

Abstract
Comparing patterns of molecular evolution between autosomes and sex chromosomes (such as X and W chromosomes) can provide insight into the forces underlying genome evolution. Here we investigate patterns of codon bias evolution on the X chromosome and autosomes in Drosophila and Caenorhabditis. We demonstrate that X-linked genes have significantly higher codon bias compared to autosomal genes in both Drosophila and Caenorhabditis. Furthermore, genes that become X-linked evolve higher codon bias gradually, over tens of millions of years. We provide several lines of evidence that this elevation in codon bias is due exclusively to their chromosomal location and not to any other property of X-linked genes. We present two possible explanations for these observations. One possibility is that natural selection is more efficient on the X chromosome due to effective haploidy of the X chromosomes in males and persistently low effective numbers of reproducing males compared to that of females. Alternatively, X-linked genes might experience stronger natural selection for higher codon bias as a result of maladaptive reduction of their dosage engendered by the loss of the Y-linked homologs.
THE sex chromosomes of most organisms are believed to derive from an ancient pair of autosomes (Charlesworth 1991). The transition of these autosomes to sex chromosomes is marked by a reduction in recombination between the proto-X and proto-Y, followed by the nearly complete degradation of the Y chromosome. This degeneration of the Y involves gene loss, and, as a result, genes on the sex chromosomes are typically present at half the copy number as compared to genes on the autosomes in the heterogametic sex (XY or XO). For the sake of simplicity we refer to the heterogametic sex as “male” and the homogametic sex as “female” for the remainder of the discussion.
This reduction in copy number of X-linked genes in males is the foundation for three major differences between the X and the autosomes, all of which have important implications for the molecular evolution of coding and noncoding sequences on these chromosomes. One difference between the X chromosome and the autosomes is that X-linked alleles are immediately visible to selection in males because of the effective haploidy of the X chromosome in those individuals. Although the X chromosome spends only one-third of its evolutionary history in males, the exposure of X-linked alleles to selection in these hemizygous individuals may lead to an accumulation of favorable mutations on the X chromosome due to an enhanced efficacy of natural selection (Charlesworth et al. 1987). This population genetic difference between the X and the autosomes is universal, and, accordingly, this difference should have similar implications for sequence evolution in all species with chromosomal sex determination.
The presence of only a single X chromosome in males may also lead to differences in the effective population sizes of X-linked and autosomal genes. Barring large differences in reproductive success between males and females, the effective population size of the X chromosome should be smaller than that of the autosomes because there are only three X chromosomes for every four autosomes. Under this scenario, the efficacy of selection acting on weakly adaptive or weakly deleterious mutations might be greater on the autosomes than on the X. However, the effective population size of the X relative to that of the autosomes will also be affected by possible differences in reproductive success between males and females. In fact, if the number of mating males is much smaller than the number of mating females, then the effective population size of the X chromosome can potentially exceed the effective population size of the autosomes (Caballero 1995; Laporte and Charlesworth 2002). These considerations suggest that the difference in effective population size between the X chromosome and autosomes is difficult to predict a priori and, furthermore, that the direction and magnitude of this difference might vary among different lineages.
The formation of the sex chromosomes from autosomes followed by the degradation of the Y chromosome also results in a universal dosage problem for X-linked genes, which are present in half the dose in males as compared to females. This reduction in dosage is likely to be deleterious for many genes and, as a consequence, many organisms have evolved elaborate dosage compensation mechanisms. In Drosophila melanogaster, for instance, dosage compensation is mediated through transcriptional upregulation of X-linked genes in the heterogametic males (for review see Baker et al. 1994; Marin et al. 2000). Alternative evolutionary responses to the dosage problem are seen in other systems; in Caenorhabditis elegans and mammalian lineages, for example, either or both copies of the X-linked gene are downregulated at the level of transcription in the homogametic sex (for review see Marin et al. 2000). Although the solutions are diverse, the dosage problem for X-linked genes is universal.
While it is clear that there are fundamental differences between the X and the autosomes, it is not clear how these differences are reflected in the evolution of coding sequences. One aspect of coding sequence evolution that may be sensitive to these differences between the X and the autosomes is codon bias. Codon bias reflects the unequal usage of synonymous codons in protein coding sequences and is thought to enhance the efficiency and/or fidelity of translation (Bulmer 1991; Akashi and Eyre-Walker 1998; Akashi et al. 1998).
Codon bias is maintained by the balance among mutation, selection, and drift (Sharp and Li 1986; Bulmer 1991; Akashi and Schaeffer 1997; McVean and Charlesworth 1999). As a result, the degree to which the codon usage of any particular gene is biased will be determined by the strength of selection on translational efficiency and the dominance of the mutations affecting codon bias, as well as effective population size. The hemizygosity of the X chromosome in males may affect codon bias by increasing the efficacy of selection on incompletely dominant mutations in X-linked genes. In addition, the presence of a single X chromosome in males may lead to differences in the effective population sizes of mutations affecting codon bias in X-linked vs. autosomal genes. Furthermore, the reduction in copy number of X-linked genes in males may alter the strength of selection on translational efficiency for genes on the X chromosome. Consequently, the differences between the X and the autosomes with respect to population genetics and dosage may have marked effects on levels of codon bias of X-linked and autosomal genes.
Here we investigate the evolution of codon bias of X-linked and autosomal genes in three distinct systems: D. melanogaster, D. pseudoobscura, and C. elegans. We present evidence that codon bias is systematically elevated on the X chromosome in all three systems and show that this increase in codon bias is due exclusively to X-linkage and not to any other property of the genes residing on these chromosomes. We further show that the evolution of higher codon bias of X-linked genes is a gradual process, with codon bias accumulating slowly over tens of millions of years.
The persistence of this pattern across taxa hints at the possibility that it is common among eukaryotes with chromosomal sex determination. Given the potential ubiquity of this elevation in codon bias on the X chromosome, it seems likely that the explanation of this pattern would be of a general nature. We suggest that the X-specific increase in codon bias evolved either as a consequence of the population genetic parameters of the X chromosome and the autosomes or in response to the universal dosage problem shared among all eukaryotes with chromosomal sex determination.
MATERIALS AND METHODS
All data are available upon request.
Coding sequences and codon usage in D. melanogaster and C. elegans:
We retrieved coding sequences for all genes in Release 3.2 (FlyBase) of the D. melanogaster genome that were not located in either telomeric (sections 1, 21, 60–61, and 100) or centromeric regions (sections 20, 40–41, and 80–81) as defined by Bridges (1935). Of these 12,444 genes, 10,356 are located on the autosomes, while 2088 are located on the X chromosome. Genes mapped to heterochromatic contigs were not included in our analysis. We also retrieved coding sequences for all genes in Release 128 (Wormbase) of the C. elegans genome. Of these 15,916 genes, 2567 are located on the X chromosome, with the remaining 13,349 genes on the autosomes.
In both D. melanogaster and C. elegans, several transcripts were listed for some genes; for these genes we included only the first transcript listed in our data set, and both the protein length and codon bias estimates are based solely on this first listed transcript. For each gene, we calculated the frequency of optimal codons (FOP) on the basis of optimal codons as defined by Duret and colleagues (Duret and Mouchiroud 1999). As optimal codons for each amino acid have been defined, we can tabulate the fraction of all amino acids with synonymous codons in a coding sequence (excluding stop codons) encoded by an optimal codon.
Duplicate gene pairs in D. melanogaster and C. elegans:
We performed an all-by-all protein BLAST of the D. melanogaster genome (Release 3.2 from FlyBase), using default parameters. We also performed an all-by-all protein BLAST of the C. elegans genome (downloaded in batch from NCBI in November 2003). Duplicate genes were conservatively defined as reciprocal best hits with an E-value of <1 × 10−10 in both directions. We obtained chromosome locations of individual genes from FlyBase (D. melanogaster) and NCBI (C. elegans) and chose the set of duplicate genes for which one member was located on an autosome and one was located on the X chromosome. We aligned the duplicate proteins using pairwise BLAST following a previously published protocol (Conery and Lynch 2001) and computed optimal codon frequencies of each duplicate using only the aligned region.
Orthologous genes between D. melanogaster and D. pseudoobscura and codon usage in D. pseudoobscura:
For each gene in the D. melanogaster genome we performed a nucleotide BLAST of the unannotated D. pseudoobscura genome (downloaded from the Baylor College of Medicine Human Genome Sequencing Center at http://www.hgsc.bcm.tmc.edu/projects/drosophila/) to identify orthologous genes. We generated protein alignment of the orthologs using methods discussed above (Conery and Lynch 2001) and retained only those genes for which >60% of both protein sequences were alignable. For all genes that had been mapped to chromosomal locations in D. pseudoobscura, we calculated optimal codon frequencies on the aligned region of the orthologous genes in D. pseudoobscura and D. melanogaster [defined optimal codons in both species are identical (Akashi and Schaeffer 1997)] provided that the coding sequence was ≥200 bp. There were 9800 coding sequences from D. pseudoobscura included in our analysis: 6180 on the autosomes, 1992 on XR, and 1628 on XL. We also calculated KA between orthologous genes using the codeml program from the PAML package (Yang 1997), letting all parameters vary.
Orthologous genes between C. elegans and C. briggsae:
In their manuscript presenting the whole-genome sequence of C. briggsae, Stein et al. (2003) identify pairs of orthologous genes between C. briggsae and C. elegans and estimate rates of protein evolution for these gene pairs as well. Orthologous gene pairs were identified using reciprocal-best-hit and synteny criteria and were aligned using the “needle” program from EMBOSS (Rice et al. 2000). KA and KS were estimated for each orthologous gene pair using a maximum-likelihood calculation in PAML (Yang 1997). We downloaded these pairs of orthologous genes from the Stein et al. data set, as well as the KA and KS estimates.
Expression estimate in D. melanogaster:
We used expressed sequence tag (EST) counts as a rough indicator of expression level, as calculated by Hey and Kliman (2002) (http://lifesci.rutgers.edu/∼heylab). These data were compiled from the Drosophila Gene Index (DGI) of the Institute of Genome Research (http://www.tigr.org/tdb/dgi), which is a catalog of multiple EST data sets. As such, these EST counts do not reflect spatial or temporal variations in expression pattern and, accordingly, should be regarded as crude estimates of overall expression pattern.
Expression estimates in C. elegans:
We used the Jones et al. (2001) serial analysis of gene expression (SAGE) data set for gene expression estimates in C. elegans. This technique uses short sequence tags unique to individual gene transcripts; quantification of the number of times a particular sequence tag is detected provides the expression level for that individual transcript. Jones et al. quantified gene expression using SAGE in a mixed population of wild-type (N2) C. elegans containing stages in the estimated ratio 20 L1:20 L2:1 L3:1 L4:1 adult. We used the SAGE data obtained from this mixed population (http://elegans.bcgsc.bc.ca/SAGE) for our expression estimates, using only transcripts that had been unambiguously mapped to specific genes.
Gonad-biased expression in D. melanogaster:
Using microarray data collected by Parisi et al. (2003), which compared relative levels of expression between testes and ovaries in adult flies, we defined gonad-biased expression as a twofold difference in the log-ratio of expression levels as suggested by the authors. We used only genes for which relative levels of expression were estimated with both the testis probe and the ovaries probe, and we averaged across experiments to calculate a measure of gonad-biased expression for each gene. Negative values mark genes that are expressed more highly in males vs. females, while positive values reflect female-biased expression patterns.
Gene density in D. melanogaster and C. elegans:
We estimated gene density using the “genes per kilobase” (GPK) metric following the example set by Hey and Kliman (2002). For each gene, we counted the number of genes included partially or completely within a 20-kb window centered on the midpoint of a given transcript. Gene coordinates for all genes were taken from header information for each gene in Release 3.2 of the D. melanogaster genome (FlyBase) and a November 2003 download of the C. elegans genome (NCBI).
Recombination estimates in D. melanogaster:
A list of all genes (615) that had been localized in both the physical and genetic maps in Release 3 of the D. melanogaster genome was kindly provided by FlyBase (D. Sutherland, personal communication). A third-order polynomial curve was fitted to the genetic distance as a function of physical distance of these genes for each chromosomal arm (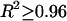 for all arms) after visually identifying and removing outliers (n = 3, 3, 3, 2, and 0 outliers on chromosome arms 2L, 2R, 3L, 3R, and X, respectively). Recombination (centimorgans per megabase) was calculated as the derivative of this polynomial at a given nucleotide coordinate. Recombination rate estimates for any locus in the D. melanogaster genome are available at http://cgi.stanford.edu/∼lipatov/recombination/recombination-rates.txt.
for all arms) after visually identifying and removing outliers (n = 3, 3, 3, 2, and 0 outliers on chromosome arms 2L, 2R, 3L, 3R, and X, respectively). Recombination (centimorgans per megabase) was calculated as the derivative of this polynomial at a given nucleotide coordinate. Recombination rate estimates for any locus in the D. melanogaster genome are available at http://cgi.stanford.edu/∼lipatov/recombination/recombination-rates.txt.
Recombination estimates in C. elegans:
Genetic and physical map locations for the 1483 genes localized on both maps were retrieved from Wormbase. There were 262, 228, 248, 220, 237, and 288 genes on chromosomes I–V and X, respectively, and all genes were included in our analysis. For all of the genes on each chromosome, genetic map position (centimorgans) was plotted as a function of physical map position (megabase pairs) and a third-order polynomial curve was fit to this relationship (R2 ≥ 0.96 for all arms). Recombination (centimorgans per megabase) was calculated as the derivative of this third-order polynomial at a given nucleotide coordinate.
Pairing autosomal and X-linked genes:
Many factors work to determine the codon bias of an individual gene such as protein length, GC content, and expression level. To gain insight into the degree to which X-linkage alone increases codon bias, we sampled autosomal genes to match the distribution of X-linked genes with respect to other gene attributes. We sampled genes according to GC content of neighboring noncoding sequences, protein length, expression level, and along all three of these parameters together. After each sampling, we then estimated the mean optimal codon frequency of this constructed set of autosomal genes (see supplementary materials and Figure S1 at http://www.genetics.org/supplemental/). Sampling was performed by pairing each X-linked gene with its most similar autosomal counterpart with respect to a particular attribute. In the event of ties, one of the tied autosomal genes was randomly selected. For the multiple-attribute sampling, for each X gene we chose an autosomal gene with the minimum root-mean-squared distance. Root-mean distances were calculated by normalizing each of the gene attributes by the mean and variance to N(0, 1) and then calculating the root-mean-squared distance using the standard formula [i.e., where 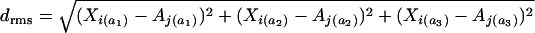 , where Xi and Aj denote particular genes on the X and autosomes, respectively, and the subscripts a1–a3 denote three gene attributes]. After each sampling procedure we confirmed that mean attribute values between X-linked and autosomal sets were not different (P ≫ 0.2, Mann-Whitney U-test all comparisons).
, where Xi and Aj denote particular genes on the X and autosomes, respectively, and the subscripts a1–a3 denote three gene attributes]. After each sampling procedure we confirmed that mean attribute values between X-linked and autosomal sets were not different (P ≫ 0.2, Mann-Whitney U-test all comparisons).
Population genetic model:
To consider potential causes of an elevation of codon bias on the X we focus on  (see text) and investigate the effect of changing effective population sizes and selection coefficients of the X and the autosomes. Assuming the model outlined in the text above, the change in frequency of a weakly selected autosomal allele
(see text) and investigate the effect of changing effective population sizes and selection coefficients of the X and the autosomes. Assuming the model outlined in the text above, the change in frequency of a weakly selected autosomal allele 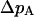 under the Wright-Fisher model is
under the Wright-Fisher model is 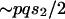 . Assuming that frequency of a weakly selected polymorphic allele is equal in the two sexes on its way to fixation and assuming that an X chromosome spends one-third of its time in males,
. Assuming that frequency of a weakly selected polymorphic allele is equal in the two sexes on its way to fixation and assuming that an X chromosome spends one-third of its time in males, 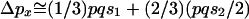 . Simplification reveals that
. Simplification reveals that 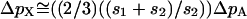 for all values of p. Thus the equivalence
for all values of p. Thus the equivalence 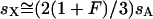 , where
, where 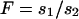 , can be used to compare the selection coefficients on the X and the autosomes. The effective population size for an autosomal locus with unequal numbers of males and females is
, can be used to compare the selection coefficients on the X and the autosomes. The effective population size for an autosomal locus with unequal numbers of males and females is 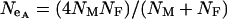 and for an X-linked locus it is
and for an X-linked locus it is 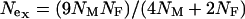 (Hartl and Clark 1989). Therefore, the equation
(Hartl and Clark 1989). Therefore, the equation 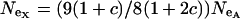 , where
, where 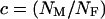 , relates the effective population size of the X and the autosomes. On the basis of these assumptions,
, relates the effective population size of the X and the autosomes. On the basis of these assumptions, 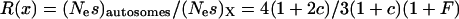 . The ridge at which
. The ridge at which 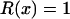 defines the solution at which levels of codon bias are equal on the X and the autosomes (Figure 6).
defines the solution at which levels of codon bias are equal on the X and the autosomes (Figure 6).
Figure 6.

Values of c (NM/NF) and F (s1/s2) for which R(x) > 1 (shaded area) and <1 (open area). The dashed line represents pairs of F and c that correspond to the observed difference in codon bias between X-linked an autosomal genes assuming the probability of mutation away from a preferred codon is three times the probability of mutation to the preferred codon (see supplementary materials at http://www.genetics.org/supplemental/).
To obtain a rough estimate for values of F and c that might explain the difference in codon bias that we observe for D. melanogaster, we solved for the relationship between these variables given values of the mean codon bias on the X and the autosomes and under the assumptions that all codons in a gene are under equal selection pressure and possess an equal ratio of mutations to and from the preferred codon. Solutions in F and c are given for several ratios of mutations to and away from a preferred codon (see supplementary materials and Figure S2 at http://www.genetics.org/supplemental/).
RESULTS AND DISCUSSION
Higher codon bias on the X chromosome in diverse eukaryotes:
We examined the potential effects of the differences between the X and the autosomes on patterns of codon usage of X-linked and autosomal genes in three taxa: D. melanogaster, D. pseudoobscura, and C. elegans. In D. melanogaster, codon bias estimated using the frequency of optimal codons (Duret and Mouchiroud 1999; Marais et al. 2001) is significantly higher for X-linked genes (0.570, n = 2088 genes) than for genes on autosomes (0.536, n = 10,356 genes) (P ≪ 0.0001, two-tailed t-test). This is not due to the strongly reduced codon bias on the nonrecombining fourth chromosome, as codon bias of X-linked genes is significantly higher than codon bias of genes on every autosomal arm (P ≪ 0.0001, two-tailed t-test, all comparisons) (Figure 1a).
Figure 1.

Optimal codon frequencies on each chromosome in (a) D. melanogaster, (b) D. pseudoobscura, and (c) C. elegans. Error bars denote standard error.
As is the case in D. melanogaster, codon bias of X-linked genes in D. pseudoobscura is higher than codon bias of autosomal genes. The two arms of the X chromosome in D. pseudoobscura have different evolutionary histories and, accordingly, we treated them separately in our analysis. The left arm of the X chromosome (XL) predates the separation of the D. melanogaster and D. pseudoobscura lineages 46 MYA (Powell and DeSalle 1995). In contrast, the right arm of the X chromosome (XR) in D. pseudoobscura arose from a translocation of an autosomal arm (corresponding to 3L in D. melanogaster) (Segarra et al. 1996) ∼6–10 MYA (Charlesworth and Charlesworth 2005), after the split of the D. melanogaster and D. pseudoobscura lineages. The frequency of optimal codons of genes on XL (0.614, n = 1628 genes) and XR (0.598, n = 1992) is significantly higher on average than it is for autosomal genes (0.580, n = 6180 genes) (P ≪ 0.0001 both comparisons, one-tailed t-test). Again, this is not due to the reduced codon bias of genes on chromosome 5 (believed to be orthologous to chromosome 4 in D. melanogaster), as codon bias of genes on XL and XR is significantly higher than codon bias of genes on every other autosomal arm (P ≪ 0.0001, one-tailed t-test, all comparisons) (Figure 1b). Interestingly, the average codon bias on XL (the older arm of the X chromosome) is significantly higher than the average codon bias on XR (the younger arm) (P≪ 0.0001, two-tailed t-test), implying that increased codon bias on the X chromosome evolves gradually.
The systematic elevation in codon bias of X-linked genes is also found in C. elegans. Overall, the frequency of optimal codons of X-linked genes in C. elegans (0.384, n = 2567 genes) is significantly higher than the frequency of optimal codons of genes on the autosomes (0.372, n = 15,916 genes) (P ≪ 0.001, one-tailed t-test). This is not due to the depressed level of codon bias on the gene-dense chromosome V of the C. elegans genome (Figure 1c), as codon bias on the X is significantly higher than codon bias on each of the five autosomes individually (P < 0.02, one-tailed t-test, all comparisons).
Elevated codon bias on the X is not a function of known determinants of codon bias:
One possible explanation for the elevation in codon bias of X-linked genes is that these genes are simply expressed more highly than autosomal genes. Expression level and codon bias are strongly positively correlated (reviewed in Akashi 2001; see also Sharp and Li 1986; Bulmer 1988; Duret and Mouchiroud 1999; Hey and Kliman 2002), which likely reflects increased selective benefits of translational efficiency for highly expressed genes. In particular, EST counts in D. melanogaster and SAGE estimates in C. elegans correlate positively with FOP on both the X and the autosomes (Singh et al. 2005).To examine the possibility that codon bias of X-linked genes is elevated because of increased expression level, we compared levels of gene expression between the X and the autosomes for the two systems for which gene expression data were available: D. melanogaster and C. elegans. Using EST counts as a metric of overall expression for genes in D. melanogaster, we found that X-linked genes are expressed at significantly lower levels than autosomal genes (EST counts are 7.5 and 11.7 for the X and the autosomes, respectively; P ≪ 0.0001, two-tailed t-test). Likewise, using SAGE estimates in C. elegans, we found that X-linked genes are also expressed at lower levels than autosomal genes (SAGE counts are 5.8 and 10.3 for the X and the autosomes, respectively; P = 0.008, two-tailed t-test).
Because the EST counts in Drosophila are compiled from a variety of sources, the ratio of males to females in the samples is not clear. Similarly, the sex ratio of the populations in which expression was analyzed in C. elegans is unknown. In addition, SAGE data for C. elegans may be GC-content biased (Margulies et al. 2001), which may also limit our ability to accurately quantify differences in expression levels of X-linked vs. autosomal genes. While the estimates of expression levels of X-linked and autosomal genes in D. melanogaster and C. elegans may not be quantitatively precise, they do suggest at least qualitatively that overall levels of expression are lower for X-linked genes than for autosomal genes in both species. Accordingly, expression levels are not sufficient to explain differences in codon usage between the X and the autosomes. In fact, X-linked genes have significantly higher codon bias than autosomal genes in spite of being expressed at significantly lower levels, making our analyses conservative.
Another possibility is that genes on the X share some other feature that would result in higher levels of codon bias relative to the autosomes. Indeed, codon bias is highly correlated with several other genic features such as protein length (Akashi 1996; Eyre-Walker 1996; Comeron et al. 1999; Duret and Mouchiroud 1999; Marais and Duret 2001), recombination rate (Kliman and Hey 1993; Comeron et al. 1999; Marais et al. 2001, 2003; Hey and Kliman 2002), rate of protein evolution (Akashi 1996; Cutter et al. 2003), gonad-biased expression (via a correlation with rate of protein evolution) (Meiklejohn et al. 2003), expression level (reviewed in Akashi 2001; see also Sharp and Li 1986; Bulmer 1988; Duret and Mouchiroud 1999; Hey and Kliman 2002), and gene density (Hey and Kliman 2002). To test this hypothesis, we partitioned our data by median values of all known correlates of codon bias in both D. melanogaster and C. elegans: protein length, recombination rate, expression level, gene density, and rate of protein evolution. Because the data were available, we were also able to partition by gonad-biased expression for genes in the D. melanogaster genome. Our data suggest that the systematic elevation in codon bias on the X chromosome is not unique to subsets of genes, as codon bias of partitioned genes on the X chromosome is still elevated relative to that of comparable genes on the autosomes in both D. melanogaster (Figure 2a) and C. elegans (Figure 2b). Moreover, the higher codon bias of the X-linked genes cannot be explained by the combined effects of all of these variables in either lineage, as partial correlation analysis reveals a significant positive association between codon bias and X-linkage (Spearman's partial correlation, r = 0.227, r = 0.058 and P ≪ 0.0001, P = 0.006 for D. melanogaster and C. elegans, respectively).
Figure 2.

Optimal codon frequencies on the autosomes (dark shading) and on the X chromosome (light shading) in (a) D. melanogaster and (b) C. elegans. For partitioned data, genes were split on the median value of a given parameter on the X chromosome; this cutoff was then also applied to the autosomal genes. Error bars denote standard error. In D. melanogaster, short proteins were those encoded by ≤394 amino acids, low recombination was defined as ≤3.48 cM/Mb, and low Ka was defined as ≤0.07945. Lowly expressed genes were those with three or fewer EST counts, and gonad-biased expression is defined as genes exceeding a twofold difference in log-ratio of expression between testes and ovaries of adult flies. Comparisons between the X and the autosomes for male- and female-biased genes are significant (P = 0.03 and 0.02, respectively, one-tailed t-test), while all other comparisons are highly significant (P ≪ 0.0001, all comparisons, one-tailed t-test). The numbers of genes included in partitions by length, recombination, Ka, gonad-biased expression, absolute expression, and gene density are 12,444, 12,444, 7335, 1783, 10,202, and 12,368, respectively. In C. elegans, short proteins were those encoded by ≤396 amino acids, low recombination was defined as ≤2.66 cM/Mb, and low Ka was defined as ≤0.113. Lowly expressed genes were those with two or fewer SAGE counts. Differences in codon usage between the X and the autosomes for genes in areas of high recombination and genes with low Ka are not statistically significant (P = 0.48 and 0.14, respectively, one-tailed t-test), while all other comparisons are highly significant (P < 0.0001, all comparisons, one-tailed t-test). The numbers of genes included in partitions by length, recombination, Ka, expression, and gene density are 18,476, 18,476, 10,157, 2988, and 18,442, respectively.
Elevated codon bias on the X is not a function of gene identity:
Another potential explanation for the increased codon bias of X-linked genes is that the gene complements of the X and the autosomes are systematically different. While controlling for all known correlates of codon bias revealed that the systematic increase in codon bias associated with X-linkage is not due to the known properties of the genes residing on the X, it remains possible that other differences between the gene complements of the X and the autosomes could result in an X-specific elevation in codon bias.
To control for this possibility we first compared patterns of codon usage for 457 pairs of duplicate genes in the D. melanogaster genome in which one member of the duplicate pair is on an autosome while the other is on the X chromosome. As predicted, the frequency of optimal codons of the X-linked member of the duplicate pair (0.615) is significantly higher than that of its autosomal paralog (0.571) (P ≪ 0.0001, paired two-tailed t-test) (Figure 3). We similarly examined levels of codon bias in duplicate gene pairs in C. elegans. As is the case in D. melanogaster, X-linked duplicate genes in C. elegans have significantly higher optimal codon frequencies than their autosomal paralogs (0.409 vs. 0.396, n = 565 duplicate pairs; P = 0.003, paired two-tailed t-test) (Figure 3). Because many gene duplications may predate the split of the D. melanogaster and D. pseudoobscura lineages, we opted not to examine codon usage in duplicate gene pairs in D. pseudoobscura.
Figure 3.

Optimal codon frequencies of duplicate gene pairs in D. melanogaster and C. elegans. Optimal codon frequencies of the autosomal members of the duplicate pair are shown with dark shading, while light shading corresponds to optimal codon frequencies in the X-linked paralog. Error bars denote standard error.
As duplicate genes may diverge in function following duplication, we controlled for the effects of changes in gene function on codon bias in an additional comparison, by taking advantage of the autosome-X translocation in D. pseudoobscura. As mentioned above, the right arm of the X chromosome (XR) in D. pseudoobscura arose from a translocation of an autosomal chromosome, what is currently 3L in D. melanogaster, ∼6–10 MYA (Charlesworth and Charlesworth 2005). Consequently, when comparing codon usage patterns of genes on XR in D. pseudoobscura with those of their orthologs located on 3L in D. melanogaster, the only apparent difference between these orthologous genes is the difference in their chromosomal location. Any changes in coding sequence evolution revealed by these orthologous gene pairs necessarily would have evolved in the last 6–10 MY and also would be associated with X-linkage.
If the systematic elevation in codon bias on the X chromosome is due solely to X-linkage, then codon bias of the genes on the right arm (XR) of the D. pseudoobscura genome should be higher than codon bias of the orthologous genes on 3L of the D. melanogaster genome. In contrast, codon bias of the genes on XL in D. pseudoobscura should be comparable to codon bias of their orthologs on the X chromosome in D. melanogaster. Our analysis reveals that this is indeed true. While codon bias of autosomal genes in D. pseudoobscura is significantly higher than codon bias of the orthologous autosomal genes in D. melanogaster (0.581 vs. 0.564, n = 6983 genes; P ≪ 0.0001, paired two-tailed t-test), the difference in codon bias between XR of the D. pseudoobscura genome and 3L of the D. melanogaster genome significantly exceeded this baseline difference (0.599 vs. 0.560, n = 1954 genes; P ≪ 0.0001, paired two-tailed t-test) (Figure 4). Also as expected, the orthologs located on the X chromosome in both species (XL and X in D. pseudoobscura and D. melanogaster, respectively) did not exceed the difference exhibited by the autosomal genes in these two species (0.615 vs. 0.602, n = 1556 genes; P = 0.12, paired two-tailed t-test) (Figure 4). Given that XR in D. pseudoobscura was formed ∼6–10 MYA (Charlesworth and Charlesworth 2005), the increase in optimal codon frequency exceeding the baseline difference between D. pseudoobscura and D. melanogaster of 0.022 suggests that the frequency of optimal codons of autosomal genes translocated to the X has increased at a rate of ∼0.0022–0.0037% per million years.
Figure 4.

Optimal codon frequencies in orthologous genes of D. melanogaster and D. pseudoobscura. Dark shading denotes genes in D. pseudoobscura and light shading shows their orthologs in D. melanogaster. “Autosome” genes are mapped to the autosomes of both species, “XL vs. X” genes are mapped to XL in D. pseudoobscura and X in D. melanogaster, and “XR vs. 3L” genes are mapped to XR in D. pseudoobscura and 3L in D. melanogaster. Error bars denote standard error.
Magnitude of the difference in codon bias between X-linked and autosomal genes:
Because the level of codon bias of an individual gene is determined by a number of factors, quantifying the magnitude of the effect of X-linkage on codon bias separately from other factors is not straightforward. Comparing levels of codon bias of X-linked vs. autosomal genes captures not only the effect of X-linkage, but also the effects of protein length, expression level, and other such factors. To gain insight into the degree to which X-linkage alone increases codon bias, we conducted a series of sampling experiments. We paired each autosomal gene in the D. melanogaster genome with its most similar X-chromosomal counterpart with respect to different gene attributes. We paired genes according to GC content of neighboring noncoding sequences, protein length, expression level, and along all three of these axes. We then estimated the mean optimal codon frequency of this new distribution of autosomal genes; in no cases were the sampled means statistically increased or decreased relative to the initial estimates (see supplementary materials and Figure S1 at http://www.genetics.org/supplemental/). These results suggest that a simple comparison of levels of codon bias between X-linked and autosomal genes estimates fairly the true magnitude of the effect.
As it is difficult to intuit the biological significance of the magnitude of the effect of X-linkage on codon bias, we performed a comparable analysis to gain insight into the scale of the difference in codon bias between X-linked and autosomal genes. Because codon bias and expression level are highly positively correlated (reviewed in Akashi 2001; see also Sharp and Li 1986; Bulmer 1988; Duret and Mouchiroud 1999; Hey and Kliman 2002) we partitioned all autosomal genes into two categories, low and high expression, for both D. melanogaster and C. elegans. We took the class of genes with the lowest expression level as well as a similarly sized class of genes representing those genes most highly expressed. In D. melanogaster, 30% of autosomal genes had EST counts of one; we compared these genes with the 29% of genes that had the highest expression levels (EST counts greater than seven). In C. elegans, 37% of genes had SAGE counts of one; we compared these genes with the 39% of genes with the highest SAGE counts (SAGE counts greater than three). We calculated optimal codon frequencies of these most highly and most lowly expressed genes for each species and compared the difference between categories to the difference in codon bias between X-linked and autosomal genes. For D. melanogaster, the difference in codon bias between X-linked vs. autosomal genes (0.029) was smaller in magnitude than the difference in codon bias of highly vs. lowly expressed genes (0.057) (Figure 5a). In C. elegans as well, highly vs. lowly expressed genes have a much greater difference in codon bias (0.071) than do X-linked vs. autosomal genes (0.024) (Figure 5b). It therefore appears that the difference in codon bias between the X and the autosomes is smaller in magnitude than the difference between the most highly and most lowly expressed genes.
Figure 5.

Frequency of optimal codons in X-linked and autosomal genes with optimal codon frequencies in highly vs. lowly expressed genes for comparison in (a) D. melanogaster and (b) C. elegans.
Population genetic model:
All of these results strongly indicate that codon bias on the X chromosome is elevated relative to that on the autosomes. This pattern is found in three separate systems, D. melanogaster, D. pseudoobscura, and C. elegans, and cannot be attributed to properties of the genes residing on these chromosomes such as expression level or protein length. In addition, this increase in codon bias of X-linked genes is not due to differing gene complements between the X and the autosomes, as we have controlled for gene function in two different ways. Finally, comparing codon usage patterns of the genes involved in the autosome-X translocation in D. pseudoobscura indicates that codon bias increases with X-linkage and does so in a gradual fashion. The persistence of this pattern in divergent taxa suggests that it may reflect a consistent evolutionary response to a universal difference between the X and the autosomes.
Since codon bias is thought to be maintained by a balance between mutation, random genetic drift, and natural selection (Sharp and Li 1986; Bulmer 1991; Akashi and Schaeffer 1997; McVean and Charlesworth 1999), differences in the population genetic parameters between the X and the autosomes could lead to the increase in codon bias of X-linked genes. Because the efficacy of positive selection on new partially recessive mutations is increased on the X chromosome because of the hemizygosity of the X in males, one might expect that X-linked genes may evolve more rapidly than autosomal genes, which could lead to increased codon bias of X-linked genes. Indeed, there is a growing body of literature devoted to “fast-X” evolution in Drosophila. While studies using duplicate genes within a single species or pairs of orthologous genes across species have found increased rates of molecular evolution for X-linked vs. autosomal genes (Thornton and Long 2002; Counterman et al. 2004), unpaired comparisons have given no indication of an increase in rates of molecular evolution of X-linked genes (Betancourt et al. 2002). With respect to the evolution of codon bias, testing for fast-X evolution has proven more challenging, as strong directional selection at amino acid sites can interfere with weak selection on codon bias at linked sites (Betancourt and Presgraves 2002; Betancourt et al. 2004; Kim 2004).
To understand how appropriate a fast-X model for codon bias evolution would be and how population genetic parameters could lead to an increase in codon bias of X-linked genes, we considered a simple population genetic model. Assuming that there are two codon states, preferred and unpreferred, and that the mutation rates from preferred to unpreferred and unpreferred to preferred are μp and μu, respectively, the proportion of codon sites fixed for the preferred codon is 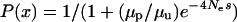 , where
, where 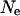 is the effective population size and s is the selection coefficient (Bulmer 1991; Kondrashov 1995). The proportion of preferred codons sampled from a population remains relatively unchanged once polymorphism is accounted for (McVean and Charlesworth 1999). If we assume that the ratio of mutation rates
is the effective population size and s is the selection coefficient (Bulmer 1991; Kondrashov 1995). The proportion of preferred codons sampled from a population remains relatively unchanged once polymorphism is accounted for (McVean and Charlesworth 1999). If we assume that the ratio of mutation rates  is the same for the X and the autosomes, then we can study the ratio
is the same for the X and the autosomes, then we can study the ratio 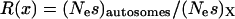 to understand which chromosome set will have higher codon bias. If
to understand which chromosome set will have higher codon bias. If 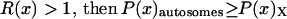 .
.
Imagine that a preferred codon in a haploid state (e.g., on the X in males) is in the following selective environment:
Table 1.
| Unpreferred codon | Preferred codon |
|---|---|
| 1 | 1+s1 |
Also assume that the preferred codon in a diploid state (e.g., on the autosomes) adheres to the following selective regime:
Table 2.
| Homozygous unpreferred | Heterozygote | Homozygous preferred |
|---|---|---|
| 1 | 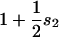 |
 |
In this simple model, 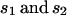 are the coefficients of selection on the X and autosomes, respectively. We assume that the numbers of males (
are the coefficients of selection on the X and autosomes, respectively. We assume that the numbers of males (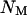 ) and females (
) and females ( ) are not necessarily equal but remain constant over generations. Alleles are assumed to be codominant. This assumption of codominance seems appropriate given that codon bias is under weak selection (Akashi 1995) and that mutations of small effect are generally codominant (Wright 1929; Greenberg and Crow 1960). Comparisons of the levels of X-linked and autosomal nucleotide polymorphism in D. melanogaster and D. simulans hint at the possibility that mutations to unpreferred codons are partially or fully recessive (McVean and Charlesworth 1999), which would change the dynamics of our population genetic model. However, these data do not provide definitive support for an alternative model of the fitness effects of codon bias, which suggests that while the dominance of mutations to and away from unpreferred codons is an open question, a simple model assuming codominance can be regarded as an appropriate starting point. For a more detailed theoretical treatment of the effects of alternative selective schemes on the evolution of codon bias see Li (1987).
) are not necessarily equal but remain constant over generations. Alleles are assumed to be codominant. This assumption of codominance seems appropriate given that codon bias is under weak selection (Akashi 1995) and that mutations of small effect are generally codominant (Wright 1929; Greenberg and Crow 1960). Comparisons of the levels of X-linked and autosomal nucleotide polymorphism in D. melanogaster and D. simulans hint at the possibility that mutations to unpreferred codons are partially or fully recessive (McVean and Charlesworth 1999), which would change the dynamics of our population genetic model. However, these data do not provide definitive support for an alternative model of the fitness effects of codon bias, which suggests that while the dominance of mutations to and away from unpreferred codons is an open question, a simple model assuming codominance can be regarded as an appropriate starting point. For a more detailed theoretical treatment of the effects of alternative selective schemes on the evolution of codon bias see Li (1987).
Given these assumptions, we can study the ratio R(x) by more closely examining Ne and s for the X and the autosomes. If we define F = s1/s2, then 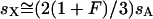 (where sX and sA are the comparable change in frequency due to selection on the X chromosome and autosomes; see materials and methods). From this equation, it is easy to see that when
(where sX and sA are the comparable change in frequency due to selection on the X chromosome and autosomes; see materials and methods). From this equation, it is easy to see that when 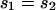 , then the effective selection coefficient on the X,
, then the effective selection coefficient on the X, 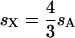 . This alone suggests that codon bias of X-linked genes could be higher than codon bias of autosomal genes since the effective strength of selection is stronger on the X. However, effective population size may also be different for the X and the autosomes. Under our assumptions,
. This alone suggests that codon bias of X-linked genes could be higher than codon bias of autosomal genes since the effective strength of selection is stronger on the X. However, effective population size may also be different for the X and the autosomes. Under our assumptions, 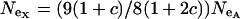 , where
, where 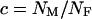 ; when the numbers of males and females are equal,
; when the numbers of males and females are equal,  .
.
Together, differences in the effective selection of preferred codons and differences in population size between the X and the autosomes will work to affect 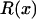 . When the numbers of males and females are equal and the coefficients of selection are equal on the X and the autosomes, R(x) = 1, suggesting that levels of codon bias on the X and the autosomes should be exactly the same. In general, we can investigate the dependence of R(x) on c and F, which reveals that codon bias of X-linked genes can exceed levels of codon bias on the autosomes [R(x) > 1] if s1 > s2 or if c < 1 (Figure 6). In other words, the effective number of males must be less than the effective number of females or the strength of selection on translational efficiency must be stronger on the X chromosome than on the autosomes to explain the increase in codon bias of X-linked genes.
. When the numbers of males and females are equal and the coefficients of selection are equal on the X and the autosomes, R(x) = 1, suggesting that levels of codon bias on the X and the autosomes should be exactly the same. In general, we can investigate the dependence of R(x) on c and F, which reveals that codon bias of X-linked genes can exceed levels of codon bias on the autosomes [R(x) > 1] if s1 > s2 or if c < 1 (Figure 6). In other words, the effective number of males must be less than the effective number of females or the strength of selection on translational efficiency must be stronger on the X chromosome than on the autosomes to explain the increase in codon bias of X-linked genes.
In addition, on the basis of several simplifying assumptions (see materials and methods) we investigated what values of c and F could explain the difference in magnitudes of codon bias that we see in D. melanogaster. Our analysis indicates that if s1 = s2, then the number of males may need to be only approximately one-half that of females to lead to the difference in codon bias that we observe (Figure 6, dashed line), although the precise fit of c and F to our data depends on the ratio of μp and μu (see supplementary materials and Figure S2 at http://www.genetics.org/supplemental/).
The dosage problem and codon bias:
Given that the variance in reproductive success is believed to be greater for males than for females, it is easy to envision how the effective number of males could be less than the effective number of females; this alone could explain the observed elevation of the codon bias on the X chromosome. It is also possible that the strength of selection on translational efficiency could be stronger on the X chromosome. Under what circumstances would the benefits of more efficient/accurate translation be greater on the X than on the autosomes?
One possible explanation is that the increases in active protein level that result from increases in codon bias are more beneficial for X-linked genes than for autosomal genes because of the dosage problem. While much of dosage compensation in Drosophila and Caenorhabditis is achieved through transcriptional regulation (for review see Baker et al. 1994; Marin et al. 2000), it is also possible that increased codon bias of X-linked genes may partially remediate the deleterious effects of the reduced dosage of these genes in males. In genes with higher codon bias, the presence of preferred codons corresponding to abundant tRNAs (Shields et al. 1988) is believed to increase the efficiency and/or fidelity of translation (Akashi and Eyre-Walker 1998; Akashi et al. 1998; Bulmer 1991). In particular, use of unpreferred codons has been empirically shown to decrease protein level in Drosophila (Carlini and Stephan 2003). The replacement of preferred codons with unpreferred ones in ADH resulted in significant decreases in protein activity (micromoles of NAD+ reduced per minute per milligram of total protein × 100), such that each substitution conferred a loss in activity of ∼2.13% (Carlini and Stephan 2003). Although these data are from only one gene, they suggest that the effects of codon usage on protein level can be substantial.
Increases in codon bias and therefore translational efficiency will necessarily affect protein levels in both sexes. Given that the deleterious effects of gene dosage reductions are generally more pronounced than those of similarly sized gene dosage increases (Lindsley et al. 1972), it seems plausible that increases in translational efficiency of X-linked genes might be advantageous overall, with the benefit in males outweighing the potential detriment of too much active protein in females. Overall, then, it seems possible that levels of codon bias can directly affect the amount of available protein, and that the dosage problem may create a selective environment in which the benefits of increases in codon bias of X-linked genes exceed the benefits of comparable increases in codon bias of autosomal genes. As a result, the consistent increase in codon bias of X-linked genes in several taxa may have evolved as a consequence of the dosage problem.
Conclusions and future work:
The evidence presented herein suggests that codon bias on the X is elevated relative to that on the autosomes. This systematic elevation in codon bias is due entirely to X-linkage and not to any other property of the genes on the X or the autosomes. In addition, we have documented this pattern in three distinct taxa, D. melanogaster, D. pseudoobscura, and C. elegans, suggesting that this X-specific increase in codon bias may be common or even universal in eukaryotes with chromosomal sex determination. That this pattern is found consistently across taxa suggests that the forces responsible should be operating similarly across species boundaries.
Population genetic analysis suggests that to explain this increase in codon bias of X-linked genes, either the strength of selection on translational efficiency is greater on the X than on the autosomes or the effective number of males is much less than the effective number of females. Although the mating systems between these two taxa differ substantially, it is certainly possible that for both Drosophila and Caenorhabditis, the number of breeding females exceeds the number of breeding males. Our population genetic model suggests that at least for Drosophila, a ratio of breeding males to females of 0.5 would be sufficient to generate the observed difference in codon bias of X-linked vs. autosomal genes; such a moderate difference in the variance of reproductive success could easily be universal across taxa.
It is similarly possible that the reduced dosage of X-linked genes in males exaggerates the benefits of increased codon bias for genes on the X chromosome. The dosage problem is universal for taxa with systems of chromosomal sex determination and thus it is capable of explaining the increase in codon bias of X-linked genes that we have documented in both Drosophila and C. elegans.
These two explanations are not mutually exclusive and both may contribute to the increased codon bias of X-linked genes. Note that the increased codon bias of the X-linked genes should have important functional consequences such as higher efficiency and fidelity of their translation, independent of the reason(s) for the increase. This in turn might have important implications with respect to the evolution of the levels of expression and the maintenance of transcriptional dosage compensation of X-linked genes.
Further investigation of the population genetics of codon usage on the X and the autosomes will likely help elucidate the ultimate cause for the differences in codon usage between the X and the autosomes in Drosophila and Caenorhabditis. Additional population genetic explanations are possible, especially if the assumption of the codominance of mutations toward codon bias proves incorrect. More sophisticated theoretical models quantifying how large the differences in reproductive success need to be between males and females to obtain the measured difference in codon usage bias between the X and the autosomes are an interesting avenue of future work. Such models can also determine whether the rate of change of codon bias that we measured in this study is consistent with weak selection generating codon bias.
In addition, the dosage compensation hypothesis requires that changes in the degree of codon bias are sufficient to cause appreciable changes in protein levels. Determining the magnitude of the change in protein level given changes in optimal codon frequency on the order of 0.01–0.03 in Drosophila and Caenorhabditis may help assess the feasibility of this explanation. Another prediction of this model is that dosage compensation through transcriptional regulation is not complete for many genes; as our ability to quantify sex-specific expression improves, this prediction may be tested directly. Species with newly formed sex chromosomes such as D. miranda should provide unique insight into the generality of this model, as dosage compensation appears to have evolved for some genes on the neo-X and not for others (Marin et al. 1996).
Acknowledgments
The authors thank Brian Charlesworth, Andrea Betancourt, Yuseob Kim, Marc Feldman, Aaron Hirsh, Bruce Baker, Mark Siegal, Guy Sellla, and members of DAP lab for enlightening discussions and feedback on this project. Comments from two anonymous reviews also substantially improved this manuscript. This work is supported in part by the Stanford Genome Training Program (funded by 5 T32 HG00044 from the NHGRI) to N.D.S., an NSF predoctoral fellowship to J.C.D., and the NSF grant 0317171 and the Alfred P. Sloan Fellowship to D.A.P.
References
- Akashi, H., 1995. Inferring weak selection from patterns of polymorphism and divergence at “silent” sites in Drosophila DNA. Genetics 139: 1067–1076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Akashi, H., 1996. Molecular evolution between Drosophila melanogaster and D. simulans: reduced codon bias. Faster rates of amino acid substitution, and larger proteins in D. melanogaster. Genetics 144: 1297–1307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Akashi, H., 2001. Gene expression and molecular evolution. Curr. Opin. Genet. Dev. 11: 660–666. [DOI] [PubMed] [Google Scholar]
- Akashi, H., and A. Eyre-Walker, 1998. Translational selection and molecular evolution. Curr. Opin. Genet. Dev. 8: 688–693. [DOI] [PubMed] [Google Scholar]
- Akashi, H., and S. W. Schaeffer, 1997. Natural selection and the frequency distributions of “silent” DNA polymorphism in Drosophila. Genetics 146: 295–307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Akashi, H., R. M. Kliman and A. Eyre-Walker, 1998. Mutation pressure, natural selection, and the evolution of base composition in Drosophila. Genetica 102/103: 49–60. [PubMed] [Google Scholar]
- Bachtrog, D., 2003. Protein evolution and codon usage bias on the neo-sex chromosomes of Drosophila miranda. Genetics 165: 1221–1232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baker, B. S., M. Gorman and I. Marin, 1994. Dosage compensation in Drosophila. Annu. Rev. Genet. 28: 491–521. [DOI] [PubMed] [Google Scholar]
- Betancourt, A. J., and D. C. Presgraves, 2002. Linkage limits the power of natural selection in Drosophila. Proc. Natl. Acad. Sci. USA 99: 13616–13620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Betancourt, A. J., D. C. Presgraves and W. J. Swanson, 2002. A test for faster X evolution in Drosophila. Mol. Biol. Evol. 19: 1816–1819. [DOI] [PubMed] [Google Scholar]
- Betancourt, A. J., Y. Kim and H. A. Orr, 2004. A pseudo-hitchhiking model of X vs. autosomal diversity. Genetics 168: 2261–2269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bridges, C. B., 1935. Salivary chromosome maps with a key to the banding of the chromosomes of Drosophila melanogaster. J. Hered. 26: 60–64. [Google Scholar]
- Bulmer, M., 1988. Are codon usage patterns in unicellular organisms determined by selection mutation balance? J. Evol. Biol. 1: 15–26. [Google Scholar]
- Bulmer, M., 1991. The selection-mutation-drift theory of synonymous codon usage. Genetics 129: 897–908. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Caballero, A., 1995. On the effective size of populations with separate sexes, with particular reference to sex-linked genes. Genetics 139: 1007–1011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carlini, D. B., and W. Stephan, 2003. In vivo introduction of unpreferred synonymous codons into the Drosophila Adh gene results in reduced levels of ADH protein. Genetics 163: 239–243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Charlesworth, B., 1991. The evolution of sex chromosomes. Science 251: 1030–1033. [DOI] [PubMed] [Google Scholar]
- Charlesworth, D., and B. Charlesworth, 2005. Sex chromosomes: evolution of the weird and wonderful. Curr. Biol. 15: R129–R131. [DOI] [PubMed] [Google Scholar]
- Charlesworth, B., J. A. Coyne and N. H. Barton, 1987. The relative rates of evolution of sex chromosomes and autosomes. Am. Nat. 130: 113–146. [Google Scholar]
- Comeron, J. M., M. Kreitman and M. Aguade, 1999. Natural selection on synonymous sites is correlated with gene length and recombination in Drosophila. Genetics 151: 239–249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Conery, J. S., and M. Lynch, 2001. Nucleotide substitutions and the evolution of duplicate genes. Pac. Symp. Biocomput., 167–178. [DOI] [PubMed]
- Counterman, B. A., C. Ortiz-Barrientos and M. A. F. Noor, 2004. Using comparative genomic data to test for fast-X evolution. Evolution 58: 656–660. [PubMed] [Google Scholar]
- Cutter, A. D., B. A. Payseur, T. Salcedo, A. M. Estes, J. M. Good et al., 2003. Molecular correlates of genes exhibiting RNAi phenotypes in Caenorhabditis elegans. Genome Res. 13: 2651–2657. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duret, L., and D. Mouchiroud, 1999. Expression pattern and, surprisingly, gene length shape codon usage in Caenorhabditis, Drosophila, and Arabidopsis. Proc. Natl. Acad. Sci. USA 96: 4482–4487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eyre-Walker, A., 1996. Synonymous codon bias is related to gene length in Escherichia coli: Selection for translational accuracy? Mol. Biol. Evol. 13: 864–872. [DOI] [PubMed] [Google Scholar]
- Greenberg, R., and J. F. Crow, 1960. A comparison of the effect of lethal and detrimental chromosomes from Drosophila populations. Genetics 45: 1153–1168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hartl, D. L., and A. G. Clark, 1989. Principles of Population Genetics. Sinauer Associates, Sunderland, MA.
- Hey, J., and R. M. Kliman, 2002. Interactions between natural selection, recombination and gene density in the genes of Drosophila. Genetics 160: 595–608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jones, S. J. M., D. L. Riddle, A. T. Pouzyrev, V. E. Velculescu, L. Hillier et al., 2001. Changes in gene expression associated with developmental arrest and longevity in Caenorbabditis elegans. Genome Res. 11: 1346–1352. [DOI] [PubMed] [Google Scholar]
- Kim, Y., 2004. Effect of strong directional selection on weakly selected mutations at linked sites: implication for synonymous codon usage. Mol. Biol. Evol. 21: 286–294. [DOI] [PubMed] [Google Scholar]
- Kliman, R. M., and J. Hey, 1993. Reduced natural selection associated with low recombination in Drosophila melanogaster. Mol. Biol. Evol. 10: 1239–1258. [DOI] [PubMed] [Google Scholar]
- Kondrashov, A. S., 1995. Contamination of the genome by very slightly deleterious mutations: Why have we not died 100 times over? J. Theor. Biol. 175: 583–594. [DOI] [PubMed] [Google Scholar]
- Laporte, V., and B. Charlesworth, 2002. Effective population size and population subdivision in demographically structured populations. Genetics 162: 501–519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li, W. H., 1987. Models of nearly neutral mutations with particular implications for nonrandom usage of synonymous codons. J. Mol. Evol. 24: 337–345. [DOI] [PubMed] [Google Scholar]
- Lindsley, D. L., L. Sandler, B. S. Baker, A. T. C. Carpenter, R. E. Denelll et al., 1972. Segmental aneuploidy and the genetic gross structure of the Drosophila genome. Genetics 71: 157–184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marais, G., and L. Duret, 2001. Synonymous codon usage, accuracy of translation, and gene length in Caenorhabditis elegans. J. Mol. Evol. 52: 275–280. [DOI] [PubMed] [Google Scholar]
- Marais, G., D. Mouchiroud and L. Duret, 2001. Does recombination improve selection on codon usage? Lessons from nematode and fly complete genomes. Proc. Natl. Acad. Sci. USA 98: 5688–5692. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marais, G., D. Mouchiroud and L. Duret, 2003. Neutral effect of recombination on base composition in Drosophila. Genet. Res. 81: 79–87. [DOI] [PubMed] [Google Scholar]
- Margulies, E. H., S. L. R. Kardia and J. W. Innis, 2001. Identification and prevention of a GC content bias in SAGE libraries. Nucleic Acids Res. 29: e60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marin, I., A. Franke, G. J. Bashaw and B. S. Baker, 1996. The dosage compensation system of Drosophila is co-opted by new evolved X chromosomes. Nature 383: 160–163. [DOI] [PubMed] [Google Scholar]
- Marin, I., M. L. Siegal and B. S. Baker, 2000. The evolution of dosage-compensation mechanisms. BioEssays 22: 1106–1114. [DOI] [PubMed] [Google Scholar]
- McVean, G. A. T., and B. Charlesworth, 1999. A population genetic model for the evolution of synonymous codon usage: patterns and predictions. Genet. Res. 74: 145–158. [Google Scholar]
- Meiklejohn, C. D., J. Parsch, J. M. Ranz and D. L. Hartl, 2003. Rapid evolution of male-biased gene expression in Drosophila. Proc. Natl. Acad. Sci. USA 100: 9894–9899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parisi, M., R. Nuttall, D. Naiman, G. Bouffard, J. Malley et al., 2003. Paucity of genes on the Drosophila X chromosome showing male-biased expression. Science 299: 697–700. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Powell, J. R., and R. DeSalle, 1995. Drosophila molecular phylogenies and their uses. Evol. Biol. 28: 87–138. [Google Scholar]
- Rice, P., I. Longden and A. Bleasby, 2000. The European molecular biology open source suite. Trends Genet. 16: 276–277. [DOI] [PubMed] [Google Scholar]
- Segarra, C., G. Ribo and M. Aguade, 1996. Differentiation of Muller's chromosomal elements D and E in the obscura group of Drosophila. Genetics 144: 139–146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sharp, P. M., and W. H. Li, 1986. An evolutionary perspective on synonymous codon usage in unicellular organisms. J. Mol. Evol. 24: 28–38. [DOI] [PubMed] [Google Scholar]
- Shields, D. C., P. M. Sharp, D. G. Higgins and F. Wright, 1988. ‘Silent’ sites in Drosophila genes are not neutral: evidence of selection among synonymous codons. Mol. Biol. Evol. 5: 704–716. [DOI] [PubMed] [Google Scholar]
- Singh, N. D., J. C. Davis and D. A. Petrov, 2005. Codon bias and noncoding GC content correlate negatively with recombination rate on the Drosophila X chromosome. J. Mol. Evol. (in press). [DOI] [PubMed]
- Stein, L. D., Z. Bao, D. Blasiar, T. Blumenthat, M. R. Brent et al., 2003. The genome sequence of Caenorhabditis briggsae: a platform for comparative genomics. PLoS Biol. 1: 166–192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thornton, K., and M. Long, 2002. Rapid divergence of gene duplicates on the Drosophila melanogaster X chromosome. Mol. Biol. Evol. 19: 918–925. [DOI] [PubMed] [Google Scholar]
- Wright, S., 1929. Fisher's theory of dominance. Am. Nat. 63: 274–279. [Google Scholar]
- Yang, Z., 1997. PAML: a program package for phylogenetic analysis by maximum likelihood. Comput. Appl. Biosci. 13: 555–556. [DOI] [PubMed] [Google Scholar]