

Abstract
The relationship of genotype, fitness components, and fitness can be complicated by genetic effects such as pleiotropy and epistasis and by heterogeneous environments. However, because it is often difficult to measure genotype and fitness directly, fitness components are commonly used to estimate fitness without regard to genetic architecture. The small bacteriophage φX174 enables direct evaluation of genetic and environmental effects on fitness components and fitness. We used 15 mutants to study mutation effects on attachment rate and fitness in six hosts. The mutants differed from our lab strain of φX174 by only one or two amino acids in the major capsid protein (gpF, sites 101 and 102). The sites are variable in natural and experimentally evolved φX174 populations and affect phage attachment rate. Within the limits of detection of our assays, all mutations were neutral or deleterious relative to the wild type; 11 mutants had decreased host range. While fitness was predictable from attachment rate in most cases, 3 mutants had rapid attachment but low fitness on most hosts. Thus, some mutations had a pleiotropic effect on a fitness component other than attachment rate. In addition, on one host most mutants had high attachment rate but decreased fitness, suggesting that pleiotropic effects also depended on host. The data highlight that even in this simple, well-characterized system, prediction of fitness from a fitness component depends on genetic architecture and environment.
IN natural populations and even in most model organisms, it is not feasible to directly measure the relationship between genotype and fitness. For most species it is still not practical to sequence genomes for many individuals, and generation times are too long and life histories too complex to directly measure fitness. Instead, fitness components are used as surrogates of fitness. Measurements of variation in fitness components are used to identify phenotypic targets of selection or make predictions about evolutionary response through analysis with quantitative genetics methods. These methods were developed on the assumptions that fitness components are indicators of fitness and that variation in fitness components is correlated to underlying genetic variation. However, few empirical studies have been able to directly examine how often or how well fitness is predictable from fitness components or how genetic and phenotypic variation correlate. To appropriately use quantitative genetics methods, it is important to gain insight about whether assumptions of the relationship between genotype, fitness components, and fitness hold, especially since genetic effects such as pleiotropy and epistasis, as well as environmental effects, can influence phenotypic expression. Questions such as Do different mutations at the same locus influence fitness through the same fitness component? and Can environment affect predictability of fitness from fitness components? are fundamental to understanding when the assumptions of quantitative genetics methods may fail, yet they remain unexplored. Since current methods in quantitative genetics are invaluable for studying evolutionary outcome in complex biological systems, it is worth investing substantial effort into understanding when or how assumptions of the tool may be inappropriate. Such data could facilitate refinement of methods or more appropriate experimental design, resulting in increased accuracy in predicting evolutionary outcome.
An explicit understanding of the effects of genetic architecture and environment on fitness can be addressed only by an integrated approach to examining the interdependencies of genotype, fitness components, and fitness. The bacteriophage φX174 is well suited for dissecting this relationship. The small genome of φX174 facilitates routine site-directed mutagenesis and makes full-genome sequencing practical. Genotypes differing at known amino acid sites can be constructed and studied. Also, the life cycle of φX174 is relatively simple and generation time is short. For example, in only 20 min a single phage can complete an infection cycle from host attachment to lysis, producing 100 progeny. Thus, it is feasible to directly measure fitness components and fitness and also to replicate measures in multiple environments (Bull et al. 1997).
Studies of φX174 and related phage have identified specific mutations with known consequences on a fitness component (Crill et al. 2000) and fitness (Bull et al. 1997, 2000; Wichman et al. 1999, 2000, 2005; Crill et al. 2000; Holder and Bull 2001). For example, during experimental evolution on particular hosts, specific substitutions occur repeatedly in the phage major capsid protein F (gpF 101/102, as well as several other sites) (Bull et al. 1997; Wichman et al. 1999, 2000, 2005; Crill et al. 2000). Since the gpF 101/102 residues have been shown to affect host attachment and fitness (Crill et al. 2000), they served as the basis for the current study. The gpF 101/102 attachment-related residues are also variable among related phages (Crill et al. 2000) and within wild φX174 populations (Rokyta et al. 2006), suggesting that they are important host switching sites in nature. They are located on the outer surface of gpF near the spike with the amino acid side chains directed outward (Figure 1) and have not been implicated in structural interactions in the mature capsid (McKenna et al. 1994). We predicted that in genotypes differing only at the gpF 101/102 attachment-related sites, fitness would be predictable from the fitness component attachment rate. Examining effects of gpF 101/102 mutations on attachment rate and fitness in multiple hosts provides a simple framework for evaluating effects of genetic and environmental factors on the predictability of fitness from a single fitness component.
Figure 1.

Location of gpF 101 and 102 on the capsid surface. The mature phage capsid is an icosahedron of 12 pentameric capsomeres. Each capsomere contains 5 units of major capsid gpF (426 aa each) and 5 units of spike gpG (138 aa each). GpF comprises most of the outer surface of the virion while each pentamer of gpG forms a large spike that protrudes. An isolated pentamer of gpG (orange) and gpF (blue) is shown from the side. Wild-type gpF 101/102 amino acids (G/Y in black/red) are indicated on each of the 5 gpF subunits. Note that functional groups protrude outward. Structure was modified from McKenna et al. (1994) by G. Daughdrill, using Insight II software from Accelerys (Cambridge, MA).
We studied the interconnection of genotype, a fitness component, and fitness by measuring attachment rate and fitness for bacteriophage mutants exposed to six hosts. We constructed mutants of gpF 101/102 sites through site-directed mutagenesis of a lab-adapted wild type. Thus, genotypes in this study differed by only one or two mutations in the genome. Using a phage life-history model that predicts effects of attachment rate on fitness, we examined whether genotype and environment affect predictability of fitness from attachment rate. In addition, because experimental evolution studies have shown that specific adaptive substitutions occur at gpF 101/102 repeatedly in response to particular hosts, we examined whether other mutations at these sites were adaptive and whether they could alter host range.
MATERIALS AND METHODS
Phage, hosts, and construction of mutants:
φX174 (GenBank accession AF176034) served as the wild type. φX174 is a small (5386 bases) lytic bacteriophage. The icosahedral capsid has a single-stranded circular DNA genome that encodes 11 genes (Sanger et al. 1977). Mutants were made by oligonucleotide-mediated mutagenesis (Sambrook et al. 1989). Briefly, the phage single-stranded genome was extracted and used as a template for constructing mismatched heteroduplexes through annealing, extension, and ligation of mutagenized oligonucleotides. Mutations were specifically introduced in gene F sites 101/102 by using a 30-nt primer that was completely randomized in the gene F 101/102 codons (5′ GGC ATG GTC AAT NNN NNN AGT AGT GTT AAC 3′). Following an extension/ligation reaction, double-stranded DNA products were electroporated into Escherichia coli C (Ec C) and Salmonella enterica (St GalE) hosts (hosts described in Bull et al. 1997 and Table 1). Electroporated cells were then mixed with 7% top agar and overlaid on LB agar plates to allow plaque formation. Plaques were screened (466 total) with radiolabeled oligonucleotide probes of the wild-type sequence. Plaques that did not hybridize with the wild-type probe were selected for sequencing in the gene F 101/102 region to confirm genotype. A total of 15 mutants (listed in Table 2A) were recovered. While our randomized primer potentially allowed for 400 possible amino acid combinations at gpF 101/102, the number of plaques screened was not intended to be exhaustive. The wild type and 15 mutants were isolated by plaque purification for phenotypic analyses that were done on six hosts (Table 1). All hosts are lipopolysaccharide (LPS) rough mutants that produce lipid A, the inner core, and outer core LPS components, but lack O-antigens. Ec C is a standard host used to propagate φX174 in the lab; the wild type is thought to be well adapted to this host. St GalE was used since gpF 101/102 sites undergo adaptive substitution in response to this host (Crill et al. 2000). Ec waaL makes the same LPS structure as Ec C but is derived from a different strain of E. coli. Ec waaV is identical to Ec waaL except for one mutation in the LPS biosynthetic pathway that results in lack of a terminal glucose (Heinrichs et al. 1998). St rfb is derived from the same strain as St GalE, but St rfb produces a full outer core LPS (Nikaido et al. 1967), while in St GalE the outer core is almost completely lacking. Shigella sonnei (Ss) phase II LPS structure is similar to Ec C except that it lacks a 3-deoxy-d-manno-2-octulosonic acid (KDO) group in the inner core (Gamian and Romanowska 1982).
TABLE 1.
Hosts used in electroporation and attachment rate and fitness assays
| Hosta | Parent strain | Derived strain | LPS mutation | Labelb |
|---|---|---|---|---|
| Escherichia coli C | Ec C | |||
| Salmonella enterica serovar typhimurium | LT2, type I restrictionless | IJ750 | GalE | St GalE |
| S. enterica serovar typhimurium | LT2, type I restrictionless | SA1627 | rfb | St rfb |
| E. coli | F470 | CGW317 | waaL | Ec waaL |
| E. coli | F470 | CGW311 | waaV | Ec waaV |
| Shigella sonnei | Phase II | Ss |
Hosts that are underlined were used for electroporation and by Crill et al. (2000).
Labels for hosts used in this study.
TABLE 2.
Mutants that were phenotypically analyzed in this study (A) and variation at gpF 101/102 seen previously (B)
| Codon
|
aa site
|
Unique codons. no. (no.)a
|
||||
|---|---|---|---|---|---|---|
| 101 | 102 | 101 | 102 | Totalb | ||
| A. | ||||||
| Single 101 only | aag | tat | K | Y | 1 (4) | 2E |
| cat | tat | H | Y | 2 (4) | 17E,S | |
| cag | tat | Q | Y | 3 (4) | 14E,S | |
| att | tat | I | Y | 1 (6) | 1E | |
| ttg | tat | L | Y | 4 (12) | 7E,S | |
| tat | tac | Y | Y | 1 (4) | 1E | |
| Double 101 and 102 | att | aat | I | N | 1 (6) | 2S |
| aag | ttg,tta | K | L | 2 (12) | 2S | |
| ctc | aat | L | N | 1 (12) | 3S | |
| atg | aat | M | N | 1 (2) | 1S | |
| ttt | aat | F | N | 1 (4) | 3E,S | |
| aat | aat | N | N | 1 (4) | 1E | |
| cag | aat | Q | N | 2 (4) | 10E,S | |
| cag | ttt | Q | F | 1 (4) | 6E,S | |
| cgt | ata | R | I | 1 (18) | 1S | |
| aa site
| |||||
|---|---|---|---|---|---|
| Sourcec | Phage (closest relative) | 101 | 102 | Hostd | Reference or GenBank accession |
| B. | |||||
| Lab/wild | φX174 | G | Y | E,S | J02482 |
| Lab/wild | G4 | S | G | E | V00657 |
| Lab | S13 | S | Y | E,S | M14428 |
| Lab | α3 | R | Y | E | X60322 |
| Lab | φK | K | Y | E,S | X60323 |
| Wild | (φX174) | R | Y | E,S | Rokyta et al. (2006) |
| Wild | (φX174) | G | S | S | |
| Wild | (W13) | R | G | E,S | |
| Exp. evol. | φX174 | D | Y | S | Crill et al. (2000) |
| φX174 | D | C | S | ||
| φX174 | H | Y | S | Wichman et al. (2000) | |
| φX174 | N | Y | S | ||
| φX174 | G | F | E | ||
| φX174 | G | H | E | ||
| φX174 | R | Y | E,S | ||
The number of unique DNA sequences recovered that encode the amino acid combination is shown. The number in parentheses represents the total number of unique codon combinations that could encode each amino acid combination.
The total number of plaques recovered from the screening process is shown. Hosts on which the mutants were isolated are indicated in superscript: E, Ec C; S, St GalE.
In the wild phages, gpF 101/102 are nonvariant within G4-like and W13-like groups; the φX174-like group has three variants. GenBank accession nos. are AY751298 and DQ079869–DQ079909.
Hosts that these genotypes can infect (lab/wild) or on which they were evolved (exp. evol.) are shown. E, Ec C; S, St GalE.
Attachment rate assays:
Attachment rates (k) for all 15 mutants and the wild type were measured on the six hosts described above. All assays were conducted in a shaking water bath at 200 rpm and 38.5°. Cultures were grown in 10 ml LB (10 g NaCl, 10 g Bacto tryptone, and 5 g yeast extract per liter and 2 mm CaCl2) in 125-ml Erlenmeyer flasks. Three to seven replicate assays for each mutant and at least seven for wild type were conducted. Each replicate was conducted on a separately prepared plaque isolate to account for possible variation arising from plaque sample preparation procedures. To minimize day effects, assays for a phage sample were done on all six different hosts within 30 min of each other. As a similar precaution, phage genotype replicates were conducted on different days. Each assay was begun with the same quantity of previously prepared host culture (stock cultures were harvested during exponential phase and stored at −80°). All assay cultures were grown for 80 min before addition of phage. Host density for each assay, measured by OD600 on a spectrophotometer, was similar across hosts. Phage were incubated for 5 min (Ss and St rfb) or 7 min (Ec C, Ec waaL, Ec waaV, and St GalE). The single-time-point attachment rate assay and times chosen for incubation were based on results from multiple-time-point attachment assays conducted on φX174 isolates in previous experiments (K. M. Pepin, unpublished data). Following incubation, 1 ml of culture was immediately spun in a 1.5-ml microcentrifuge tube at 15,000 rpm for 4 min to pellet free cells and cells with attached phage. The supernatant was titered to determine the number of unattached phage. Attachment rate was expressed as k = −ln(Nf/No)/Ct, where Nf is the number of unattached phage, No is the total number of phage added, C is the host density, and t is the incubation time in minutes.
Fitness assays:
Conditions and levels of replication in fitness assays were the same as those in attachment assays except that phage were incubated for 40 min before being sampled. Phage were sampled by transferring 1 ml of culture to 50 μl CHCl3, vortexing for 10 sec, spinning at 15,000 rpm for 4 min, and immediately titering the supernatant. Growth rates (fitness = w) were calculated as the number of population doublings per hour, w = [log2(Nt) − log2(No)]/t, where Nt is the total number of phage after incubation, No is the number of phage added to the assay, and t is the incubation time (Bull et al. 1997).
Model of w–k relationship:
Empirical data and theoretical work have produced a framework for predicting fitness from three phage life-history parameters: attachment rate, latent period, and burst size (Levin et al. 1977; Wang et al. 1996; Abedon et al. 2001; Bull et al. 2005). Attachment rate to hosts, which is the fitness component measured in our study, is a fitness component analogous to food handling efficiency in animals or nutrient uptake efficiency in plants. The relationship of attachment rate and fitness is predicted to be logarithmic such that much greater gains in fitness occur from increases in low attachment rates relative to increases in high attachment rates (Figure 2). We used the following equation, derived in Bull et al. (2006), to predict fitness from observed attachment rates,
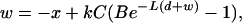 |
(1) |
where w is the long-term growth rate in phage per minute, x is the phage death rate, k is the attachment rate of phage per milliliter per cell per minute, C is the host density per milliliter, B (burst size) is the number of progeny each phage produces, L (latent period) is the time from adsorption to lysis, and d is the cell death rate. Average optical density at the start of attachment assays was used to calculate C for each host where 8 × 108 cells are OD600 = 1. B and L parameters were measured by growth curve assays for the wild type on all six hosts. These parameter values are shown in Table 3. Under our assay conditions, which include a 40-min growth period in a constant environment, the values of x and d are zero. While phage binding has been described as a multistep process, the parameter k refers only to the rate at which phage attach to hosts. Subsequent events related to the initial interaction of the phage with a host are considered part of parameter L (latent period). L includes most events in the phage life cycle: the eclipse reaction where attached phage undergo a conformational change to eject genomic DNA into the host, replication of genomic DNA, production of phage proteins, assembly of mature virions, and release of progeny virus from the host. Since the complete life cycle includes the time for released progeny to encounter new hosts, Equation 1 was derived by treating the growth process as an exponential random variable.
Figure 2.

Attachment rate–fitness relationship for genotypes on each host. The host is shown in each top left corner. Fitness (w) is the number of population doublings per hour as described in materials and methods. Attachment rate (k) is per milliliter per cell per phage per minute and has been multiplied by 108 for visual simplicity. The solid line indicates the attachment rate–fitness relationship predicted by the model of Bull et al. (2006). Using this model, expected mutant fitnesses were estimated from observed mutant attachment rates and observed wild-type L and B parameters (Table 3). Points indicate observed fitness–attachment rate relationships for each genotype. Large shaded diamonds are the wild type. Error bars are 99.948% confidence intervals for least-squares means of fitness and attachment rates [GLM: w or k = host + genotype + (host × genotype)]. Genotypes and error bars are shown only where observed and predicted fitnesses are significantly different from those of the model (as indicated in Table 4).
TABLE 3.
Parameters used in calculating model predictions
| Host
|
||||||
|---|---|---|---|---|---|---|
| St rfb | St GalE | Ss | Ec C | Ec waaL | Ec waaV | |
| C × 108 | 2.1 | 1.5 | 1.7 | 2.7 | 2.6 | 2.1 |
| B | 98 | 138 | 106 | 175 | 74 | 24 |
| L | 16 | 19.5 | 17 | 17.5 | 17.5 | 20 |
| T | 60 | 60 | 60 | 60 | 60 | 60 |
Data are from single-step growth curves for the wild type on each host.
C, cell density (cells per milliter); B, burst size (number of progeny per infection); L, latent period (mean time to burst in minutes); T, duration of assay (minutes).
Equation 1 cannot be solved analytically. We used a C++ program written by J. J. Bull to get numerical approximations of fitness and attachment rates on each host from wild-type B and L parameters and host density C. For each set of B, L, and C parameters, the program generates a list of w–k values over a specified range. The lists were used to assign a predicted fitness value to each observed attachment rate. The data were interpreted as follows. For genotypes that do not follow the predicted w–k relationship, fitness is influenced by a fitness component other than k. That is, the mutation affects a trait that influences B or L parameters instead of, or in addition to, k. Due to measurement error, some values of k were measured to be negative. Since the model does not act similarly for negative and positive values of k, these data were taken to be zero.
Statistical analyses:
Attachment rate–fitness relationship:
We examined predictability of fitness from attachment rate by testing whether the observed and predicted fitnesses for each genotype × host combination were significantly different. This was done by examining overlap in the confidence intervals of the observed and predicted fitness values. Since this approach involves 96 comparisons, we used a Bonferroni correction of α = 0.05/96 = 0.00052, which are 99.948% confidence intervals (C.I.'s). If the 99.948% C.I.'s for a given genotype × host combination did not overlap, then we concluded that there was a significant difference in observed and predicted fitness for that genotype × host combination. We obtained the C.I.'s of least-squares means for fitness and attachment data by using a two-factor general linear model (GLM) in SAS (Proc GLM), where w or k = host + genotype + (host × genotype). The estimates of least-squares means for k, and their upper and lower 99.948% confidence limits, were then used in the phage life-history model to obtain estimates of predicted mean fitness with upper and lower 99.948% confidence limits. This approach is suitable in this case for two reasons. First, since there were different numbers of w- and k-values, it was not possible to associate a predicted fitness value with each observed fitness. Second, the k data were much more variable then the w data (Figure 2). Calculating predicted w from the mean of observed k would not account for these differences in variation. Our method accounts for both effects from unbalanced replication and differences in variation between w and k data.
Effects of mutations:
Effects of mutations were expressed as the difference between mutant and wild-type w or k (d = mutant − wild type). The d data were analyzed using a GLM (SAS, Proc GLM) with host, genotype, and host × genotype as factors. Since replication across genotypes and hosts was unbalanced, we used the least-squares means and error levels from the GLM to calculate mean d for each host × genotype combination and test whether each mean d was significantly different from zero. This involved 90 t-tests. To account for inflated type I error rates from the numerous tests, we used a Bonferroni correction to adjust significance levels (α = 0.05/90 = 0.00056).
Host range:
To test if mutants were more likely to include St or Ec hosts in their host range, we conducted a likelihood-ratio chi-square test (SAS, Proc FREQ). We also used this method to test if single and double mutants were equally likely to include Ec hosts in their host range. To assess levels of variation in fitness across hosts, we first estimated the coefficients of variation in fitness across hosts for each mutant. We then used a t-test to assess if single and double mutants showed the same levels of variability in fitness across hosts.
RESULTS
Recovery of mutants:
We obtained 15 mutants of gpF 101/102 by site-directed PCR mutagenesis of a wild-type phage and electroporation to St GalE and Ec C hosts (see Table 1 for host description). There were 9 double mutants, 6 single mutants of gpF 101, and no single mutants of gpF 102 (Table 2). Two mutants have been observed previously: HY has evolved during experimental evolution in a chemostat on St GalE at 42°, and KY occurs commonly in wild phage populations (Table 2). While the degenerate primers theoretically could allow for all possible combinations of gpF 101/102 amino acids, our protocol for obtaining mutants was not exhaustive and probably biased toward particular mutations by virtue of the need to grow on the hosts used for screening. It is not clear whether the lack of single gpF 102 mutations was due to a bias in mutant construction, exclusion by chance, or selection against single substitutions at gpF 102. However, in a subsequent study, we were able to construct three viable single mutants of gpF 102 (GN, GL, and GF; our unpublished data).
Interconnection of genotype, a fitness component, and fitness:
To evaluate predictability of fitness from the fitness component attachment rate, fitnesses were predicted from measured attachment rates and compared to observed fitness values. Comparisons were done separately for each genotype × host combination (96 total). Fitness was predictable from attachment rate in 72 of the 96 genotype × host treatments (Figure 2, Table 4). In the 24 cases where observed fitness was significantly different from predicted fitness, 17 were large significant differences while 7 others were only marginally different. Most of the cases of large significant differences (15/17) occurred in genotypes IY, LY, and YY (Figure 2, Table 4). These genotypes had much lower fitness than predicted on most hosts. The observation that IY, LY, and YY genotypes attached rapidly but had low fitness across hosts indicates that their fitness is decreased through a fitness component other than attachment rate. Thus, the I, L, and Y mutations at site gpF 101 had pleiotropic effects on fitness. Also, by comparing differences in observed and predicted fitness for genotypes across hosts, we found that host influenced the predictability of fitness from attachment rate. On the St rfb host, there were five genotypes with large differences in observed and predicted fitness (IY, LY, YY, IN, and FN) and three more genotypes with smaller significant differences (KY, HY, and RI). On the other hosts, significant differences were mainly attributable to the pleiotropic mutants IY, LY, and YY (Figure 2, Table 4).
TABLE 4.
Fitness difference between upper 99.948% confidence limit (CL) of observed fitness and lower 99.948% CL of predicted fitness
| St rfb | St galE | Ss | Ec C | Ec waaL(a) | Ec waaV | |
|---|---|---|---|---|---|---|
| GY | 0 | 0 | 0 | 0 | 1.3 (0) | 0 |
| KY | 2.0 | 0 | 0 | 0.7 | 1.6 (0.3) | 0 |
| HY | 0.9 | 0 | 0 | 0 | 0.6 (0) | 0 |
| QY | 0 | 0 | 0 | 0 | 0 | 0 |
| IY | 11.0 | 0 | 16.9 | 9.1 | 14.4 (13.1) | 0 |
| LY | 11.4 | 8.7 | 17.5 | 8.5 | 9.2 (7.9) | 0 |
| YY | 11.6 | 7.5 | 15.8 | 6.5 | 16.6 (15.3) | 4.7 |
| IN | 5.6 | 0 | 0 | 0 | 0 | 0 |
| LN | 0 | 0 | 0 | 0 | 0 | 0 |
| FN | 6.3 | 0 | 0 | 0 | 0 | 0 |
| MN | 0 | 0 | 0 | 0 | 0 | 0 |
| NN | 0 | 0 | 0 | 0 | 0 | 0 |
| QN | 0 | 0 | 0 | 0 | 0 | 0 |
| QF | 0 | 0 | 0 | 0 | 0 | 0 |
| KL | 0 | 0 | 0 | 0 | 0 | 0 |
| RI | 1.7 | 0 | 0 | 0 | 0 | 0 |
Zero indicates overlap. Genotypes are indicated in the first column (top row, wild type; next six rows, gpF 101 single mutants; bottom nine rows, gpF 101/102 double mutants). Hosts are listed at the top.
There was a difference between observed and predicted fitness in the wild type due to measurement error in determination of B and L parameters. Numbers in parentheses show the adjusted difference between 99.948% CLs of observed and predicted fitness after the measurement error has been accounted for.
Effects of mutations on attachment rate and fitness:
To assess whether artificially induced mutations at gpF 101/102 host recognition sites are adaptive, we compared attachment rates and fitness of the 15 mutants to those of the wild type on six hosts. Within the limits of detection of our assays, there were no significant beneficial effects of gpF 101 or 102 substitutions on any of the six hosts (Figure 3). Although not statistically significant, some of the double mutants show higher fitness and/or attachment rates on St rfb hosts (Figure 3, open circles). It is possible that these differences are biologically significant on a scale more sensitive than our assay could measure.
Figure 3.

Difference in mean fitness (top) and attachment rate (bottom) relative to wild type for each genotype on six hosts. Single mutants (gpF 101) are shown on the left, and double mutants (gpF 101/102) are on the right. Genotypes are shown encased between top and bottom parts. The horizontal lines indicate equivalence to wild type (GY). Symbols indicating hosts are shown across the top. Statistical comparisons were of least-squares means using a GLM with genotype and host × genotype factors. Solid symbols indicate significant difference from wild type at P < 0.00056 (calculated from a Bonferroni correction of α = 0.05/90 tests = 0.00056), shaded symbols indicate P < 0.01, and open symbols indicate P > 0.01.
Effects on host range:
To investigate if the gpF 101/102 host recognition sites can alter host range, we compared the infectivity patterns for the 15 mutants across the six hosts. Eleven of 15 mutants lost infectivity on at least one host (Figure 4). A likelihood-ratio chi-square test found that the mutants are more likely to include St hosts over Ec hosts in their host range (G2 = 15.72, P < 0.0001). Single and double mutants are equally likely to include St hosts (all mutants from both groups include St hosts). While there was no significant difference between single and double mutants in the likelihood of including Ec hosts in their host range, the result was marginal (G2 = 2.66, P = 0.056). This suggests that single mutants may be more likely than double mutants to include Ec hosts. In general, mutations at gpF 101 had similar effects across the six hosts, while double mutants had high fitness on Salmonella and very low fitness or lost infectivity on Escherichia and Shigella hosts (tested by comparing coefficients of variation for mean fitness across hosts in single vs. double mutants, t = 3.49, P = 0.004).
Figure 4.

Effects on host range. Hosts are listed across the top and genotypes in the first column. Wild type is shown first, single mutants second, and then double mutants. Open blocks indicate that mean fitness is between 12.1 and 24 population doublings per hour for that genotype–host combination. The shaded area is 0.1–12 and the solid area indicates that the SE of the mean encompasses 0. When the standard error encompassed two fitness classes, the block color of the lower-fitness class was used.
DISCUSSION
We used a simple model to assess effects of genetic architecture and environment on the predictability of fitness from a fitness component. Our system is advantageous for this application because it enables an integrated approach to studying the relationship of genotype, fitness components, and fitness. We expected that fitness would be predictable from the fitness component attachment rate because genotypes differed at only one or two amino acid sites that are known to affect attachment rate and host-specific fitness. While fitness was indeed predictable from attachment rate in many cases, there were significant genotype- and host-specific deviations from the life-history model. Our data show that even in a simple and well-characterized system, prediction of fitness from a fitness component can be hampered by pleiotropic genetic effects and environment.
Interconnection of genotype, a fitness component, and fitness:
Predictability of fitness from attachment rate was complicated by pleiotropic effects at a single amino acid position (gpF 101). For example, some gpF 101 mutants had high fitness and high attachment rates, while other mutants (I, L, and Y) had high attachment rates but low fitness. Phenotypic effects of mutations also depended on host; fitness was predictable by attachment rate on five of the six hosts, but not on host St rfb. Our results show that both pleiotropy and environment affect prediction of fitness from a fitness component. In both laboratory and natural populations, fitness components known to be strongly correlated with fitness are frequently used as surrogates for fitness because fitness may be difficult to measure directly. Yet even when studying genotypes that differ at a single locus, genetic effects and environment can hamper estimating fitness from fitness components. It is not clear how often there is a discord between fitness components and fitness due to pleiotropic loci in natural populations. The fact that we found that this discord can occur from pleiotropic effects in genomes differing by single mutations emphasizes that further empirical attention to factors influencing the interdependencies of genotype, fitness components, and fitness is strongly warranted. These empirical data, combined with theoretical investigation of the evolutionary consequences of variable phenotypic effects from pleiotropic loci in heterogeneous environments, will help in refining methods for studying natural diversification patterns and evolutionary processes.
Our conclusions assume that the theoretical relationship we used to predict fitness from attachment rate is accurate. While empirical data do agree with the theoretical model (Abedon et al. 2001; Bull et al. 2004a,b), the model was not previously tested for icosahedral phages such as φX174. From our data it appears that the model works well for icosahedral phages since 72 of 96 genotype–host measurements were consistent with the model, and some of the others differed only slightly. We argue that because the observed fitnesses of LY, IY, and YY genotypes are in extreme discord with predicted fitnesses (Figure 2, Table 4), yet the model accurately predicts fitness from attachment rate in most cases, a pleiotropic effect of these mutations at gpF 101 accounts for the inconsistent predictability. Our data clearly show distinct clustering of genotypes that have high attachment rates but very different fitnesses (Figure 2). It would be difficult to describe the attachment rate–fitness relationship of all these genotypes by a single mathematical function that makes biological sense unless other (pleiotropic) factors are taken into account.
Eclipse rate is another fitness component that is affected by phage–LPS interaction and possibly by gpF 101/102 residues. Once attached, the phage coat undergoes a conformational change, known as the eclipse reaction, to eject DNA into the host. The reaction has a high energy of activation, making it sensitive to small changes in activation entropy (Hayashi et al. 1988). Residues at gpF 101/102 have side chains that protrude outward near spike protein G (Figure 1). The pleiotropic residues (gpF 101; L, I, and Y) have large side chains relative to that of the wild-type G residue. However, considering that the high-fitness gpF 101 mutants K, H, and Q also have large side chains, size must not be the only factor responsible for decreased fitness in the pleiotropic mutants. One difference is that the high-fitness mutants are more hydrophilic than wild type and L, I, and Y (although Y is more hydrophilic than L and I and the wild-type G). A combination of bulkiness and hydrophobicity level in the L, I, and Y pleiotropic mutations could interfere with movement of the gpF 102 Y residue or other neighboring residues in a manner that increases the energy of activation of the eclipse reaction, thus decreasing eclipse rate.
On the other hand, virus assembly could also be affected by the pleiotropic mutations. Although gpF residues 101/102 are not involved in structural interactions in the mature virus particle, they could affect protein interactions during assembly. During assembly in φX174, scaffolding proteins bind to pentamers of gpF subunits, bringing them together to form the virus capsid (Dokland et al. 1997, 1999; Burch et al. 1999). The immature virus particle is externally coated by scaffolding protein D (Dokland et al. 1997, 1999). Thus, the bulky hydrophobic gpF 101 residues could interfere with association to specific sites in the external scaffolding proteins and decrease the efficiency of procapsid assembly (Fane and Hayashi 1991; Fane et al. 1993; Burch and Fane 2003; Uchiyama and Fane 2005).
Effects of mutations:
Attachment rates and fitness for the 15 gpF 101/102 mutants were similar to or lower than those of a lab-adapted strain on six different hosts. This result was unexpected since these sites undergo adaptive substitution in response to hosts that were assayed in our study (Ec C and St galE) and because these sites are variable among wild phages (Wichman et al. 2000; Rokyta et al. 2006). The lack of beneficial effects may be due to several factors. First, if only a few gpF 101/102 mutations could potentially improve fitness of the wild-type phage under our assay conditions, then it is possible that we failed to recover beneficial mutations by chance. For example, there are 400 possible amino acid combinations at sites gpF 101/102. Our screening sample size was only 466, most of which were wild type. A second reason for the lack of beneficial mutations may be that we used only two hosts for screening. There may be genotypes that are adaptive on one of the other four hosts, but that could not grow on the St GalE or Ec C hosts well enough to be sampled. Finally, previously documented variation at gpF 101/102 sites occurred in phage grown in chemostats or isolated from sewage. These gpF 101/102 amino acids may be adaptive in host recognition only in the chemostat or sewage environments and not under our assay conditions.
We did isolate two genotypes that have been observed previously. HY occurred in experimental evolution in a chemostat and KY occurs in a closely related phage (φK) and has been isolated from sewage. It is possible that these mutations were not beneficial in our experiment because: (1) as mentioned above, our assay conditions are very different from a chemostat or sewage environment, and (2) our mutations were inserted in the wild-type genome while the previously observed HY and KY genotypes occurred in other genetic backgrounds. The wild type is well adapted to the assay conditions used in our study; it may be at a local fitness optimum under these conditions. Thus, although HY and KY genotypes had no detectable beneficial effects in our experiment, the fact they have been observed in other environments and genetic backgrounds suggests that effects of gpF 101/102 amino acids are moderated by environment and epistasis. In support of this, Crill et al. (2000) found that the degree of beneficial effect of the gpF G101D substitution depends on the genetic background. Negative epistatic interactions of gpF 101/102 amino acids with other amino acids in the wild-type phage capsid could dampen the degree of potentially host-specific beneficial effects of gpF 101/102 amino acids. Our data cannot distinguish whether genotype × environment interaction, epistasis, or an interaction of the two is responsible for the patterns of genetic variation at gpF 101/102.
Effects on host range:
Infectivity profiles of the 15 mutants among six hosts support previous findings that one or two point mutations can alter virus host range by both host-specific and nonspecific mechanisms (Llamas-Saiz et al. 1996; Truyen et al. 1996; Taplitz and Coffin 1997; Hanley et al. 2003; Rainey et al. 2003). For example, in an avian retrovirus, one substitution in the coat protein extended host range through a non-host-specific mechanism; the mutant showed no improved attachment to hosts relative to wild type. Likewise, some gpF 101 substitutions decreased fitness through a fitness component other than attachment rate. Also, the infectivity profiles and low variability in fitness across hosts for gpF 101 mutations suggest that in the wild-type genome, single mutations at gpF 101 have unconditional effects on fitness. This finding was contrary to our expectations since substitution of the gpF 101 residue occurred in response to every host switch during experimental evolution on alternating hosts (Crill et al. 2000). However, we did find host-specific effects when mutations at gpF 101 and 102 were inserted in the wild-type genome. Thus, host-specific effects of mutations at gpF 101 may be conditional on the amino acid at gpF 102 and on other mutations in the genome.
We also found that mutations at both gpF 101/102 sites tend to be more detrimental to Escherichia hosts relative to Salmonella hosts. This supports results from experimental evolution where wild-type GY evolves to be DC on St GalE (Bull et al. 1997; Wichman et al. 1999; Crill et al. 2000). While GY grows relatively well on both Ec C and St GalE, the St GalE-adapted DC genotype has very low fitness and attachment on Ec C. The St rfb and St GalE hosts in our study have very different outer core LPS structures, but a similar inner core structure that differs from that of the Ec hosts. Since the double gpF 101/102 mutants were more likely to exclude Ec while including St hosts, and since these sites have an effect on attachment, gpF 101/102 sites may be sensitive to the inner core LPS structural differences between Ec and St. On the other hand, the LPS structural differences are not the only factor differentiating Ec and St hosts. Thus, it is also possible that gpF 101/102 sites respond to more general differences between Ec and St hosts. In general, some feature of St hosts appears to accommodate a wider range of amino acids at gpF 101/102 sites (at least in the wild-type genome).
Conclusions:
Even in this simple, well-characterized biological system, predictability of fitness from a fitness component is complicated by underlying genetic architecture and environment. A single amino acid mutation in a genome can have pleiotropic and environmentally dependent effects that decrease the predictability of fitness from fitness components. This suggests that genetic and phenotypic variation may often not be correlated. When the genetic determinants of fitness components involve pleiotropic loci, it is important to measure numerous fitness components and analyze covariance matrices carefully before drawing conclusions about evolutionary outcome. A challenge in this pursuit is that the commonality of pleiotropic loci and degree to which genetic architecture influences phenotypic variation remain unknown in most systems. These parameters merit substantial empirical investigation. A second significant implication of our results is that phenotypes can rarely be generalized across environments, yet environmental variation is unavoidable in nature. To predict evolutionary outcome in heterogeneous environments, methods need to be refined such that environmental variation can be explained more accurately. This will require a combination of controlled empirical studies that examine effects of environment on the interconnection of genotype, fitness components, and fitness in coordination with theoretical development of quantitative genetics methods. A final implication of our results pertains to the application of microbes as biological controls and disease therapies, as well as models for furthering our understanding of the processes of evolution. The complex genetics of our simple system highlight that further empirical and theoretical studies of the genetic architecture of specific fitness components, the interaction of genotype and environment, and the mapping of fitness components onto fitness are needed before we can predict, or even explain, adaptation.
Acknowledgments
We thank Zaid Abdo, David Althoff, Jim Bull, Christina Burch, Gary Daughdrill, Bentley Fane, Scott Nuismer, Chris Smith, LuAnn Scott, and an anonymous reviewer for insightful comments on the manuscript. We thank Jim Bull for writing and providing a program to analyze the phage life-history model, LuAnn Scott for help with constructing the mutants, Brandi Lund for help with sequencing, Chris Williams for statistical advice, and Gary Daughdrill for making Figure 1. Also, we thank Christina Burch for her suggestion that the data be analyzed in the framework of the theoretical model. This project was supported by National Institutes of Health grant RR16448.
References
- Abedon, S. T., T. D. Herschler and D. Stopar, 2001. Bacteriophage latent-period evolution as a response to resource availability. Appl. Environ. Microbiol. 67: 4233–4241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bull, J. J., M. R. Badgett, H. A. Wichman, J. P. Huelsenbeck, D. M. Hillis et al., 1997. Exceptional convergent evolution in a virus. Genetics 147: 1497–1507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bull, J. J., M. R. Badgett and H. A. Wichman, 2000. Big-benefit mutations in a bacteriophage inhibited with heat. Mol. Biol. Evol. 17: 942–950. [DOI] [PubMed] [Google Scholar]
- Bull, J. J., M. R. Badgett, R. Springman and I. J. Molineux, 2004. a Genome properties and the limits of adaptation in bacteriophages. Evol. Int. J. Org. Evol. 58: 692–701. [DOI] [PubMed] [Google Scholar]
- Bull, J. J., D. W. Pfennig and I.-N. Wang, 2004. b Genetic details, optimization and phage life histories. TREE 19: 76–82. [DOI] [PubMed] [Google Scholar]
- Bull, J. J., J. Millstein, J. Orcutt and H. A. Wichman, 2006. Evolutionary feedback mediated through population density, illustrated with viruses in chemostats. Am. Nat. 167: E39–E51. [DOI] [PubMed] [Google Scholar]
- Burch, A. D., and B. A. Fane, 2003. Genetic analyses of putative conformation switching and cross-species inhibitory domains in Microviridae external scaffolding proteins. Virology 310: 64–71. [DOI] [PubMed] [Google Scholar]
- Burch, A. D., J. Ta and B. A. Fane, 1999. Cross-functional analysis of the Microviridae internal scaffolding protein. J. Mol. Biol. 286: 95–104. [DOI] [PubMed] [Google Scholar]
- Crill, W. D., H. A. Wichman and J. J. Bull, 2000. Evolutionary reversals during viral adaptation to alternating hosts. Genetics 154: 27–37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dokland, T., R. McKenna, L. L. Ilag, B. R. Bowman, N. L. Incardona et al., 1997. Structure of a viral procapsid with molecular scaffolding. Nature 389: 308–313. [DOI] [PubMed] [Google Scholar]
- Dokland, T., R. A. Bernal, A. Burch, S. Pletnev, B. A. Fane et al., 1999. The role of scaffolding proteins in the assembly of the small, single-stranded DNA virus φX174. J. Mol. Biol. 288: 595–608. [DOI] [PubMed] [Google Scholar]
- Fane, B. A., and M. Hayashi, 1991. Second-site suppressors of a cold-sensitive prohead assembly protein of bacteriophage φX174. Genetics 128: 663–671. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fane, B. A., S. Shien and M. Hayashi, 1993. Second-site suppressors of a cold sensitive external scaffolding protein of bacteriophage φX174. Genetics 134: 1003–1011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gamian, A., and E. Romanowska, 1982. The core structure of Shigella sonnei lipopolysaccharide and the linkage between O-specific polysaccharide and the core region. Eur. J. Biochem. 129: 105–109. [DOI] [PubMed] [Google Scholar]
- Hanley, K. A., L. R. Manlucu, L. E. Gilmore, J. E. Blaney, Jr., C. T. Hanson et al., 2003. A trade-off in replication in mosquito versus mammalian systems conferred by a point mutation in the NS4B protein of dengue virus type 4. Virology 312: 222–232. [DOI] [PubMed] [Google Scholar]
- Hayashi, M., A. Aoyama, D. L. Richardson, Jr. and M. N. Hayashi, 1988. Biology of the bacteriophage φX174, pp. 1–71 in The Bacteriophages, edited by R. Calendar. Plenum Press, New York.
- Heinrichs, D. E., M. A. Monteiro, M. B. Perry and C. Whitfield, 1998. The assembly system for the lipopolysaccharide R2 core-type of Escherichia coli is a hybrid of those found in Escherichia coli K-12 and Salmonella enterica. Structure and function of the R2 WaaK and WaaL homologs. J. Biol. Chem. 273: 8849–8859. [DOI] [PubMed] [Google Scholar]
- Holder, K. K., and J. J. Bull, 2001. Profiles of adaptation in two similar viruses. Genetics 159: 1393–1404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Levin, B. R., F. M. Stewart and L. Chao, 1977. Resource-limited growth, competition, and predation: a model and experimental studies with bacteria and bacteriophage. Am. Nat. 111: 3–24. [Google Scholar]
- Llamas-Saiz, A. L., M. Agbandje-McKenna, J. S. Parker, A. T. Wahid, C. R. Parrish et al., 1996. Structural analysis of a mutation in canine parvovirus which controls antigenicity and host range. Virology 225: 65–71. [DOI] [PubMed] [Google Scholar]
- McKenna, R., L. L. Ilag and M. G. Rossmann, 1994. Analysis of the single-stranded DNA bacteriophage φX174, refined at a resolution of 3.0 A. J. Mol. Biol. 237: 517–543. [DOI] [PubMed] [Google Scholar]
- Nikaido, H., M. Levinthal, K. Nikaido and K. Nakane, 1967. Extended deletions in the histidine-rough-B region of the Salmonella chromosome. Proc. Natl. Acad. Sci. USA 57: 1825–1832. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rainey, G. J., A. Natonson, L. F. Maxfield and J. M. Coffin, 2003. Mechanisms of avian retroviral host range extension. J. Virol. 77: 6709–6719. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rokyta, D., C. B. Burch, S. B. Caudle and H. A. Wichman, 2006. Horizontal gene transfer and the evolution of microvirid coliphage genomes. J. Bacteriol. 188: 1134–1142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sambrook, J., E. F. Fritsch and T. Maniatis, 1989. Molecular Cloning: A Laboratory Manual. Cold Spring Harbor Laboratory Press, Cold Spring Harbor, NY.
- Sanger, F., G. M. Air, B. G. Barrell, N. L. Brown, A. R. Coulson et al., 1977. Nucleotide sequence of bacteriophage φX174 DNA. Nature 265: 687–695. [DOI] [PubMed] [Google Scholar]
- Taplitz, R. A., and J. M. Coffin, 1997. Selection of an avian retrovirus mutant with extended receptor usage. J. Virol. 71: 7814–7819. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Truyen, U., J. F. Evermann, E. Vieler and C. R. Parrish, 1996. Evolution of canine parvovirus involved loss and gain of feline host range. Virology 215: 186–189. [DOI] [PubMed] [Google Scholar]
- Uchiyama, A., and B. A. Fane, 2005. Identification of an interacting coat-external scaffolding protein domain required for both the initiation of φX174 procapsid morphogenesis and the completion of DNA packaging. J. Virol. 79: 6751–6756. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang, I.-N., D. E. Dykhuizen and L. B. Slobodkin, 1996. The evolution of phage lysis timing. Evol. Ecol. 10: 545–558. [Google Scholar]
- Wichman, H. A., M. R. Badgett, L. A. Scott, C. M. Boulianne and J. J. Bull, 1999. Different trajectories of parallel evolution during viral adaptation. Science 285: 422–424. [DOI] [PubMed] [Google Scholar]
- Wichman, H. A., L. A. Scott, C. D. Yarber and J. J. Bull, 2000. Experimental evolution recapitulates natural evolution. Philos. Trans. R. Soc. Lond. B Biol. Sci. 355: 1677–1684. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wichman, H. A., J. Millstein and J. J. Bull, 2005. Adaptive molecular evolution for 13,000 phage generations: a possible arms race. Genetics 170: 19–31. [DOI] [PMC free article] [PubMed] [Google Scholar]