

Abstract
The use of linkage disequilibrium to localize the genes underlying quantitative traits has received considerable attention in the livestock genetics community over the past few years. This has resulted in the investigation of linkage disequilibrium structures of several domestic livestock populations to assess their potential use in fine-mapping efforts. However, the linkage disequilibrium structure of free-living populations has been less well investigated. As the direct evaluation of linkage disequilibrium can be both time consuming and expensive the use of simulations that include as many aspects of population history as possible is advocated as an alternative. A simulation of the linkage disequilibrium structure of the Soay sheep population of St. Kilda, Scotland, is provided as an example. The simulated population showed significant decline of linkage disequilibrium with genetic distance and low levels of background linkage disequilibrium, indicating that the Soay sheep population is a viable resource for linkage disequilibrium fine mapping of quantitative trait loci.
RECENTLY, investigations into the linkage disequilibrium (LD) structures of domestic livestock have shown the potential for the use of LD to localize the genes underlying quantitative traits (Farnir et al. 2000; McRae et al. 2002; Tenesa et al. 2003; Nsengimana et al. 2004). This potential has been realized in at least two cases, with the fine mapping of a twinning locus to a region of <1 cM (Meuwissen et al. 2002) and the identification and subsequent characterization of a gene for milk yield and composition (Grisart et al. 2002).
Wild populations provide a valuable resource for the understanding of the genetics of quantitative traits. Not only can the genetics of traits such as birth weight be compared to domestic populations, providing an insight into the effects of population management on the expression of quantitative traits, but also other traits that cannot be studied in managed populations, such as traits associated with fitness (survival, lifetime breeding success), can be considered. However, before attempting to use LD for fine mapping genes in any population, it is important to have an understanding of the underlying LD structure to enable an appropriate choice of study design.
Direct evaluation of the LD structure of a population is both time consuming and expensive. However, the LD structure of a population should not be evaluated only as a secondary objective of a LD mapping experiment. LD could extend over prohibitively short distances given the density of markers available in the studied species. Alternatively, large amounts of LD between markers that are unlinked or separated by large distances will result in an increased number of false positive results and a lack of power to localize the regions of interest. In these scenarios, this may result in any generated marker data being unusable for its intended purpose. Theoretical estimation of the levels of LD for realistic populations is complex due to the large number of interrelated factors involved in the formation of LD, including genetic drift, population admixture, and natural selection. Any LD formed is broken down in a subsequent generation by recombination, which generally results in a pattern of LD decreasing with distance. With respect to LD mapping, the important factors in the LD structure are a significant decay with genetic distance and the amount of variation about the average decay.
Simulation studies provide a useful alternative to the direct evaluation of LD structure. This approach has been used in humans (Kruglyak 1999) and these predictions for the level of LD are reasonably close to those observed from experimental data, despite the very simple model applied. However, significant deviations are observed in populations where the model's assumptions about population structure are not applicable (reviewed in Pritchard and Przeworski 2001; Wall and Pritchard 2003). In this article, a simulation of a well-studied wild sheep population is performed. The large amount of data available on the population history allows this simulation to include the major aspects of the population dynamics, providing a model that should predict the overall structure of LD in the population with a reasonable accuracy.
MATERIALS AND METHODS
A three-step strategy is used to model the LD in the Soay sheep population. First, census data are used to model various aspects of population dynamics, including population size, adult sex ratio, variation in reproductive success, and survival. These models are then used to simulate the population given the current understanding of its history. Finally, LD is measured from the loci simulated in the population.
Population data:
The Soay sheep is a primitive domestic sheep that has lived on the islands of St. Kilda, Scotland (54°49′N, 08°34′W), for many centuries (Figure 1). The exact date of their introduction is unknown but is likely to have been between 1000 and 2000 years ago (Boyd and Boyd 1990). The Soay sheep were eventually restricted to the island of Soay after which they were named. In 1932 a flock of 107 Soay sheep was moved from Soay to Hirta. The flock consisted of 20 rams, 44 ewes, and 43 lambs, of which 21 were female and the remaining were castrated males (Boyd 1953).
Figure 1.

Outline map of part of the St. Kilda archipelago. The Soay sheep were restricted to the island of Soay until 1932 when a small group was moved to Hirta, which now holds the majority of the population. The Village Bay study area is shaded.
A census of Hirta is performed each year by three teams using designated census routes that have been followed since 1959 (Boyd et al. 1964). An intensive study of the sheep in the Village Bay area began in 1985, this being the largest concentration of sheep on the island, comprising ∼30% of the total number. In the spring, lambs are caught when a few days old, weighed, tagged, and blood sampled. Each year during the summer, an extensive effort is made to catch all the sheep in the Village Bay area, with all animals caught being recorded and any new animals tagged and blood sampled. Mortality is recorded by searches during late winter and early spring, with additional searches being performed in years of high mortality.
Statistical modeling:
Population size:
The population size appears to fluctuate randomly (see Figure 2), with local maxima occurring at irregular intervals. Boyd (1974) showed a weak upward trend in population size in the years until 1973 and this increase appears to have continued until today. This increase may be an artifact of improved census counts. This explanation suggests modeling population size using the log of the data, as the size of fluctuations will be confounded with the proportion of sheep being counted. Using the log of the data also allows for the indication of density-dependent mortality in the populations (Grenfell et al. 1992). The standardized residuals of the linear regression of the log of the population size on the year of the count are used in all following models as these allow for the interpretation of population dynamics in terms of deviations from the average population trend, thus removing any potential effects of improving census quality. For convenience, these standardized residuals are referred to as residual population size.
Figure 2.

Census population size of the Soay sheep population on Hirta for the years 1955–2001. The population size is characterized by its frequent crashes when population size decreases by up to 60% in 1 year. The weak upward trend in population size with time is shown (dashed line).
The changing residual population size was modeled using a self-exciting threshold autoregressive (SETAR) model (Tong 1990), with model selection based on the improved cross-validation criterion, Cu, proposed by De Gooijer (2001). SETAR models are useful in modeling data in which future values depend on previous values but this dependence varies with the current value of the system. For example, in years where the population size is small a model showing a rapid increase in population size may be appropriate. Once the population size becomes larger, this rate of increase may be smaller than that for small population sizes and then become negative for large population sizes. Each region where a different model is applied in predicting future values is called a regime, with its boundaries known as thresholds. For the above example three regimes are given by thresholds that define what constitutes small and large population sizes. In each regime an autoregressive (AR) model is used in predicting future values. This type of model has previously been applied to the Soay sheep population (Grenfell et al. 1998) but without removing the trend and using a different model selection criterion.
Model selection:
A modified model selection procedure was used for modeling the proportion of lambs, adult sex ratio, and survival. All models are based on linear regressions on residual population sizes of the current and previous years. Initial models used residual population sizes from the previous 5 years, with this being successively reduced when the earliest year in the model was not significant at the 5% level. This modified selection procedure allows effects for years after the earliest significant year to be nonsignificant. As a delayed response to population size is unlikely, this type of model selection is useful, especially when the power to detect small effects is low. One disadvantage of this selection regime is the potential of false positive results that lead to a highly overparameterized model, but choosing reasonable starting models reduces the likelihood of this occurring.
Lamb proportion and adult sex ratio:
The proportion of lambs in the population in year i, denoted pi, was modeled as
 |
where rj is the residual population size in year j and  is the normally distributed error term. The delay parameter d was successively reduced from an initial value of 5 in the model selection procedure. The modeling of neonatal mortality is prevented as the model is based on census data that are collected several months after lambing but before the mating season. The ratio of number of male to female adult sheep in year i, denoted si, was modeled as
is the normally distributed error term. The delay parameter d was successively reduced from an initial value of 5 in the model selection procedure. The modeling of neonatal mortality is prevented as the model is based on census data that are collected several months after lambing but before the mating season. The ratio of number of male to female adult sheep in year i, denoted si, was modeled as
 |
with model selection again reducing d from an initial value of 5.
Survival:
For the modeling of survival, the data set was restricted to all sheep with known birth date and known fate (either a known death date or known to be still alive). Separate models for the effect of residual population size on survival were made for each age and sex. For each year, the hazard function was evaluated using the Nelson-Aalen estimator
 |
where di,j,s is the number of observed deaths in the ni,j,s sheep of age i and sex s alive in year j and followed to year j + 1. The effect of residual population size on the hazard function was modeled using a weighted regression as
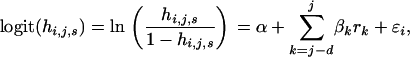 |
where d was selected using the model selection procedure described above starting from an initial value of 4 and weights were given by ni,j,s. In older age groups, where data were available from only a few years and there was no observable effect of residual population size on the hazard function, the hazard was modeled as a beta distribution with parameters 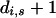 and
and 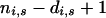 , where di,s and ni,s are obtained from the summation over j of di,j,s and ni,j,s, respectively. As no observations of males older than 12 years or females older than 16 years were made, these were considered the upper limit for the lifespan of the Soay sheep.
, where di,s and ni,s are obtained from the summation over j of di,j,s and ni,j,s, respectively. As no observations of males older than 12 years or females older than 16 years were made, these were considered the upper limit for the lifespan of the Soay sheep.
Simulation model:
The Soay sheep population was modeled to reflect the population's history since the introduction to Hirta in 1932. For each of the 1000 simulation replicates, the initial haplotypes, which contain a pair of loci separated by a random distance between 0 and 40 cM, were simulated using a coalescent process (see below). The ages of the adults in the original sheep were sampled from the equilibrium age structure given by the pooled survival estimates for each sex. The castrated ram lambs that were initially transferred to Hirta were not included in the simulation, as these would not leave any descendants. The initial population growth on Hirta was modeled by the doubling of population size each year until this exceeded the population size trend. From this point, until island census data were available, the population size was given by a realization from the model for population size. For each year that the simulation iterated, realizations from models for sex ratio, proportion of lambs, and survival were taken, conditioned on the realized residual population size.
Given the yearly pattern of mortality of the Soay sheep after the rut but before the birth of lambs that is observed in the Soay sheep and the fact that rams can have offspring born after their death but ewes cannot, the following steps were cycled through in each year of the simulation: (1) death of ewes, (2) creation of lambs from remaining sheep, (3) death of rams, and (4) aging of sheep that remain from the previous year. The number of sheep that were to die at each step of the simulation model was determined by the required adult sex ratio and proportion of lambs given the residual population size. The survival model was used only to give an appropriate age structure to the population. This was achieved by choosing a random sheep and comparing a random number to the realized hazard for sheep of that age, hi. If the random number was less than hi, the sheep was removed from the population, otherwise it was returned to the sampling pool. This was repeated until the required population size was achieved. The efficiency of the steps involved was greatly increased by replacing hi with 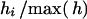 . The sampling process used in choosing parents for the generated lambs is described below. Haplotypes passed on to lambs were generated assuming recombination rates given by Haldane's map function.
. The sampling process used in choosing parents for the generated lambs is described below. Haplotypes passed on to lambs were generated assuming recombination rates given by Haldane's map function.
Generation of initial haplotypes:
Although the population fluctuations on Hirta and its surrounding islands have been shown to be in synchrony (Grenfell et al. 1998) and estimates about the period of time that the Soay sheep have been present on Soay are available, it is difficult to extend these concepts to generating appropriate haplotypes for the initial sheep moved to Hirta. However, some of the properties of the current Soay sheep population can be used to provide an appropriate benchmark for the simulation of these haplotypes.
An estimate for the genome-wide heterozygosity of the Soay sheep population of 0.385 (SE = 0.009) was obtained by considering a panel of 144 microsatellite markers with a high polymorphic information content covering the majority of the genome of five rams from the current population chosen because of their high prolificacy (our unpublished observations). Simulating the population starting with completely heterozygous founders indicates that heterozygosity in the Soay sheep has decreased by 7.1% since their introduction to Hirta (10,000 replicates, data not shown). Thus the average heterozygosity of the microsatellite markers examined in the founding sheep is inferred to be 0.414. Under a neutral model, the heterozygosity at a locus equals 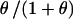 ,
, 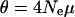 , where
, where 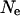 is the effective population size and μ is the mutation rate at the locus. A reasonable estimate for μ will be in the high end of the observed mutation rate in mammals given the highly polymorphic loci chosen to measure the average heterozygosity. The microsatellite markers used in paternity analysis are similarly chosen and have mutation rates ranging from 0.02 to 0.0001 (Ellegren 2004). Using
is the effective population size and μ is the mutation rate at the locus. A reasonable estimate for μ will be in the high end of the observed mutation rate in mammals given the highly polymorphic loci chosen to measure the average heterozygosity. The microsatellite markers used in paternity analysis are similarly chosen and have mutation rates ranging from 0.02 to 0.0001 (Ellegren 2004). Using 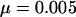 gives
gives 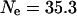 , which is ∼15% of the average population size on Soay. This estimate is consistent with the observed decrease in heterozygosity given above and with estimates of
, which is ∼15% of the average population size on Soay. This estimate is consistent with the observed decrease in heterozygosity given above and with estimates of 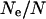 in other wild populations (Frankham 1995).
in other wild populations (Frankham 1995).
For each replicate, 40 Mb of sequence was simulated with a neutral coalescent model using the program “ms” (Hudson 2002). A mutation rate of 10−8 was used in the simulation as ms uses an infinite-site model of mutation and thus generates loci with histories similar to SNP markers. Recombination was assumed to occur at a rate of 10−8 between base pairs, giving a 1 cM/Mb ratio as approximately observed on average in the sheep genome. This generated a large number of polymorphic sites (mean 320) of which the leftmost and another random site were chosen for use in the remaining simulation, giving an approximately uniform distribution of allele separation across the simulation replicates.
Modeling of reproduction:
The reproductive success of female Soay sheep is relatively uniform, with adult females only rarely not giving birth to lambs. However, the proportion of births producing twins decreases with population size. This association does not need to be modeled due to being intrinsically modeled through changes in population size. When the number of lambs to be generated was less than the number of ewes, the lambs were assigned randomly to individual ewes. If the number of lambs was greater than the number of ewes, all ewes were assigned one lamb with the remaining lambs being distributed to individual ewes.
Modeling of male reproductive success is more difficult as the promiscuous nature of the Soay sheep requires paternities to be inferred by molecular methods. From the inferred paternities (at 80% confidence) of 44% of the Village Bay lambs born between 1986 and 1996, the distribution of male lifetime breeding success was highly skewed with mean of 1.05 and variance 4.41 (Pemberton et al. 2004). Although these data are not complete, they provide information on the coefficient of variation of male breeding success. Male breeding success was modeled by assigning each male individual a random “success” variable from a two-parameter gamma distribution with mean and variance as given above. As with survival, the selection of male parents was achieved using rejection sampling methodology. For ewes having a single lamb, a male parent was chosen by randomly selecting a male and comparing its success variable (scaled to have maximum value one) to a uniform random number. The male was selected when the random number was less than the scaled success variable; otherwise the sampling process was repeated. When a ewe was assigned as having twins, first the number of male parents was randomly determined. The proportion of twins that are fathered by the same sire has been estimated as 26% (Pemberton et al. 1999) and any effect of population size on this value is likely to be small. The sampling of male parents for twins with different fathers was achieved using the same methodology as for single lambs. For sampling a single father for twin lambs, the square of the scaled success variable was compared to the random uniform number to correct for the fact that the selected individual was being assigned two paternities.
Measuring linkage disequilibrium:
LD was measured using Lewontin's (1964) standardized measure of LD, 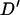 . This measure was chosen to enable comparisons with LD observed in previous studies of domestic livestock that have used Hedrick's (1987) multiallelic extension of this measure.
. This measure was chosen to enable comparisons with LD observed in previous studies of domestic livestock that have used Hedrick's (1987) multiallelic extension of this measure. 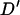 is calculated as
is calculated as
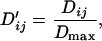 |
where
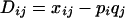 |
and
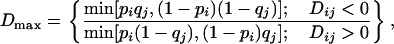 |
where xij is the frequency of haplotypes with allele i at the first marker and allele j at the second marker. The absolute value of 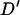 is reported as the sign of the
is reported as the sign of the 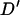 -statistic depends on the arbitrary choice of allele at each locus. As
-statistic depends on the arbitrary choice of allele at each locus. As 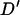 can be skewed when one or both markers contain rare alleles (Eyre-Walker 2000), replicates giving final allele frequencies <0.1 were repeated.
can be skewed when one or both markers contain rare alleles (Eyre-Walker 2000), replicates giving final allele frequencies <0.1 were repeated.
Stability of estimates:
The effect of model parameters on LD estimates was examined by replicating the simulation with varied parameters. Three areas need to be examined for their effect on the final LD estimates: the value of 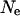 used in the construction of the original haplotypes, the parameter estimates in the models for population dynamics, and the model of male breeding success. The effect of the initial value of
used in the construction of the original haplotypes, the parameter estimates in the models for population dynamics, and the model of male breeding success. The effect of the initial value of 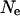 is examined by both reducing and increasing the assumed value of
is examined by both reducing and increasing the assumed value of 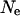 by a factor of two. For the examination of the effects of population model parameter estimates, an ∼95% confidence interval was constructed for each coefficient of residual population size. These were then categorized into groups of high and low effect of residual population size. The simulation was run using these groupings representing extremes of possible variation caused through modeling population dynamics. The effect of the model of male breeding structure was examined by simulating with the coefficient of variation of the success variable reduced and increased by a factor of two.
by a factor of two. For the examination of the effects of population model parameter estimates, an ∼95% confidence interval was constructed for each coefficient of residual population size. These were then categorized into groups of high and low effect of residual population size. The simulation was run using these groupings representing extremes of possible variation caused through modeling population dynamics. The effect of the model of male breeding structure was examined by simulating with the coefficient of variation of the success variable reduced and increased by a factor of two.
RESULTS
Figure 3 compares aspects of the Village Bay population with that of the entire island. The census counts in the Village Bay area show a strong correlation with the island census counts (see Figure 3, a and b, r = 0.962, P < 0.001), highlighting that a fluctuation in population size is not area specific but affects the whole island. The proportions of lambs in the two populations are similar (see Figure 3c), although a slightly higher proportion of lambs is given by the island census. The adult sex ratio is higher in the Village Bay population (Figure 3d), but this is likely to be due to the increased accuracy of the Village Bay count with the island census, as rams are undercounted compared to ewes in the island census (Grubb 1974). This indicates that the use of the Village Bay data is preferential in the modeling of aspects of population structure, despite having fewer data points, as this is likely to give less biased results. However, the strong correlation between island census counts and the Village Bay population counts suggests that the use of the larger data set would be beneficial in modeling the population fluctuations as it is unlikely to bias the results.
Figure 3.

Comparison between Village Bay and Hirta population data. (a) Plot of census population sizes of Hirta (solid line) and the Village Bay population (dashed line). (b) Scatter plot showing the strong correlation between the census population sizes of Hirta and the Village Bay (r = 0.962, P < 0.0001). (c) Comparison of the proportion of lambs observed in Hirta and the Village Bay. The dashed line indicates a perfect relationship. (d) Comparison of the adult sex ratio (male/female) in the two populations.
The trend for increase in population size was significant (P = 0.018) and gave residual population sizes, r, as
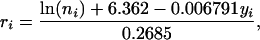 |
where ni is the population size of year yi. The SETAR model for changing residual population size found no evidence for the existence of more than one regime. Furthermore, no autocorrelation or partial autocorrelation is observed between successive residual population sizes and these show no deviation from normality. This indicates that a simpler model of normally distributed noise is appropriate for residual population size.
The model selection procedure found no evidence for a relationship between residual population size and the proportion of lambs in the population. As the untransformed proportions showed no significant deviation from normality, a simpler model was used with the proportion of lambs being normally distributed with mean and standard deviation estimated from the data as μ = 0.3305 and σ = 0.0442. The adult sex ratio was significantly related to the residual population size, with residual population sizes from 3 years previous being included in the model. The model selected for sex ratio, s, was
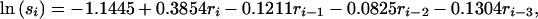 |
with the error term being normally distributed with standard deviation σ = 0.2385. The coefficients for the linear models for survival are given in Table 1. Males aged 7 years and older and females aged 11 years and above showed no significant effect of residual population size on survival and were modeled using a beta distribution with parameters given in Table 2.
TABLE 1.
Parameters for regression survival models used for younger sheep ages
| Sex | Age | α | βj | βj−1 | βj−2 | σ |
|---|---|---|---|---|---|---|
| Male | 0 | −1.4770 | — | — | — | 1.7724 |
| 1 | −0.3618 | −1.2913 | 1.5353 | — | 2.5560 | |
| 2 | −1.4943 | −1.3316 | 0.8181 | — | 2.7048 | |
| 3 | −1.5097 | −1.4484 | 0.4840 | −0.9484 | 1.5289 | |
| 4 | −1.2173 | −0.6971 | — | — | 1.3955 | |
| 5 | −1.1331 | −0.7749 | — | — | 1.5302 | |
| 6 | −0.8604 | −0.8639 | — | — | 1.0594 | |
| Female | 0 | −1.7764 | — | — | — | 4.2702 |
| 1 | −0.6938 | −0.7917 | 1.0860 | — | 5.0143 | |
| 2 | −2.3327 | −1.0169 | 0.3643 | — | 3.2646 | |
| 3 | −2.9098 | −0.7836 | 0.3779 | −0.7582 | 2.3816 | |
| 4 | −2.3903 | −0.4918 | 0.3524 | — | 1.7974 | |
| 5 | −2.6413 | −0.5388 | — | — | 3.7839 | |
| 6 | −2.3169 | −0.5115 | — | — | 2.9303 | |
| 7 | −2.0225 | −0.6870 | — | — | 2.1175 | |
| 8 | −1.9015 | −0.9580 | — | — | 2.0272 | |
| 9 | −0.8832 | −1.0156 | — | — | 2.0590 | |
| 10 | −1.1700 | −0.9479 | — | — | 3.2613 |
TABLE 2.
Values used to calculate parameters for beta-distribution survival models for older sheep ages
| Sex | Age | di,s | ni,s |
|---|---|---|---|
| Male | 7 | 32 | 77 |
| 8 | 14 | 41 | |
| 9 | 6 | 21 | |
| 10 | 4 | 11 | |
| 11 | 3 | 6 | |
| Female | 11 | 20 | 81 |
| 12 | 12 | 44 | |
| 13 | 11 | 30 | |
| 14 | 6 | 14 | |
| 15 | 1 | 4 |
Figure 4 plots the simulated LD against distance for 1000 locus pairs. As expected from population genetic theory, LD was significantly negatively correlated with marker distance (r = −0.4188, P < 0.0001). There is a large amount of variation in the amount of linkage disequilibrium between loci separated by a small distance. As with the average LD value, this decreases as the distance marker pairs are separated by increases, with no absolute 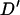 -value >0.2 being observed for marker pairs separated by >15 cM. As markers separated by >20 cM showed no significant relationship with distance (P = 0.444), these can be used to estimate nonsyntenic LD. These 490 marker pairs have a mean
-value >0.2 being observed for marker pairs separated by >15 cM. As markers separated by >20 cM showed no significant relationship with distance (P = 0.444), these can be used to estimate nonsyntenic LD. These 490 marker pairs have a mean 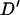 -statistic of 0.026 and standard deviation 0.026. This indicates low levels of LD between nonsyntenic marker pairs. Among marker pairs separated by <10 cM, 79% (219/276) have
-statistic of 0.026 and standard deviation 0.026. This indicates low levels of LD between nonsyntenic marker pairs. Among marker pairs separated by <10 cM, 79% (219/276) have 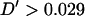 and 22% (62/276) have
and 22% (62/276) have 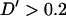 .
.
Figure 4.

Simulated linkage disequilibrium structure of the Soay sheep population. Each point is the absolute value of Lewontin's 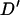 -statistic from a random pair of loci from one simulation (1000 replicates). The mean
-statistic from a random pair of loci from one simulation (1000 replicates). The mean 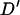 -value over successive intervals of 2.5 cM is given by a horizontal line.
-value over successive intervals of 2.5 cM is given by a horizontal line.
The stability of the LD estimates with respect to variation in the parameters used to simulate the population is examined in Figure 5. The main effect of variation of the parameters used in the simulation is the changing of the average amount of LD at closely linked loci. The distance over which the linkage disequilibrium decays and the amount of LD at more distantly linked loci remain similar with all parameter values. The variation of the estimated population dynamics parameters has the least effect on the LD structure (Figure 5, c and d). Altering parameter values of the effective population size used for generating the initial haplotypes (Figure 5, a and b) and variation in male breeding success (Figure 5, e and f) result in similar changes in the overall LD structure.
Figure 5.

Analysis of the stability of simulated linkage disequilibrium structure to variation in population parameters. Each plot shows the linkage disequilibrium structure as given by 1000 simulated replicates using altered parameter values: (a) decreasing the effective population size used to simulate the initial haplotypes, (b) increasing the initial effective population size by a factor of two, (c) using population dynamics parameters at the “low” end of their 95% confidence interval, (d) using population dynamics parameters at the “high” end of their confidence interval, (e) decreasing the coefficient of variation of male reproductive success by a factor of two, and (f) increasing the variation of male reproductive success by a factor of two. Horizontal bars give the mean 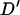 -value over successive intervals of 2.5 cM.
-value over successive intervals of 2.5 cM.
DISCUSSION
Wild populations provide novel opportunities for the understanding of the genetics of quantitative traits. However, the approach used to locate the genes underlying quantitative traits will depend on the amount of information available from the population. If a detailed pedigree is available, linkage analyses can be performed using the two-step approach proposed by George et al. (2000). This approach has been used to map QTL for birth weight in a wild population of red deer (Slate et al. 2002). When pedigree information is unavailable LD mapping methods that rely upon associations among marker haplotypes to infer historical relationships in the population can be used. However, before such an experiment is started, it is important to know the structure of LD in the population to determine the necessary marker distances for such a scan. In this article, the use of simulations is advocated as an inexpensive alternative to the direct evaluation of LD and this is applied to a wild sheep population, the Soay sheep of St. Kilda, Scotland.
The simulated Soay sheep population showed considerable amounts of LD between tightly linked loci. As expected there is a significant decline in the amount of LD with distance. Perhaps the most striking feature about this decline is the low level of LD between effectively unlinked marker pairs. Previous studies of LD in domestic livestock have observed a mean nonsyntenic LD of 0.211 in Coopworth sheep (McRae et al. 2002), 0.12–0.20 in Dutch black and white dairy cattle (Farnir et al. 2000), 0.39 in the United Kingdom dairy cattle population (Tenesa et al. 2003), and between 0.11 and 0.22 in domestic pig lines (Nsengimana et al. 2004). The simulated Soay sheep population gave a mean D′-value of 0.026 for markers separated by >60 cM. McRae et al. (2003) showed that using a small number of haplotypes inflated the value of 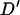 . Here a total of 3778 haplotypes were used in the estimation of
. Here a total of 3778 haplotypes were used in the estimation of 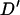 , resulting in a very low inflation of
, resulting in a very low inflation of 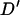 compared to Farnir et al. (2000), who used 581–1254 haplotypes, McRae et al. (2003), 276 haplotypes, Tenesa et al. (2003), <100 haplotypes, and Nsengimana et al. (2004), 184–302 haplotypes. Correcting the mean D′-values based on the model given by McRae et al. (2002) accounts for most of the variation in the mean values. However, the range of LD for marker pairs separated by >60 cM is lower in the simulated Soay sheep population, giving a maximum of 0.25, which is under half of the value given by nonsyntenic markers in domestic livestock studies (Farnir et al. 2000; McRae et al. 2002; Tenesa et al. 2003). In this study, marker genotypes were simulated to represent SNP loci and restricted to have minor allele frequency of >0.1. This is likely to account for some of the differences observed in LD structure as the previous studies have used microsatellite loci and Hedrick's (1987) multiallele
compared to Farnir et al. (2000), who used 581–1254 haplotypes, McRae et al. (2003), 276 haplotypes, Tenesa et al. (2003), <100 haplotypes, and Nsengimana et al. (2004), 184–302 haplotypes. Correcting the mean D′-values based on the model given by McRae et al. (2002) accounts for most of the variation in the mean values. However, the range of LD for marker pairs separated by >60 cM is lower in the simulated Soay sheep population, giving a maximum of 0.25, which is under half of the value given by nonsyntenic markers in domestic livestock studies (Farnir et al. 2000; McRae et al. 2002; Tenesa et al. 2003). In this study, marker genotypes were simulated to represent SNP loci and restricted to have minor allele frequency of >0.1. This is likely to account for some of the differences observed in LD structure as the previous studies have used microsatellite loci and Hedrick's (1987) multiallele 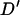 extension, which is susceptible to bias by small allele frequencies. A further explanation for the differences in the patterns of background linkage disequilibrium observed between domestic livestock populations and the simulated Soay sheep population is concurrent selection at unlinked loci. This will increase the range of linkage disequilibrium at unlinked loci without dramatically increasing mean levels and will not be observed in the simulated population as selection was not included in the population models. However, Farnir et al. (2000) found no significant evidence for selection causing an increase in LD between unlinked loci and showed that drift accounted for most of the variation in linkage disequilibrium observed in the Dutch black and white dairy cattle population. Thus, the cause of the difference between background LD observed in domestic livestock and the simulated Soay sheep population requires further investigation, possibly with direct measurement of LD in the Soay sheep population.
extension, which is susceptible to bias by small allele frequencies. A further explanation for the differences in the patterns of background linkage disequilibrium observed between domestic livestock populations and the simulated Soay sheep population is concurrent selection at unlinked loci. This will increase the range of linkage disequilibrium at unlinked loci without dramatically increasing mean levels and will not be observed in the simulated population as selection was not included in the population models. However, Farnir et al. (2000) found no significant evidence for selection causing an increase in LD between unlinked loci and showed that drift accounted for most of the variation in linkage disequilibrium observed in the Dutch black and white dairy cattle population. Thus, the cause of the difference between background LD observed in domestic livestock and the simulated Soay sheep population requires further investigation, possibly with direct measurement of LD in the Soay sheep population.
From the structure of LD given by the simulated Soay sheep an estimate of the required marker density for LD mapping can be obtained. Considering regions with consecutive LD statistics that are overall significantly greater than the mean LD statistic for nonlinked markers and single LD statistics outside the range of LD for nonlinked markers as indicators of linkage, a minimum marker density for LD mapping in the Soay sheep of ∼2 cM can be obtained. The latest linkage map of the sheep genome has a marker density on average of 3.4 cM (Maddox et al. 2001). Slate et al. (1998) found that only 42.5% of the bovine markers that amplified in the Soay sheep were polymorphic, indicating a higher-density linkage map will be required. However, this density is likely to be achievable with the use of SNP markers. This indicates that the Soay sheep population is a viable resource for linkage disequilibrium fine mapping of quantitative trait loci.
One potential pitfall of using a simulation study to assess the LD structure of a population is an incorrect specification of population dynamics and history. As the demography of the historical Soay sheep population is not recorded, a coalescent model was used to simulate the initial chromosomes used. This requires an estimate of effective population size that was derived from the heterozygosity observed in the current population. From Figure 5, a and b, it can be seen that changing this value has little effect on the overall conclusions of this study; LD still decays rapidly from its initial value and the amount of background LD is small. The main difference is the average LD between markers separated by small differences, which increases when 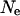 decreases and decreases otherwise. A decrease in the average LD value at small differences causes the density of loci needed to fine map QTL using LD to increase due to a smaller overall decay of LD with distance. However, given the population dynamics of the Soay sheep population, a value of
decreases and decreases otherwise. A decrease in the average LD value at small differences causes the density of loci needed to fine map QTL using LD to increase due to a smaller overall decay of LD with distance. However, given the population dynamics of the Soay sheep population, a value of 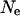 double the estimate (as used in Figure 5b) is unlikely. Similarly, the parameter values used to test the effect of the estimation of population dynamics models and variation of male breeding success are extreme values. Even using these, the overall LD structure does not deviate extremely from that observed using the average parameter values, indicating that the estimated LD structure provides an accurate representation of the actual LD structure.
double the estimate (as used in Figure 5b) is unlikely. Similarly, the parameter values used to test the effect of the estimation of population dynamics models and variation of male breeding success are extreme values. Even using these, the overall LD structure does not deviate extremely from that observed using the average parameter values, indicating that the estimated LD structure provides an accurate representation of the actual LD structure.
In conclusion, the use of simulation studies provides a useful alternative to the direct evaluation of LD in wild populations. This can provide an indication of the population's potential for LD mapping and a first estimate of the required marker density. Although the accuracy of the simulated LD structure will depend on the inclusion of all major aspects of population dynamics, the simulated LD structure can be readily confirmed in the early stages of genotyping.
Acknowledgments
We are grateful to the National Trust for Scotland and Scottish Natural Heritage for permission to work on St. Kilda and their assistance in the field. We also thank the Ministry of Defence, Defence Evaluation and Research Agency, Eurest, and QinetiQ staff stationed on St. Kilda and Benbecula for their assistance and support and all the field assistants and volunteers helping with the field work, especially J. Pilkington. The collection of the data used in this study is funded by the United Kingdom Natural Environmental Research Council and the Wellcome Trust. A.M. is funded by a Commonwealth Scholarship and P.V. is funded by a Biotechnology and Biological Sciences Research Council research development fellowship.
References
- Boyd, J. M., 1953. The sheep population of Hirta. St. Kilda. Scott. Nat. 65: 25–28. [Google Scholar]
- Boyd, J. M., 1974. Introduction, pp. 1–7 in Island Survivors: The Ecology of the Soay Sheep of St. Kilda, edited by P. A. Jewell, C. Milner and J. M. Boyd. Athlone Press, London.
- Boyd, J. M., and I. L. Boyd, 1990. The Hebrides. Collins, London.
- Boyd, J. M., J. M. Doney, R. G. Gunn and P. A. Jewell, 1964. The Soay sheep of the island of Hirta, St. Kilda. A study of a feral population. Proc. Zool. Soc. Lond. 142: 129–163. [Google Scholar]
- De Gooijer, J. G., 2001. Cross-validation criteria for SETAR model selection. J. Time Ser. Anal. 22: 267–281. [Google Scholar]
- Ellegren, H., 2004. Microsatellites: simple sequences with complex evolution. Nat. Rev. Genet. 5: 435–445. [DOI] [PubMed] [Google Scholar]
- Eyre-Walker, A., 2000. Do mitochondria recombine in humans? Philos. Trans. R. Soc. Lond. Ser. B 355: 1573–1580. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Farnir, F., W. Coppetiers, J.-J. Arranz, P. Berzi, N. Cambisano et al., 2000. Extensive genome-wide linkage disequilibrium in cattle. Genome Res. 10: 907–920. [DOI] [PubMed] [Google Scholar]
- Frankham, R., 1995. Effective population size/adult population size ratios in wildlife: a review. Genet. Res. 66: 95–107. [DOI] [PubMed] [Google Scholar]
- George, A. W., P. M. Visscher and C. S. Haley, 2000. Mapping quantitative trait loci in complex pedigrees: a two-step variance components approach. Genetics 156: 2081–2092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grenfell, B. T., O. F. Price, S. D. Albon and T. H. Clutton-Brock, 1992. Overcompensation and population cycles in an ungulate. Nature 355: 823–826. [DOI] [PubMed] [Google Scholar]
- Grenfell, B. T., K. Wilson, B. F. Finkenstadt, T. N. Coulson, S. Murray et al., 1998. Noise and determinism in synchronized sheep dynamics. Nature 394: 674–677. [Google Scholar]
- Grisart, B., W. Coppieters, F. Farnir, L. Karim, C. Ford et al., 2002. Positional candidate cloning of a QTL in dairy cattle: identification of a missense mutation in the bovine DGAT1 gene with major effect on milk yield and composition. Genome Res. 12: 222–231. [DOI] [PubMed] [Google Scholar]
- Grubb, P., 1974. Population dynamics of the Soay sheep, pp. 242–272 in Island Survivors: The Ecology of the Soay Sheep of St. Kilda, edited by P. A. Jewell, C. Milner and J. M. Boyd. Athlone Press, London.
- Hedrick, P., 1987. Gametic disequilibrium measures: proceed with caution. Genetics 117: 331–341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hudson, R. R., 2002. Generating samples under a Wright-Fisher neutral model of genetic variation. Bioinformatics 18: 337–338. [DOI] [PubMed] [Google Scholar]
- Kruglyak, L., 1999. Prospects for whole genome linkage disequilibrium mapping of common disease genes. Nat. Genet. 22: 139–144. [DOI] [PubMed] [Google Scholar]
- Lewontin, R. C., 1964. The interaction between selection and linkage. I. General considerations; heterotic models. Genetics 49: 49–67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maddox, J., K. P. Davies, A. M. Crawford, D. J. Hulme, D. Vaiman et al., 2001. An enhanced linkage map of the sheep genome comprising more than 1000 loci. Genome Res. 11: 1275–1289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McRae, A. F., J. C. Mcewan, K. G. Dodds, T. Wilson, A. M. Crawford et al., 2002. Linkage disequilibrium in domestic sheep. Genetics 160: 1113–1122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meuwissen, T. H. E., A. Karlsen, S. Lien, I. Olsaker and M. E. Goddard, 2002. Fine mapping of a quantitative trait locus for twinning rate using combined linkage and linkage disequilibrium mapping. Genetics 161: 373–379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nsenimana, J., P. Baret, C. S. Haley and P. M. Visscher, 2004. Linkage disequilibrium in the domesticated pig. Genetics 166: 1395–1404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pemberton, J. M., D. W. Coltman, J. A. Smith and J. G. Pilkington, 1999. Molecular analysis of a promiscuous, fluctuating mating system. J. Linn. Soc. 68: 289–301. [Google Scholar]
- Pemberton, J. M., D. W. Coltman, J. A. Smith and D. R. Bancroft, 2004. Mating patterns and male breeding success, pp. 166–189 in Soay Sheep: Dynamics and Selection in an Island Population, edited by T. Clutton-Brock and J. M. Pemberton. Cambridge University Press, Cambridge, UK.
- Pritchard, J. K., and M. Przeworski, 2001. Linkage disequilibrium in humans: models and data. Am. J. Hum. Genet. 69: 1–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Slate, J., D. W. Coltman, S. J. Goodman, I. Maclean, J. M. Pemberton et al., 1998. Bovine microsatellite loci are highly conserved in red deer (Cervus elaphus), sika deer (Cervus nippon) and Soay sheep (Ovis aries). Anim. Genet. 29: 307–315. [DOI] [PubMed] [Google Scholar]
- Slate, J., P. M. Visscher, S. Macgregor, D. Stevens, M. L. Tate et al., 2002. A genome scan for quantitative trait loci in a wild population of red deer (Cervus elaphus). Genetics 162: 1863–1873. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tenesa, A., S. A. Knott, D. Ward, D. Smith, J. L. Williams et al., 2003. Estimation of linkage disequilibrium in a sample of the United Kingdom dairy cattle population using unphased genotypes. J. Anim. Sci. 81: 617–623. [DOI] [PubMed] [Google Scholar]
- Tong, H., 1990. Non-Linear Time Series: A Dynamical System Approach. Clarendon Press, Oxford.
- Wall, J. D., and J. K. Pritchard, 2003. Haplotype blocks and linkage disequilibrium in the human genome. Nat. Rev. Genet. 4: 587–597. [DOI] [PubMed] [Google Scholar]