed] [Google Scholar]
- Kampa, D., J. Cheng, P. Kapranov, M. Yamanaka, S. Brubaker et al., 2004. Novel RNAs identified from an in-depth analysis of the transcriptome of human chromosomes 21 and 22. Genome Res. 14: 331–342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koch, A. L., 1972. Enzyme evolution. I. Importance of untranslatable intermediates. Genetics 72: 297–316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kondrashov, F. A., and E. V. Koonin, 2003. Evolution of alternative splicing: deletions, insertions and origin of functional parts of proteins from intron sequences. Trends Genet. 19: 115–119. [DOI] [PubMed] [Google Scholar]
- Kriventseva, E. V., I. Koch, R. Apweiler, M. Vingron, P. Bork et al., 2003. Increase of functional diversity by alternative splicing. Trends Genet. 19: 124–128. [DOI] [PubMed] [Google Scholar]
- Liang, H., A. R. O. Cavalcanti and L. F. Landweber, 2005. Conservation of tandem stop codons in yeasts. Genome Biol. 6: R31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu, J. J., and S. Lindquist, 1999. Oligopeptide-repeat expansions modulate ‘protein-only’ inheritance in yeast. Nature 400: 573–576. [DOI] [PubMed] [Google Scholar]
- Lund, P. M., and B. S. Cox, 1981. Reversion analysis of [psi−] mutations in Saccharomyces cerevisiae. Genet. Res. 37: 173–182. [DOI] [PubMed] [Google Scholar]
- Lynch, M., 2005. Simple evolutionary pathways to complex proteins. Protein Sci. 14: 2217–2225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lynch, M., and J. S. Conery, 2003. The origins of genome complexity. Science 302: 1401–1404. [DOI] [PubMed] [Google Scholar]
- Masel, J., 2005. Evolutionary capacitance may be favored by natural selection. Genetics 170: 1359–1371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Masel, J., and A. Bergman, 2003. The evolution of the evolvability properties of the yeast prion [PSI+]. Evolution 57: 1498–1512. [DOI] [PubMed] [Google Scholar]
- Modrek, B., and C. J. Lee, 2003. Alternative splicing in the human, mouse and rat genomes is associated with an increased frequency of exon creation and/or loss. Nat. Genet. 34: 177–180. [DOI] [PubMed] [Google Scholar]
- Nakayashiki, T., K. Ebihara, H. Bannai and Y. Nakamura, 2001. Yeast [PSI+] “prions” that are crosstransmissible and susceptible beyond a species barrier through a quasi-prion state. Mol. Cell 7: 1121–1130. [DOI] [PubMed] [Google Scholar]
- Paushkin, S. V., V. V. Kushnirov, V. N. Smirnov and M. D. Ter-Avanesyan, 1996. Propagation of the yeast prion-like [PSI+] determinant is mediated by oligomerization of the SUP35-encoded polypeptide chain release factor. EMBO J. 15: 3127–3134. [PMC free article] [PubMed] [Google Scholar]
- Queitsch, C., T. A. Sangster and S. Lindquist, 2002. Hsp90 as a capacitor of phenotypic variation. Nature 417: 618–624. [DOI] [PubMed] [Google Scholar]
- Resch, A., Y. Xing, A. Alekseyenko, B. Modrek and C. Lee, 2004. Evidence for a subpopulation of conserved alternative splicing events under selection pressure for protein reading frame preservation. Nucleic Acids Res. 32: 1261–1269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rutherford, S. L., and S. Lindquist, 1998. Hsp90 as a capacitor for morphological evolution. Nature 396: 336–342. [DOI] [PubMed] [Google Scholar]
- Schmalhausen, I. I., 1949. Factors of Evolution. Blakiston, Philadelphia.
- Sorek, R., R. Shamir and G. Ast, 2004. How prevalent is functional alternative splicing in the human genome? Trends Genet. 20: 68–71. [DOI] [PubMed] [Google Scholar]
- Stansfield, I., K. M. Jones, V. V. Kushnirov, A. R. Dagkesamanskaya, A. I. Poznyakovski et al., 1995. The products of the sup45 (eRF1) and sup35 genes interact to mediate translation termination in Saccharomyces cerevisiae. EMBO J. 14: 4365–4373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- True, H. L., and S. L. Lindquist, 2000. A yeast prion provides a mechanism for genetic variation and phenotypic diversity. Nature 407: 477–483. [DOI] [PubMed] [Google Scholar]
- True, H. L., I. Berlin and S. L. Lindquist, 2004. Epigenetic regulation of translation reveals hidden genetic variation to produce complex traits. Nature 431: 184–187. [DOI] [PubMed] [Google Scholar]
- Waddington, C. H., 1942. Canalization of development and the inheritance of acquired characters. Nature 150: 563–565. [Google Scholar]
- Waddington, C. H., 1953. Genetic assimilation of an acquired character. Evolution 7: 118–126. [Google Scholar]
- Waddington, C. H., 1956. Genetic assimilation of the bithorax phenotype. Evolution 10: 1–13. [Google Scholar]
- Waddington, C. H., 1957. The Strategy of the Genes. George Allen & Unwin, London.
- Wagner, A., 2005. Energy constraints on the evolution of gene expression. Mol. Biol. Evol. 22: 1365–1374. [DOI] [PubMed] [Google Scholar]
- Wickner, R. B., D. C. Masison and H. K. Edskes, 1995. [PSI] and [URE3] as yeast prions. Yeast 11: 1671–1685. [DOI] [PubMed] [Google Scholar]
- Williams, G. C., 1966. Adaptation and Natural Selection. Princeton University Press, Princeton, NJ.
- Wilson, M. A., S. Meaux, R. Parker and A. van Hoof, 2005. Genetic interactions between [PSI+] and nonstop mRNA decay affect phenotypic variation. Proc. Natl. Acad. Sci. USA 102: 10244–10249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xing, Y., and C. J. Lee, 2004. Negative selection pressure against premature protein truncation is reduced by alternative splicing and diploidy. Trends Genet. 20: 472–475. [DOI] [PubMed] [Google Scholar]
- Xing, Y., and C. Lee, 2005. Evidence of functional selection pressure for alternative splicing events that accelerate evolution of protein subsequences. Proc. Natl. Acad. Sci. USA 102: 13526–13531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhouravleva, G., L. Frolova, X. Legoff, R. Leguellec, S. Inge-Vechtomov et al., 1995. Termination of translation in eukaryotes is governed by two interacting polypeptide-chain release factors, eRF1 and eRF3. EMBO J. 14: 4065–4072. [DOI] [PMC free article] [PubMed] [Google Scholar]