

Abstract
Saccharomyces cerevisiae RAM is a conserved signaling network that regulates maintenance of polarized growth and daughter-cell-specific transcription, the latter of which is critical for septum degradation. Consequently, cells defective in RAM function (designated ramΔ) are round in morphology, form feeble mating projections, and fail to separate following cytokinesis. It was recently demonstrated that RAM genes are essential in strains containing functional SSD1 (SSD1-v), which encodes a protein of unknown function that binds the RAM Cbk1p kinase. Here we investigated the essential function of RAM in SSD1-v strains and identified two functional groups of dosage suppressors for ramΔ lethality. We establish that all ramΔ mutants exhibit cell integrity defects and cell lysis. All dosage suppressors rescue the lysis but not the cell polarity or cell separation defects of ramΔ cells. One class of dosage suppressors is composed of genes encoding cell wall proteins, indicating that alterations in cell wall structure can rescue the cell lysis in ramΔ cells. Another class of ramΔ dosage suppressors is composed of ZRG8 and SRL1, which encode two unrelated proteins of unknown function. We establish that ZRG8 and SRL1 share similar genetic interactions and phenotypes. Significantly, Zrg8p coprecipitates with Ssd1p, localizes similarly to RAM proteins, and is dependent on RAM for localization. Collectively, these data indicate that RAM and Ssd1p function cooperatively to control cell integrity and suggest that Zrg8p and Srl1p function as nonessential inhibitors of Ssd1p.
THE conserved Saccharomyces cerevisiae regulation of Ace2p transcription factor and polarized morphogenesis (RAM) signaling network controls two genetically distinct cellular processes. One function of RAM is to regulate the daughter-cell-specific nuclear localization and activation of Ace2p transcription factor during mitotic exit (Weiss et al. 2002). In wild-type cells, Ace2p localizes specifically to the daughter nucleus during mitotic exit and induces the expression of several genes encoding septum degradation proteins (Colman-Lerner et al. 2001; Weiss et al. 2002). In addition to regulating cell separation, Ace2p also regulates a G1 delay in daughter cells (Laabs et al. 2003). In RAM mutant cells (designated throughout as ramΔ cells), Ace2p localizes to both nuclei and is not active as a transcription factor. Thus, ramΔ cells accumulate as large clusters of unseparated cells, similar to ace2Δ cells (O'Conallain et al. 1998; Bidlingmaier et al. 2001). RAM is also required to maintain the polarity of the actin cytoskeleton during polarized growth, which is important for bud site selection, cell morphogenesis, and mating projection formation (Weiss et al. 2002; Nelson et al. 2003). Consequently, ramΔ cells are spherical in morphology, exhibit random bud site selection, and form feeble mating projections. Notably, ace2Δ cells do not display defects in polarized morphogenesis, indicating that the morphogenetic function of RAM is independent of Ace2p.
RAM is composed of two protein kinases, Cbk1p and Kic1p, and several associated proteins: Mob2p, Hym1p, Tao3p, and Sog2p (Jorgensen et al. 2002; Nelson et al. 2003). All RAM proteins are conserved from yeast to human and localize to the sites of polarized growth, which include the bud cortex during bud emergence and growth, the tips of mating projections, and the bud neck region during mitotic exit. In addition, Cbk1p and Mob2p colocalize with Ace2p in the daughter cell nucleus during mitotic exit (Colman-Lerner et al. 2001; Weiss et al. 2002). Cbk1p kinase belongs to the LATS/NDR protein kinase family, which includes the S. cerevisiae mitotic exit network (MEN) protein Dbf2p and the Schizosaccharomyces pombe proteins Orb6p and Sid2p (Dorland et al. 2000; Hou et al. 2000, 2003; Racki et al. 2000; Bidlingmaier et al. 2001; Tamaskovic et al. 2003). Mob2p is a Cbk1p-binding protein that is related to the S. cerevisiae MEN protein Mob1p (Colman-Lerner et al. 2001; Luca et al. 2001; Weiss et al. 2002). Mob1p and Mob2p are MOB family proteins, which function as regulatory subunits of LATS/NDR family kinases (Luca and Winey 1998; Hou et al. 2000, 2003; Weiss et al. 2002). Kic1p is a GCK-II family kinase related to the MEN kinase Cdc15p (Dan et al. 2001). The molecular functions of Hym1p, Tao3p, and Sog2p are unknown. Hym1p interacts with Kic1p, and Cbk1p and is an ortholog of the Aspergillus nidulans hyphal growth protein hymA and the uncharacterized mouse protein MO25 (Miyamoto et al. 1993; Karos and Fischer 1999; Dorland et al. 2000; Bidlingmaier et al. 2001). Tao3p (aka Pag1p) is a conserved 270-kDa protein that also interacts with both Cbk1p and Kic1p (Du and Novick 2002; Nelson et al. 2003). Sog2p is a leucine-rich-repeat-containing protein that binds Hym1p, Kic1p, and Cbk1p (Nelson et al. 2003). Homologs to Cbk1p, Mob2, Tao3, and Kic1p were identified in S. pombe and were shown to play a role in polarized growth (Verde et al. 1998; Hirata et al. 2002; Hou et al. 2003; Huang et al. 2003).
Mob2p and Cbk1p appear to function late in RAM signaling (Nelson et al. 2003). Mob2p binds Cbk1p throughout the cell cycle and is required for Cbk1p kinase activity, which is critical for all known RAM functions (Weiss et al. 2002). Nevertheless, Mob2p binding is not sufficient for Cbk1p activation in kic1Δ, hym1Δ, sog2Δ, or tao3Δ cells (Nelson et al. 2003). The daughter-cell-specific nuclear localization of Mob2p, Cbk1p, and Ace2p is also dependent on all other RAM proteins (Nelson et al. 2003). These data suggest that Kic1p, Hym1p, Sog2p, and Tao3p function upstream of Mob2p-Cbk1p. By analogy to MEN, it is likely that Mob2p-Cbk1p is directly activated by Kic1p kinase, since Mob1p-Dbf2p is phosphorylated and activated by the Kic1p-related kinase Cdc15p (Mah et al. 2001). Thus far, substrates of Mob2p-Cbk1p are unknown.
Intriguingly, the RAM signaling network is essential for cell viability in strains that contain wild-type SSD1 (designated SSD1-v), such as S288C-derived strains from the S. cerevisiae deletion consortium (Du and Novick 2002; Jorgensen et al. 2002). In contrast, none of the RAM genes is essential for viability in strains that express a nonfunctional allele of SSD1 (designated ssd1-d), such as W303 and our laboratory's S288C-derived strains (Luca and Winey 1998; Dorland et al. 2000; Racki et al. 2000; Bidlingmaier et al. 2001; Du and Novick 2002; Jorgensen et al. 2002; Nelson et al. 2003). Thus, ssd1-d serves as an extragenic suppressor of ramΔ lethality. In contrast, SSD1 does not exhibit the same genetic relationship with ACE2, since ace2Δ mutations are viable in SSD1-v and ssd1-d strains. These data suggest that RAM regulation of Ace2p is independent of SSD1.
Insight into the essential function of RAM may come from understanding SSD1 function and regulation. Ssd1p is a conserved, nonessential protein of unknown function that localizes to punctate spots or patches in the cytoplasm (Sutton et al. 1991; Uesono et al. 1997). It contains an RNB domain that binds poly(A) RNA in vitro. Cells lacking SSD1 have higher concentrations of chitin and mannan in the cell wall and decreased concentrations of 1,3-β-glucan and 1,6-β-glucan, suggesting a role for SSD1 in cell wall integrity (Wheeler et al. 2003). Genetic interactions suggest that SSD1 participates in a cell integrity pathway that functions in parallel to the protein kinase C (PKC) pathway (Kaeberlein and Guarente 2002). In agreement, ssd1-d is synthetic lethal with a variety of mutations in the PKC, cAMP/PKA, TOR signaling, and secretory pathways (Sutton et al. 1991; Costigan et al. 1992; Uesono et al. 1997; Li and Warner 1998; Kosodo et al. 2001; Rosenwald et al. 2002; Vincent et al. 2003; Reinke et al. 2004). Significantly, Ssd1p interacts with Cbk1p in two-hybrid assays, suggesting that Ssd1p and RAM are functionally interconnected (Racki et al. 2000). Moreover, recent data suggest that RAM functions in parallel to PKA signaling, as observed for SSD1, perhaps to control cell proliferation via regulation of Rho1p (Bogomolnaya et al. 2004; Schneper et al. 2004).
Several lines of evidence point to a role of Cbk1p, Tao3p, and Kic1p in maintenance of cell wall integrity. Two-hybrid assays revealed that Cbk1p interacts with Lre1p, a protein that affects cell wall chitinase and trehalose accumulation and functions antagonistically to protein kinase A (Racki et al. 2000; Versele and Thevelein 2001). Moreover, the lethality of cbk1Δ and tao3Δ mutations in SSD1-v strains is suppressed by overexpression of the cell wall protein Sim1p (Du and Novick 2002). It was also demonstrated that, in some strains, kic1 mutations lead to severe cell damages and eventually to cellular lysis (Sullivan et al. 1998; Vink et al. 2002). Furthermore, diminished Kic1p expression reduces 1,6-β-glucan levels in the cell wall (Vink et al. 2002). Despite these data, it was not known if the apparent cell integrity function of Cbk1p, Kic1p, and Tao3p was universal for all RAM proteins. Nor was it known if the essential function of RAM was genetically distinct from its function in polarized growth.
To investigate the essential function of RAM, we analyzed the phenotypes of ramΔ SSD1-v cells and employed a dosage suppressor screen. Here, we present evidence that RAM functions in an SSD1-dependent cell integrity pathway that is genetically distinct from its Ace2p regulation and polarized growth. Furthermore, we identified several components of the SSD1-dependent cell integrity pathway, including two cell wall proteins and a novel Ssd1p-binding protein, Zrg8p, which localizes similarly to RAM proteins. Our data suggest that Zrg8p and another dosage suppressor, Srl1p, function in a RAM- and Ssd1p-dependent cell integrity pathway.
MATERIALS AND METHODS
Yeast strains and cultures:
Yeast strains used in this study are listed in Table 1. Yeast were grown and manipulated using standard methods (Guthrie and Fink 1991). Yeast proteins were GFP-, HA-, and Myc-tagged by integration of PCR-based cassettes, as described (Longtine et al. 1998).
TABLE 1.
Yeast strains
| Name | Genotype | Source |
|---|---|---|
| FLY849 | MATaACE2-GFP∷KANMX mob2Δ∷HIS3 ssd1-d | Weiss et al. (2002) |
| FLY858 | MATamob2Δ SSD1-v [pRS316-MOB2] | This work |
| FLY1279 | MATaZRG8-GFP∷HIS3 | This work |
| BY4742 | MATα SSD1-v | Deletion Consortium |
| FLY1306 | MATα ccw12Δ SSD1-v | Deletion Consortium |
| FLY1307 | MATα sim1Δ SSD1-v | Deletion Consortium |
| FLY1308 | MATα srl1Δ SSD1-v | Deletion Consortium |
| FLY1309 | MATα zrg8Δ SSD1-v | Deletion Consortium |
| FLY1310 | MATα zap1Δ SSD1-v | Deletion Consortium |
| FLY1313 | MATα cbp3Δ SSD1-v | Deletion Consortium |
| FLY1327 | MATaZRG8-GFP∷HIS3 cbk1Δ∷KANMX ssd1-d | This work |
| FLY1632 | MATα ace2Δ SSD1-v | Deletion Consortium |
| FLY1662 | MATα cbk1Δ SSD1-v [pRS316-CBK1] | This work |
| FLY1663 | MATaACE2-GFP∷KANMX mob2Δ SSD1-v [YEp13-CBP3] | This work |
| FLY1665 | MATaACE2-GFP∷KANMX mob2Δ SSD1-v [YEp13-ZRG8] | This work |
| FLY1667 | MATaACE2-GFP∷KANMX mob2Δ SSD1-v [YEp13-SRL1] | This work |
| FLY1669 | MATaACE2-GFP∷KANMX mob2Δ SSD1-v [YEp13-SIM1] | This work |
| FLY1671 | MATaACE2-GFP∷KANMX mob2Δ SSD1-v [YEp13-CCW12] | This work |
| FLY1680 | MATα ZRG8-GFP∷HIS3 ace2Δ∷KANMX ssd1-d | This work |
| FLY1687 | MATahym1Δ SSD1-v [pRS316-HYM1] | This work |
| FLY1692 | MATasog2Δ SSD1-v [pYES-SOG2] | This work |
| FLY1718 | MATaZRG8-myc∷KANMX SSD1-HA∷HIS3 SSD1-v | This work |
| FLY1641 | MATazrg8Δ∷KANMX ccw12Δ∷KANMX SSD1-v | This work |
| FLY1722 | MATasrl1Δ∷KANMX ccw12Δ∷KANMX SSD1-v | This work |
| FLY1741 | MATamob2Δ∷KANMX srl1Δ∷KANMX SSD1-v [pRS316-MOB2] | This work |
| FLY1745 | MATamob2Δ∷KANMX zrg8Δ∷KANMX SSD1-v [pRS316-MOB2] | This work |
| FLY1739 | MATα srl1Δ∷KANMX zrg8Δ∷KANMX SSD1-v | This work |
| FLY1735 | MATα ZRG8-Myc∷KANMX SSD1-v | This work |
| FLY1737 | MATα SSD1-HA∷HIS3 SSD1-v | This work |
Dosage suppressor screen:
Strain Y25654 (mob2Δ∷KANMX/MOB2 SSD1-v/SSD1-v) was obtained from Open Biosystems and transformed with pRS316-MOB2, which contains URA3 as a selectable marker. Upon sporulation and tetrad dissection, a KANMX URA3 segregant (FLY858) was obtained and transformed with a YEp13-based genomic S. cerevisiae library (DeMarini et al. 1997). Transformants were replica plated on 5-FOA to select for cells that lost the URA3-containing pRS316-MOB2 plasmid. Plasmids were rescued and retransformed into FLY858 to confirm the mob2Δ suppressor activity. Suppressor plasmids containing MOB2 were identified by PCR and discarded. We isolated 23 dosage suppressor plasmids from ∼100,000 transformants. The genomic DNA was identified in each plasmid by DNA sequencing. Eleven plasmids contained SIM1, which was previously described as a multicopy suppressor of cbk1Δ and tao3Δ (Du and Novick 2002). The SIM1 plasmid used in this analysis, YEp13-SIM1, contains a genomic fragment of chromosome IX (125,951–133,378). Of the 23 dosage suppressor plasmids, 5 contained SRL1, 2 contained CCW12, 3 contained ZAP1 and YJL055w, 1 contained ZRG8, 1 contained CBP3, and 1 contained FSP2-HXT9. CCW12, CPB3, SRL1, and ZRG8 were subcloned as described below.
Plasmid construction:
A SacI-ClaI fragment (2.6 kb) of YEp13-CCW12 containing ORFs AHP1, CCW12, and YLR111 was subcloned into pRS423. From this plasmid, the EcoRI-KpnI fragment containing CCW12 was subcloned into pRS424 to yield pRS424-CCW12. A DraI fragment (2.7 kb) of YEp13-CBP3 containing CBP3 was subcloned into SmaI of pRS425 to yield pRS425-CBP3. YEp13-SRL1 contains the fragment of chromosome XV with APC5 and SRL1. An EcoRI-ApaI fragment (2.2 kb) of YEp13-SRL1 was subcloned into pRS423 to yield pRS423-SRL1. The PstI-HindIII fragment (4.1 kb) of YEp13-ZRG8 was subcloned into pRS425 to produce pRS425-ZRG8.
Microscopy and image analysis:
Differential interference contrast (DIC) and fluorescence microscopy was conducted as described (Luca et al. 2001; Weiss et al. 2002).
Calcofluor white sensitivity assays:
Calcofluor white sensitivity assays were performed as described using medium supplemented with 10, 50, and 100 μg/ml of Calcofluor white (Ram et al. 1994). Cells were grown at 22°, unless otherwise stated.
Immunoprecipitation and immunoblotting:
Co-immunoprecipitation and immunoblot analyses were conducted using monoclonal anti-HA antibody (12CA5, Covance) and monoclonal anti-Myc antibody (9E10, Covance), as described (Weiss et al. 2002).
RESULTS
RAM signaling is required for maintaining cell integrity:
To determine the essential function of RAM, we analyzed the phenotypes of RAM deletion mutations in SSD1-v cells. We sporulated diploid cells that were heterozygous for RAM gene deletions and homozygous for SSD1-v and assayed the phenotypes of the meiotic products. We confirmed that cbk1Δ, sog2Δ, and hym1Δ cells were inviable in the presence of SSD1-v (data not shown). Most of the cbk1Δ SSD1-v spores germinated and lysed after several cell divisions. Some cells could form colonies of <100 cells before completely dying out (data not shown). In rare instances, some cbk1Δ SSD1-v segregants formed microcolonies after 5 days (data not shown). The viable cbk1Δ SSD1-v cells exhibited severe cell lysis phenotypes, which diminished in severity when cells were continually grown on rich media. Some kic1Δ cells also progressively improve over time (Vink et al. 2002), suggesting that both kic1Δ and cbk1Δ cells can readily obtain suppressor mutations. In contrast to cbk1Δ SSD1-v cells, all mob2Δ SSD1-v segregants formed slowly growing microcolonies on YPD plates. Many mob2Δ SSD1-v cells contained broken cell walls and exhibited varying degrees of cellular lysis (Figure 1). The remaining cells appeared very sick and contained wide bud necks. Similar phenotypes were described for conditional kic1 mutants (Sullivan et al. 1998). The cellular lysis and lethality of ramΔ SSD1-d strains were not suppressed by supplementing the growth media with 1 m sorbitol (data not shown). Collectively, these data indicate that RAM is required for maintaining cell integrity and suggest a role for RAM in cell wall maintenance.
Figure 1.

mob2Δ SSD1-v cells exhibit severe morphology and lysis defects. DIC images of mob2Δ SSD1-v cells (segregants of strain Y25654, Open Biosystems) show cell separation, morphology, and cell lysis defects. Arrowheads point to abnormally wide bud necks.
High-copy suppressors of the SSD1-dependent function of RAM:
To elucidate the role of RAM in maintaining cell integrity and possibly identify important regulators or targets of RAM or Ssd1p, we conducted a dosage suppressor screen in mob2Δ SSD1-v cells (see materials and methods). We introduced a yeast genomic DNA library of high-copy plasmids into mob2Δ SSD1-v cells that also contained a counterselectable MOB2 plasmid. Upon counterselection for the MOB2 plasmid on 5-FOA plates (see materials and methods), we selected for robustly growing colonies and recovered the dosage suppressor plasmids. We identified the corresponding dosage suppressor genes by DNA sequencing and confirmed their suppressor activities by subcloning and retransforming them into mob2Δ SSD1-v cells.
We identified several dosage suppressors of mob2Δ SSD1-v cells, including SIM1, CCW12, SRL1, ZRG8, CBP3, and a DNA fragment encoding truncated ZAP1 and YJL055w. Intriguingly, none of these genes is essential for viability (http://www.yeastgenome.org/). High-copy plasmids of SIM1, CCW12, SRL1, and ZRG8 allowed mob2Δ SSD1-v cells to grow robustly on plates, although not as well as MOB2-containing cells (Figure 2A; supplementary Figure S1 at http://www.genetics.org/supplemental/). In contrast, CBP3 plasmids provided much more modest growth improvement for mob2Δ cells. The cell lysis phenotypes of mob2Δ SSD1-v cells were greatly diminished by the high-copy SIM1, CCW12, SRL1, ZRG8, and CBP3 plasmids (Figure 2B). The plasmid-containing mob2Δ SSD1-v cells appeared healthy and were nearly indistinguishable in morphology from mob2Δ ssd1-d cells. The cells were spherical in morphology and persisted as clusters of connected cells. These data indicate that overexpression of SIM1, CCW12, SRL1, CBP3, and ZRG8 suppresses the cellular lysis phenotypes, but not other phenotypes of mob2Δ cells.
Figure 2.

Dosage suppressors of mob2Δ SSD1-v lethality suppress the cell lysis but not the cell separation defect of ramΔ SSD1-v cells. (A) mob2Δ SSD1-v cells (FLY858) containing pRS316-MOB2 were transformed with empty vector (pRS425) or with high-copy plasmids containing MOB2 (pMOB2), CBP3 (pCBP3), CCW12 (pCCW12), SIM1 (pSIM1), SRL1 (pSRL1), or ZRG8 (pZRG8). All of the high-copy plasmids contained LEU2 and thus were selectable on leucine deficient (Leu−) medium. The cells were serially diluted (10-fold) and spotted onto Leu− and 5-FOA plates. 5-FOA selects for cells that do not contain pRS316-MOB2. Note that CBP3 is a weak suppressor and that none of the high-copy suppressors rescue the lethality of mob2Δ as well as that of MOB2. (B) DIC images of the suppressed mob2Δ SSD1-v cells. High-copy CBP3, CCW12, SIM1, SRL1, and ZRG8 plasmids suppress the cell lysis defects but not the cell separation defects of mob2Δ SSD1-v cells (FLY858). mob2Δ SSD1-v cells containing pMOB2 are indistinguishable from wild-type cells. (C) High-copy CCW12, SIM1, SRL1, and ZRG8 plasmids suppress the lethality of cbk1Δ SSD1-v (FLY1662), hym1Δ SSD1-v (FLY1687), and sog2Δ SSD1v (FLY1692) cells. The rescued cells display cell separation defects that are identical in phenotype to mob2Δ ssd1-d and cbk1Δ ssd1-d cells (FLY168 and FLY757), which were previously described in Weiss et al. (2002). cbk1Δ SSD1-v, hym1Δ SSD1-v, and sog2Δ SSD1-v cells containing cognate CBK1, HYM1, and SOG2 plasmids are indistinguishable from wild-type cells (data not shown). All high-copy suppressor plasmids are derived from a YEp13-based genomic library (DeMarini et al. 1997).
Most of the mob2Δ dosage suppressor genes were not well characterized; however, previous data implicate SIM1, CCW12, and SRL1 in cell wall biogenesis. SIM1 encodes a protein that is noncovalently bound to the cell wall and is highly glycosylated (Velours et al. 2002). SIM1 overexpression was demonstrated to suppress the lethality of tao3Δ SSD1-v and cbk1Δ SSD1-v cells (Du and Novick 2002). CCW12 encodes a glycosylphosphatidylinositol (GPI)-anchored mannoprotein and SRL1 encodes a putative structural mannoprotein that localizes to the cortex of small buds (Mrsa et al. 1999; Terashima et al. 2002; Shepard et al. 2003; Hagen et al. 2004; Teparic et al. 2004). Recent work suggests that deletion of GPI-anchored proteins, such as Ccw12p, activates an SRL1-dependent compensatory pathway for cell wall integrity (Hagen et al. 2004). Thus, the dosage suppressors SIM1, CCW12, and SRL1 implicate RAM signaling in cell wall maintenance and suggest a cell-wall-dependent mechanism for suppressing the lethality of ramΔ mutations.
Prior to this study ZRG8, ZAP1, and CBP3 were not implicated in cell wall maintenance or function. Both ZRG8 and ZAP1 are zinc-regulated genes whose expression is increased during zinc deficiency 1.9- and 5.7-fold, respectively (Yuan 2000). ZRG8 encodes an uncharacterized 1076-amino-acid protein that contains no obvious protein motifs. ZAP1 encodes a transcription factor that regulates expression of proteins involved in zinc homeostasis (Zhao et al. 1998) and CBP3 encodes a putative mitochondrial chaperonin that is required for cytochrome-c reductase assembly (Shi et al. 2001). It is possible that ZRG8, ZAP1, and CBP3 overexpression suppresses the cell wall defects of mob2Δ cells indirectly or via a cell-wall-independent mechanism.
To determine if the mob2Δ dosage suppressors could suppress the lethality of other ramΔ mutations, we introduced each SIM1, CCW12, SRL1, ZRG8, and CBP3 multicopy plasmids into cbk1Δ, sog2Δ, and hym1Δ cells and analyzed their viability. We found that high-copy SIM1, CCW12, SRL1, and ZRG8 plasmids suppressed the lethality and slow-growth defects of cbk1Δ, sog2Δ, and hym1Δ cells (Figure 2C), indicating that the dosage suppression is not specific to mob2Δ SSD1-v cells. Thus, SIM1, CCW12, SRL1, and ZRG8 overexpression can bypass the essential function of the RAM signaling network. Intriguingly, high-copy CBP3 plasmids, which provided only modest growth improvement for mob2Δ cells, did not rescue the lethality of cbk1Δ, sog2Δ, or hym1Δ.
Overexpression of SIM1, CCW12, SRL1, and ZRG8 suppress the cell lysis defects but not the cell separation and polarized growth defects of ramΔ cells:
It was established that RAM signaling is important for (1) maintenance of polarized growth and (2) regulating the daughter-cell-specific localization and activity of Ace2p transcription factor, which controls the expression of cell separation proteins (Weiss et al. 2002). We therefore tested if the dosage suppressor plasmids could suppress the cell polarity and cell separation defects of ramΔ SSD1-v and ramΔ ssd1-d cells. As noted above, high-copy SIM1, CCW12, SRL1, and ZRG8 plasmids suppressed the cell lysis phenotypes of mob2Δ SSD1-v, cbk1Δ SSD1-v, hym1Δ SSD1-v, and sog2Δ SSD1-v cells. When we analyzed the cell morphology, we found that the cells were round and persisted in clusters of unseparated cells (Figure 2, B and C). The round cell shape suggests that, despite the presence of the dosage suppressor plasmids, ramΔ SSD1-v cells fail to maintain apical growth after bud emergence. It was previously demonstrated that ramΔ cells also fail to maintain polarized growth during mating projection formation (Nelson et al. 2003). Thus, we assayed whether SIM1, CCW12, SRL1, ZRG8, and CBP3 plasmids could restore proper mating projection formation in mob2Δ cells. As can be observed in Figure 3, none of the dosage suppressor plasmids allowed mob2Δ cells to form robust mating projections in response to mating pheromone.
Figure 3.

mob2Δ SSD1-v cells containing high-copy CBP3, CCW12, SIM1, SRL1, and ZRG8 plasmids do not form robust mating projections. mob2Δ SSD1-v cells containing high-copy CBP3, CCW12, SIM1, SRL1, and ZRG8 plasmids were treated with α-factor for 3 hr. All mob2Δ SSD1-v cells containing pRS316-MOB2 (top left) formed normal mating projections. In contrast, cells lacking pRS316-MOB2 formed feeble mating projections and remained connected.
The persistent cell separation defects in plasmid-containing ramΔ cells indicate that Ace2p function is not restored by SIM1, CCW12, SRL1, ZRG8, or CBP3 overexpression. In agreement, none of the dosage suppressor plasmids rescued the cell separation defects of ace2Δ cells (Figure 4). Nevertheless, it was possible that one or more of the dosage suppressors could influence Ace2p localization, which is normally controlled by RAM. We therefore monitored the effects of high-copy SIM1, CCW12, SRL1, ZRG8, and CBP3 plasmids on the localization of Ace2-GFP in both mob2Δ ssd1-d and mob2Δ SSD1-v cells. As previously observed, Ace2-GFP localizes to the daughter cell nucleus at the end of mitosis and mislocalizes to both mother and daughter cell nuclei in ramΔ ssd1-d cells. We found that Ace2p localizes to the daughter cell nucleus in SSD1-v cells (Figure 5). High-copy SIM1, CCW12, SRL1, ZRG8, and CBP3 plasmids did not restore proper daughter-specific localization of Ace2p in any mob2Δ cells (Figure 5). These data indicate that SIM1, CCW12, SRL1, ZRG8, and CBP3 are not involved in Ace2p regulation. Furthermore, these results demonstrate that RAM performs genetically distinct functions in maintaining cell integrity (via SSD1) and Ace2p regulation.
Figure 4.
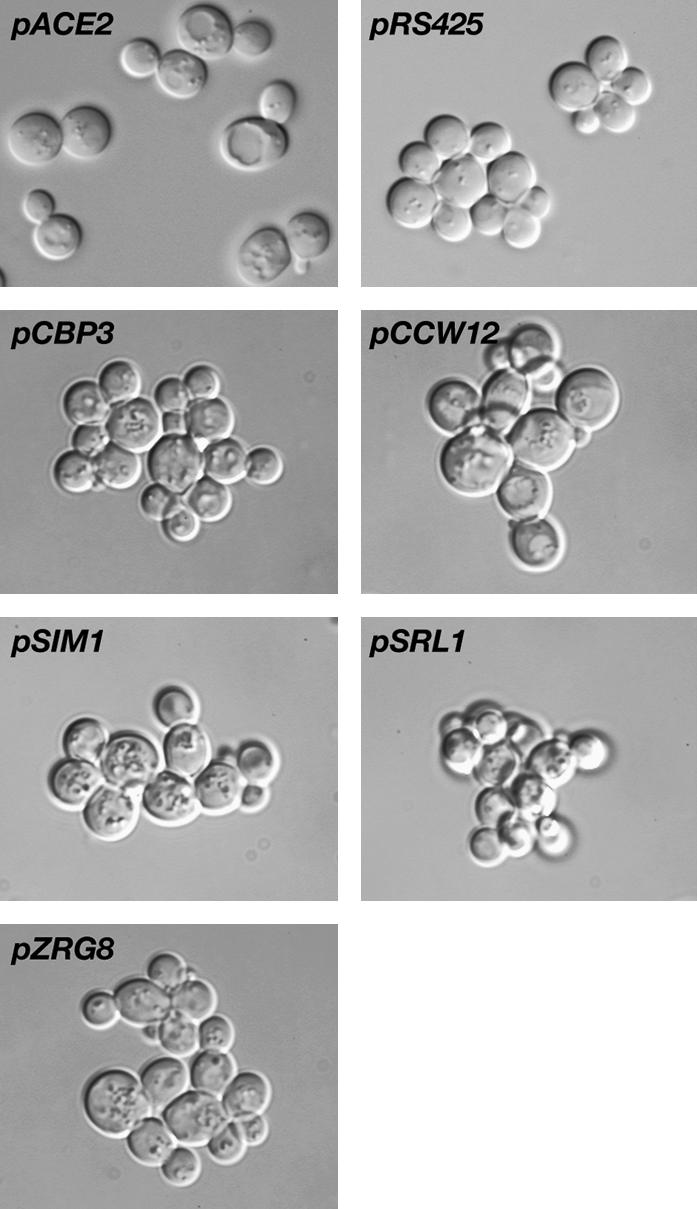
High-copy CBP3, CCW12, SIM1, SRL1, and ZRG8 plasmids do not suppress the cell separation defects of ace2Δ cells. High-copy CBP3, CCW12, SIM1, SRL1, and ZRG8 (YEp13-based plasmids) and low-copy ACE2 plasmids were introduced into ace2Δ SSD1-v cells (FLY1632). Cells were sonicated and analyzed for morphology. Only pACE2 rescued the cell separation defects of ace2Δ cells. Cells containing the empty high-copy vector pRS425 are shown as a negative control.
Figure 5.
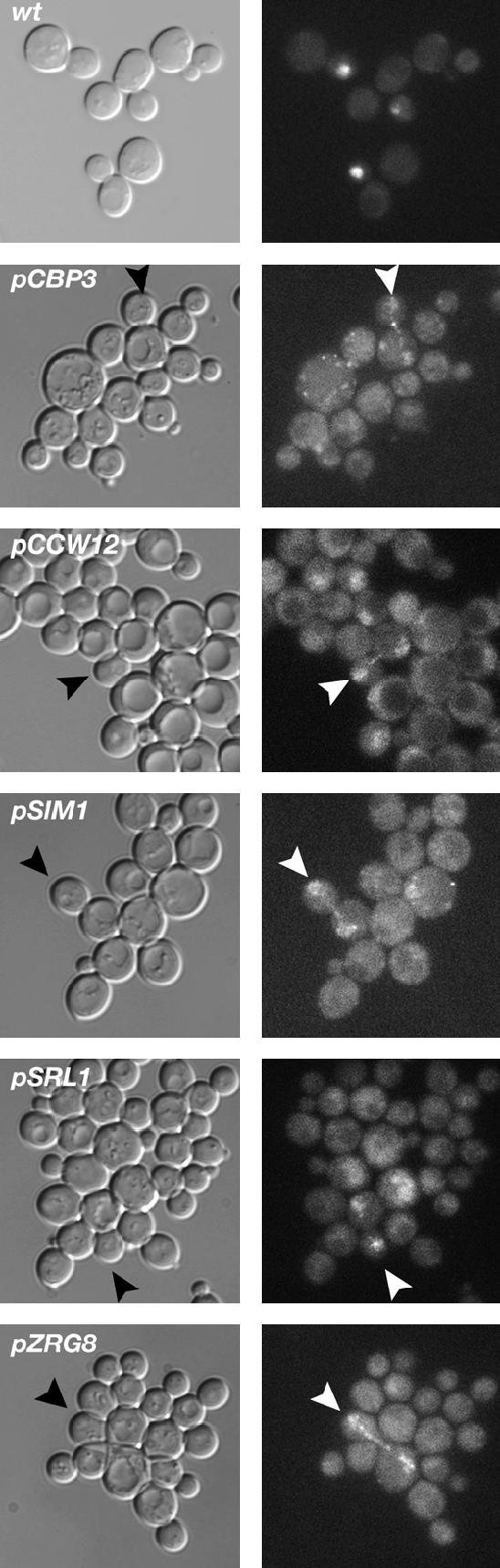
High-copy CBP3, CCW12, SIM1, SRL1, and ZRG8 plasmids do not restore the daughter-specific localization of Ace2p in mob2Δ SSD1-v cells. High-copy plasmids containing CBP3, CCW12, SIM1, SRL1, or ZRG8 were introduced into mob2Δ SSD1-v cells expressing Ace2-GFP. The localization of Ace2p-GFP was analyzed by fluorescence microscopy. Wild-type cells (wt) are shown as a control to illustrate the daughter-specific nuclear localization of Ace2p in SSD1-v cells, as previously observed for ssd1-d cells (Weiss et al. 2002). Arrowheads point to late mitotic cells where Ace2p can be detected in both the mother and the daughter cell nucleus. The Ace2-GFP nuclear fluorescence is consistently weaker in mob2Δ cells than in wild-type cells. One hundred percent of the wild-type cells with detectable Ace2-GFP displayed the daughter-cell-specific localization in contrast to 0% of the mob2Δ cells (n = 50).
SIM1, CCW12, SRL1, and ZRG8 influence cell wall biogenesis:
It is possible that overexpression of SIM1, CCW12, SRL1, ZRG8, and CBP3 suppresses the cellular lysis phenotypes of ramΔ SSD1-v cells by inducing a compensatory change in cell wall architecture. If this model is correct, then high-copy SIM1, CCW12, SRL1, ZRG8, and CBP3 plasmids might induce detectable changes in cell wall architecture in wild-type cells (such as BY4742). Thus, we introduced the high-copy plasmids into SSD1-v cells and assayed for Calcofluor white sensitivity. We found that cells containing SIM1, CCW12, SRL1, or ZRG8 plasmids were more sensitive to Calcofluor white than control cells that contained empty vector (Figure 6A). In contrast, CBP3 overexpression caused no observable effect on Calcofluor sensitivity. These results are consistent with the model that overexpression of SIM1, CCW12, SRL1, or ZRG8 suppresses ramΔ SSD1-v cells by inducing a compensatory change in cell wall architecture.
Figure 6.

Calcofluor white sensitivity assays for mob2Δ dosage suppressors. (A) Wild-type cells (BY4742) containing multicopy CBP3, CCW12, SIM1, SRL1, and ZRG8 plasmids were assayed for Calcofluor white sensitivity on selective media. (B) Cells deleted for CBP3 (FLY1313), CCW12 (FLY1306), SIM1 (FLY1307), SRL1 (FLY1308), ZAP1 (FLY1310), and ZRG8 (FLY1309) were analyzed for their ability to grow on plates containing 10 or 50 μg/ml Calcofluor white. Serial dilutions (10-fold) of cells were spotted onto each plate and grown at 22°.
CCW12 and SIM1 were shown to encode cell wall proteins. Srl1p is a serine- and threonine-rich protein, which is tightly associated with the cell wall (Terashima et al. 2002). If ZRG8, ZAP1, and CBP3 are also important for cell wall biosynthesis, then deletion of those genes may induce cell wall defects that are detectable by assaying Calcofluor sensitivity. Indeed, it was previously established that ccw12Δ cells are more sensitive to Calcofluor than control cells (Mrsa et al. 1999). Thus, we assayed isogenic sim1Δ, srl1Δ, zrg8Δ, zap1Δ, and cbp3Δ strains for increased Calcofluor sensitivity. We found that ccw12Δ, srl1Δ, and zap1Δ cells were much more sensitive to Calcofluor than control SSD1-v cells (Figure 6B). At 22°, zrg8Δ cells were slightly more Calcofluor sensitive than wild-type cells, but sim1Δ cells were as Calcofluor sensitive as wild-type cells (Figure 6B). Calcofluor sensitivity of ccw12Δ, srl1Δ, and zrg8Δ cells is significantly enhanced at 37° (see Figure 8B). Intriguingly, ssd1Δ cells are also more sensitive to Calcofluor than SSD1-v cells and have elevated levels of chitin and mannan in their cell wall, indicating that SSD1 is required for normal cell wall biosynthesis (Wheeler et al. 2003). Collectively, these data indicate that both overexpression and loss of function of CCW12, SRL1, and ZRG8 alter cell wall biosynthesis. Thus, it is possible that SIM1, CCW12, SRL1, or ZRG8 overexpression suppresses the lethality of ramΔ cells by altering cell wall integrity.
Figure 8.

SRL1 and ZRG8 exhibit synthetic genetic interactions with CCW12. (A) The morphologies of srl1Δ, zrg8Δ, ccw12Δ single- and double-mutant cells were monitored at 22° and 37°. srl1Δ (FLY1308), zrg8Δ (FLY1309), ccw12Δ (FLY1306) single-mutant cells and srl1Δ zrg8Δ (FLY1739) double-mutant cells appear normal in morphology at 22° and 37° (top). In contrast, many zrg8Δ ccw12Δ double-mutant cells (FLY1641) are aberrant in morphology and budding patterns at 22° and 37° (middle). The presence of cellular chains indicates that budding occurred in a polar fashion, as opposed to axial budding that is typical for haploid cells. In addition, zrg8Δ ccw12Δ cells fail to separate efficiently at 22° and 37°. The clusters of zrg8Δ ccw12Δ cells are resistant to disruption by sonication (S) at 37° or EDTA treatment (data not shown). srl1Δ ccw12Δ cells (FLY1722) exhibit severe morphology defects at 22°, but not at 37°. At 22°, srl1Δ ccw12Δ and some zrg8Δ ccw12Δ cells resemble mating-pheromone-treated wild-type cells. Seventy-nine percent of srl1Δ ccw12Δ and 68% of zrg8Δ ccw12Δ cells displayed aberrant morphologies at 22° (n > 260). (B) srl1Δ, zrg8Δ, ccw12Δ single- and double-mutant cells were assayed for Calcofluor white sensitivity at 22° and 37°. At 22°, srl1Δ ccw12Δ and zrg8Δ ccw12Δ cells exhibit enhanced Calcofluor white sensitivity in comparison to the corresponding single-mutant cells. At 37°, all single- and double-mutant strains are hypersensitive to Calcofluor white.
Zrg8p localizes similarly to RAM proteins:
One or more of the mob2Δ dosage suppressors might function as a RAM substrate or regulator. A RAM substrate or regulator is likely to localize similarly to RAM proteins at the bud cortex, bud neck, or daughter cell nucleus. Indeed, Srl1p was shown to localize to the cortex of small buds (Shepard et al. 2003). In contrast, Ccw12p, Sim1p, and Cbp3p may not directly interact with RAM proteins because they localize to the cell wall (Ccw12p, Sim1p) and mitochondria (Cbp3p), where RAM proteins are not known to localize (Mrsa et al. 1999; Shi et al. 2001; Velours et al. 2002). To determine if Zrg8p localizes similarly to RAM proteins, we investigated the distribution of GFP-tagged Zrg8p in live cells. Zrg8-GFP localized to the cortex of small and large buds during bud growth and to the bud neck during mitotic exit (Figure 7A). Moreover, in pheromone-treated cells, Zrg8-GFP localized to the tips of mating projections (Figure 7B).
Figure 7.

Localization of Zrg8p-GFP. (A) The localization of Zrg8p-GFP was analyzed in wild type (wt; FLY1279), cbk1Δ (FLY1327), and ace2Δ (FLY1680) cells by fluorescence microscopy. Zrg8p-GFP localizes to small and large buds and to the bud neck at the end of mitosis (top right) in wild-type cells. In cbk1Δ cells, Zrg8p-GFP is absent from the cortex and bud neck of most large budded cells and is greatly diminished on the cortex of most small buds. In contrast, Zrg8p localization appears normal in ace2Δ cells. Zrg8-GFP was detectable on the bud cortex in 63% (n = 60), 21% (n = 79), and 54% (n = 65) of small budded wild-type, cbk1Δ, and ace2Δ cells, respectively. Arrowheads in ace2Δ indicate Zrg8p on the bud neck and the cortex of large budded cells. (B) Zrg8p-GFP was analyzed in cells that were treated with mating pheromone for 2 hr. In wild-type cells (wt) and ace2Δ cells, Zrg8p-GFP localizes to the tip of all mating projections. Zrg8p-GFP is undetectable at the cortex of most (64%, n = 133) pheromone-treated cbk1Δ cells. In the remaining cbk1Δ cells, Zrg8p localizes to the tips of mating projections or to cortical spots or patches (arrowhead) that are not associated with mating projections.
To test if Zrg8p localization is dependent on RAM, we monitored Zrg8-GFP in cbk1Δ and ace2Δ cells. We found that Zrg8p localization was greatly diminished or absent from the bud cortex and bud neck in cbk1Δ cells (Figure 7A). Zrg8p-GFP was detectable on the cortex in 61% (n = 65) and 54% (n = 60) of the small budded wild-type and ace2Δ cells, but was detectable only in 21% (n = 79) of the small budded cbk1Δ cells. Zrg8p localization was also aberrant in cbk1Δ cells that were treated with mating pheromone (Figure 7B). Zrg8p was completely absent from mating projections and cell cortexes in 64% (n = 133) of the pheromone-treated cbk1Δ cells (Figure 7). In 22% of the pheromone-treated cbk1Δ cells, Zrg8p localized aberrantly to spots or patches on the sides of the mating projection or cell cortex. The remaining 14% of cells contained Zrg8p at the tips of mating projections. Moreover, some Zrg8-GFP appeared to localize to vacuoles in many of the mating-pheromone-treated cbk1Δ cells, suggesting that in the absence of Cbk1p, some Zrg8p is targeted for degradation. Zrg8p localization appeared normal in ace2Δ cells and in srl1Δ cells (Figure 7, A and B, and data not shown). These results indicate that Zrg8p localizes similarly to all RAM proteins throughout the cell cycle and is RAM dependent, but Ace2p independent, for localization. The RAM-like cortical localizations establish Zrg8p and Srl1p as viable candidates for RAM substrates or regulators.
zrg8Δ and srl1Δ mutations do not cause additive phenotypes:
The similar (albeit not identical) localization patterns of Zrg8p and Srl1p suggest that ZRG8 and SRL1 function in the same genetic pathway. If this model is correct, then the phenotypes of zrg8Δ srl1Δ cells should be similar to zrg8Δ and srl1Δ single-mutant cells. Alternatively, if Zrg8p and Srl1p function in different or parallel pathways, zrg8Δ srl1Δ cells may exhibit more severe phenotypes than the single-mutant cells. Thus, we crossed zrg8Δ and srl1Δ strains to construct a zrg8Δ srl1Δ double-mutant strain and assayed for synthetic phenotypes. The zrg8Δ srl1Δ cells did not exhibit any obvious growth or morphological differences from the zrg8Δ and srl1Δ single-mutant cells (Figure 8A). Likewise, zrg8Δ srl1Δ cells exhibited the same degree of Calcofluor sensitivity as zrg8Δ and srl1Δ single-mutant cells (Figure 8B). These data are consistent with the model that ZRG8 and SRL1 function in the same genetic pathway.
SRL1 and ZRG8 are not required for dosage suppression of mob2Δ SSD1-v cells:
If Zrg8p and Srl1p function together, then they may be dependent on each other to suppress the lethality of mob2Δ SSD1-v cells. Thus, we conducted a plasmid shuffle strategy, similar to that described for the dosage suppressor screen, to determine if dosage suppression of mob2Δ requires both SRL1 and ZRG8. We introduced high-copy ZRG8 or SRL1 plasmids into mob2Δ srl1Δ and mob2Δ zrg8Δ double-mutant cells that also contained a counterselectable MOB2 plasmid. We then selected for cells that lost the MOB2 plasmid and assayed cell viability. We observed that high-copy SRL1 and ZRG8 plasmids could restore the viability of mob2Δ srl1Δ double-mutant cells (Figure 9). Similarly, SRL1 and ZRG8 plasmids could suppress the lethality of mob2Δ zrg8Δ double-mutant cells. We performed similar experiments to determine if CBP3, CCW12, or SIM1 overexpression could suppress mob2Δ lethality in the absence of ZRG8 or SRL1. We found that CCW12 and SIM1, but not CBP3, plasmids suppressed the lethality of mob2Δ zrg8Δ and mob2Δ srl1Δ cells (Figure 9). Collectively, these data suggest that suppression of the cell integrity defects of ramΔ cells by CCW12 and SIM1 overexpression is independent of ZRG8 and SRL1. These data also support the model that ZRG8 and SRL1 function independently. Alternatively, ZRG8 and SRL1 may function redundantly when overexpressed.
Figure 9.

Dosage suppression of mob2Δ lethality does not require SRL1 or ZRG8. mob2Δ srl1Δ SSD1-v and mob2Δ zrg8Δ SSD1-v cells containing pRS316-MOB2 (FLY1741 and FLY1745, respectively) were transformed with YEp13-based plasmids containing CBP3, CCW12, SIM1, SRL1, or ZRG8. Cells were serially diluted (10-fold) and spotted onto leucine deficient (Leu−) and 5-FOA plates to counterselect for pRS316-MOB2. Each dosage suppressor plasmid, with the exception of pCBP3, rescued the lethality of mob2Δ srl1Δ SSD1-v and mob2Δ zrg8Δ SSD1-v cells.
ZRG8 and SRL1 function in parallel to CCW12:
To elucidate the mechanism of ramΔ suppression, we investigated the relationship between CCW12, ZRG8, and SRL1. We assayed zrg8Δ ccw12Δ and srl1Δ ccw12Δ double-mutant cells for enhanced morphological and cell wall defects. In contrast to zrg8Δ, srl1Δ, and ccw12Δ single-mutant cells, which were wild type in morphology (Figure 8A), srl1Δ ccw12Δ and zrg8Δ ccw12Δ double-mutant cells exhibited aberrant cellular morphologies. At 22°, 79% (n = 311) of the srl1Δ ccw12Δ cells displayed aberrant cell shapes that were similar in morphology to pheromone-treated cells. Intriguingly, the aberrant morphology of srl1Δ ccw12Δ cells was completely abolished at 37°. Most (68%, n = 268) zrg8Δ ccw12Δ cells also appeared swollen or misshapen. Moreover, they persisted in cell clusters at 22°, which could be partially separated by brief sonication. At 37°, the cell clusters were more resistant to sonication (Figure 8A). The cell clusters were not diminished by addition of 20 mm EDTA (data not shown). The haploid srl1Δ ccw12Δ and zrg8Δ ccw12Δ cells displayed apparent defects in bud site selection, since many buds emerged at opposite poles of cells, which is typical for diploid cells but not for haploid cells.
We also assayed srl1Δ ccw12Δ and zrg8Δ ccw12Δ cells for Calcofluor sensitivity. As shown in Figures 6B and 8B, zrg8Δ, srl1Δ, and ccw12Δ single-mutant cells are sensitive to low concentrations of Calcofluor, indicating cell wall defects. Intriguingly, all three single-mutant strains were more sensitive to Calcofluor at 37° than at 22° (Figure 8B). At 22°, both the srl1Δ ccw12Δ and zrg8Δ ccw12Δ double-mutant cells display enhanced Calcofluor sensitivity in comparison to the single mutants. Indeed, even the most densely plated aliquot of srl1Δ ccw12Δ cells died in the presence of 10 μg/ml Calcofluor. At 37°, srl1Δ ccw12Δ and zrg8Δ ccw12Δ cells displayed the same degree of Calcofluor sensitivity as the single mutant cells. Collectively, these data are consistent with the model that ZRG8 and SRL1 function in the same genetic pathway and that both ZRG8 and SRL1 function in parallel to CCW12. Recent results suggest that deletion of CCW12 and other genes encoding GPI-anchored cell wall proteins activates a novel SRL1-dependent cell integrity pathway (Hagen et al. 2004). Thus, our data may suggest that ZRG8 and SRL1 function together in this novel cell integrity pathway, which is probably SSD1 dependent for function.
Zrg8p coprecipitates with Ssd1p:
It was demonstrated that SSD1 inactivation rescues the lethality of ramΔ cells (Du and Novick 2002; Jorgensen et al. 2002). Thus, it is possible that one or more of the dosage suppressors restore the viability of ramΔ SSD1-v cells by regulating Ssd1p function. For example, Zrg8p overexpression might suppress the lethality of ramΔ SSD1-v cells by inhibiting Ssd1p or preventing Ssd1p from interacting with other RAM-associated proteins. If this is true, then Zrg8p might physically associate with Ssd1p. Indeed, large-scale affinity precipitation methods suggest that Zrg8p associates in a complex with Ssd1p (Krogan et al. 2004). Thus, to determine if Zrg8p interacts with Ssd1p, we conducted coprecipitation experiments in strains expressing Myc-tagged Zrg8p and HA-tagged Ssd1p. We established that Zrg8-Myc and Ssd1-HA are functional in vivo because Zrg8-Myc does not cause morphology defects in ccw12Δ cells and Ssd1-HA expression is lethal in mob2Δ ssd1-d cells (data not shown). We immunoprecipitated Ssd1-HA from extracts of asynchronously growing cells and assayed for the presence of coprecipitated Zrg8-Myc by immunoblot and vice versa. We found that a significant portion of Zrg8p coprecipitated Ssd1p (and vice versa) (Figure 10). These data indicate that Zrg8p and Ssd1p associate as a complex and are consistent with the model that Zrg8p functions as an inhibitor or cofactor of Ssd1p. We therefore hypothesize that Zrg8p dosage suppression of ramΔ SSD1-v cells occurs via inhibition of Ssd1p function.
Figure 10.

Zrg8p and Ssd1p coprecipitate. Lysates of cells expressing Zrg8p-Myc (FLY1735), Ssd1p-HA (FLY1735), or both Zrg8p-Myc and Ssd1p-HA (FLY1718) were immunoprecipitated with anti-Myc (Myc IP) or anti-HA (HA IP) antibodies. Immunoprecipitated material was loaded onto a protein gel, immunoblotted, and probed with anti-Myc (Myc blot) or anti-HA (HA blot). Immune complexes of Zrg8-Myc contain Ssd1p-HA and immune complexes of Ssd1p-HA contain Zrg8-Myc (right lanes). Note that Zrg8p-Myc migrates on protein gels as three bands. This could be caused by post-translational modification to Zrg8p or by partial proteolysis.
DISCUSSION
The S. cerevisiae RAM signaling network is critical for maintaining polarized growth and for regulating Ace2p transcription factor, which controls cell separation at the end of mitosis. In this article, we demonstrate that RAM has a third genetically separable function in maintaining cell integrity that involves SSD1. Deletion of RAM genes in SSD1-v cells causes severe cell lysis, which ultimately leads to cell death. The lethality of ramΔ cells is suppressed by ssd1-d, which encodes a nonfunctional form of SSD1 (Sutton et al. 1991; Du and Novick 2002; Jorgensen et al. 2002; McDonald et al. 2002). Although the cell lysis defects are suppressed by a loss of SSD1 function, all ramΔ ssd1-d cells exhibit cell polarity and cell separation defects (Racki et al. 2000; Bidlingmaier et al. 2001; Colman-Lerner et al. 2001; Du and Novick 2002; Weiss et al. 2002; Nelson et al. 2003). Interestingly, deletion of SSD1 does not cause severe defects in otherwise wild-type cells (Wheeler et al. 2003). Given the two-hybrid interactions between Cbk1p and Ssd1p (Racki et al. 2000), it is likely that RAM and Ssd1p function in a common biochemical pathway to maintain cell integrity; however, it remains a mystery why cells lacking both RAM and SSD1 exhibit no cellular lysis defects.
In an effort to elucidate the essential function of RAM, we identified several dosage suppressors of ramΔ lethality, including SIM1, CCW12, SRL1, and ZRG8. Although each dosage suppressor rescued the severe cell wall defects of ramΔ cells, none suppressed the polarized growth or Ace2p regulatory defects. These data establish that the maintenance of cell integrity function of RAM is genetically distinct from its polarized growth and Ace2p regulatory functions. The dosage suppressors could rescue ramΔ lethality by several possible mechanisms. In one mechanism, overexpression of the suppressor gene may alter or strengthen the cell wall and thereby prevent cellular lysis of ramΔ SSD1-v cells. In another mechanism, overexpression of an Ssd1p-binding protein or regulator might functionally inhibit Ssd1p and thus restore cell viability to ramΔ cells. Specific inhibition of Ssd1p in ramΔ SSD1-v cells would yield cells that are phenotypically indistinguishable from ramΔ ssd1-d cells. Alternatively, ramΔ SSD1-v cells might be suppressed by overexpression of a downstream target of RAM or Ssd1p that bypasses the essential cell integrity function of RAM.
Genetic and cytological experiments provide insight into the mechanism of ramΔ suppression. We found that although none of the identified ramΔ suppressor genes is essential for viability, most of them are required for proper cell wall biosynthesis, as detected by increased Calcofluor sensitivity in deletion mutants. Furthermore, we established that the ramΔ dosage suppressors fall into at least two functional groups, with CCW12 and SIM1 constituting one group and SRL1 and ZRG8 constituting a second group. CCW12 and SIM1 encode cell wall proteins, which implies that the suppression of the lethality of ramΔ cells occurs via altering, and possibly strengthening, cell wall architecture. Ccw12p is a GPI-anchored cell wall protein and Sim1p is a member of the SUN protein family (Mrsa et al. 1999; Velours et al. 2002). SUN family proteins share a common stretch of 258 aa at their C terminus, but appear to be involved in different cellular processes (Velours et al. 2002). Sim1p was recently shown to be a noncovalently linked cell wall protein (Velours et al. 2002) that was previously identified as a dosage suppressor of tao3Δ and cbk1Δ. It is possible that Sim1p and Ccw12p are downstream targets of RAM signaling; however, thus far, there is no evidence that RAM proteins directly interact with the cell wall. Thus, it may be more likely that CCW12 and SIM1 overexpression suppresses the lethality of ramΔ cells via indirect mechanisms that alter cell wall structure.
Several lines of evidence suggest that SRL1 and ZRG8, which encode proteins of unknown function, belong to the same pathway. First, Srl1p and Zrg8p localize similarly to the cortex of small buds (Figure 7; Shepard et al. 2003). Moreover, the phenotypes of srl1Δ and zrg8Δ mutations are not additive. As shown in Figure 8, srl1Δ zrg8Δ double-mutant cells do not exhibit more severe phenotypes than the corresponding single-mutant cells, as would be expected if Srl1p and Zrg8p function in separate or parallel pathways. SRL1 and ZRG8 also display similar genetic interactions with CCW12. Specifically, deletion of SRL1 or ZRG8 in ccw12Δ cells enhances Calcofluor sensitivity and morphological defects. These data suggest that SRL1 and ZRG8 function in parallel or cooperating pathways with CCW12 to maintain cell integrity. Nevertheless, the morphological differences between srl1Δ ccw12Δ and zrg8Δ ccw12Δ double-mutant cells indicate that SRL1 and ZRG8 are not functionally redundant. Thus, collectively these data support the model that SRL1 and ZRG8 function in a RAM- and SSD1-dependent cell integrity pathway.
ZRG8 may interact directly with RAM. In support of this, Zrg8p localizes similarly to RAM proteins throughout the cell cycle. ZRG8 was first identified as a zinc-regulated gene that encodes a serine-rich protein of unknown function. Its expression is enhanced 1.9-fold when cells are grown in zinc-deficient media (Yuan 2000), although the significance of this phenomenon is unknown. We established that Zrg8p localizes to the cortex of small and large buds, to the bud neck region during mitotic exit, and to the tips of mating projections. Significantly, these localizations are dependent on the RAM kinase Cbk1p, but are not aberrant in zinc-limiting medium (data not shown) or in ace2Δ cells (Figure 7). These findings suggest that Zrg8p is involved in the cell polarity or integrity functions of RAM, but not in the cell separation functions of RAM. Evidence that Zrg8p coprecipitates with Ssd1p supports a role for Zrg8p in regulating cell integrity (Figure 10) and is consistent with the model that Zrg8p overexpression disrupts SSD1 function and thereby suppresses the lethality of ramΔ SSD1-v cells. Similar coprecipitation approaches did not reveal an interaction between Zrg8p and Cbk1p in ssd1-d cells (data not shown). Nevertheless, since Ssd1p interacts with Cbk1p (Racki et al. 2000), it is likely that Zrg8p associates with at least one of the RAM proteins at the cell cortex. Thus, Zrg8p may functionally link Ssd1p with RAM activity.
Although genetically related, SRL1 is probably not functionally redundant to ZRG8. SRL1 encodes a 210-amino-acid mannoprotein that may associate with membranes or cell walls (Terashima et al. 2002; Hagen et al. 2004). SRL1 expression is regulated by Swi4p during G1, as was shown by direct binding of Swi4p to the promoter region of SRL1 (Baetz et al. 2001). Like Zrg8p, Srl1p localizes to the cortex of small buds (Shepard et al. 2003); however, unlike Zrg8p, Srl1p is undetectable in large buds or on the bud neck (our unpublished observations). These localization patterns suggest that Srl1p functions at the cortex from late G1 to early S phase, whereas Zrg8p functions at the sites of polarized growth throughout the cell cycle. Recent data suggest that Srl1p is required for a cell integrity pathway that is triggered in the absence of multiple GPI-anchored cell wall proteins, such as Ccw12p (Hagen et al. 2004). Moreover, SRL1 expression is enhanced by cell-wall-damaging agents and in ramΔ ssd1-d strains (Jorgensen et al. 2002; Garcia et al. 2004). These data suggest that cell wall and/or polarized growth defects (such as those caused by ramΔ mutations) enhance the activation of a cell integrity pathway via SRL1 activation. Thus, RAM may regulate cell integrity by indirectly controlling SRL1 expression and/or by regulating Srl1p function at the bud cortex. Alternatively, Srl1p may function as an inhibitor of Ssd1p function, as discussed below.
It is probable that Zrg8p and Srl1p contribute to cell integrity via Ssd1p and/or RAM. In support of this, Zrg8p coprecipitates with Ssd1p and both Zrg8p and Srl1p localize similarly to RAM proteins. Nevertheless, the bulk of Zrg8p does not appear to associate with Ssd1p in vivo. Ssd1p localizes to poorly defined punctate spots in the cytoplasm, but does not accumulate at the bud cortex or bud neck, as do Zrg8p and RAM proteins (Uesono et al. 1997). Thus, a fraction of Ssd1p is likely to function independently of RAM and Zrg8p and vice versa. In agreement, RAM functions independently of Ssd1p for its cell polarity and cell separation function. Curiously, ZRG8 and SRL1 are not essential for viability, indicating that they are not critical for Ssd1p- or RAM-dependent roles in maintenance of cell integrity (Figure 8). Moreover, deletion of ZRG8 or SRL1 does not rescue the lethality of mob2Δ SSD1-v cells. To resolve these observations, we propose that Zrg8p and Srl1p function as nonessential inhibitors of Ssd1p. In accordance with this model, Ssd1p would still be able to function with RAM to ensure cell integrity in the absence of these nonessential Ssd1p inhibitors.
Although the specific molecular role that SSD1 plays in RAM signaling is unknown, insight has come from analyzing its genetic interactions. Ssd1p contains a RNase-II-like RNA-binding domain that can bind RNA in vitro (Uesono et al. 1997), although the physiological relevance of RNA binding has not been demonstrated in vivo. SSD1 is linked genetically to several important cellular processes. Mutations in SSD1 are synthetic lethal with mutations in genes involved in mRNA splicing, cell wall integrity, and vesicle transport (Luukkonen and Seraphin 1999; Kaeberlein and Guarente 2002; Rosenwald et al. 2002). SSD1 exhibits a different type of genetic relationship with RAM genes and RVS167. Mutations in SSD1, such as ssd1-d, suppress the lethality of ramΔ and rvs167Δ mutations (Breton et al. 2001; Du and Novick 2002; Jorgensen et al. 2002). Intriguingly, RVS167 encodes a BAR adapter protein that interacts with actin and participates in regulation of endocytosis and vesicle transport (Breton et al. 2001). The similarity of the genetic interactions suggests that RAM and RVS167 are functionally linked. In agreement, both RVS167 and the RAM gene KIC1 share genetic interactions with KRE6, which encodes a putative β-1,6-glucan synthase whose activity is dependent on vesicle transport (Breton et al. 2001; Vink et al. 2002). Furthermore, the S. pombe Kic1p homolog was recently shown to associate with the S. pombe Rvs167p homolog (Huang et al. 2005). It is therefore possible that SSD1 and RAM function cooperatively to regulate vesicle transport and/or endocytosis, which is critical for polarized growth, cell wall biosynthesis and ultimately cell integrity. Further work is necessary to test this model for RAM and SSD1 function and establish the specific molecular roles of Zrg8p and Srl1p in RAM signaling and Ssd1p regulation.
Acknowledgments
We thank Charlie Boone, Eric Weiss, Erfei Bi, Jan Stoepel, and members of the Luca lab for many insightful discussions. We are indebted to Jack Greenblatt and Nevan Krogan for sharing unpublished observations regarding Ssd1p protein complexes. We also thank Aparna Iyer and Manali Mody for technical assistance. This work was supported by a grant from the National Institutes of Health (GM60575 to F.C.L.).
References
- Baetz, K., J. Moffat, J. Haynes, M. Chang and B. Andrews, 2001. Transcriptional coregulation by the cell integrity mitogen-activated protein kinase Slt2 and the cell cycle regulator Swi4. Mol. Cell. Biol. 21: 6515–6528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bidlingmaier, S., E. L. Weiss, C. Seidel, D. G. Drubin and M. Snyder, 2001. The Cbk1p pathway is important for polarized cell growth and cell separation in Saccharomyces cerevisiae. Mol. Cell. Biol. 21: 2449–2462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bogomolnaya, L. M., R. Pathak, J. Guo, R. Cham, R. Aramayo et al., 2004. Hym1p affects cell cycle progression in Saccharomyces cerevisiae. Curr. Genet. 46: 183–192. [DOI] [PubMed] [Google Scholar]
- Breton, A. M., J. Schaeffer and M. Aigle, 2001. The yeast Rvs161 and Rvs167 proteins are involved in secretory vesicles targeting the plasma membrane and in cell integrity. Yeast 18: 1053–1068. [DOI] [PubMed] [Google Scholar]
- Colman-Lerner, A., T. E. Chin and R. Brent, 2001. Yeast Cbk1 and Mob2 activate daughter-specific genetic programs to induce asymmetric cell fates. Cell 107: 739–750. [DOI] [PubMed] [Google Scholar]
- Costigan, C., S. Gehrung and M. Snyder, 1992. A synthetic lethal screen identifies SLK1, a novel protein kinase homolog implicated in yeast cell morphogenesis and cell growth. Mol. Cell. Biol. 12: 1162–1178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dan, I., N. M. Watanabe and A. Kusumi, 2001. The Ste20 group kinases as regulators of MAP kinase cascades. Trends Cell Biol. 11: 220–230. [DOI] [PubMed] [Google Scholar]
- DeMarini, D. J., A. E. Adams, H. Fares, C. De Virgilio, G. Valle et al., 1997. A septin-based hierarchy of proteins required for localized deposition of chitin in the Saccharomyces cerevisiae cell wall. J. Cell Biol. 139: 75–93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dorland, S., M. L. Deegenaars and D. J. Stillman, 2000. Roles for the Saccharomyces cerevisiae SDS3, CBK1 and HYM1 genes in transcriptional repression by SIN3. Genetics 154: 573–586. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Du, L. L., and P. Novick, 2002. Pag1p, a novel protein associated with protein kinase Cbk1p, is required for cell morphogenesis and proliferation in Saccharomyces cerevisiae. Mol. Biol. Cell 13: 503–514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garcia, R., C. Bermejo, C. Grau, R. Perez, J. M. Rodriguez-Pena et al., 2004. The global transcriptional response to transient cell wall damage in Saccharomyces cerevisiae and its regulation by the cell integrity signaling pathway. J. Biol. Chem. 279: 15183–15195. [DOI] [PubMed] [Google Scholar]
- Guthrie, C., and G. R. Fink, 1991. Guide to Yeast Genetics and Molecular Biology (Methods in Enzymology, Vol. 194). Academic Press, San Diego. [PubMed]
- Hagen, I., M. Ecker, A. Lagorce, J. M. Francois, S. Sestak et al., 2004. Sed1p and Srl1p are required to compensate for cell wall instability in Saccharomyces cerevisiae mutants defective in multiple GPI-anchored mannoproteins. Mol. Microbiol. 52: 1413–1425. [DOI] [PubMed] [Google Scholar]
- Hirata, D., N. Kishimoto, M. Suda, Y. Sogabe, S. Nakagawa et al., 2002. Fission yeast Mor2/Cps12, a protein similar to Drosophila Furry, is essential for cell morphogenesis and its mutation induces Wee1-dependent G(2) delay. EMBO J. 21: 4863–4874. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hou, M. C., J. Salek and D. McCollum, 2000. Mob1p interacts with the Sid2p kinase and is required for cytokinesis in fission yeast. Curr. Biol. 10: 619–622. [DOI] [PubMed] [Google Scholar]
- Hou, M. C., D. J. Wiley, F. Verde and D. McCollum, 2003. Mob2p interacts with the protein kinase Orb6p to promote coordination of cell polarity with cell cycle progression. J. Cell Sci. 116: 125–135. [DOI] [PubMed] [Google Scholar]
- Huang, T. Y., N. A. Markley and D. Young, 2003. Nak1, an essential germinal center (GC) kinase regulates cell morphology and growth in Schizosaccharomyces pombe. J. Biol. Chem. 278: 991–997. [DOI] [PubMed] [Google Scholar]
- Huang, T. Y., M. Renaud-Young and D. Young, 2005. Nak1 interacts with Hob1 and Wsp1 to regulate cell growth and polarity in Schizosaccharomyces pombe. J. Cell Sci. 118: 199–210. [DOI] [PubMed] [Google Scholar]
- Jorgensen, P., B. Nelson, M. D. Robinson, Y. Chen, B. Andrews et al., 2002. High-resolution genetic mapping with ordered arrays of Saccharomyces cerevisiae deletion mutants. Genetics 162: 1091–1099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaeberlein, M., and L. Guarente, 2002. Saccharomyces cerevisiae MPT5 and SSD1 function in parallel pathways to promote cell wall integrity. Genetics 160: 83–95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karos, M., and R. Fischer, 1999. Molecular characterization of HymA, an evolutionarily highly conserved and highly expressed protein of Aspergillus nidulans. Mol. Gen. Genet. 260: 510–521. [DOI] [PubMed] [Google Scholar]
- Kosodo, Y., K. Imai, A. Hirata, Y. Noda, A. Takatsuki et al., 2001. Multicopy suppressors of the sly1 temperature-sensitive mutation in the ER-Golgi vesicular transport in Saccharomyces cerevisiae. Yeast 18: 1003–1014. [DOI] [PubMed] [Google Scholar]
- Krogan, N. J., W. T. Peng, G. Cagney, M. D. Robinson, R. Haw et al., 2004. High-definition macromolecular composition of yeast RNA-processing complexes. Mol. Cell 13: 225–239. [DOI] [PubMed] [Google Scholar]
- Laabs, T. L., D. D. Markwardt, M. G. Slattery, L. L. Newcomb, D. J. Stillman et al., 2003. ACE2 is required for daughter cell-specific G1 delay in Saccharomyces cerevisiae. Proc. Natl. Acad. Sci. USA 100: 10275–10280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li, B., and J. R. Warner, 1998. Genetic interaction between YPT6 and YPT1 in Saccharomyces cerevisiae. Yeast 14: 915–922. [DOI] [PubMed] [Google Scholar]
- Longtine, M. S., A. McKenzie, III, D. J. Demarini, N. G. Shah, A. Wach et al., 1998. Additional modules for versatile and economical PCR-based gene deletion and modification in Saccharomyces cerevisiae. Yeast 14: 953–961. [DOI] [PubMed] [Google Scholar]
- Luca, F. C., and M. Winey, 1998. MOB1, an essential yeast gene required for completion of mitosis and maintenance of ploidy. Mol. Biol. Cell 9: 29–46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luca, F. C., M. Mody, C. Kurischko, D. M. Roof, T. H. Giddings et al., 2001. Saccharomyces cerevisiae Mob1p is required for cytokinesis and mitotic exit. Mol. Cell. Biol. 21: 6972–6983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luukkonen, B. G., and B. Seraphin, 1999. A conditional U5 snRNA mutation affecting pre-mRNA splicing and nuclear pre-mRNA retention identifies SSD1/SRK1 as a general splicing mutant suppressor. Nucleic Acids Res. 27: 3455–3465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mah, A. S., J. Jang and R. J. Deshaies, 2001. Protein kinase Cdc15 activates the Dbf2-Mob1 kinase complex. Proc. Natl. Acad. Sci. USA 98: 7325–7330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McDonald, H. B., A. H. Helfant, E. M. Mahony, S. K. Khosla and L. Goetsch, 2002. Mutational analysis reveals a role for the C terminus of the proteasome subunit Rpt4p in spindle pole body duplication in Saccharomyces cerevisiae. Genetics 162: 705–720. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miyamoto, H., A. Matsushiro and M. Nozaki, 1993. Molecular cloning of a novel mRNA sequence expressed in cleavage stage mouse embryos. Mol. Reprod. Dev. 34: 1–7. [DOI] [PubMed] [Google Scholar]
- Mrsa, V., M. Ecker, S. Strahl-Bolsinger, M. Nimtz, L. Lehle et al., 1999. Deletion of new covalently linked cell wall glycoproteins alters the electrophoretic mobility of phosphorylated wall components of Saccharomyces cerevisiae. J. Bacteriol. 181: 3076–3086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nelson, B., C. Kurischko, J. Horecka, M. Mody, P. Nair et al., 2003. RAM: a conserved signaling network that regulates Ace2p transcriptional activity and polarized morphogenesis. Mol. Biol. Cell 14: 3782–3803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- O'Conallain, C., T. Doolin and G. Butler, 1998. Inappropriate expression of the yeast transcription factor Ace2p affects cell growth. Biochem. Soc. Trans. 26: S78. [DOI] [PubMed] [Google Scholar]
- Racki, W. J., A. M. Becam, F. Nasr and C. J. Herbert, 2000. Cbk1p, a protein similar to the human myotonic dystrophy kinase, is essential for normal morphogenesis in Saccharomyces cerevisiae. EMBO J. 19: 4524–4532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ram, A. F., A. Wolters, R. Ten Hoopen and F. M. Klis, 1994. A new approach for isolating cell wall mutants in Saccharomyces cerevisiae by screening for hypersensitivity to calcofluor white. Yeast 10: 1019–1030. [DOI] [PubMed] [Google Scholar]
- Reinke, A., S. Anderson, J. M. McCaffery, J. Yates, III, S. Aronova et al., 2004. TOR complex 1 includes a novel component, Tco89p (YPL180w), and cooperates with Ssd1p to maintain cellular integrity in Saccharomyces cerevisiae. J. Biol. Chem. 279: 14752–14762. [DOI] [PubMed] [Google Scholar]
- Rosenwald, A. G., M. A. Rhodes, H. Van Valkenburgh, V. Palanivel, G. Chapman et al., 2002. ARL1 and membrane traffic in Saccharomyces cerevisiae. Yeast 19: 1039–1056. [DOI] [PubMed] [Google Scholar]
- Schneper, L., A. Krauss, R. Miyamoto, S. Fang and J. R. Broach, 2004. The Ras/protein kinase A pathway acts in parallel with the Mob2/Cbk1 pathway to effect cell cycle progression and proper bud site selection. Eukaryot. Cell 3: 108–120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shepard, K. A., A. P. Gerber, A. Jambhekar, P. A. Takizawa, P. O. Brown et al., 2003. Widespread cytoplasmic mRNA transport in yeast: identification of 22 bud-localized transcripts using DNA microarray analysis. Proc. Natl. Acad. Sci. USA 100: 11429–11434. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shi, G., M. D. Crivellone and B. Edderkaoui, 2001. Identification of functional regions of Cbp3p, an enzyme-specific chaperone required for the assembly of ubiquinol-cytochrome c reductase in yeast mitochondria. Biochim. Biophys. Acta 1506: 103–116. [DOI] [PubMed] [Google Scholar]
- Sullivan, D. S., S. Biggins and M. D. Rose, 1998. The yeast centrin, cdc31p, and the interacting protein kinase, Kic1p, are required for cell integrity. J. Cell Biol. 143: 751–765. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sutton, A., D. Immanuel and K. T. Arndt, 1991. The SIT4 protein phosphatase functions in late G1 for progression into S phase. Mol. Cell. Biol. 11: 2133–2148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tamaskovic, R., S. J. Bichsel and B. A. Hemmings, 2003. NDR family of AGC kinases—essential regulators of the cell cycle and morphogenesis. FEBS Lett. 546: 73–80. [DOI] [PubMed] [Google Scholar]
- Teparic, R., I. Stuparevic and V. Mrsa, 2004. Increased mortality of Saccharomyces cerevisiae cell wall protein mutants. Microbiology 150: 3145–3150. [DOI] [PubMed] [Google Scholar]
- Terashima, H., S. Fukuchi, K. Nakai, M. Arisawa, K. Hamada et al., 2002. Sequence-based approach for identification of cell wall proteins in Saccharomyces cerevisiae. Curr. Genet. 40: 311–316. [DOI] [PubMed] [Google Scholar]
- Uesono, Y., A. Toh-e and Y. Kikuchi, 1997. Ssd1p of Saccharomyces cerevisiae associates with RNA. J. Biol. Chem. 272: 16103–16109. [DOI] [PubMed] [Google Scholar]
- Velours, G., C. Boucheron, S. Manon and N. Camougrand, 2002. Dual cell wall/mitochondria localization of the ‘SUN’ family proteins. FEMS Microbiol. Lett. 207: 165–172. [DOI] [PubMed] [Google Scholar]
- Verde, F., D. J. Wiley and P. Nurse, 1998. Fission yeast orb6, a ser/thr protein kinase related to mammalian rho kinase and myotonic dystrophy kinase, is required for maintenance of cell polarity and coordinates cell morphogenesis with the cell cycle. Proc. Natl. Acad. Sci. USA 95: 7526–7531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Versele, M., and J. M. Thevelein, 2001. Lre1 affects chitinase expression, trehalose accumulation and heat resistance through inhibition of the Cbk1 protein kinase in Saccharomyces cerevisiae. Mol. Microbiol. 41: 1311–1326. [DOI] [PubMed] [Google Scholar]
- Vincent, K., Q. Wang, S. Jay, K. Hobbs and B. C. Rymond, 2003. Genetic interactions with CLF1 identify additional pre-mRNA splicing factors and a link between activators of yeast vesicular transport and splicing. Genetics 164: 895–907. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vink, E., J. H. Vossen, A. F. Ram, H. Van Den Ende, S. Brekelmans et al., 2002. The protein kinase Kic1 affects 1,6-beta-glucan levels in the cell wall of Saccharomyces cerevisiae. Microbiology 148: 4035–4048. [DOI] [PubMed] [Google Scholar]
- Weiss, E. L., C. Kurischko, C. Zhang, K. Shokat, D. G. Drubin et al., 2002. The Saccharomyces cerevisiae Mob2p-Cbk1p kinase complex promotes polarized growth and acts with the mitotic exit network to facilitate daughter cell-specific localization of Ace2p transcription factor. J. Cell Biol. 158: 885–900. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wheeler, R. T., M. Kupiec, P. Magnelli, C. Abeijon and G. R. Fink, 2003. A Saccharomyces cerevisiae mutant with increased virulence. Proc. Natl. Acad. Sci. USA 100: 2766–2770. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yuan, D. S., 2000. Zinc-regulated genes in Saccharomyces cerevisiae revealed by transposon tagging. Genetics 156: 45–58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao, H., E. Butler, J. Rodgers, T. Spizzo, S. Duesterhoeft et al., 1998. Regulation of zinc homeostasis in yeast by binding of the ZAP1 transcriptional activator to zinc-responsive promoter elements. J. Biol. Chem. 273: 28713–28720. [DOI] [PubMed] [Google Scholar]