

Abstract
The initiation of growth cessation and dormancy represents a critical ecological and evolutionary trade-off between survival and growth in most forest trees. The most important environmental cue regulating the initiation of dormancy is a shortening of the photoperiod and phytochrome genes have been implicated in short-day-induced bud set and growth cessation in Populus. We characterized patterns of DNA sequence variation at the putative candidate gene phyB2 in 4 populations of European aspen (Populus tremula) and scored single-nucleotide polymorphisms in an additional 12 populations collected along a latitudinal gradient in Sweden. We also measured bud set from a subset of these trees in a growth chamber experiment. Buds set showed significant clinal variation with latitude, explaining ∼90% of the population variation in bud set. A sliding-window scan of phyB2 identified six putative regions with enhanced population differentiation and four SNPs showed significant clinal variation. The clinal variation at individual SNPs is suggestive of an adaptive response in phyB2 to local photoperiodic conditions. Three of four SNPs showing clinal variation were located in regions with excessive genetic differentiation, demonstrating that searching for regions of high genetic differentiation can be useful for identifying sites putatively involved in local adaptation.
THE initiation of growth cessation and dormancy late in the growing season represents a critical ecological and evolutionary trade-off between survival and growth in most forest trees (Horvath et al. 2003; Howe et al. 2003). Many forest trees therefore show latitudinal clines in important phenological traits related to the annual development cycle and this adaptive population differentiation has occurred despite high levels of gene flow among populations as evidenced by low levels of population differentiation at neutral molecular markers in many forest trees (Wright 1976; Adams et al. 1992; Dvornyk et al. 2002; Garcia-Gil et al. 2003; Howe et al. 2003). Dormancy is a prerequisite for the development of cold hardiness and the developmental processes leading up to complete endodormancy take several weeks to complete, thereby reducing the length of the season during which active growth can take place (Horvath et al. 2003; Howe et al. 2003). Although several factors play a role, evidence suggests that the most important environmental cues regulating the initiation of dormancy in perennial plants are a shortening of the photoperiod and exposure to extended periods of low, nonfreezing temperatures (Howe et al. 1995; Li et al. 2002; Horvath et al. 2003).
Dormancy-related traits have been shown to be under strong genetic control (e.g., Eriksson et al. 1978; Faust et al. 1997; Frewen et al. 2000; Howe et al. 2000, 2003) and the genetic architecture of dormancy traits has been investigated in many forest trees, using pedigree studies (e.g., Bradshaw and Stettler 1995; Frewen et al. 2000; Howe et al. 2000; Chen et al. 2002). However, functional dissection of adaptive traits through the development of segregating mapping populations is hampered by the long generation times of trees (e.g., Brunner et al. 2004; Neale and Savolainen 2004). An alternative approach is to investigate patterns of genetic variation in natural populations to detect possible statistical associations between phenotypic traits and genetic markers segregating in a population, so-called association mapping (Gaut and Long 2003; Neale and Savolainen 2004). Association studies involve detecting coinheritance between phenotypes and QTL that have persisted over many generations and the resolution of such studies is therefore primarily determined by the extent of linkage disequilibrium (LD) and the level of polymorphism in the regions surveyed (Gaut and Long 2003). In outbreeding species, such as most forest trees, LD usually extends only a couple of hundred to a few thousand bases, whereas in selfing organisms, such as Arabidopsis thaliana, LD can extend up to 250 kb or more (Flint-García et al. 2003; Brown et al. 2004; Ingvarsson 2005). Because of low LD in most tree populations, associations will be detected only when a marker locus is tightly linked to a QTL of interest (Gaut and Long 2003). Genomewide association studies therefore would require a substantial number of markers to reliability detect most of the important QTL for a given trait and such approaches would thus be both time consuming and prohibitively expensive in most forest tree species. This is particularly true for conifers that are hampered by extremely large genomes (Garcia-Gil et al. 2003). An alternative approach is to search for phenotype-marker associations in a set of candidate genes, i.e., genes with functions, positions, and/or patterns of gene expression that are known or suspected to be associated with the trait of interest.
In poplars (Populus spp.), phytochrome genes have been implicated in short-day-induced bud set and growth cessation (Populus trichocarpa) (Howe et al. 1996, 1998). Phytochromes are photoreceptor proteins that respond to red (R) and far-red (FR) light and are known to play an important role in detecting photoperiod cues (Smith 2000). Individual phytochromes appear to have adopted different functions and mutational analysis of the five phytochrome genes of Arabidopsis has shown that although some of these genes have overlapping functions, some (in particular, phyA) regulate processes that are not affected by other phytochromes (Smith 2000). Populus has three phytochrome genes, phyA, phyB1, and phyB2 (Howe et al. 1998, confirmed by inspection of the complete genome sequence of P. trichocarpa). One of these, phyB2, has been mapped to a linkage group containing QTL for bud set and bud flush in several replicated experiments using independent mapping populations (Frewen et al. 2000; Chen et al. 2002). We consider QTL data from natural populations to be a strong argument for causal relationships and have therefore chosen phyB2 as the most likely candidate for regulating dormancy-related traits in Populus. To further elucidate the genetic basis of dormancy-related traits and their possible roles in adaptation to the length of the growing season in Populus, we have characterized patterns of DNA sequence variation at phyB2 in samples from four populations of European aspen (P. tremula). Single-nucleotide polymorphisms (SNPs) identified from the complete phyB2 sequences were scored in an expanded set of populations collected along a latitudinal gradient in Sweden. In addition, we measured day-length-induced growth cessation and bud set in a subset of these trees in a growth chamber experiment.
We have two aims with our study. First, we are interested in estimating levels of nucleotide diversity and LD in phyB2. Populus has become the de facto tree model species (Wullschleger et al. 2002) and extensive genomic resources are available [complete genome sequence, extensive EST resources (Sterky et al. 2004), and high-density microarrays (Andersson et al. 2004)]. However, to utilize these resources in analyses of natural variation, more data are needed on the amount of sequence diversity and linkage disequilibrium in natural populations of Populus. An earlier study (Ingvarsson 2005) has shown that P. tremula is highly polymorphic and that LD declines rapidly with physical distance but it is not known how general these observations are. Second, we are interested in searching for signs of adaptive differentiation among populations originating from different latitudes. Local selection is expected to enhance genetic diversity at neutral sites surrounding a site maintained by local selection (Charlesworth et al. 1997) and it is conceivable that regions involved in local adaptation can be identified by having exceptionally high population differentiation. We therefore scanned the phyB2 gene for signs of enhanced genetic differentiation among populations and searched for clinal variation at individual SNPs.
MATERIALS AND METHODS
Plant material:
Leaf material was sampled from 24 naturally occurring trees of P. tremula at four different sites throughout Europe in 2002 and 2003. Samples were taken within a few kilometers of Besacon in eastern France, Klagenfurt in southern Austria, Färjestaden in southeastern Sweden, and Umeå in northern Sweden. Three to four young and undamaged leaves were collected from each tree, dried in a silica gel, and stored at room temperature until DNA extraction.
In 2003 we established a common garden consisting of trees collected from 12 sites sampled along a latitudinal cline (55.9°N–66.0°N) in Sweden. From each site, 10 unique tree genotypes were collected (with the exception of one site from which only 6 genotypes were collected), for a total of 116 trees. Since aspen has clonal growth, sampled trees were separated by at least 2 km. Trees were also marked in the field to allow future verification and additional collection of materials. The full data set describing the sampled populations is found in supplemental Table 1 at http://www.genetics.org/supplemental/ and a map of the geographic origin of the populations is shown in Figure 1. Root stocks were dug up from each tree and brought to the Forestry Research Institute of Sweden's (Skogforsk) research station Ekebo in Skåne, southern Sweden. The root stocks were placed in peat moss and allowed to sprout new shoots. Leaf material was collected from all trees, flash-frozen in liquid nitrogen, and stored at −80° until DNA extraction. At least 10–15 shoots per genotype were planted individually in pots and overwintered in a cold greenhouse. We refer collectively to these tree genotypes as the Swedish Aspen (SwAsp) collection.
Figure 1.

Map showing the location of the 12 populations in the SwAsp collection.
DNA extraction, PCR amplification, and sequencing:
Total genomic DNA was extracted from 24 individuals collected at four sites throughout Europe. DNA was extracted from dried or frozen leaf tissue, using the DNeasy plant mini prep kit (QIAGEN, Valencia, CA). Primers to amplify the P. tremula phyB2 gene were designed on the basis of BLAST searches of PopulusDB (http://poppel.fysbot.umu.se; Sterky et al. 2004), using homologs from P. trichocarpa (GenBank accession no. AF309807). All primer sequences are available from the authors upon request. All PCR products were cloned into the pCR2.1 vector, using a TA-cloning kit from Invitrogen (Carlsbad, CA). Fragments were sequenced using BigDye chemistry (Applied Biosystems, Foster City, CA) on an ABI377 automated sequencer at the Umeå Plant Science Centre sequencing facility. Several clones of each fragment were sequenced (at least three per allele) to identify the presence of multiple haplotypes within individuals and to control for Taq polymerase errors. Sequences were verified manually and contigs were assembled using the computer program Sequencher v 4.0. Multiple sequence alignments were made using Clustal W (Thompson et al. 1994) and adjusted manually using Bio Edit (http://www.mbio.ncsu.edu/BioEdit/bioedit.html). All sequences described in this article have been deposited in the EMBL database (accession nos. AM072290–AM072337).
Population genetic data analyses:
Estimates of nucleotide polymorphism and statistical tests of neutrality were obtained using the computer program DnaSP v4.00.5 (Rozas et al. 2003, http://www.ub.es/dnasp/) or Jody Hey's SITES program (http://lifesci.rutgers.edu/∼heylab/HeylabSoftware.htm#SITES). Linkage disequilibrium was scored between pairs of polymorphic sites, using the squared allele frequency correlations, r2 (Weir 1990). We calculated total genetic diversity, πT and the absolute levels of population differentiation, πT−S (Charlesworth 1998) across phyB2, using a sliding-window approach in an attempt to identify regions with enhanced levels of population subdivision. We chose πT−S as our measure of genetic differentiation since it is not influenced by within-population genetic diversity to the same degree as FST is (Charlesworth 1998). Local selection is expected to yield a peak in the between-population component of genetic diversity, located at or close to the sites(s) that are under local selection (Charlesworth et al. 1997). To determine which window segments showed enhanced levels of genetic differentiation, we used coalescent simulation of an infinite-island model, to estimate the expected range of πT−S for each window. We simulated the same number of alleles that were present in the four populations sampled (10, 12, 12, and 14, for a total of 48). Simulations were run conditional on the number of segregating sites in each window. The migration parameter, M = 4Nm, was set so that the expected FST matched the estimate of FST across the entire phyB2 gene. We also included recombination, by including the per base pair recombination rate estimated using the method of Hey and Wakeley (1997) (ρ = 0.076). This estimator of the recombination rate is known to be downwardly biased (Hudson 2001) and thus provides a conservative estimate of the recombination rate. Since LD extends only a few hundred base pairs in P. tremula (Ingvarsson 2005), signals of diversifying selection are expected to affect only small genomic regions of phyB2. In our sliding-window analyses we used a fixed window size of 250 with a step size of 20 bp; smaller window sizes yielded too little polymorphism in many windows and larger windows run the risk of averaging over too many sites with independent evolutionary histories, thereby reducing the power to detect regions with enhanced genetic differentiation. We regard the sliding-window analysis as exploratory and as a way to identify regions worth studying in greater detail and we therefore do not correct for multiple testing in the analysis.
Clinal variation at individual SNPs:
Five amino acid polymorphisms having a minor allele frequency >0.1 and an additional noncoding SNP were selected for further analysis. The frequencies of these six SNPs were scored in the SwAsp collection either by developing cleaved amplified polymorphism sequence (CAPS) markers (Konieczny and Ausubel 1993) or by direct sequencing of fragments containing the putative SNPs. We developed CAPS markers for two nonsynonymous mutations located in exon 1 (cut by BseYI and BccI), one nonsynonymous mutation in exon 2 (cut by NcoI), and one site in intron 3 (cut by ApaLI). The latter site differentiated between the two divergent haplotypes found in intron 3 (see results). The remaining two nonsynonymous SNPs were scored by complete sequencing of exon 4 (237 bp). In the process of scoring these SNPs we also detected an additional three SNPs that were segregating at low frequency (<0.1) in the SwAsp collection and these SNPs were included in the later analyses. Since SNPs in exon 4 were scored from PCR amplifications of genomic DNA, heterozygous sites were hence visible as double peaks in the chromatograms generated by the automated sequencer. To test for clinal variation we estimated the statistical fit of a linear regression of arcsine-transformed allele frequencies against latitude of origin for the 12 populations in the SwAsp collection and the 4 populations from which complete sequences were available using the statistical package R 2.0.1 (R Development Core Team 2004).
Growth chamber experiments:
We scored critical day-length-induced growth cessation for a subset of trees in the SwAsp collection. Following a period of growth in the greenhouse a subset of potted trees from the SwAsp collection was kept under “winter” conditions in a cold chamber (4° for 30 days followed by 30 days at −5°). They were then placed in a greenhouse under “spring” conditions (20° temperature, 23-hr photoperiod) to induce bud flush. After 28 days the plants were transferred to four growth chambers with a 23-hr photoperiod, 20° constant temperature, and irradiance between 400 and 600 mol m−2 sec−1. Clones from different areas were equally divided between the chambers and randomized inside each chamber. Sixty-nine different genotypes were included in the experiment: 47 were represented by a single tree and 22 by two replicas (91 trees in total). Some trees were excluded from the experiment because of a spider mite infestation in two of the growth chambers. After 12 days in the chamber, the photoperiod was reduced by 1 hr per week until all the healthy branches of all clones had set terminal buds. The photoperiod inducing bud set and the time until bud set (expressed as days since the plants were placed under spring conditions in the greenhouse) were recorded for each clone. Variation among clones and populations in the timing of growth cessation was analyzed using a linear mixed-effects model,
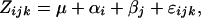 |
where Zijk is the phenotype of the kth individual from the jth clone and the ith population. The grand mean is denoted by μ and αi is the fixed population effect, βi is the random clone effect, and ɛijk is the residual error term. The model was fitted using a restricted maximum-likelihood method implemented in R 2.0.1 (R Development Core Team 2004).
RESULTS
Nucleotide polymorphism and linkage disequilibrium:
We obtained the complete coding sequence and all intervening introns of phyB2 from 48 haplotypes. The total aligned region, including indels, covered 6236 bp and we identified a total of 245 segregating sites. In addition, we identified 28 short (1–2 bp) and 5 longer (21–54 bp) indels that were polymorphic in the four study populations. All indels except one were located in introns; indels are ignored in all further analyses. Sequence diversity at phyB2 (πtot = 6.1 × 10−3 and θW = 9.3 × 10−3) was similar to that at other genes characterized from the same set of individuals (Ingvarsson 2005). Synonymous diversity was substantially higher than nonsynonymous diversity (πsil = 8.5 × 10−3 and πrepl = 3.0 × 10−3, respectively, Table 1), suggesting purifying selection at most codons. Synonymous and nonsynonymous site divergence from P. trichocarpa was 26.1 × 10−3 and 9.2 × 10−3, respectively, and there was no deviation from neutrality in the ratio of polymorphism to divergence at synonymous and nonsynonymous sites as judged by the McDonald–Kreitman test (P < 0.45) (McDonald and Kreitman 1991).
TABLE 1.
Estimates of nucleotide variation at phyB2
| No. of sites
|
Polymorphism (×10−3) (±SE)
|
|||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| Population | Alleles | Total | Synonymous | Nonsynonymous | S | Singletons | θW | πtot | πsil | πrepl |
| FRA | 10 | 6234 | 708.7 | 2726.5 | 116 | 66 | 6.74 | 5.96 (0.64) | 9.26 (0.80) | 1.82 (0.35) |
| AUT | 12 | 6235 | 710.3 | 2733.7 | 118 | 58 | 6.66 | 5.71 (0.56) | 8.42 (0.68) | 2.53 (0.37) |
| SWE S | 12 | 6236 | 709.0 | 2732.0 | 133 | 66 | 7.44 | 6.53 (0.60) | 10.09 (0.75) | 2.10 (0.34) |
| SWE N | 14 | 6236 | 707.0 | 2727.5 | 148 | 38 | 7.55 | 7.24 (0.58) | 9.29 (0.65) | 4.42 (0.45) |
| All | 48 | 6236 | 706.4 | 2725.0 | 245 | 75 | 9.31 | 6.08 (0.27) | 8.54 (0.32) | 3.03 (0.19) |
FRA, France; AUT, Austria; SWE S, southern Sweden; SWE N, northern Sweden.
Local populations contained a sizable fraction of the total variation (average πwithin = 6.36 × 10−3). This is also evident from the low genetic differentiation among populations (FST = 0.045), which is comparable to estimates from several other genes in P. tremula (Ingvarsson 2005). The frequency spectrum of segregating sites showed an excess of low-frequency polymorphisms as evidenced by negative DTajima and FFu&Li (Table 2). Interestingly, DTajima and FFu&Li calculated for the total sample are significantly negative where as DTajima and FFu&Li in the local populations are not, implying a greater excess of low-frequency polymorphisms in the total sample. This is also apparent from the larger number of segregating sites found in the pooled sample than in the within-population samples (Table 1). Even though the number of segregating sites is a function of sample size, the number of segregating sites in the total sample is greater than expected in a sample of that size.
TABLE 2.
Statistical tests of neutrality
| Population | DTajima | FFu&Li | ZnS |
|---|---|---|---|
| FRA | −0.574 | −0.916 | 0.153 |
| AUT | −0.565 | −0.852 | 0.127 |
| SWE S | −0.536 | −0.854 | 0.128 |
| SWE N | −0.176 | 0.296 | 0.118 |
| All | −1.259 | −1.162 | 0.038 |
See Table 1 legend for definitions of abbreviations.
Because of the length of the phyB2 gene and a high recombination rate (CHW = 0.076), all haplotypes in our sample are unique. The high recombination rate is also evident from low levels of LD (Table 2). However, average LD is substantially higher within local populations (Table 2), confirming earlier observations from other P. tremula genes (Ingvarsson 2005). The greater LD observed within local populations remains even if low-frequency polymorphisms are filtered out (data not shown), suggesting that the low LD seen in the specieswide sample is not simply an artifact of a larger number of low-frequency polymorphisms. Less extreme excesses of low-frequency polymorphisms and higher LD within local populations have also been observed at other loci in P. tremula (Ingvarsson 2005). The cause of these patterns remains unclear, although demographic processes, such as population expansion or population subdivision, are likely explanations (Ingvarsson 2005).
Sliding-window analyses:
Even though genetic differentiation was low at phyB2 on average, there is substantial variation across the gene region in how genetic diversity is partitioned within and among populations. The sliding-window analysis identified six putative regions where genetic differentiation appeared to be enhanced compared to neutral expectations (Figure 2, asterisks). All peaks are quite narrow and do not extend more than a few hundred base pairs. Most notable is a region in intron 3 that contains a very distinct peak of high genetic differentiation. This region contains two major haplotypes that differ at ∼20 bp. These haplotypes vary in frequency in the sampled populations with one haplotype being common in the two Swedish populations and the other haplotype being common in France and Austria. This peak also corresponds to a region showing high divergence from P. trichocarpa (Figure 2). The association between the degree of divergence between species and the excess population differentiation in this region is puzzling. The high divergence suggests an elevated mutation rate in this region and it is possible that the power of detecting genetic differentiation in this region is greater, simply because populations are more likely to have picked up mutations that differentiate them within this region.
Figure 2.

Sliding-window plot of genetic differentiation, measured as πT−S = πT − πS (Charlesworth 1998) (top), Tajima's D (middle), and divergence from P. trichocarpa and nucleotide diversity, πT (bottom). A window of width 250 bp was moved along the sequence in 20-bp increments and statistics were calculated for each window segment. In the top, the horizontal dashed line indicates the upper 95% confidence limit for πT−S obtained through coalescent simulations (see text for further details). Putative regions harboring significantly enhanced genetic differentiation are indicated by asterisks. Exon–intron structure of phyB2 is indicated by solid boxes (exons) and thin lines (introns) at the top. Locations of SNPs scored in the SwAsp collection are marked by solid circles.
We also performed sliding-window analyses on the five loci from Ingvarsson (2005). Data from these loci were collected from the same set of individuals from which phyB2 was sequenced. These loci are not thought to be involved in regulating bud set and are therefore not likely to be under diversifying selection. We did not find any sites that showed excess genetic differentiation at these five loci (supplemental Figure 2 at http://www.genetics.org/supplemental/), increasing the likelihood that the results seen at phyB2 are the result of diversifying selection in response to photoperiod.
Clinal variation in polymorphic sites in phyB2:
Frequencies of four SNPs (T608N, L789M, Int3, and L1078P) showed significant clinal variation with latitude (Table 3, Figure 3). If the data are restricted to the SwAsp populations only, two sites, L789M and L1078P, show significant clinal variation where as the other two approach significance (P < 0.1). Of the SNPs that show clinal variation, three (L789M, Int3, and L1078P) are located in regions we had previously identified as having significantly increased genetic differentiation in the original four-population sample. The only exception is the T608N mutation, which is located in a region (nucleotide site 1823) that appears to have a very low level of population differentiation. The SNP in intron 3 and the five SNPs in exon 4 all occur within 500 bp of each other. It is therefore not clear whether LD between these sites can explain why two sites in this region show significant latitudinal clines. We elaborate on this point in the discussion. We also inferred the ancestral state of each SNP, using a phyB2 sequence from P. trichocarpa. Interestingly, in all four SNPs with significant latitudinal clines it is the derived allele that increased in frequency with latitude (Figure 3).
TABLE 3.
Population frequencies of SNPs and test for clinal variation
| Polymorphisms
|
||||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| Populationa | Latitude | T608N | D645E | L789M | Int3 | L1078P | V1089I | M1090K | S1116N | F1117L |
| Klagenfurt | 46.4 | 0.333 | 0.417 | 0.167 | 0.083 | 0.083 | 1.000 | 0.250 | 1.000 | 0.000 |
| Besancon | 47.3 | 0.300 | 0.400 | 0.200 | 0.000 | 0.300 | 1.000 | 0.100 | 1.000 | 0.000 |
| Svalöv (1) | 55.9 | 0.833 | 0.278 | 0.444 | 0.167 | 0.500 | 0.778 | 0.500 | 0.889 | 0.111 |
| Ronneby (2) | 56.2 | 0.813 | 0.611 | 0.444 | 0.438 | 0.556 | 0.889 | 0.556 | 1.000 | 0.111 |
| Färjestaden | 56.7 | 0.500 | 0.333 | 0.750 | 0.500 | 0.500 | 1.000 | 0.333 | 1.000 | 0.167 |
| Ydre (4) | 57.4 | 0.650 | 0.400 | 0.389 | 0.400 | 0.800 | 0.900 | 0.450 | 1.000 | 0.100 |
| Vårgårda (3) | 58.0 | 0.700 | 0.350 | 0.450 | 0.350 | 0.650 | 0.750 | 0.500 | 1.000 | 0.100 |
| Brunnsberg (5) | 59.4 | 0.700 | 0.500 | 0.500 | 0.500 | 0.850 | 1.000 | 0.350 | 1.000 | 0.000 |
| Uppsala (6) | 59.8 | 0.850 | 0.150 | 0.550 | 0.250 | 0.650 | 0.900 | 0.300 | 0.850 | 0.100 |
| Älvdalen (7) | 61.2 | 0.800 | 0.350 | 0.611 | 0.350 | 0.750 | 0.850 | 0.600 | 1.000 | 0.000 |
| Delsbo (8) | 61.8 | 0.944 | 0.389 | 0.611 | 0.389 | 0.611 | 0.833 | 0.778 | 0.778 | 0.000 |
| Umeå (10) | 63.4 | 0.778 | 0.500 | 0.611 | 0.444 | 0.778 | 0.889 | 0.667 | 0.889 | 0.000 |
| Umeå | 63.5 | 0.786 | 0.286 | 0.786 | 0.857 | 0.857 | 0.929 | 0.286 | 1.000 | 0.000 |
| Dorotea (9) | 64.4 | 0.944 | 0.438 | 0.833 | 0.444 | 0.833 | 0.889 | 0.556 | 0.889 | 0.000 |
| Luleå (12) | 65.5 | 0.714 | 0.500 | 0.643 | 0.571 | 0.857 | 0.929 | 0.571 | 1.000 | 0.000 |
| Arjeplog (11) | 66.0 | 0.850 | 0.500 | 0.650 | 0.400 | 0.850 | 0.900 | 0.450 | 0.900 | 0.000 |
| FST | 0.181 | 0.052 | 0.143 | 0.170 | 0.219 | 0.065 | 0.122 | 0.109 | 0.085 | |
| ρlat | 0.802*** | 0.033 | 0.890*** | 0.724** | 0.895*** | 0.327 | 0.357 | 0.291 | 0.211 | |
**P < 0.01, ***P < 0.001.
Numbers in parentheses refer to locations shown in Figure 1.
Figure 3.

Population frequencies for nine SNPs scored in the complete SwAsp collection and from four populations with completely sequenced haplotypes. Dashed lines are regression lines of allele frequency on latitude.
Clinal variation in bud set in the growth chamber experiment:
We obtained data on growth arrest from a total of 91 trees, with the number of clones scored per population ranging from 4 to 11. Despite the modest data there was significant variation among populations in the critical photoperiod that induced bud set and growth cessation in the SwAsp collection (Table 4). Days to bud set (measured from the start of the experiment) ranged from 101.5 days in the southernmost population to only 53.6 days in the northernmost population, a difference of almost 48 days. The population variation in bud set was clearly organized in a latitudinal cline and a linear regression of population mean critical photoperiod on latitude of origin was highly significant (F1,10 = 111.9, P < 0.001, R2 = 0.918, Figure 4). In addition to the population component of variation, there was also a small, but statistically significant, variation among clones within populations in the time to bud set (Table 4).
TABLE 4.
Linear mixed-effects model of bud set in greenhouse experiment
| Variable | d.f. | SS | MS | F | P |
|---|---|---|---|---|---|
| Population | 11 | 21,864.7 | 1,987.7 | 91.97 | <0.001 |
| Clone | 57 | 3,231.0 | 56.7 | 2.62 | 0.0074 |
| Error | 22 | 475.5 | 21.6 |
Figure 4.

Time to bud set (population mean ± SE) as a function of latitude of origin for the 12 populations in the SwAsp collection.
DISCUSSION
Knowledge about natural genetic variation is rapidly becoming a principal tool for understanding biological systems (Alonso-Blanco and Koornneef 2000). However, levels of nucleotide polymorphism and extent of LD vary dramatically across the genome of a species (Nordborg et al. 2002) and it is therefore important to characterize patterns of variation at loci of interest prior to performing association studies. This is particularly true in Populus, where patterns of genetic diversity at the nucleotide level have thus far been characterized only in a limited number of genomic regions. Here we have studied a phenology trait that is known to be under tight genetic control (e.g., Frewen et al. 2000; Howe et al. 2000, 2003) and show latitudinal clines with the aim of determining the feasibility of linking variation at the sequence level to phenotypic variation at the population level.
Bud set showed significant clinal variation across the original latitudinal gradient in our growth chamber experiment and latitude explained >90% of the variation among populations in the timing of bud set (Figure 4). Such steep latitudinal clines have been demonstrated in many other forest trees and have been shown to represent critical adaptive responses to the length of the growing season (Howe et al. 2003). For instance, trees that were late setting buds and that grew late into the season suffered both greater fall frost damage and lower winter survival in an F2 population of a P. trichocarpa × P. deltoides cross (Howe et al. 2000). Similar observations have been made in several other forest trees [Betula (Li et al. 2002) and conifers (Clapham et al. 2001)].
Divergent selection is expected to enhance levels of genetic differentiation not only for the sites that are the direct target of selection but also for neutral sites in the vicinity of the site(s) under selection (Charlesworth et al. 1997; Nordborg and Innan 2003). Such windows of high among-population genetic differentiation are a hallmark of divergent selection and should be detectable using a sliding-window approach to examine patterns of between-population diversity across a gene region. One problem is to determine what actually constitutes high genetic differentiation, because even under a neutral model diversity can vary across a gene region because of the stochastic nature of the coalescent process (Nordborg 2001). The approach we have taken here is to use coalescent simulations to compare the amount of genetic diversity found among populations across the phyB2 gene with that expected under a neutral model of population subdivision. Interestingly, of the seven SNPs that we screened in the SwAsp collection and that were located in one of the regions with excessive genetic differentiation, three showed significant clinal variation. This suggests that sliding-window scans can be useful for detecting gene regions that are involved in local adaptation.
The width of peaks generated by local selection depends on specieswide population size, recombination rates, and the strength of selection (Charlesworth et al. 1997; Nordborg and Innan 2003). Peaks are expected to be quite narrow in species such as P. tremula, with high levels of both gene flow and recombination, and the likelihood of detecting such peaks can therefore be quite low (Charlesworth et al. 1997; Nordborg and Innan 2003). In addition, even if divergent selection occurs, the stochastic nature of the coalescent process means that even when a peak of high diversity is expected on average, any given realization of the evolutionary process leading to the current sample may or may not show such a pattern. In line with this, individual loci contributing to quantitative traits are expected to show substantial heterogeneity, with a few loci showing strong allelic differentiation while other loci behave as neutral markers (Le Corre and Kremer 2003). These could be possible explanations for why the T608N mutation is found in a region with very low genetic differentiation, despite showing significant clinal variation.
The sites showing clinal variation are spread out over ∼4 kb of the phyB2 region. Unfortunately, we do not have access to haplotype data for these sites and hence we cannot test whether these four clines are truly independent or whether they represent parallel clines generated by LD (e.g., Berry and Kreitman 1993). The T608N, L789M, and Int3 mutations are separated by 1.8 and 2.0 kb, making LD a very unlikely cause of the clines at these sites. Int3 and L1078P, on the other hand, are separated by 396 bp and this is sufficiently close that LD could be a factor in explaining the clines at these two sites.
We classified the mutations showing significant clinal variation as either ancestral or derived on the basis of comparisons with the outgroup species P. trichocarpa. Surprisingly, the derived alleles increase in frequency with latitude at all sites that show clines in phyB2. Similar observations have been made in Drosophila melanogaster and it has been suggested that this is the result of adaptation to temperate environments following the recent spread of D. melanogaster from sub-Saharan Africa (Sezgin et al. 2004). This phenomenon clearly deserves further study in P. tremula.
T608N, L789M, and L1078P are all conservative amino acid substitutions, based on the chemical properties of the ancestral and substituted amino acids, but it is currently not known how these mutations might affect the phyB2 protein. The location of these mutations in the phyB2 protein can, however, shed some light on their possible functional significance. T608N is located in a region of phyB2 that is involved in determining the spectral integrity and photosensory specificity whereas L798M and L1078P are located in a region specifying the regulatory activity of phyB2 (Smith 2000). It is thus possible that if these mutations influence bud set, they do so in very different ways. In addition, intron 3 contained two major haplotypes that differed at ∼20 bp. These two haplotypes varied in frequency with one haplotype common in the two southern populations (France and Austria) and the other haplotype common in the two Swedish populations. This is also reflected in the quite distinct peak of genetic differentiation seen in this region (Figure 2). We can only speculate about the functional significance of this region, but it is, for instance, possible that intron 3 contains cis-regulatory elements involved in regulating the expression of phyB2. Such regulatory elements have been identified in introns of other plant genes (e.g., Fiume et al. 2004).
We have shown that a search for signs of adaptive divergence in phyB2 through a sliding-window approach led to the identification of several sites that show clinal variation and of a pattern that is suggestive of an adaptive response in phyB2 to local photoperiodic conditions. However, this does not exclude the possibility that other phytochrome genes (phyA and phyB1) have related functions. Photoperiod regulates not only bud set but also growth arrest, initiation of leaf senescence and probably leaf abscission, hardiness development, and cambial dormancy and variation in the genes regulating all these traits could therefore be expected to show similar latitudinal clines. The ultimate question is how all these phenological traits are controlled. Is there a “master calendar” set by a single photoreceptor gene that initiates these processes in a predefined manner and with downstream signal transduction pathways that branch off to induce various processes or do several photoreceptor genes feed information into the master calendar? Alternatively, are the processes regulated independently, thereby allowing for variation in the different developmental processes initiated by the end of the growing season? These questions will be addressed in future evolutionary and ecological genomics studies where Populus promises to be an excellent model system.
Acknowledgments
We are indebted to the staff at Skogforsk in Ekebo and Sävar (in particular, Bo Karlsson and Lars-Göran Stener) and to the local “Skogsvårdsstyrelser” that have been instrumental in the creation of the SwAsp collection. We are also grateful to Carin Olofsson for help with SNP scoring and to Barbara Giles for linguistic help. This study was funded by grants from the Swedish Research Council to P.K.I. and by grants from the Swedish Research Council and the Foundation for Strategic Research to S.J.
References
- Adams, W. T., S. H. Strauss, S. L. Sopes and A. R. Griffin, 1992. Population Genetics of Forest Trees. Kluwer, Dordrecht, The Netherlands.
- Andersson, A., J. Keskitalo, A. Sjödin, R. Bhalerao, F. Sterky et al., 2004. A transcriptional timetable of autumn senescence. Genome Biol. 5: R24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alonso-Blanco, C, and M. Koornneef, 2000. Naturally occurring variation in Arabidopsis: an underexploited resource for plant genetics. Trends Plant Sci. 5: 22–29. [DOI] [PubMed] [Google Scholar]
- Bradshaw, H. D., and R. F. Stettler, 1995. Molecular genetics of growth and development in Populus. IV. Mapping QTL with large effects on growth, form and phenology traits in a forest tree. Genetics 139: 963–973. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berry, A. J., and M. Kreitman, 1993. Molecular analysis of an allozyme cline: alcohol dehydrogenase in Drosophila melanogaster on the east coast of North America. Genetics 134: 869–893. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brown, G. R., G. P. Gill, R. J. Kuntz, C. H. Langley and D. B. Neale, 2004. Nucleotide diversity and linkage disequilibrium in loblolly pine. Proc. Natl. Acad. Sci. USA 101: 15255–15260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brunner, A. M., V. B. Busov and S. H. Strauss, 2004. Poplar genome sequence: functional genomics in an ecologically dominant plant species. Trends Plant Sci. 9: 49–56. [DOI] [PubMed] [Google Scholar]
- Charlesworth, B., 1998. Measures of divergence between populations and the effects of forces that reduce variability. Mol. Biol. Evol. 15: 538–542. [DOI] [PubMed] [Google Scholar]
- Charlesworth, B., M. Nordborg and D. Charlesworth, 1997. The effects of local selection, balanced polymorphism and background selection on equilibrium patterns of genetic diversity in subdivided populations. Genet. Res. 70: 155–174. [DOI] [PubMed] [Google Scholar]
- Chen, T. H. H., G. T. Howe and H. D. Bradshaw, 2002. Molecular genetic analysis of dormancy-related traits in poplars. Weed Sci. 50: 232–240. [Google Scholar]
- Clapham, D., I. Ekberg, C. H. A. Little and O. Savolainen, 2001. Molecular biology of conifer frost tolerance and potential applications to tree breeding, pp. 187–219 in Conifer Cold Hardiness, edited by F. J. Brigas and S. J. Colombo. Kluwer Academic, Dordrecht, The Netherlands.
- Dvornyk, V., A. Sirviö, M. Mikkonen and O. Savalainen, 2002. Low nucleotide diversity at the pal1 locus in the widely distributed Pinus sylvestris. Mol. Biol. Evol. 19(2): 179–188. [DOI] [PubMed] [Google Scholar]
- Eriksson, G., I. Ekberg, I. Dormling, B. Matérn and D. von Wettstein, 1978. Inheritance of bud-set and bud-flushing in Picea abies (L.). Karst. Theor. Appl. Genet. 52: 3–19. [DOI] [PubMed] [Google Scholar]
- Faust, M., A. Erez, L. J. Rowland, S. Y. Wang and H. A. Norman, 1997. Bud dormancy in perennial fruit trees: physiological basis for dormancy induction, maintenance and release. Hortscience 32: 623–629. [Google Scholar]
- Fiume, E., P. Christou, S. Giani and D. Breviaro, 2004. Introns are key regulatory elements of rice tubulin expression. Planta 218: 693–703. [DOI] [PubMed] [Google Scholar]
- Flint-García, S. A., J. M. Thornsberry and E. S. Buckler, 2003. Structure of linkage disequilibrium in plants. Annu. Rev. Plant Biol. 54: 357–374. [DOI] [PubMed] [Google Scholar]
- Frewen, B. E., T. H. H. Chen, G. T. Howe, J. Davis, A. Rohde et al., 2000. Quantitative trait loci and candidate gene mapping of bud set and bud flush in Populus. Genetics 154: 837–845. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garcia-Gil, M. R., M. Mikkonen and O. Savolainen, 2003. Nucleotide diversity at two phytochrome loci along a latitudinal cline in Pinus sylvestris. Mol. Ecol. 12: 1195–1206. [DOI] [PubMed] [Google Scholar]
- Gaut, B. S., and A. D. Long, 2003. The lowdown on linkage disequilibrium. Plant Cell 15: 1502–1506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hey, J., and J. Wakeley, 1997. A coalescent estimator of the population recombination rate. Genetics 145: 833–846. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Horvath, D. P., J. V. Anderson, W. S. Chao and M. E. Foley, 2003. Knowing when to grow: signals regulating bud dormancy. Trends Plant Sci. 8: 534–540. [DOI] [PubMed] [Google Scholar]
- Howe, G. T., W. P. Hackett, G. R. Furnier and R. E. Klevorn, 1995. Photoperiodic responses of a northern and southern ecotype of black cottonwood. Physiol. Plant. 93: 695–708. [Google Scholar]
- Howe, G. T., G. Gardner, W. P. Hackett and G. R. Furnier, 1996. Phytochrome control of short day induced bud set in black cottonwood. Physiol. Plant. 97: 95–103. [Google Scholar]
- Howe, G. T., P. A. Bucciaglia, W. P. Hackett, G. R. Furnier, M. M. Cordonnier-Pratt et al., 1998. Evidence that the phytochrome gene family in black cottonwood has one PHYA locus and two PHYB loci but lacks members of the PHYC/F and PHYE subfamilies. Mol. Biol. Evol. 15(2): 160–175. [DOI] [PubMed] [Google Scholar]
- Howe, G. T., P. Saruul, J. Davis and T. H. H. Chen, 2000. Quantitative genetics of bud phenology, frost damage and winter survival in an F2 family of hybrid poplars. Theor. Appl. Genet. 101: 632–642. [Google Scholar]
- Howe, G. T., S. N. Aitken, D. B. Neale, K. D. Jermstad, N. C. Wheeler et al., 2003. From genotype to phenotype: unraveling the complexities of cold adaptation in forest trees. Can. J. Bot. 81: 1247–1266. [Google Scholar]
- Hudson, R. R., 2001. Two-locus sampling distributions and their application. Genetics 159: 1805–1817. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ingvarsson, P. K., 2005. Nucleotide polymorphism and linkage disequilibrium within and among natural populations of European aspen (Populus tremula L., Salicaceae). Genetics 169: 945–953. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Konieczny, A., and F. M. Ausubel, 1993. A procedure for mapping Arabidopsis mutations using co-dominant ecotype-specific PCR-based markers. Plant J. 4: 403–410. [DOI] [PubMed] [Google Scholar]
- Le Corre, V., and A. Kremer, 2003. Genetic variability at neutral markers, quantitative trait loci and trait in a subdivided population under selection. Genetics 164: 1205–1219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li, C., T. Puhakainen, A. Welling, A. Viherä-Aarnio, A. Ernstsen et al., 2002. Cold acclimatization in silver birch (Betula pendula). Development of freezing tolerance in different tissues and climatic ecotypes. Physiol. Plant. 116: 478–488. [Google Scholar]
- McDonald, J. H., and M. Kreitman, 1991. Adaptive protein evolution at the Adh locus in Drosophila. Nature 351: 652–654. [DOI] [PubMed] [Google Scholar]
- Neale, D. B., and O. Savolainen, 2004. Association genetics of complex traits in conifers. Trends Plant Sci. 9: 325–330. [DOI] [PubMed] [Google Scholar]
- Nordborg, M., 2001. Coalescent theory, pp. 179–212 in Handbook of Statistical Genetics, edited by D. J. Balding, M. Bishop and C. Cannings. John Wiley & Sons, Chichester, UK.
- Nordborg, M., and H. Innan, 2003. The genealogy of sequences containing multiple sites subject to strong selection in a subdivided population. Genetics 163: 1201–1213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nordborg, M., J. O. Borevitz, J. Bergelson, C. C. Berry, J. Chory et al., 2002. The extent of linkage disequilibrium in Arabidopsis thaliana. Nat. Genet. 30: 190–193. [DOI] [PubMed] [Google Scholar]
- R Development Core Team, 2004. R: A Language and Environment for Statistical Computing. R Foundation for Statistical Computing, Vienna, Austria (http://www.R-project.org).
- Rozas, J., J. C. Sánchez-DelBarrio, X. Messeguer and R. Rozas, 2003. DnaSP, DNA polymorphism analyses by the coalescent and other methods. Bioinformatics 19: 2496–2497. [DOI] [PubMed] [Google Scholar]
- Sezgin, E., D. D. Duvernell, L. M. Matzkin, Y. Duan, C. T. Zhu et al., 2004. Single-locus latitudinal clines and their relationship to temperature adaptation in metabolic genes and derived alleles in Drosophila melanogaster. Genetics 168: 923–931. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith, H., 2000. Phytochromes and light signal perception by plants—an emerging synthesis. Nature 407: 585–591. [DOI] [PubMed] [Google Scholar]
- Sterky, F., R. R. Bhalerao, P. Unneberg, B. Segerman, P. Nilsson et al., 2004. A Populus EST resource for plant functional genomics. Proc. Natl. Acad. Sci. USA 101: 13951–13956. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thompson, J. D., D. G. Higgins and T. J. Gibson, 1994. CLUSTAL W: improving the sensitivity of progressive multiple sequence alignment through sequence weighting, positions-specific gap penalties and weight matrix choice. Nucleic Acids Res. 22: 4673–4680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weir, B. S., 1990. Genetic Data Analysis. Sinauer Associates. Sunderland, MA.
- Wright, J., 1976. An Introduction to Forest Genetics. Academic Press, London.
- Wullschleger, S. D., S. Jansson and G. Taylor, 2002. Genomics and forest biology: Populus emerges as the perennial favorite. Plant Cell 14: 2651–2655. [DOI] [PMC free article] [PubMed] [Google Scholar]