

Abstract
The purpose of this study was to investigate the efficacy of a novel steroid, fluasterone (DHEF, a dehydroepiandrosterone (DHEA) analog), at improving functional recovery in a rat model of traumatic brain injury (TBI). The lateral cortical impact model was utilized in two studies of efficacy and therapeutic window. DHEF was given (25 mg/kg, intraperitoneally) at the initial time point and once a day for 2 more days. Study A included four groups: sham injury, vehicle treated (n = 22); injured, vehicle treated (n = 30); injured, pretreated (5–10 min prior to injury, n = 24); and injured, posttreated (initial dose 30 min postinjury, n = 15). Study B (therapeutic window) included five groups: sham injury, vehicle treated (n = 17); injured, vehicle treated (n = 26); and three posttreatment groups: initial dose at 30 min (n = 18), 2 h (n = 23), or 12 h (n = 16) postinjury. Three criteria were used to grade functional recovery. In study A, DHEF improved beam walk performance both with pretreatment (79%) and 30-min posttreatment group (54%; p < 0.01, Dunnett vs. injured vehicle). In study B, the 12-h posttreatment group showed a 97% improvement in beam walk perfomance (p < 0.01, Dunnett). The 30-min and 12-h posttreatment groups showed a decreased incidence of falls from the beam, which reached statistical significance (p < 0.05, Dunnett). Tests of memory (Morris water maze) and neurological reflexes both revealed significant improvements in all DHEF treatment groups. In cultured rat mesangial cells, DHEF (and DHEA) potently inhibited interleukin-1β–induced cyclooxygenase-2 (COX2) mRNA and prostaglandin (PGE2) production. In contrast, DHEF treatment did not alter injury-induced COX2 mRNA levels in the cortex or hippocampus. However, DHEF (and DHEA) relaxed ex vivo bovine middle cerebral artery preparations by about 30%, with an IC50 ≈ 40 μM. This was a direct effect on the vascular smooth muscle, independent of the endothelial cell layer. Fluasterone (DHEF) treatments improved functional recovery in a rat TBI model. Possible mechanisms of action for this novel DHEA analog are discussed. These findings suggest an exciting potential use for this agent in the clinical treatment of traumatic brain injury.
Keywords: cyclooxygenase-2, neurosteroid, rat traumatic brain injury
INTRODUCTION
The primary injury of brain trauma sets into motion cascades of responses which may be beneficial or lead to secondary injury. The acute release of cytokines (Morganti-Kossman et al., 1997, Soares et al., 1995; Yan et al., 1992) and vasoactive factors (Ellis et al., 1989) alters cerebrovascular reactivity (Robertson, 1996; Sioutos et al., 1995) and initiates the central inflammatory response. However, if not properly regulated, these initially beneficial responses may ultimately become harmful (Strauss, 1998). No pharmacotherapy has been proven effective for treatment of the primary and secondary insults of traumatic brain injury (TBI).
The goal of the present study is to investigate a new class of drugs, the dehydroepiandrosterone (DHEA) analogs, for the treatment of experimental TBI. Lowered endogenous DHEA levels in humans and lab animals have been associated with a variety of undesirable physiological processes including aging, cognitive decline, aggression, obesity, cardiovascular disease, immune dysfunction, and changes in basal metabolism (Majewska, 1995). Exogenous DHEA steroids were shown to reduce the severity of symptoms in adjuvant-induced arthritis (Williams, 1997) and block the interleukin (IL)-1β–mediated rise in prostaglandin production in rat mesangial cells (A.G. Schwartz et al., unpublished results). However, DHEA is androgenic which may limit its usage in the treatment of patients. This prompted the development (by Dr. A.G. Schwartz, Dr. J.R. Williams, both at Temple University, and Dr. M. Lewbart) of a nonandrogenic fluorinated analog, 16α-fluoro-5-androsten-17-one, fluasterone or DHEF (Fig. 1). DHEF demonstrates similar biological activity to DHEA, without the undesirable side effects linked to androgenic steroids, glucocorticoids, or nonsteroidal antiinflammatory drugs (NSAIDs). DHEF did not demonstrate the androgenic, estrogenic, or peroxisome-proliferating effects of DHEA in rodents and humans, yet did retain the antineoplastic and anti-diabetic effects of DHEA (Ratko, 1991; Schwartz and Pashko, 1993; Schwartz et al., 1989). Fluasterone is currently in Phase I/II clinical trials for autoimmune (e.g., psoriasis) and metabolic (e.g., Syndrome X hypertriglyceridemia) diseases.
FIG. 1.

Dehydroepiandrosterone (DHEA) and its analogs function via pleiotropic mechanisms that are independent of androgen and other steroid receptors. These include glucose-6-phosphate dehydrogenase enzyme inhibition, alteration of cytokine-induced gene expression (e.g., attenuation of IL-1β-induced COX2), and allosteric modulation of neurotransmitter receptors (e.g., GABAA receptor antagonism).
There is much evidence that free radicals exacerbate tissue damage and inflammation after brain injury (Hall, 1996). The DHEA class of steroids inhibits production of reactive oxygen and nitrogen intermediates, possibly by uncompetitively inhibiting glucose-6-phosphate dehydrogenase (G6PDH), thereby suppressing the formation of NADPH, a critical reductant in free radical formation (Gordon et al., 1995; Pashko et al., 1991; Raineri and Levy, 1970; Schwartz and Pashko 1993, 1994). DHEA also effectively inhibits nitric oxide (NO) production (Mei et al., 1995); presumably by limiting NADPH production, a required co-factor for nitric oxide synthase activity (Marletta, 1994). The pentose phosphate pathway, of which G6PDH is the rate-limiting step, is also a primary source of nucleic acid backbones, necessary for cellular proliferation. In models of peripheral and central inflammation, as well as in human subjects, DHEA, DHEF, and other analogs have proven beneficial in reducing cellular proliferation, attenuating free radical formation, and altering inflammatory-mediated gene expression (Aragno et al., 2000; Boccuzzi et al., 1997; Dersken, 1998; Morales et al., 1994; Van Vollenhoven, 1994, 1995, 1998). These findings are consistent with the hypothesis that DHEA steroids suppress inflammation via G6PDH inhibition. In addition, DHEA and related pregnane and androstane steroids can act as neuroactive neurosteroids (Baulieu and Robel, 1996). As allosteric inhibitors of GABAA receptor function, and weak agonists at the glutamate receptor sigma subunit (Majewska, 1995; Maurice et al., 1997) these compounds may modulate neuronal excitability. DHEAs block pharmacologically induced amnesia in mice (Flood et al., 1988a,b), and improve memory on automatized tasks in humans (Wolkowitz et al., 1995). This concept is supported by earlier findings that GABA agonists (barbiturates, benzodiazepines) produce amnesia, whereas GABA antagonists (picrotoxin, pentylenetetrazole) reverse amnesia and improve memory (Breen, 1961; Irwin, 1966). For example, chronic administration of the benzodiazepine antagonist flumazenil can prolong the life span and improve memory in rats (Marczynski et al., 1994).
In addition, DHEAs may increase hypothalamic and possibly cortical levels of serotonin as well as potentiate NMDA-induced noradrenaline release from hippocampal slices, apparently by stimulating glutamatergic σ receptors (Majewska, 1995; Maurice et al., 1997). These latter mechanisms, may account for the reported antidepressant effects of DHEA in humans (Wolkowitz et al., 1995), and may additionally contribute to its potentiating effect on memory, in which noradrenaline plays a pivotal role (Majewska, 1995; Sara et al., 1994; Stanton and Sarvey, 1985). Neurosteroids may play a dynamic role in shaping neuronal architecture and, as such, may have critical functions in learning, memory and neuronal survival during or after injury to the brain. Thus, DHEF may have pleiotropic mechanisms of action in recovery from TBI. Our current work is focused on determining the efficacy of DHEF in experimental head injury, and characterizing its therapeutic window.
MATERIALS AND METHODS
Traumatic Brain Injury Model
The rat lateral controlled cortical impact (LCI) model of TBI was used to induce moderate levels of injury. LCI results in a temporary loss of strength and coordination in the limbs contralateral to the injury, significant retrograde amnesia, a loss of cortical and subcortical neurons, and induction of inflammatory responses. LCI produces focal cortical and subcortical damage similar to human closed head injury, with little involvement of the brainstem. The relevance of this rat model of TBI to clinical head trauma has been validated by several groups, reproducing common cognitive-behavioral outcome measures and histopathological findings (Dixon et al., 1991; Hamm et al., 1992; McIntosh et al., 1989).
Animals
Male Sprague-Dawley rats (Harlan), weighing 300–400 g, were studied. Animals were cared for in accordance with U.S. Public Health Service regulations. All protocols were approved by the Temple University Institutional Animal Care and Use Committee.
Surgical preparation
Rats were preanesthetized (∼2% isoflurane), scalp shaved, and given oxygen with 0.75% isoflurane through a fixed nose cone on a stereotaxic platform. The cranium was exposed and a craniectomy made laterally (left side), midway between lambda and bregma, between the central suture and the left temporal ridge using a 6-mm trephine. The intact dura was subjected to a piston impact of 3.0 mm depth, 4 m/sec velocity, and 100 msec duration. The animal was taken off anesthesia and assessed for exclusion criteria. To be included in the study, animals needed to regain the pinna and corneal reflexes within 2–4 min, awaken within 4–5 min, and regain their righting response within 9 min after removal from anesthesia.
Experimental treatment groups
For these studies, animals were treated immediately before or at various times after LCI TBI (Table 1). The DHEF and the Emulphor-saline vehicle were provided by Dr. Arthur Schwartz. Sham/vehicle and injured/vehicle groups received intraperitoneal injections 10 min before injury, followed by doses at 24 and 48 h postinjury. The injured/DHEF groups received DHEF (25 mg/kg, 8.75 mg/mL) at the initial time point (10-min pretreatment, or 30-min, 2-h, or 12-h posttreatment) followed by doses at 24 and 48 h postinjury.
Table 1.
DHEF Treatments Groups: Additional Weight Loss and Improved Beam Walk Performance at 3 Days Postinjury
| Group name | Study (n) | Treatmenta | Description | Weight loss (g) | Falls from the beam (n) |
|---|---|---|---|---|---|
| Sham | A (22) | Sham/vehicle | Surgery, no injury, vehicle 10 min prior | 0.1 ± 1.9 | n.d. |
| Injured | A (30) | Injured/vehicle | LCI injury, vehicle 10 min prior | 5.8 ± 1.4* | n.d. |
| DHEF | A (24) | Injured/DHEF | DHEF started 10 min before LCI injury | 15.2 ± 2.3*§ | n.d. |
| DHEF/0.5 | A (15) | Injured/DHEF/0.5 | LCI, DHEF started 0.5 h after LCI injury | 14.5 ± 4.5*§ | n.d. |
| Sham | B (17) | Sham/vehicle | Surgery, no injury, vehicle 10 min prior | 0.7 ± 2.2 | 0 |
| Injured | B (26) | Injured/vehicle | LCI injury with vehicle | 7.7 ± 2.1* | 9 |
| DHEF/0.5 | B (24) | Injured/DHEF/0.5 | LCI, DHEF started 0.5 h after LCI injury | 17.8 ± 2.8*§ | 1† |
| DHEF/2 | B (23) | Injured/DHEF/2 | LCI, DHEF started 2 h after LCI injury | 15.8 ± 1.4*§ | 4 |
| DHEF/12 | B (16) | Injured/DHEF/12 | LCI, DHEF started 12 h after LCI injury | 17.1 ± 3.1*§ | 1† |
All groups received initial intraperitoneal injections of vehicle or DHEF (25 mg/kg) at the indicated time, then at 24 h and 48 h postinjury.
n.d., not done.
p < 0.05, Dunnett versus sham
p < 0.05, Scheffé versus injured/vehicle
p < 0.05, χ2 analysis.
Functional Evaluations
All neurological and behavioral assessments were performed before and 3 days after injury. The observer was blinded to the treatment group information. Improvements were calculated as the difference between injured/DHEF-treated and injured/vehicle groups divided by the difference between sham and injured/vehicle groups (×100%).
Behavioral assessments
Behavioral assessments included an open field test and the Morris water maze. To test exploratory behavior, animals were placed in an open field (30 cm × 61 cm × 20 cm deep) and observed for 2 min. The number of rearings and transits to each corner were summed for an activity score. The water maze was used to assess declarative memory function. Pre-training and training consisted of 120-sec trials, starting at four alternating compass points in a 2-m circular tank with opaque water (white tempera paint) and a submerged platform halfway from the wall to the center of the maze. After reaching the platform, animals remained there for 60 sec to become familiar with the extra-maze cues placed on the surrounding walls. Animals were pretrained (nine trials) the day before injury, and trained (10 trials) the day of injury, finishing 1.5 h before the anesthesia. Water maze scoring used two concentric zones around the platform site (40 and 100 cm diameter); time spent in each zone was measured during a 120-sec videotaped probe trial (with the platform removed). The water maze score was calculated as the sum of the weighted times in each zone, adjusted using the inverse proportion of the zone area compared to the whole tank (multipliers: inner zone = 24, outer zone = 4). This approach weights the time in a particular concentric zone by the (inverse) probability that the animal would be found in that area of the maze by chance. Thus, the smaller the zone, the greater the weighting factor, and vice versa. This nonarbitrary weighting should yield more sensitive and accurate results when access to a computerized analysis package is limited.
Neurological reflexes
Neuroscores were based on three tests of limb reflexes (McIntosh et al., 1989). The tests were contraflexion (forelimb and head flexion in response to anticipation of falling), hind limb extension (in response to repetitive raising and lowering by the tail), and lateral pulsion (test of strength and coordination upon attempts to roll the animal onto its back). Scores of 0–4 (4 being best) were given for left and right limbs separately for a total of 24 points on the three tests.
Beam walk test
The beam walk is a balance beam task with both cognitive and motor components. Animals were trained to escape an unpleasant pair of stimuli (200-W light bulb and 90-dB white noise) by running 1 m along a 2.5-cm beam with several posts mounted on alternating sides, and entering a black box (the stimuli were turned off once the animal entered the box). The time taken to enter the box was the outcome measure (0–120 sec). In addition, in Study B, if animals were unable to complete the task or fell from the beam, these were recorded and studied separately.
Cerebral vessel reactivity
We optimized an ex vivo preparation of bovine middle cerebral arteries (bMCA) to test DHEA and DHEF vasoactivity (Wendling and Harakal, 1987, 1991; Wendling et al., 1994). With prior approval of the US. Dept. of Agriculture Meat Inspection Program, bMCA were obtained from freshly slaughtered animals. The arteries were isolated, immersed in physiologic saline solutions (PSS) at 1°C, transported to the laboratory, and cleaned of fat and debris. For isometric tension experiments, arteries were cut into rings of uniform width (2.5 mm) and mounted vertically on two L-shaped wire holders between a wire anchor and a Grass isometric transducer. The rings were maintained in 40–50 mL of oxygenated PSS ([CaCl2] = 1.2 mM, pH 7.4, 37°C) over a 2–3-h equilibration period. Constrictors were used to gradually increase resting tension to optimal (0.5 g) for the bovine cerebral arteries. Isometric tension was recorded simultaneously on Radnoti Model TRN005 or TRN006 instrumentation amplifiers and on a Kipp and Zonen Model BD112 recorder, linked to the amplifiers via an 8-channel multiplexer (Instech Laboratories). For endothelial stripping experiment, arteries were opened into strips approximately 1–1.5 cm long. The endothelial layer was carefully stripped, and each tissue was weighed, mounted on two wire holders, and equilibrated at 0.5 g tension, as above. After a 3-h equilibration in PSS at 37°C, the strips were treated with vehicle, DHEA, or DHEF for 45 min. In all experiments, arterial rings or strips were selected at random to receive a treatment. Results were expressed as mean + SEM (n ≥ 6 rings or strips for each variable). The inter group comparisons were examined using analysis of variance, and the Student's t-test or the Newman-Keuls multiple range t-test.
In vitro inflammation model
Kidney-derived rat mesangial cells (kind gift of Dr. Jeffrey Kreisberg) were cultured in RPMI medium containing 20% fetal bovine serum. After reaching confluence, cells were incubated in serum-free medium for 48 h. The inflammatory cytokine IL-1β (1 ng/mL) was added (with or without DHEA steroids), and the cells were harvested after 4 h by scraping into 5 M guanidine thiocyanate, 0.1 M EDTA, pH 8.
Measurement of COX2 mRNA levels
COX2 gene expression was measured in a lysate ribonuclease protection assay (Strauss et al., 2000). The cell lysates were either from cultured rat mesangial cells (above) or from frozen rat brain micropunches sonicated in 6 M guanidine thiocyanate, 0.1 M EDTA, pH 8. Aliquots (40 μL, ∼2 × 105 cells) were analyzed using cRNA probes to COX2 and cyclophilin (Strauss et al., 2000). Comparisons were made on mRNA levels normalized for protein or cyclophilin in each sample. Results were expressed as mean ± SEM (n = 4–6 per group).
Statistical analyses
Statistical evaluation of data was performed using chi-square, and analysis of variance with Dunnett and Scheffé post hoc tests. For all analyses, the injured/vehicle group was used as the positive control. Probability values less than 0.05 were considered statistically significant to reject the null hypothesis that the group means were equal.
RESULTS
Fluasterone (DHEF, 25 mg/kg ip., 3 days) treatments initiated before or up to 12 h after TBI improved functional recovery at 3 days postinjury. The open field test was used as a negative control, to ensure that the injury did not alter the animals' exploratory behavior. No injury-induced changes in open field behavior were observed among any of the groups. This treatment induced weight loss greater than that caused by the cortical contusion injury alone (Table 1). DHEF administered at lower doses (8.3 and 2.5 mg/kg ip., initiated 0.5 h postinjury) did not improve behavioral recovery and did not produce additional weight loss (data not shown).
Improved Beam Walk Performance
DHEF treatments, initially given before or after injury, improved beam walk performance measured 3 days postinjury. In Study A, DHEF produced 79% and 54% improvements in beam walk latency in the pretreatment and 0.5-h posttreatment groups, respectively (p < 0.01, Dunnett, Fig. 2A). In Study B, both latency and ability to complete the beam walk were examined. DHEF initially administered 0.5 h and 12 h (but not at 2 h) postinjury markedly improved performance 3 days after TBI compared to the injured, vehicle treated littermate controls (Fig. 2B). The 12-h posttreatment group was the only group that showed no significant difference from sham animals on this task. DHEF posttreatments (0.5 and 12 h) also reduced the number of falls on the beam walk task, compared to injured controls (Table 1, p , 0.05, χ2 test).
FIG. 2.
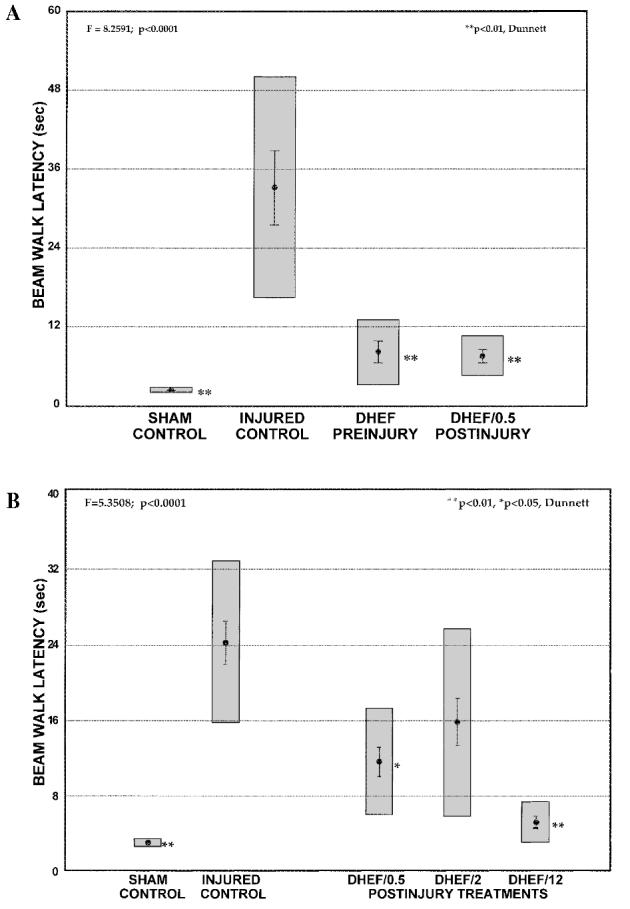
DHEF treatment improved beam walk performance tested at 3 days after traumatic brain injury. (A) Reduced latency to complete the beam walk task in brain injured animals treated before (DHEF preinjury) or one half hour after (DHEF/0.5 postinjury) TBI, as compared to injured animals treated with vehicle. (B) DHEF treatments initiated 0.5 h (DHEF/0.5) and 12 h (DHEF/12) after TBI reduce the mean latency to complete the beam walk 3 days postinjury. Sham animals were surgerized but not injured. Results are presented as mean ± SEM, gray boxes represent the 99% confidence interval. *p < 0.05, **p < 0.01, Dunnett versus injured controls.
Improved Neurological Reflexes Measured Using Neuroscore
Neurological reflexes improved significantly in both treatment groups of Study A (29%, Fig. 3A). All treatment groups in Study B also showed improved recovery compared to injured, vehicle-treated controls (37–63%). The group neuroscores for Study B were as follows: sham, 19.9 ± 1.8; injured, 17.3 ± 1.3; DHEF/0.5, 19.0 ± 1.2; DHEF/2, 19.1 ± 1.2; DHEF/12, 14.5 ± 0.6. The raw data revealed a trend toward intergroup differences before injury (p = 0.0623), as well as a trend toward intergroup differences at 3 days postinjury (p = 0.0896). Thus, to specifically extract the injury-induced changes, the difference between pre- and postinjury neuroscores were evaluated as the dependent variable (p = 0.0259, Fig. 3B). This was not the case in Study A; preinjury differences were not significant (p = 0.7283) and both raw (Fig. 3A) and difference neuroscores showed statistically significant treatment effects at 3 days postinjury (p = 0.0001). The 12-h posttreatment group seemed to improve more than the other groups, showing no significant difference from shams.
FIG. 3.
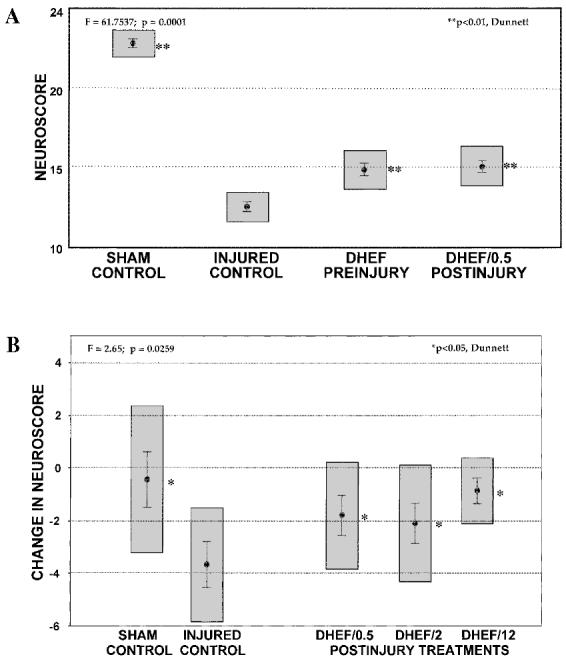
DHEF treatments improved neurological reflexes tested 3 days postinjury. (A) Improved recovery of neurological reflexes in injured animals treated before or one half hour after TBI, compared to vehicle-treated injured controls. Neuroscores are presented as mean ± SEM. (B) Posttreatment with DHEF (0.5, 2, and 12 h postinjury) revealed a small but statistically significant improvement in neurological reflexes. Results are mean difference (postinjury – preinjury) ± SEM. Gray boxes represent the 99% confidence interval. *p < 0.05, **p < 0.01, Dunnett versus injured controls.
Declarative Memory Measured in the Morris Water Maze
Water maze performance similarly revealed statistically significant improvements (45%) in the pretreatment and 0.5-h posttreatment groups of Study A (p < 0.05, Scheffé, Fig. 4A). In Study B, all treatment groups performed better than injured/vehicle (8–36%, p < 0.05, Dunnett), with the 12-h posttreatment group again improving most (p < 0.01, Dunnett, Fig. 4B). To dissociate possible motor deficits at this time, the swim speed (time to cross the maze diameter) was determined at 3 days postinjury during the testing regime. There were no differences in swim speed between sham and any of the injured groups.
FIG. 4.
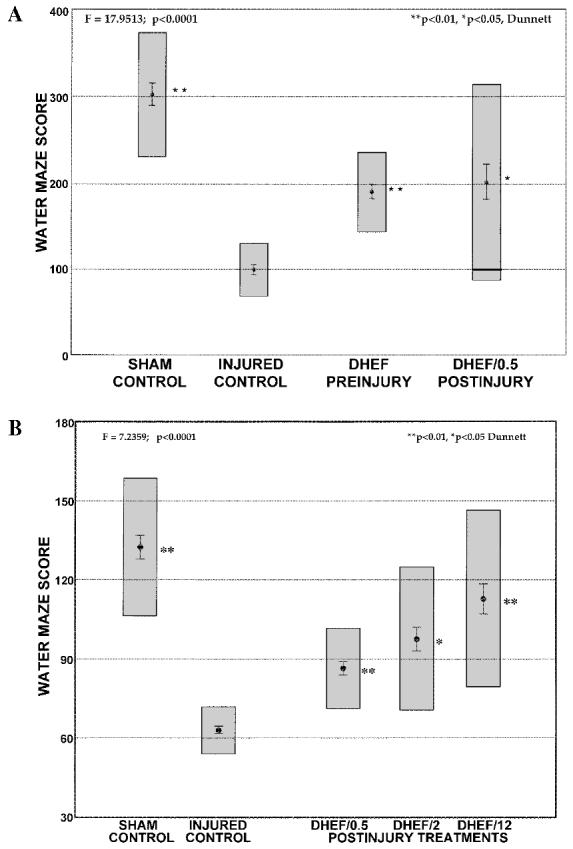
DHEF improves declarative memory function tested 3 days postinjury. (A) Improvements were observed in Morris water maze performance in brain-injured rats treated with DHEF (10 min before, 0.5 h after injury) compared to vehicle-treated injured controls 3 days postinjury. (B) Posttreatment with DHEF (0.5, 2, and 12 h postinjury) also revealed statistically significant improvements in memory function. Water maze score calculated as described in Methods. Results presented are mean ± SEM; gray boxes represent the 99% confidence interval. **p < 0.01, *p < 0.05, Dunnett versus injured controls.
Modulation of COX2 mRNA Induction
COX2 is an early effector in the central inflammatory pathway and is induced by cytokines. To test the antiinflammatory properties of DHEF, an in vitro culture of rat mesangial cells was utilized, as previously described (Feng, 1995). Interleukin-1β (1 ng/mL) increased the level of COX2 mRNA by threefold in these cells. Treatment with DHEF (2.5 μM) or DHEA (data not shown) completely attenuated interleukin-1β–mediated induction of COX2 mRNA (Fig. 5).
FIG. 5.
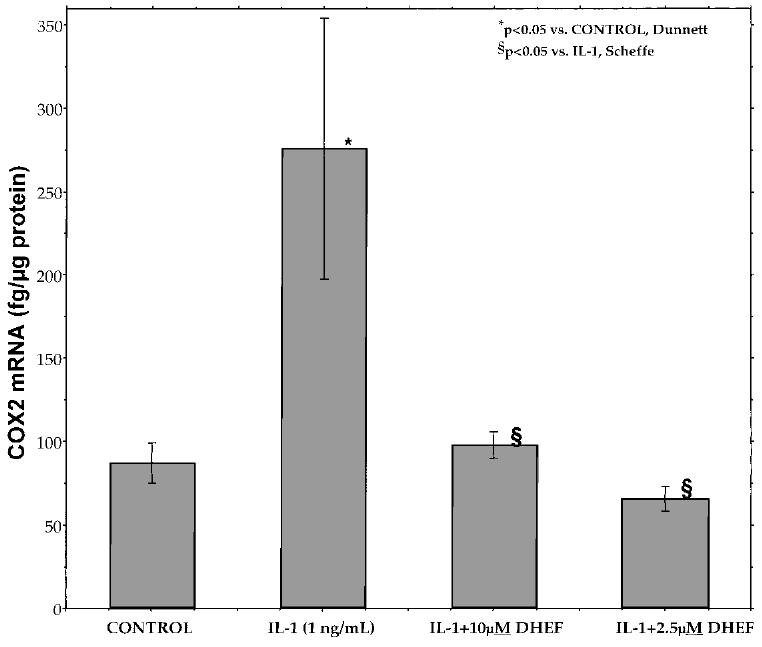
DHEF attenuates the induction of COX2 mRNA by interleukin-1β in rat mesangial cells. The rat mesangial cell model of in vitro inflammation normally shows a robust induction of COX2 gene expression following a 4-h exposure to IL-1β in the absence of serum. DHEF (2.5 and 10 μM) completely blocks this, indicating a role for DHEA steroids in restricting inflammatory gene expression. Cell cultures and mRNA measurements performed as described in Methods. Results are mean 6 SEM (n = 5 per group). *p < 0.05, Dunnett versus control; §p < 0.05, Scheffé versus IL-1 group.
In rats, cerebral COX2 mRNA and protein are induced within 2 h of TBI, and continue to be elevated for at least 3–7 days postinjury (Strauss et al. 2000). Surprisingly, DHEF did not modulate the in vivo expression of COX2 in the injured cortex or hippocampus at any time following TBI (Fig. 6).
FIG. 6.

DHEF does not alter COX2 mRNA levels in injured cortex and hippocampus after lateral cortical impact TBI. Rats were injured and sacrificed at various times after TBI. Fresh frozen brain tissue was sectioned (300 μm) and micropunch ribonuclease protection assays were performed on cortex and hippocampus ipsilateral (and contralateral, not shown) to injury. No inhibition of COX2 induction was observed in these brain areas. Sham controls from multiple time points were grouped (SHAMS). Dashed lines represent injured DHEF pretreatment; solid lines are injured controls. Results are mean ± SEM (n = 4–6 per group). *p < 0.05, Dunnett versus SHAMS.
Modulation of Cerebral Vasoreactivity
To investigate possible mechanisms of action, we tested whether DHEF or DHEA affected cerebral blood vessel constriction or dilation. Serial addition of DHEF or DHEA to K1 constricted bovine middle cerebral artery preparation (bMCA) caused direct vasodilatation with IC50 ≈ 40 μM (Fig. 7). The site of action of this vasoactivity was further investigated by removal of the endothelial cell layer from the bMCA preparations. At their IC50, the relaxation produced by DHEF and DHEA was preserved, indicating a direct effect on the vascular smooth muscle cells (data not shown).
FIG. 7.
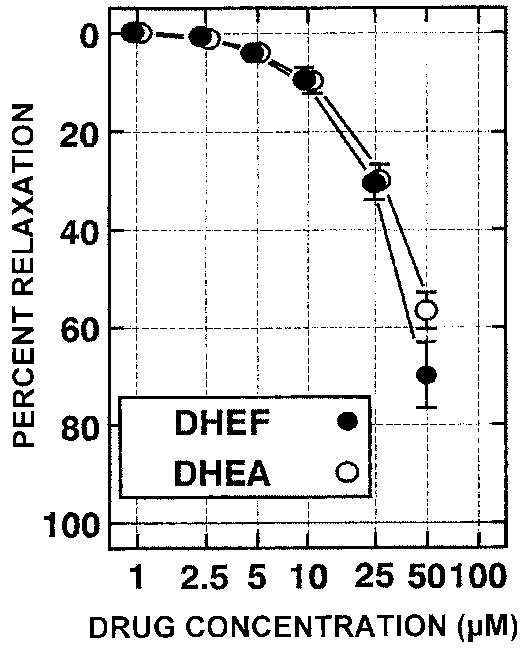
DHEF and DHEA demonstrate vasodilatory effects on bovine middle cerebral artery. Serial addition of DHEA or DHEF to K+ constricted bovine middle cerebral artery preparation (bMCA) causes vasodilatation with IC50 ≈ 40 μM.
DISCUSSION
The DHEA analog fluasterone (DHEF) improved neurological and cognitive recovery in a rat model of lateral cortical contusion traumatic brain injury. No changes were observed in the open field test, a control to confirm that the injury was limited to the somatosensory cortex and underlying structures. Changes were most pronounced on the beam walk test which is a complex coordination task with cognitive components. Not only did the time latency improve in the treatment groups but the animals also revealed a decreased incidence of falls from the beam compared to their vehicle treated littermates. Improvements were significant for declarative memory scores (Morris water maze) and neurological reflexes, as well. Interestingly, delaying the initiation of treatment for 12 h did not diminish the effect of this treatment, and appeared to improve recovery on the beam walk and water maze tasks.
Certain steroids have been shown to benefit the injured brain (Stein and Fulop, 1990). For example, progesterone was found to reduce lipid peroxidation (8-isoprostaglandin-F2α levels) in injured brain (Roof et al., 1997). Antiinflammatory glucocorticoids have been shown to down-regulate COX2 expression (Herschman et al., 1995; Masferrer et al., 1992; Yamagata et al., 1993). DHEA steroids have been shown to benefit the aging brain in a number of laboratory studies, and to reduce prostaglandin production in models of peripheral inflammation (Hastings et al., 1988). In cultured primary astrocytes, DHEA (∼30 μM) virtually abolished inflammatory-mediated TNF-α and IL-6 production, while not affecting NO or PGE2 synthesis (Kipper-Galperin et al., 1999). Interestingly, whereas glucocorticoids had their maximal effect when given 18 h prior to mycoplasma or LPS challenge, “DHEA exerted its optimal effect when added together with the activating agents. When added 18 hours prior to activation, it was ineffective” (Kipper-Galperin et al., 1999). This not only indicates a distinct antiinflammatory mechanism from glucocorticoids, but suggests a time dependence for maximal activity that may relate to our in vivo results. DHEF was less effective on the beam walk and water maze tasks when the initial dose was administered at 2 h postinjury, compared to earlier or later initial doses (Figs. 2B and 3).
COX2 expression in the brain can be induced by inflammatory cytokines (Moore et al., 1999; Seibert et al., 1994) and by glutamatergic excitation (Hewett et al., 2000; Strauss and Marini, 2002). Increases in patient plasma cytokine levels (IL-1β, IL-6, IL-8, TNF) in the acute response (Ott et al., 1994) to brain injury might be responsible for prolonged COX2 induction in the injured brain. Although DHEF blocked IL-1–induced COX2 in cell cultures, DHEF did not affect injury-induced brain COX2 mRNA levels. Thus, the induction of brain COX2 following TBI may be mediated primarily via excitotoxic rather than inflammatory mechanisms (Strauss et al., 2000). This does not rule out the possibility that DHEF affects IL-1–mediated COX2 expression in other regions.
The DHEA class of steroids inhibit inflammation through pleiotropic mechanisms distinct from both NSAIDs and glucocorticoids. They may act by inhibiting glucose-6-phosphate dehydrogenase (G6PDH) and suppressing oxygen-free radical formation (Babior, 1982; Imlay, 1988; Sadowski, 1985; Schwartz and Pashko, 1993, 1994). DHEA steroids are also very effective inhibitors of NO production. DHEA (10 and 100 μM) reduces mouse macrophage NO production by 50% and 95%, respectively (Mei et al., 1995). DHEF is even more potent; 1 μM DHEF lowers NO production by 60%. DHEF has a much higher affinity for G6PDH than DHEA (Ki = 0.51 μM and 18.7 μM, respectively; Schwartz et al., 1988). If DHEF improves behavioral recovery via G6PDH inhibition, it should be more potent than DHEA. However, the observation that the ex vivo vasoactivity of these two compounds is virtually identical (Fig. 7) indicates a G6PDH-independent mechanism, at least in the cerebrovascular response. DHEA and DHEF may inhibit the responsiveness of the cerebral vasculature to increased concentrations of arachidonic acid metabolites (e.g., produced by phospholipase activation) or free radicals (e.g., produced by COX2 and/or NO synthase).
Treatment with DHEA or related steroids might protect neurons against excitatory amino acid induced damage. In vivo, DHEA protected hippocampal CA1/2 neurons against unilateral infusion of NMDA (Kimonides et al., 1998), and reduced CA1 neuronal death in a transient forebrain ischemia model (Li et al., 2001). DHEA and DHEA-sulfate (DHEAs) reduced the neurotoxic actions of NMDA, AMPA, and kainic acid induced toxicity in primary hippocampal cultures (Kimonides et al., 1998). In addition, DHEAs (10 nM) enhanced neuronal and glial survival and differentiation in vitro (Bologa et al., 1987; Roberts et al., 1987). These results suggest that DHEAs have a generally protective action against glutamate neurotoxicity. Further studies are underway to characterize the effect of DHEF treatment on injury-induced neuronal survival.
In response to brain injury, reactive astrocytes and microglia proliferate, secrete growth factors and cytokines, and participate in the repair of the disrupted blood–brain barrier. However, excessive proliferation of glia forms a physiological barrier that may impede plasticity and repair (Schumacher et al., 1996). In addition, microglia may contribute to the release of reactive oxygen species, nitric oxide, and glutamate. As a potent G6PDH inhibitor, DHEF may behave as an antioxidant and antiproliferative agent, retarding glial scar formation and enhancing neurite regrowth. Recent studies have shown that the progesterone, 17β-estradiol, testosterone, pregnenolone, and DHEA regulate gliosis in male rat brain (Cerro et al., 1995; García-Estrada et al., 1999). DHEA was the most potent neurosteroid in reducing cellular proliferation and gliotic scar formation in a penetrating brain injury model. However, no alterations in cerebral glial fibrillary acidic protein immunostaining were observed in DHEF-treated injured animals in this study (data not shown).
The GABA-active neurosteroids are modulators of synaptic events and may play a vital role in neuronal plasticity. DHEAs at low micromolar concentrations are allosteric antagonists of the GABAA receptors, and non-competitively modulate GABA-induced chloride currents in neurons by accelerating desensitization of GABAA receptors (Balieu and Robel, 1996; Imamura and Prasad, 1998; Majewska, 1995; Majewska et al., 1990). DHEAs reduce the amplitudes of GABA receptor-mediated inhibitory postsynaptic potentials (Spivak, 1994), implying a possible enhancement of depolarization in neurons. DHEA injected intraperitoneally in high doses (100–150 mg/kg) produces tonic-clonic seizures in mice (Heuser, 1961; Reddy and Kulkarni, 1998), but at lower doses causes sedation (Majewska et al., 1990). Neither sedation nor seizures were observed in these studies (DHEF, 25 mg/kg). Because different neurosteroids (as well as different doses of a single neurosteroid) modulate GABAA receptor function in opposing directions, the ratio between excitatory and inhibitory inputs may be the critical factor in reshaping synaptic activity (Majewska, 1995).
Finally, Reddy and Kulkarni (1998) have recently shown that DHEAs facilitated memory in a modified-passive avoidance memory retention task, and suggested a role for central glutamate receptors in the memory modulating effects of neurosteroids. Thus, DHEA and its analogs may counteract some of the effects of injury or aging on neurons, thus promoting neuronal survival and improving memory via multiple mechanisms of action (Flood et al., 1988a,b; Roberts et al., 1987).
Previous studies have demonstrated androgen cerebral vasoactivity in humans and animals. Hata et al. (1996) showed a transient reduction in the pulsatile index (a measure of vascular resistance) of fetal middle cerebral artery in full term women receiving 200 mg intravenous DHEA-sulfate, with no changes in umbilical artery or fetal heart rate (Hata et al., 1996). The direct vasodilatory activity of DHEA and DHEF on bovine middle cerebral artery smooth muscle demonstrates yet another potential avenue for improving recovery, although the mechanism is as yet unknown. While the IC50 is about four-fold the plasma level achieved with an oral dosage form in rats (200 mg/kg), and within an order of magnitude of those produced in patients (1600 mg/day, p.o.) in a phase I study, several-fold higher peak blood concentrations can be achieved with intraperitoneal administration, as in this study (A. Schwartz, personal communication). Development of a parenteral formulation will be necessary for clinical utility of this class of compounds.
There are no drugs available today that improve the behavioral deficits (changes in affect, memory, coordination, and loss of motor function) suffered by many patients with TBI. In our animal model, DHEF given preor up to 12 h postinjury significantly improved behavioral recovery at 3 days. Possible mechanisms of actions of DHEA and related steroids include attenuation of free radical production via G6PDH inhibition, allosteric modulation of GABAA receptors, and inhibition of NMDA, AMPA and kainic acid receptor toxicity via modulation of central σ receptors. Further work is underway to elucidate which of these mechanisms may contribute to the behavioral improvements seen in our model of TBI. Nevertheless, these results support further development of DHEF in the treatment of TBI.
ACKNOWLEDGMENTS
This research was supported in part by the Joint Section on Neurotrauma and Critical Care (AANS/CNS) Codman Fellowship to (A.S.M.), the Irving and Felicia Rubin Family Award to (K.I.S., R.K.N.), and NINDS R01 NS38654 (to K.I.S., R.K.N.).
REFERENCES
- ARAGNO M, PAROLA S, TAMAGNO E, et al. Oxidative derangement in rat synaptosomes induced by hyperglycaemia: restorative effect of dehydroepiandrosterone treatment. Biochem. Pharmacol. 2000;60:389–395. doi: 10.1016/s0006-2952(00)00327-0. [DOI] [PubMed] [Google Scholar]
- BABIOR B. The enzymatic basis for O·2− production by human neutrophils. Can. J. Physiol. Pharmacol. 1982;60:1353–1358. doi: 10.1139/y82-202. [DOI] [PubMed] [Google Scholar]
- BAULIEU E, ROBEL P. Dehydroepiandrosterone and dehydroepiandrosterone sulfate as neuroactive neurosteroids. J. Endocrinol. 1996;150:S221–S239. [PubMed] [Google Scholar]
- BOCCUZZI G, ARAGNO M, SECCIA M, et al. Protective effect of dehydroepiandrosterone against copper-induced lipid peroxidation in the rat. Free Radic. Biol. Med. 1997;22:1289–1294. doi: 10.1016/s0891-5849(96)00543-6. [DOI] [PubMed] [Google Scholar]
- BOLOGA L, SHARMA J, ROBERTS E. Dehydroepiandrosterone and its sulfated derivative reduce neuronal death and enhance astrocytic differentiation in brain cell cultures. J. Neurosci. Res. 1987;17:225–234. doi: 10.1002/jnr.490170305. [DOI] [PubMed] [Google Scholar]
- BREEN R, McGAUGH J. Facilitation of maze learning with post trial injections of picrotoxin. J. Comp. Physiol. Psychol. 1961;54:498–501. doi: 10.1037/h0046436. [DOI] [PubMed] [Google Scholar]
- CERRO S, GARCIA-ESTRADA J, GARCIA-SEGURA L. Neuroactive steroids regulate astroglial morphology in hippocampal cultures from adult rats. Glia. 1995;14:65–71. doi: 10.1002/glia.440140109. [DOI] [PubMed] [Google Scholar]
- DERSKEN RH. Dehydroepiandrosterone (DHEA) and systemic lupus erythematosus. Semin. Arthritis Rheum. 1998;27:335–347. doi: 10.1016/s0049-0172(98)80013-9. [DOI] [PubMed] [Google Scholar]
- DIXON C, CLIFTON GL, LIGHTHALL JW, et al. A controlled cortical impact model of traumatic brain injury in the rat. J. Neurosci. Methods. 1991;39:253–262. doi: 10.1016/0165-0270(91)90104-8. [DOI] [PubMed] [Google Scholar]
- ELLIS EF, POLICE RJ, RICE LY, et al. Increased plasma pge2, 6–keto-pgf1 alpha, and 12–hete levels following experimental concussive brain injury. J. Neurotrauma. 1989;6:31–37. doi: 10.1089/neu.1989.6.31. [DOI] [PubMed] [Google Scholar]
- FENG L, XIA Y, GARCIA GE, et al. Involvement of reactive oxygen intermediates in cyclooxygenase-2 expression induced by interleukin-1, tumor necrosis factor–alpha, and lipopolysaccharide. J. Clin. Invest. 1995;95:1669–1675. doi: 10.1172/JCI117842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- FLOOD J, ROBERTS E. Dehydroepiandrosterone sulfate improves memory in aging mice. Brain Res. 1988a;448:178–181. doi: 10.1016/0006-8993(88)91116-x. [DOI] [PubMed] [Google Scholar]
- FLOOD J, SMITH GE, ROBERTS E. Dehydroepiandrosterone and its sulfate enhance memory retention in mice. Brain Res. 1988b;447:269–278. doi: 10.1016/0006-8993(88)91129-8. [DOI] [PubMed] [Google Scholar]
- GARCÍA-ESTRADA J, LUQUÍN S, FERNÁNDEZ AM, et al. Dehydroepiandrosterone, pregnenolone and sex steroids down-regulate reactive astroglia in the male rat brain after a penetrating brain injury. Int. J. Dev. Neurosci. 1999;17:145–151. doi: 10.1016/s0736-5748(98)00065-3. [DOI] [PubMed] [Google Scholar]
- GORDON G, MACKOW MC, LEVY HR. On the mechanism of interaction of steroids with human glucose 6–phosphate dehydrogenase. Arch. Biochem. Biophys. 1995;318:25–29. doi: 10.1006/abbi.1995.1199. [DOI] [PubMed] [Google Scholar]
- HALL E. Free radicals and lipid peroxidation. In: Narayan R, Wilberger J, Povlishock J, editors. Neurotrauma. McGraw-Hill; New York: 1996. pp. 1405–1419. [Google Scholar]
- HAMM RJ, DIXON CE, GBADEBO DM, et al. Cognitive deficits following traumatic brain injury produced by controlled cortical impact. J. Neurotrauma. 1992;9:11–20. doi: 10.1089/neu.1992.9.11. [DOI] [PubMed] [Google Scholar]
- HASTINGS LP, LEWBART ML, SCHWARTZ AG. Dehydroepiandrosterone and two structural analogs inhibit 12-o-tetradecanoylphorbol-13–acetate stimulation of prostaglandin E2 content in mouse skin. Carcinogenesis. 1988;9:1099–1102. doi: 10.1093/carcin/9.6.1099. [DOI] [PubMed] [Google Scholar]
- HATA T, SENOH D, HATA K, et al. Effect of dehydroepiandrosterone sulfate on fetal middle cerebral artery flow velocity waveforms in term pregnancy. Acta Obstet. Gynecol. Scand. 1996;75:343–346. doi: 10.3109/00016349609033328. [DOI] [PubMed] [Google Scholar]
- HERSCHMAN H, XIE W, REDDY S. Inflammation, reproduction, cancer and all that.… The regulation and role of the inducible prostaglandin synthase. Bioessays. 1995;17:1031–1037. doi: 10.1002/bies.950171207. [DOI] [PubMed] [Google Scholar]
- HEUSER G, EIDELBERG E. Steroid-induced convulsions in experimental animals. Endocrinology. 1961;69:915–924. doi: 10.1210/endo-69-5-915. [DOI] [PubMed] [Google Scholar]
- HEWETT S, ULIASZ TF, VIDWANS AS, et al. Cyclooxygenase-2 contributes to n-methyl-d-aspartate–mediated neuronal cell death in primary cortical cell culture. J. Pharm. Exp. Ther. 2000;293:417–425. [PubMed] [Google Scholar]
- IMAMURA M, PRASAD C. Modulation of GABA-gated chloride ion influx in the brain by dehydroepiandrosterone and its metabolites. Biochem. Biophys. Res. Commun. 1998;243:771–775. doi: 10.1006/bbrc.1998.8177. [DOI] [PubMed] [Google Scholar]
- IMLAY J, LINN S. DNA damage and oxygen radical toxicity. Science. 1988;240:1302–1309. doi: 10.1126/science.3287616. [DOI] [PubMed] [Google Scholar]
- IRWIN S, BENUAZIZI A. Pentylenetetrazol enhances memory function. Science. 1966;152:100–102. doi: 10.1126/science.152.3718.100. [DOI] [PubMed] [Google Scholar]
- KIMONIDES VG, KHATIBI NH, SVENDSEN CN, et al. Dehydroepiandrosterone (DHEA) and dhea-sulfate (DHEAS) protect hippocampal neurons against excitatory amino acid–induced neurotoxicity. Proc. Natl. Acad. Sci. USA. 1998;95:1852–1857. doi: 10.1073/pnas.95.4.1852. [DOI] [PMC free article] [PubMed] [Google Scholar]
- KIPPER-GALPERIN M, GALILLY R, DANENBERG HD, et al. Dehydroepiandrosterone selectively inhibits production of tumor necrosis factor–α and interlukin-6 in astrocytes. Int. J. Dev. Neurosci. 1999;17:765–775. doi: 10.1016/s0736-5748(99)00067-2. [DOI] [PubMed] [Google Scholar]
- KOSSMANN T, STAHEL PF, LENZLINGER PM, et al. Interleukin-8 released into the cerebrospinal fluid after brain injury is associated with blood–brain barrier dysfunction and nerve growth factor production. J. Cereb. Blood Flow Metab. 1997;17:280–289. doi: 10.1097/00004647-199703000-00005. [DOI] [PubMed] [Google Scholar]
- LI H, KLEIN GM, SUN P, et al. Dehydroepiandrosterone (DHEA) reduces neuronal injury in a rat model of global cerebral ischemia. Brain Res. 2001;888:263–266. doi: 10.1016/s0006-8993(00)03077-8. [DOI] [PubMed] [Google Scholar]
- MAJEWSKA MD. Neuronal actions of dehydroepiandrosterone. Possible roles in brain development, aging, memory, and affect. Ann. N.Y. Acad. Sci. 1995;774:111–120. doi: 10.1111/j.1749-6632.1995.tb17375.x. [DOI] [PubMed] [Google Scholar]
- MAJEWSKA M, DEMIRGOREN S, SPIVAK CE, et al. The neurosteroid dehydroepiandrosterone sulfate is an allosteric antagonist of the GABAA receptor. Brain Res. 1990;526:143–146. doi: 10.1016/0006-8993(90)90261-9. [DOI] [PubMed] [Google Scholar]
- MARCZYNSKI T, ARTWOHL J, MARCZYNSKA B. Chronic administration of flumazenil increases life span and protects rats from age-related loss of cognitive functions: a benzodiazepine/GABAergic hypothesis of brain aging. Neurobiol. Aging. 1994;15:69–84. doi: 10.1016/0197-4580(94)90146-5. [DOI] [PubMed] [Google Scholar]
- MARLETTA M. Nitric oxide synthase: aspects concerning structure and catalysis. Cell. 1994;78:927–930. doi: 10.1016/0092-8674(94)90268-2. [DOI] [PubMed] [Google Scholar]
- MASFERRER JL, SEIBERT K, ZWEIFEL B, et al. Endogenous glucocorticoids regulate an inducible cyclooxygenase enzyme. Proc. Natl. Acad. Sci. USA. 1992;89:3917–3921. doi: 10.1073/pnas.89.9.3917. [DOI] [PMC free article] [PubMed] [Google Scholar]
- MAURICE T, JUNIEN JL, PRIVAT A. Dehydroepiandrosterone sulfate attenuates dizocilpine-induced learning impairment in mice via sigma 1–receptors. Behav. Brain Res. 1997;83:159–164. doi: 10.1016/s0166-4328(97)86061-5. [DOI] [PubMed] [Google Scholar]
- McINTOSH T, VINK R, NOBLE L, et al. Traumatic brain injury in the rat: characterization of a lateral fluid-percussion model. Neuroscience. 1989;28:233–244. doi: 10.1016/0306-4522(89)90247-9. [DOI] [PubMed] [Google Scholar]
- MEI J, HURSTING S, PHANG L. Inhibitory effects of DHEA and 16b-flouro-5–androsten-17–one on nitric oxide generation in vitro and in vivo mouse macrophages. Proc. Am. Assoc. Cancer Res. 1995;36:585. [Google Scholar]
- MOORE SA, YODER E, RICH G, et al. Regulation of cerebrovascular cyclooxygenase-2 by pro- and anti-inflammatory cytokines. Adv. Exp. Med. Biol. 1999;469:125–129. doi: 10.1007/978-1-4615-4793-8_19. [DOI] [PubMed] [Google Scholar]
- MORALES A, NOLAN JJ, NELSON JC, et al. Effects of replacement dose of dehydroepiandrosterone in men and women of advancing age. J. Clin. Endocrinol. Metab. 1994;78:1360–1367. doi: 10.1210/jcem.78.6.7515387. [DOI] [PubMed] [Google Scholar]
- MORGANTI-KOSSMAN MC, LENZLINGER PM, HANS V, et al. Production of cytokines following brain injury: beneficial and deleterious for the damaged tissue. Mol. Psychiatry. 1997;2:133–136. doi: 10.1038/sj.mp.4000227. [DOI] [PubMed] [Google Scholar]
- OTT L, McCLAIN CJ, GILLESPIE M, et al. Cytokines and metabolic dysfunction after severe head injury. J. Neurotrauma. 1994;11:447–472. doi: 10.1089/neu.1994.11.447. [DOI] [PubMed] [Google Scholar]
- PASHKO L, LEWBART ML, SCHWARTZ AG. Inhibition of 12–o-tetradecanoylphorbol-13–acetate-promoted skin tumor formation in mice by 16 alpha-fluoro-5–androsten-17–one and its reversal by deoxyribonucleo-sides. Carcinogenesis. 1991;12:2189–2192. doi: 10.1093/carcin/12.11.2189. [DOI] [PubMed] [Google Scholar]
- RAINERI R, LEVY HR. On the specificity of steroid interaction with mammary gland glucose-6–phosphate dehydrogenase. Biochemistry. 1970;9:2233–2243. doi: 10.1021/bi00813a003. [DOI] [PubMed] [Google Scholar]
- RATKO T, DETRISAC CJ, MEHTA RG, et al. Inhibition of rat mammary gland chemical carcinogenesis by dietary dehydroepiandrosterone or a fluorinated analogue of dehydroepiandrosterone. Cancer Res. 1991;51:481–486. [PubMed] [Google Scholar]
- REDDY DS, KULKARNI SK. The effects of neurosteroids on acquisition and retention of a modified passive-avoidance learning task in mice. Brain Res. 1998;791:108–116. doi: 10.1016/s0006-8993(98)00085-7. [DOI] [PubMed] [Google Scholar]
- ROBERTS E, BOLOGA L, FLOOD JF, et al. Effects of dehydroepiandrosterone and its sulfate on brain tissue in culture and on memory in mice. Brain Res. 1987;406:357–362. doi: 10.1016/0006-8993(87)90807-9. [DOI] [PubMed] [Google Scholar]
- ROBERTSON C. Nitric oxide saturation technique for CBF measurement. In: Narayan R, Wilberger J, Povlishock J, editors. Neurotrauma. McGraw-Hill; New York: 1996. pp. 487–501. [Google Scholar]
- ROOF R, HOFFMAN SW, STEIN DG. Progesterone protects against lipid peroxidation following traumatic brain injury in rats. Mol. Chem. Neuropathol. 1997;31:1–11. doi: 10.1007/BF02815156. [DOI] [PubMed] [Google Scholar]
- SADOWSKI I, WRIGHT JA, ISRAELS LG. A permeabilized cell system for studying regulation of aryl hydrocarbon hydroxylase: NADPH as rate limiting factor in benzópyrene metabolism. Int. J. Biochem. 1985;17:1023–1025. doi: 10.1016/0020-711x(85)90250-2. [DOI] [PubMed] [Google Scholar]
- SARA S, VANKOV A, HERVE A. Locus coeruleus-evoked responses in behaving rats: a clue to the role of noradrenaline in memory. Brain Res. Bull. 1994;35:457–465. doi: 10.1016/0361-9230(94)90159-7. [DOI] [PubMed] [Google Scholar]
- SCHUMACHER M, ROBEL P, BAULIEU EE. Development and regeneration of the nervous system: a role for neurosteroids. Dev. Neurosci. 1996;18:6–21. doi: 10.1159/000111391. [DOI] [PubMed] [Google Scholar]
- SCHWARTZ A, PASHKO LL. Cancer chemo-prevention with the adrenocortical steroid dehydroepiandrosterone and structural analogs. J. Cell Biochem. 1993;17G:73–79. doi: 10.1002/jcb.240531114. [DOI] [PubMed] [Google Scholar]
- SCHWARTZ A, PASHKO LL. Role of adrenocortical steroids in mediating cancer-preventive and age-retarding effects of food restriction in laboratory rodents. J. Gerontol. 1994;49:B37–B41. doi: 10.1093/geronj/49.2.b37. [DOI] [PubMed] [Google Scholar]
- SCHWARTZ A, LEWBART ML, PASHKO LL. Novel dehydroepiandrosterone analogues with enhanced biological activity and reduced side-effects in mice and rats. Cancer Res. 1988;48:4817–4822. [PubMed] [Google Scholar]
- SCHWARTZ A, FAIRMAN DK, POLANSKY M, et al. Inhibition of 7,12–dimethyl benz(a)anthracene–initiated and tetradecanoylphorbol-13-acetate–promoted skin papilloma formation in mice by dehydroepiandrosterone and two synthetic analogs. Carcinogenesis. 1989;10:1809–1813. doi: 10.1093/carcin/10.10.1809. [DOI] [PubMed] [Google Scholar]
- SEIBERT K, ZHANG Y, LEAHY K, et al. Pharmacological and biochemical demonstration of the role of cyclooxygenase-2 in inflammation and pain. Proc. Natl. Acad. Sci. USA. 1994;91:12013–12017. doi: 10.1073/pnas.91.25.12013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- SIOUTOS P, OROZCO JA, CARTER LP, et al. Continuous regional cerebral cortical blood flow monitoring in head-injured patients. Neurosurgery. 1995;36:943–950. doi: 10.1227/00006123-199505000-00009. [DOI] [PubMed] [Google Scholar]
- SOARES HD, HICKS RR, SMITH D, et al. Inflammatory leukocytic recruitment and diffuse neuronal degeneration are separate pathological processes resulting from traumatic brain injury. J. Neurosci. 1995;15:8223–8233. doi: 10.1523/JNEUROSCI.15-12-08223.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- SPIVAK CE. Desensitization and noncompetitive blockade of GABAA receptors in ventral midbrain neurons by a neurosteroid dehydroepiandrosterone sulfate. Synapse. 1994;16:113–122. doi: 10.1002/syn.890160205. [DOI] [PubMed] [Google Scholar]
- STANTON P, SARVEY JM. Depletion of norepinephrine, but not serotonin, reduces long-term potentiation in the dentate gyrus of rat hippocampal slices. J. Neurosci. 1985;5:2169–2176. doi: 10.1523/JNEUROSCI.05-08-02169.1985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- STEIN D, FULOP Z. Progesterone and recovery after traumatic brain injury: an overview. Neuroscientist. 1990;4:435–422. [Google Scholar]
- STRAUSS K. Recent advances in understanding the pathophysiology of neurotrauma. Trauma Q. 1998;13:353–372. [Google Scholar]
- STRAUSS KI, MARINI AM. Cyclooxygenase-2 inhibition protects cultured cerebellar granule neurons from glutamate-mediated excitotoxic cell death. J. Neurotrauma. 2002;19:627–638. doi: 10.1089/089771502753754091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- STRAUSS KI, BARBE MF, MARSHALL R, et al. Prolonged cyclooxygenase-2 induction in neurons and glia following traumatic brain injury in the rat. J. Neurotrauma. 2000;17:695–711. doi: 10.1089/089771500415436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- VAN VOLLENHOVEN R, ENGLEMAN EG, McGUIRE JL. An open study of dehydroepiandrosterone in systemic lupus erythematosus. Arthritis Rheum. 1994;37:1305–1310. doi: 10.1002/art.1780370906. [DOI] [PubMed] [Google Scholar]
- VAN VOLLENHOVEN R, ENGLEMAN EG, McGUIRE JL. Dehydroepiandrosterone in systemic lupus erythematosus. Results of a double-blind, placebo-controlled, randomized clinical trial. Arthritis Rheum. 1995;38:1826–1831. doi: 10.1002/art.1780381216. [DOI] [PubMed] [Google Scholar]
- VAN VOLLENHOVEN R, MORABITO LM, ENGLE-MAN EG, et al. Treatment of systemic lupus erythematosus with dehydroepiandrosterone: 50 patients treated up to 12 months. J. Rheumatol. 1998;25:285–289. [PubMed] [Google Scholar]
- WENDLING W, HARAKAL C. Effects of calcium antagonists on isolated bovine cerebral arteries: inhibition of constriction and calcium-45 uptake induced by potassium or serotonin. Stroke. 1987;18:591–598. doi: 10.1161/01.str.18.3.591. [DOI] [PubMed] [Google Scholar]
- WENDLING W, HARAKAL C. Effects of prostaglandin F2 alpha and thromboxane A2 analogue on bovine cerebral arterial tone and calcium fluxes. Stroke. 1991;22:66–72. doi: 10.1161/01.str.22.1.66. [DOI] [PubMed] [Google Scholar]
- WENDLING W, DANIELS FB, CHEN D, et al. Ketamine directly dilates bovine cerebral arteries by acting as a calcium entry blocker. J. Neurosurg. Anesthesiol. 1994;6:186–192. doi: 10.1097/00008506-199407000-00007. [DOI] [PubMed] [Google Scholar]
- WILLIAMS P, JONES RH, RADEMACHER TW. Reduction in the incidence and severity of collagen-induced arthritis in dba/1 mice, using exogenous dehydroepiandrosterone. Arthritis Rheum. 1997;40:907–911. doi: 10.1002/art.1780400519. [DOI] [PubMed] [Google Scholar]
- WOLKOWITZ O, REUS VI, ROBERTS E, et al. Antidepressant and cognition-enhancing effects of DHEA in major depression. Ann. N.Y. Acad. Sci. 1995;774:337–339. doi: 10.1111/j.1749-6632.1995.tb17403.x-i1. [DOI] [PubMed] [Google Scholar]
- YAMAGATA K, ANDREASSON KI, KAUFMANN WE, et al. Expression of a mitogen-inducible cyclooxygenase in brain neurons: Regulation by synaptic activity and glucocorticoids. Neuron. 1993;11:371–386. doi: 10.1016/0896-6273(93)90192-t. [DOI] [PubMed] [Google Scholar]
- YAN HQ, BANOS MA, HERREGODTS P, et al. Expression of interleukin (IL)–1 beta, IL-6 and their respective receptors in the normal rat brain and after injury. Eur. J. Immunol. 1992;22:2963–2971. doi: 10.1002/eji.1830221131. [DOI] [PubMed] [Google Scholar]