

Abstract
In female mammalian cells, the inactive X chromosome is replicated late in S phase while the active X chromosome is replicated earlier. The replication times of the X chromosomes reflect a general trend in which late replication is associated with gene repression and earlier replication with transcriptional competence. The X-linked Xist gene is expressed exclusively from the inactive X chromosome where it is involved in the initiation and maintenance of X-inactivation. In contrast, no biological activity has been assigned to the Xist locus of the active X chromosome where the Xist gene is transcriptionally silenced. Here, we provide evidence that the element(s) at the nontranscribed Xist locus of the active X chromosome controls chromosomal replication timing in cis.
X-INACTIVATION in female mammals is the formation of heterochromatin throughout one of two X chromosomes (Gartler and Riggs 1983). The X-linked Xist gene plays a central role in this process (Penny et al. 1996) and Xist knockout mice die early in embryogenesis due to a failure to undergo X-inactivation (Marahrens et al. 1997). Xist encodes an untranslated RNA that is expressed from the inactive X chromosome (Xi) but not from the active X chromosome (Xa) (Brockdorff et al. 1991; Brown et al. 1991). The Xist RNA is quite stable and colocalizes exclusively with the Xi (Brown et al. 1992; Clemson et al. 1996).
Another feature that distinguishes the Xi from other chromosomes is that the Xi is replicated very late in S phase while the Xa is replicated earlier (Taylor 1968; Taylor and Miner 1968). In this regard, the replication timing of the X chromosomes reflects a general trend where late replication times are associated with gene repression and early replication with transcriptional competence (Gilbert 2002). The available evidence indicates that the source of the replication-timing differences between the two X chromosomes are the times in S phase that their origins are activated, with the same replication origins being utilized on the active and inactive X chromosome (Cohen et al. 2003; Gomez and Brockdorff 2004). It has been estimated that the human genome is replicated by ∼30,000 origins (Todorovic et al. 1999). The X chromosome constitutes ∼5% of the human (Lander et al. 2001) and is therefore estimated to be replicated by ∼1500 origins. The mouse X chromosome is very similar in size to the human X (Waterston et al. 2002) and therefore may by replicated by comparable numbers of origins. We expect many of these origins to be activated earlier on the Xa than on the Xi. However, in view of evidence that at least a subset of adjacent origins are activated at approximately the same time in S phase in mammalian cells (Huberman and Riggs 1968), we expect considerably <1500 replication-timing domains on the Xa and on the Xi.
The elements responsible for maintaining the replication timing of the Xa and Xi have not been identified. Excision of 21 kb from the transcribed Xist allele from the Xi, after X-inactivation has been completed, led to a partial destabilization of X chromosomal gene silencing (Csankovszki et al. 2001) but did not disrupt late replication (Csankovszki et al. 1999). The destabilization of X-inactivation was accompanied by the loss of the Xist RNA and macroH2A from the Xi (Csankovszki et al. 1999). The transcribed Xist allele on the Xi, therefore, functions in cis to maintain certain features of the Xi. In contrast, no biological activity has been assigned to the nontranscribed Xist locus on the Xa. Here we show that the excision of 21 kb from the Xist locus on the Xa alters the timing of Xa replication relative to the replication-timing program of the rest of the genome.
MATERIALS AND METHODS
Fibroblasts and growth conditions:
All of the mice used in this study had a 129 genetic background. Mouse primary fibroblasts were obtained from crosses involving previously described mouse strains (Csankovszki et al. 1999, 2001) by trypsinization of 13-day embryos, culture, and immortalization with SV40 T-antigen (Jat et al. 1986). Inherited Xist-Δ21kb alleles were always from the mother, since paternal inheritance of the Xist-Δ21kb allele leads to embryonic lethality (not shown) and in this regard resembles a Xist-Δ16kb allele (Marahrens et al. 1997). Female mice carrying Xist-Δ21kb alleles were maintained by breeding to 129 male mice. Subsequent to random X-inactivation, individually transformed fibroblast lines were obtained by plating cells at less than one cell per well of a multi-well plate (limiting dilution). To obtain XaXist-Δ21kbXiWT fibroblasts, mice that were homozygous for an X-linked GFP transgene (Hadjantonakis et al. 1998) and the Xist-2Lox allele were mated to 129 mice and fibroblasts were prepared from E13.5 embryos. Female fibroblasts were identified using PCR and subjected to cell sorting using flow cytometry to purify GFP-expressing cells (inferred to be 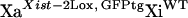 ) from cells that do not express GFP (inferred to be
) from cells that do not express GFP (inferred to be 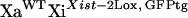 ).
). 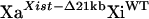 fibroblasts were derived from the GFP-expressing
fibroblasts were derived from the GFP-expressing 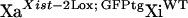 fibroblasts by infection with adenovirus carrying cre recombinase (AdCre) (Tan et al. 1999). The infection was performed in Dulbecco's Modified Eagle Medium (DMEM) with 5% fetal bovine serum for 1 hr. Primary and transformed mice fibroblast cell lines were grown in DMEM supplemented with 10% fetal bovine serum (GIBCO, Gaithersburg, MD), penicillin (100 μg/ml), and streptomycin (100 μg/ml). The PCR primers 5′-LoxF (5′-TTT CTG GTC TTT GAG GGC AC-3′), 5′-LoxR (5′-ACC CTT GCC TTT TCC ATT TT-3′), and Xint3R (5′-CAC TGG CAA GGT GAA TAG CA-3′) were used to identify the Xist-2Lox (612-bp PCR product), Xist-Δ21kb (513-bp), and Xist-WT (427-bp) alleles. XaXist-Δ21kbXiWT fibroblasts were distinguished from XaWTXiXist-Δ21kb fibroblasts using RNA FISH against the Xist RNA (see below).
fibroblasts by infection with adenovirus carrying cre recombinase (AdCre) (Tan et al. 1999). The infection was performed in Dulbecco's Modified Eagle Medium (DMEM) with 5% fetal bovine serum for 1 hr. Primary and transformed mice fibroblast cell lines were grown in DMEM supplemented with 10% fetal bovine serum (GIBCO, Gaithersburg, MD), penicillin (100 μg/ml), and streptomycin (100 μg/ml). The PCR primers 5′-LoxF (5′-TTT CTG GTC TTT GAG GGC AC-3′), 5′-LoxR (5′-ACC CTT GCC TTT TCC ATT TT-3′), and Xint3R (5′-CAC TGG CAA GGT GAA TAG CA-3′) were used to identify the Xist-2Lox (612-bp PCR product), Xist-Δ21kb (513-bp), and Xist-WT (427-bp) alleles. XaXist-Δ21kbXiWT fibroblasts were distinguished from XaWTXiXist-Δ21kb fibroblasts using RNA FISH against the Xist RNA (see below).
RNA FISH:
Fibroblasts were grown on coverslips for 24 hr and then fixed in 4% formaldehyde for 15 min at RT. The cells were permeabilized in PBS containing 0.5% Triton-X for 5 min on ice and washed in PBS and 2× SSC. RNA FISH hybridization was carried out as previously described (Spector and Goldman 1998.). The Xist probe was labeled by nick translation with biotin-21-dUTP. After overnight hybridization at 37°, posthybridization washes were done as previously described (Spector and Goldman 1998). The probe was detected with 500-fold dilution of avidin-FITC (Jackson ImmunoResearch, West Grove, PA) at RT for 1 hr. The nuclei were counterstained with 4′,6-diamidino-2-phenylindole (DAPI).
Assay for late replication:
Medium containing 30 μm of bromodeoxyuridine (BrdU) was added to actively growing fibroblasts at 80% confluence. When lymphocytes were used, 1 × 106 B lymphocytes/ml from mouse spleens were cultivated in 10% fetal bovine serum/10 mm HEPES/RPMI 1640 under stimulation of 10 μg/ml of lipopolysaccharide for 36 hr at 37° and 5% CO2 prior to adding 30 μm of BrdU. A time course of BrdU pulse length was performed on multiple cell lines and these data were used to determine that 4.5 hr is the appropriate length of BrdU incorporation for each replication-timing experiment for both fibroblasts and activated lymphocytes (data not shown). After exponentially growing primary fibroblasts or activated lymphocytes were cultured for 4.5 hr in the presence of BrdU, 0.050 μg/ml colcemid (Life Technologies, Grand Island, NY) was added 1 hr before harvesting. Cell suspensions were incubated 13 min in 0.4% KCl at 37° followed by multiple changes of 3:1 methanol:acetic acid. Metaphase spreads were prepared by dropping the BrdU-treated cells onto coverslips followed by DNA denaturation in 70% formamide/2× SSC at 73° for 2 min. Following preincubation with blocking buffer (1× PBS, 10% goat serum, 0.2% Tween 20), incorporated BrdU was detected using monoclonal anti-BrdU antibody (Sigma, St. Louis) followed by Texas-Red anti-mouse antibody (Jackson Immunoresearch) in blocking buffer. Images were captured with Quips mFISH software (Vysis). For a recorded image, the individual colors were stored separately by the Vysis Quips mFISH software and the representation of each color in the final image was determined by the software setting of the gain for that color. Xist RNA FISH was not done simultaneously with the BrdU incorporation studies because the Xist RNA is lost from the Xi during mitosis. The Xist RNA that can be seen on the Xi in mitotic cells is very fragile and the treatments entailed by the BrdU incorporation assay caused the Xist RNA to be lost from the Xi.
X chromosome paint:
Ten microliters of mouse X chromosome-specific biotinylated probe (Cambio, Cambridge, UK) was used to detect the X chromosomes by in situ hybridization to ethanol-dehydrated cells according to the manufacturer's instructions. The probe was detected using the biotin (FITC) detection kit (Cambio). The chromosomes were counterstained with DAPI and viewed with a Leica DMR fluorescent microscope.
Quantitation:
The proportions of BrdU signal in wild-type and mutant X chromosomes were compared using Fisher's exact test. The active and inactive X chromosomes were identified using X chromosome paints, and their relative levels of BrdU incorporation determined where the chromosome displaying the higher level of BrdU incorporation was always assumed to be the inactive one. Images were transferred from Quips mFISH to the National Institutes of Health (NIH) Image and the number of pixels occupied by the X chromosomes and the number of pixels occupied by fluorescently labeled BrdU were then calculated from images of mitotic chromosomes using NIH Image (http://rsb.info.nih.gov/nih-image). In Figures 1–4, “% area BrdU” represents the number of pixels of BrdU signal divided by the number of pixels of DAPI signal multiplied by 100. NIH Image also measured the intensity of each pixel. “BrdU area × intensity” represents the number of pixels of BrdU signal multiplied by the average intensity of these pixels. BrdU quantitation was performed on several autosomes that displayed no BrdU according to visual inspection and also on five mitotic chromosomes from BrdU-untreated cells; in both cases we failed to record a single pixel of BrdU using the NIH Image program, indicating that the program was not recording a BrdU signal that could not be detected by eye nor was the program recording background noise. Differences in measurements were tested across categorical groupings by using the Kruskal-Wallis test (Kruskal 1964) and listed as P-values above the corresponding boxplots. Distributions of measurements across categorical groupings were visualized with boxplots. The statistical analyses were conducted using the software package R (Ihaka and Gentleman 1996), which can be downloaded from http://cran.r-project.org/.
Figure 1.

BrdU incorporation in late S phase in transformed female mouse embryonic fibroblasts that are wild type with respect to the Xist locus (XaXist-WTXiXist-WT) or carry a 21-kb deletion in the Xist locus on the active X chromosome (XaXist-Δ21kbXiXist-WT). In all mitotic chromosome spreads, the active X chromosome is always assumed to be the X chromosome displaying a lower proportion of BrdU incorporation. Following exposure of actively growing cultures to BrdU for 4.5 hr, mitotic chromosome spreads were prepared and immunostained using anti-BrdU (red), hybridized to X chromosome paint (green), and counterstained using DAPI (blue). The late-replicating inactive X chromosome in a mitotic chromosome spread may be marked by an arrow. (A–D) Diploid wild-type transformed fibroblast (A); possible times in the cell cycle that the indicated fibroblasts were inferred to be exposed to BrdU (B); diploid wild-type transformed fibroblast (C and D). (E) Simplified cartoon of a hypothetical mitotic telocentric chromosome (blue) with the centromere at the left end of the chromosome ( “Cent.”) and late replication units along that chromosome (vertical red lines). Above the chromosome is a graph representing the hypothetical intensities of BrdU signal along the X chromosome from a replication-timing assay where BrdU had been incorporated into the chromosomal DNA late in the S phase and then subjected to red fluorescent immunostaining in the following mitosis. The graph illustrates that only the highest concentrations of late replication units on the chromosome would be visible at a low gain, while regions with lower concentrations of late replication units would also display a visible fluorescent signal at an intermediate gain, and even lower concentrations of late-replicating units would display a fluorescent signal at a high gain. (F–I) Mitotic chromosome spread from a diploid primary XaWTXiWT fibroblast displaying X chromosome paint (F), BrdU incorporation at a higher gain (G), an intermediate gain (H), and a lower gain where only four autosomes display BrdU and one autosome displays one pixel of BrdU (I). (J–M) Mitotic chromosome spread from a transformed XaXist-Δ21kbXiWT fibroblast displaying X chromosome paint (J), BrdU incorporation at a higher gain (K), an intermediate gain (L), and a lower gain (M). Transformed XaWTXiWT (N and O) and XaXist-Δ21kbXiWT (P and Q) fibroblasts subjected to the BrdU replication-timing assay and recorded at a gain where five autosomes display BrdU incorporation. Transformed XaWTXiWT (R and S) and XaXist-Δ21kbXiWT (T and U) fibroblasts subjected to the BrdU replication-timing assay and recorded at a gain where all but five autosomes display BrdU incorporation. (V–X) Boxplots representing the quantitation of the proportion of area of the Xa or Xi displaying BrdU signal from the mitotic spreads of 17 XaWTXiWT and 12 XaXist-Δ21XiWT transformed cells at a gain where five autosomes display BrdU signal and the BrdU signal in the Xa was quantitated (V) and at a gain where all but five autosomes display BrdU signal and the signal in the Xa (W) or the Xi (X) was quantitated. In V and W, the median of each set of measurements is indicated to the right of the boxplots. In V–X, P-values represent the significance of the difference between pairs of boxplots and were obtained using the Kruskal-Wallis test.
Figure 2.

Late S-phase BrdU incorporation into the X chromosomes in primary mouse embryonic fibroblasts. Mitotic chromosome spreads were prepared from exponentially growing cultures of primary mouse embryonic fibroblasts exposed to BrdU for 4.5 hr prior to immunostaining. Each spread is from a different embryo and each spread was subjected to X chromosome paint and fluorescent immunostaining using antibodies that recognize BrdU. Within each spread, the active X chromosome is always assumed to be the X chromosome displaying the lower level BrdU incorporation. (A–G) Data obtained from mitotic chromosome spreads of primary fibroblasts where the gain is adjusted such that, for each spread, all but five autosomes display BrdU signal; XaXist-WTXiXist-WT spread displaying X chromosome paint (A) and BrdU incorporation (B); XaXist-Δ21XiXist-WT spread displaying X chromosome paint (C) and BrdU incorporation (D); quantitation of BrdU signal using NIH Image from 120 XaXist-WTXiXist-WT and 120 XaXist-Δ21XiXist-WT spreads from six embryos (40 spreads from each embryo) summarized in boxplots that display the percentage of BrdU signal for the Xa (E), the percentage of BrdU signal multiplied by the average intensity of the BrdU signal for the Xa (F), and the percentage of BrdU signal for the Xi (G). (H–R) Data obtained from mitotic chromosome spreads where the gain is adjusted such that, for each spread, five autosomes display BrdU signal and one autosome displays one pixel of BrdU signal; XaXist-WTXiXist-WT spread displaying X chromosome paint (H) and BrdU incorporation (I); XaXist-WTXiXist-WT spread displaying X chromosome paint (J) and BrdU incorporation (K); XaXist-Δ21XiXist-WT spread displaying X chromosome paint (L) and BrdU incorporation (M); XaXist-Δ21XiXist-WT spread displaying X chromosome paint (N) and BrdU incorporation (O); quantitation of BrdU signal between 120 XaXist-Δ21 cells and 120 XaXist-Δ21 cells (40 spreads from each of six embryos) using boxplots that display the percentage of BrdU signal for the Xa (P), the percentage of BrdU signal multiplied by average intensity of the BrdU signal for the Xa (Q), and the percentage of BrdU signal for the Xi (R). P-values measure the significance of differences in measurements across the two categorical groupings using the Kruskal-Wallis test.
Figure 3.

BrdU signal observed in mitotic chromosome spreads from exponentially growing cultures of lymphocytes from adult mice. Following exposure of cultures to BrdU for 4.5 hr, mitotic chromosome spreads were prepared and immunostained using anti-BrdU (red), hybridized to X chromosome paint (green), and counterstained using DAPI (blue). Images were analyzed with the gain adjusted such that, for each spread, five autosomes display at least one pixel of BrdU signal (“low gain”). Each spread is from a different embryo. XaXist-WTXiXist-WT spread displaying X chromosome paint (A) and BrdU incorporation (B); XaXist-WTXiXist-WT spread displaying X chromosome paint (C) and BrdU incorporation (D); XaXist-Δ21XiXist-WT spread displaying X chromosome paint (E) and BrdU incorporation (F); XaXist-Δ21XiXist-WT spread displaying X chromosome paint (G) and BrdU incorporation (H); boxplots comparing the amount of BrdU signal between 80 XaXist-Δ21 cells and 80 XaXist-WT cells (40 mitotic chromosome spreads from each embryo) when considering the proportion of the area of the active X chromosomes occupied by BrdU (I), the area of the Xa occupied by BrdU signal multiplied by the intensity of the BrdU signal (J), and the area of the Xi occupied by BrdU (K). P-values measure the significance of differences in measurements across categorical groupings using the Kruskal-Wallis test.
Figure 4.

BrdU signal observed in mitotic chromosome spreads from exponentially growing cultures of primary embryonic XO and XY fibroblasts. Following exposure of cultures to BrdU for 4.5 hr, mitotic chromosome spreads were prepared and immunostained using anti-BrdU (red), hybridized to X chromosome paint (green), and counterstained using DAPI (blue). Each spread is from a different embryo. XXist-WTO spread displaying X chromosome paint (A) and BrdU incorporation (B); XXist-Δ21O spread displaying X chromosome paint (C) and BrdU incorporation (D); boxplots of the data from 40 XXist-WTO and 40 XXist-Δ21O spreads (E); XXist-WTY spread displaying X chromosome paint (F) and BrdU incorporation (G); XXist-Δ21Y spread displaying X chromosome paint (H) and BrdU incorporation (I); XXist-Δ21Y spread displaying X chromosome paint (J) and BrdU incorporation (K); boxplots comparing the proportion of the area of the active X chromosomes occupied by BrdU signal between 80 XXist-WTY cells and 80 XXist-Δ21Y cells (L) (40 mitotic chromosome spreads from each of four embryos). The P-value measures the significance of differences in measurements across categorical groupings using the Kruskal-Wallis test.
RESULTS
Altered replication times in transformed fibroblast line with 21 kb excised from the active X chromosome:
DNA replication timing was examined in transformed mouse fibroblasts by pulse labeling growing cultures of wild-type female fibroblasts for 4.5 hr with BrdU and thereafter detecting the incorporated BrdU by fluorescent immunostaining. Cells that were in early or mid S phase at the beginning of the pulse failed to reach mitosis in 4.5 hr and were expected to display a decondensed nucleus with high levels of BrdU incorporation similar to the image in Figure 1, A and B. However, cells in late S phase at the onset of the 4.5-hr incubation period reached mitosis and had their incorporated BrdU represented in mitotic chromosome spreads (Figure 1, B and C). Consequently, only BrdU incorporation in late S phase was detected in mitotic chromosome spreads using this procedure. The X chromosomes were identified using X chromosome paint (Cambio) (Figure 1D). This revealed that the latest replicating portions of the genome were disproportionately on one of the two X chromosomes in diploid cells (Figure 1, C and D). In female human cells the Xi is replicated considerably later in S phase than the Xa (Priest et al. 1967). In female mouse cells, the Xi is not replicated as late in S phase as it is in human cells (Evans et al. 1965; Galton and Holt 1965; Tiepolo et al. 1967). This has led to the murine Xi being distinguished by its absence of label incorporation in early S phase rather than by its being disproportionately replicated late in S phase (Nesbitt and Gartler 1970). Although there was always a consistent trend of the Xi displaying more BrdU incorporation late in S phase than the other chromosomes in both primary and transformed fibroblasts, the amount of incorporated BrdU observed on both X chromosomes varied somewhat among mitotic chromosome spreads when using this assay (not shown). One source of variability is that the mitotic cells were not all in the same position in S phase at the time of BrdU addition. A possible second source of variability is that the X chromosomes may not be replicated at the exact same time in every S phase but may be replicated in a distribution of times. Third, the number of fluorescently labeled antibody molecules binding a given amount of incorporated BrdU may not be perfectly even throughout a preparation of mitotic chromosome spreads. This potential source of variability can be reduced by carefully adjusting the setting of the gain used to obtain each image and by using the autosomes as a control.
The amount of fluorescent signal representing incorporated BrdU is dependent on the setting of the gain used to obtain the image. As mentioned in the Introduction, the X chromosomes are likely replicated by >1000 replication units and, at the low resolution afforded by mitotic chromosome spreads, differences in BrdU signal along the chromosome likely reflect differences in the concentration of late replicating units (Figure 1E, bottom). At low gain, only the most intense fluorescent signal emanating from the chromosome is recorded; this represents the regions on the X chromosome with the greatest amounts of BrdU incorporation and, most likely, the regions of the chromosome with the highest concentration of late replication units (Figure 1E). At intermediate gain, intermediate densities of BrdU incorporation also are recorded. At high gain, smaller amounts of fluorescent signal are also recorded, which means that BrdU incorporation will be detected in chromosomal regions where late replicating units are too few in number to be detected at intermediate or low gain (Figure 1E). Other possible sources of BrdU signal in late S phase regions are elongating replication forks derived from early activated origins and possibly even scatter from neighboring fluorescence because of optical resolution issues. Another possibility is that the anti-BrdU antibody has uneven access to the BrdU-labeled DNA because of differences in chromatin structure or other variables. In primary mouse embryonic fibroblasts subjected to the aforementioned replication-timing assay and photographed at a relatively high gain, a considerable amount of BrdU incorporation was detected on both X chromosomes and most autosomes displayed BrdU incorporation late in S phase (Figure 1, F and G). In a photograph taken at a lower gain, BrdU incorporation could be detected in only roughly half of the chromosomes (Figure 1H). At a still lower gain, BrdU was detectable on the later-replicating X chromosomes and on four autosomes (Figure 1I). To reduce unwanted variation in BrdU signal among samples, all photographs were taken at “high gain” such that, in each photograph, BrdU signal is visible in all but five autosomes. In addition, all mitotic chromosome spreads were recorded at “low gain” such that, in each photograph, BrdU signal is visible in only five autosomes and only one pixel of BrdU is detected in one of these five autosomes. Note that even at disparate settings, the same X chromosome displays a greater amount of BrdU incorporation than the other X chromosome (compare Figure 1, G and I). Thus at either high or low gain the X chromosome with the greater amount of BrdU incorporation can be assumed to be the inactive one.
To determine whether the Xist locus on the Xa influences chromosomal replication timing, we examined a TAg-transformed fibroblast cell line derived from mouse embryos containing a Xist allele in which 21 kb of the gene was flanked by Lox sites (Csankovszki et al. 1999). The 21-kb Xist sequence was excised using cre recombinase and clonal cell lines carrying the resulting Xist-Δ21 allele on the Xa were established. Mitotic chromosome spreads prepared from one such cell line and from a wild-type fibroblast line were subjected to X chromosome paint to distinguish the X chromosomes from autosomes. The level of BrdU incorporation in the Xa appeared to be higher in mutant than in wild-type cells and rivaled the level of incorporation observed in the Xi across various gain settings (Figure 1, J–M). The levels of BrdU incorporation were compared at a low gain where BrdU signal is visible in only five autosomes. Using this methodology, little or no BrdU incorporation was detected on the earlier-replicating (active) X chromosome from XaXist-WTXiXist-WT cells (Figure 1, N and O, 17 diploid mitotic chromosome spreads examined). In contrast, BrdU signal was observed on the earlier-replicating X chromosome from XaXist-Δ21XiWTcells (Figure 1, P and Q, 12 diploid mitotic chromosome spreads examined). The level of BrdU incorporation in each X chromosome was quantitated using NIH Image software and signals between the 17 XaXist-WTXiXist-WT and 12 XaXist-Δ21XiXist-WT mitotic chromosome spreads were compared using boxplots. This revealed that significantly higher levels of BrdU incorporation were detected on the active X chromosome in the XaXist-Δ21XiXist-WT mitotic chromosome spreads than in the XaXist-WTXiXist-WT spreads (Figure 1V) while little or no significant difference was seen for the Xi in the same spreads (P = 0.0344) (not shown). In the same mitotic chromosome spreads, the levels of BrdU signal were also compared at high gain where the fluorescent signal representing BrdU incorporation was visible in all but five autosomes. At the high gain, the Xa also displayed significantly higher levels of BrdU signal in XaXist-Δ21XiXist-WT (Figure 1, T and U) than in XaXist-WTXiXist-WT chromosome spreads (Figure 1, R and S). At the high gain, the BrdU signal was quantitated using NIH Image software, revealing a significantly higher level of BrdU incorporation on the Xa in XaXist-Δ21XiXist-WT spreads (Figure 1W), while little or no significant difference was seen in the level of BrdU incorporation on Xi in these same spreads (P = 0.48) (Figure 1X). This suggested that the 21-bp region of the Xa may be needed to maintain earlier replication of the Xa.
Altered replication timing in primary female fibroblasts:
The higher levels of BrdU incorporation in the Xa in a XaXist-Δ21XiXist-WT cell line than in a XaXist-WTXiXist-WT cell line raised the issue of whether this difference in replication timing was caused by the 21-kb deletion on the Xa or by changes that occurred during the transformation of the two cell lines that were independent of the deletion. To distinguish between these two possibilities, primary fibroblasts were obtained from three XaXist-Δ21XiXist-WT and three XaXist-WTXiXist-WT embryos. Inheritance of the Xist-Δ21kb allele results in nonrandom X-inactivation with the wild-type X chromosome being the inactive X chromosome (XiWT) and the mutant X chromosome being the active one (XaXist-Δ21kb) in all cells (not shown) and in this regard resembles the nonrandom X-inactivation that had previously been reported for embryos that had inherited a 16-kb deletion in the Xist gene (Marahrens et al. 1998). The six primary fibroblast lines were subjected to the aforementioned replication-timing assay. At a high gain setting where all but five autosomes displayed BrdU signal, the primary XaXist-WTXiXist-WT mitotic chromosome spreads displayed less BrdU signal on the Xa (Figure 2, A and B) than did the primary XaXist-Δ21XiXist-WT spreads (Figure 2, C and D). At these settings, the signal representing BrdU incorporation was quantitated from 40 mitotic chromosome spreads from each of three primary XaXist-WTXiXist-WT cell lines (120 mitotic chromosome spreads total) and three primary XaXist-Δ21XiXist-WT fibroblast cell lines (120 mitotic chromosome spreads total) and the medians and distributions were compared using boxplots. A significantly larger proportion of the area of the Xa displayed BrdU incorporation in XaXist-Δ21XiXist-WT than in XaXist-WTXiXist-WT primary fibroblasts (Figure 2E). When, for each individual Xa, the area displaying BrdU signal was multiplied by the average intensity of BrdU signal, the significance of the difference between the XaXist-Δ21 and the XaXist-WT in the primary fibroblasts increased (Figure 2F). In contrast, there was no significant difference in the level of BrdU signal on the XiXist-WT in the same mitotic chromosome spreads (P = 0.32) (Figure 2G). Fluorescent signal representing incorporated BrdU was also quantitated from the same 240 mitotic chromosome spreads at a low gain setting where only five autosomes display signal. Under these conditions, incorporated BrdU was undetectable on the Xa in most chromosome spreads from XaXist-WTXiXist-WT cells (Figure 2, H–K) while all mitotic chromosome spreads from XaXist-Δ21XiXist-WT cells displayed BrdU signal on the Xa (Figure 2, L–O). The increase in BrdU signal on the Xa in primary XaXist-Δ21XiXist-WT over XaXist-WTXiXist-WT cells was even more significant at these settings: compare P = 1.40 × 10−16 (Figure 2P) and P = 2.24 × 10−15 (Figure 2Q) at low gain to P = 1.09 × 10−7 (Figure 2E) and P = 1.29 × 10−10 (Figure 2F) at high gain. A slight difference in BrdU signal on the Xi between the XaXist-Δ21XiXist-WT and the XaXist-WTXiXist-WT chromosome spreads was detected at low gain settings (Figure 2R); however, this difference disappeared when BrdU area × intensity was considered (P = 0.0920) (not shown). At both high and low gain our results showed that change in replication timing of the Xa in relation to the rest of the genome was caused by the 21-kb deletion. Because in primary fibroblasts the differences between the XaXist-Δ21 and the XaXist-WT were most striking at a lower gain where BrdU incorporation can be detected on only five autosomes, all subsequent measurements were performed using only this approach.
Shift in replication timing is not unique to fibroblasts:
We next set out to determine whether the increase in BrdU incorporation on the Xa in response to the Xist-Δ21kb allele was unique to embryonic fibroblasts or also occurred in other cell types. To this end, activated lymphocytes were obtained from the spleens of two XaXist-Δ21XiXist-WT mice and from two age-matched XaXist-WTXiXist-WT 129 mice. The four lymphocyte preparations were subjected to the BrdU replication-timing assay and 80 mitotic chromosome spreads were photographed from each preparation at a gain where five autosomes display BrdU signal. The results for the activated lymphocytes closely resembled the earlier results for primary fibroblasts: significantly lower levels of incorporated BrdU were observed on the Xa in mitotic chromosome spreads from activated XaXist-WTXiXist-WT lymphocytes (Figure 3, A–D, I–J) than from activated XaXist-Δ21XiXist-WT lymphocytes (Figure 3, E–J) while measurements on the Xi in the same spreads were not significantly different (P = 0.61) (Figure 3K). Thus, the relatively late replication of the Xa in XaXist-Δ21XiXist-WT cells was not unique to fibroblasts.
The finding that the XaXist-Δ21 displays more BrdU incorporation than the XaXist-WT in an assay for late replication raised the possibility that the 21-kb deletion causes S phase and/or G2 phase to be compressed. If the S + G2 phase interval is compressed, then a low proportion of an asynchronous culture will be in these phases at any given time and the proportion of cells labeled by a pulse of BrdU will be lower. One hundred XaXist-Δ21XiXist-WT and 100 XaXist-WTXiXist-WT nonmitotic fibroblasts were scored for presence or absence of BrdU. No significant difference in the proportion of BrdU-containing cells was observed using Fisher's exact test (P = 0.45). One hundred XaXist-Δ21XiXist-WT and 100 XaXist-WTXiXist-WT nonmitotic lymphocytes also did not produce a significant difference (P = 0.75).
The deletion at the Xist locus alters replication timing in cells with one X chromosome:
The finding that a deletion in the Xist locus on the Xa alters chromosomal replication timing raised the issue of whether a Xi was needed to be present in the cell to achieve this effect. The BrdU incorporation assay for late replication timing was performed on primary fibroblasts from a XXist-WTO (Figure 4, A and B) and a XXist-Δ21O (Figure 4, C and D) mouse (40 mitotic chromosome spreads from each). The X chromosome was found to display significantly more BrdU signal in the XXist-Δ21O cells than in the XXist-WTO cells (Figure 4E). The assay was also performed on mitotic chromosome spreads from primary fibroblasts from two XXist-WTY (Figure 4, F and G) and two XXist-Δ21Y (Figure 4, H–K) embryos (40 spreads from each embryo). The XY chromosome spreads displayed significantly higher levels of BrdU signal on the X chromosome when in the presence of the Xist-Δ21 allele (Figure 4L).
DISCUSSION
We show that the active X chromosome displayed increased BrdU incorporation in an assay for late replication when 21 kb was deleted from the Xist locus of the Xa. This effect was observed in transformed fibroblasts, primary embryonic fibroblasts, and in activated lymphocytes. The presence of the 21-kb deletion also led to increased levels of BrdU incorporation in the late replication assay in male cells and in female XO cells. The element(s) residing at the Xist locus on the Xa therefore influences the timing at which regions on the Xa are replicated relative to the rest of the genome. The element(s) influences replication time independently of the presence of an inactive X chromosome. Regions located far away from the Xist locus were affected by the 21-kb deletion. For example, one or more regions located very close to the centromere, and therefore >90 Mb away from the Xist locus, which is 98 Mb distal to the centromere, frequently displayed a BrdU signal at low gain in XaXist-Δ21kbXiWT cells (Figure 1, M and Q; Figure 2O; Figure 3H; Figure 4D) but not at low gain in XaWTXiWT cells (Figure 1, I and O; Figure 2, I and K; Figure 3, B and C; Figure 4, B and G).
The replication-timing data are most easily explained by a cis-acting model:
Two ways in which our results can be interpreted are as follows. First, a cis-acting element at the Xist locus of the Xa may influence the replication timing of the Xa. A second explanation is that an element at the Xist locus on the Xa controls the time that it takes a cell to traverse S phase and/or G2 phase of the cell cycle. In the absence of this element, S phase and/or G2 phase are compressed. However, compression of S and/or G2 phase would be expected to result in an increase in BrdU incorporation on the Xi. Since no significant difference in BrdU incorporation was observed on the Xi between XaXist-Δ21XiXist-WT and XaXist-WTXiXist-WT cells, our data are more consistent with cis-regulation of replication timing on the Xa. Triplet-repeat expansions at the fragile X locus on the Xa in humans have previously been shown to influence replication time over megabase distances (Hansen et al. 1997) and long-distance control of replication timing has also been reported on autosomes (Forrester et al. 1990; Cimbora et al. 2000; Singh et al. 2003; Ensminger and Chess 2004). Since the Xist locus of the Xa is not transcribed, and there is precedent for long-distance cis influence of replication timing by a nontranscribed locus (Hansen et al. 1997), we consider cis-regulation of the Xa replication timing an attractive explanation for our results. It was previously proposed that LINE-1 elements play a role in X-inactivation (Lyon 1998; Marahrens 1999; Bailey et al. 2000; Hansen 2003) and we previously proposed that the Xist locus controls the chromatin structure of the Xi via physical looping interactions with LINE elements (Marahrens 1999). We speculate that elements at the nontranscribed Xist locus of the Xa control replication at distant sites on the Xa by similar looping interactions with LINE-1 elements and other repetitive sequences.
Since late replication times are associated with gene repression, our findings raise the question of whether gene expression is affected by the later replication of the XaXist-Δ21. The altered gene expression levels that may occur on the XaXist-Δ21 are presumably not very severe since XaXist-Δ21XiXist-WT mice are viable. We are currently using gene expression arrays to search for gene expression changes on the XaXist-Δ21.
Acknowledgments
We thank Sui Huang and Chen Wang for valuable technical help. Adenovirus AdCre (Tan et al. 1999) and adenovirus expressing GFP were kindly provided by Carol Eng and Arnold J. Berk. We thank Jennifer Salstrom and Elaine Wong for generating some of the primary fibroblasts and Tao Shi for performing some statistical analyses. This work was supported by March of Dimes Basil O'Connor Starter Scholar research award 5-FY99-819 (Y.M.) and National Institutes of Health grant R01 HD41451:01 (Y.M.).
References
- Bailey, J. A., L. Carrel, A. Chakravarti and E. E. Eichler, 2000. Molecular evidence for a relationship between LINE-1 elements and X chromosome inactivation: the Lyon repeat hypothesis. Proc. Natl. Acad. Sci. USA 97: 6634–6639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brockdorff, N., A. Ashworth, G. F. Kay, P. Cooper, S. Smith et al., 1991. Conservation of position and exclusive expression of mouse Xist from the inactive X chromosome. Nature 351: 329–331. [DOI] [PubMed] [Google Scholar]
- Brown, C. J., A. Ballabio, J. L. Rupert, R. G. Lafreniere, M. Grompe et al., 1991. A gene from the region of the human X inactivation centre is expressed exclusively from the inactive X chromosome. Nature 349: 38–44. [DOI] [PubMed] [Google Scholar]
- Brown, C. J., B. D. Hendrich, J. L. Rupert, R. G. Lafreniere, Y. Xing et al., 1992. The human XIST gene: analysis of a 17 kb inactive X-specific RNA that contains conserved repeats and is highly localized within the nucleus. Cell 71: 527–542. [DOI] [PubMed] [Google Scholar]
- Cimbora, D. M., D. Schubeler, A. Reik, J. Hamilton, C. Francastel et al., 2000. Long distance control of origin choice and replication timing in the human b-globin locus are independent of the locus control region. Mol. Cell. Biol. 20: 5581–5591. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clemson, C. M., J. A. McNeil, H. F. Willard and J. B. Lawrence, 1996. XIST RNA paints the inactive X chromosome at interphase: evidence for a novel RNA involved in nuclear/chromosome structure. J. Cell Biol. 132: 259–275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cohen, S. M., B. P. Brylawski, M. Cordeiro-Stone and D. G. Kaufman, 2003. Same origins of DNA replication function on the active and inactive human X chromosomes. J. Cell. Biochem. 88: 923–931. [DOI] [PubMed] [Google Scholar]
- Csankovszki, G., B. Panning, B. Bates, J. R. Pehrson and R. Jaenisch, 1999. Conditional deletion of Xist disrupts histone macroH2A localization but not maintenance of X inactivation. Nat. Genet. 22: 323–324. [DOI] [PubMed] [Google Scholar]
- Csankovszki, G., A. Nagy and R. Jaenisch, 2001. Synergism of Xist RNA, DNA methylation, and histone hypoacetylation in maintaining X chromosome inactivation. J. Cell Biol. 153: 773–784. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ensminger, A. W., and A. Chess, 2004. Coordinated replication timing of monoallelically expressed genes along human autosomes. Hum. Mol. Genet. 13: 651–658. [DOI] [PubMed] [Google Scholar]
- Evans, H. J., C. E. Ford, M. F. Lyon and J. Gray, 1965. DNA replication and genetic expression in female mice with morphologically distinguishable X chromosomes. Nature 206: 900–903. [DOI] [PubMed] [Google Scholar]
- Forrester, W. C., E. Epner, M. C. Driscoll, T. Enver, M. Brice et al., 1990. A deletion of the human b-globin locus activation region causes a major alteration in chromatin structure and replication across the entire b-globin locus. Genes Dev. 4: 1637–1649. [DOI] [PubMed] [Google Scholar]
- Galton, M., and S. F. Holt, 1965. Asynchronous replication of the mouse sex chromosomes. Exp. Cell Res. 37: 111–116. [DOI] [PubMed] [Google Scholar]
- Gartler, S. M., and A. D. Riggs, 1983. Mammalian X-chromosome inactivation. Annu. Rev. Genet. 17: 155–190. [DOI] [PubMed] [Google Scholar]
- Gilbert, D. M., 2002. Replication timing and transcriptional control: beyond cause and effect. Curr. Opin. Cell Biol. 14: 377–383. [DOI] [PubMed] [Google Scholar]
- Gomez, M., and N. Brockdorff, 2004. Heterochromatin on the inactive X chromosome delays replication timing without affecting origin usage. Proc. Natl. Acad. Sci. USA 101: 6923–6928. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hadjantonakis, A. K., M. Gertsenstein, M. Ikawa, M. Okabe and A. Nagy, 1998. Non-invasive sexing of pre-implantation mammalian embryos. Nat. Genet. 19: 220–222. [DOI] [PubMed] [Google Scholar]
- Hansen, R. S., 2003. X inactivation-specific methylation of LINE-1 elements by DNMT3B: implications for the Lyon repeat hypothesis. Hum. Mol. Genet. 12: 2559–2567. [DOI] [PubMed] [Google Scholar]
- Hansen, R. S., T. K. Canfield, A. D. Fjeld, S. Mumm, C. D. Laird et al., 1997. A variable domain of delayed replication in FRAXA fragile X chromosomes: X inactivation-like spread of late replication. Proc. Natl. Acad. Sci. USA 94: 4587–4592. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huberman, J. A., and A. D. Riggs, 1968. On the mechanism of DNA replication in mammalian chromosomes. J. Mol. Biol. 32: 327–341. [DOI] [PubMed] [Google Scholar]
- Ihaka, R., and R. Gentleman, 1996. R: a language for data analysis and graphics. J. Comput. Graph. Stat. 5: 299–314. [Google Scholar]
- Jat, P. S., C. L. Cepko, R. C. Mulligan and P. A. Sharp, 1986. Recombinant retroviruses encoding simian virus 40 large T antigen and polyomavirus large and middle T antigens. Mol. Cell. Biol. 6: 1204–1217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kruskal, J. B., 1964. Multidimensional scaling by optimizing goodness of fit to a nonmetric hypothesis. Psychometrika 29: 1–27. [Google Scholar]
- Lander, E. S., L. M. Linton, B. Birren, C. Nusbaum, M. C. Zody et al., 2001. Initial sequencing and analysis of the human genome. Nature 409: 860–921. [DOI] [PubMed] [Google Scholar]
- Lyon, M. F., 1998. X-chromosome inactivation: a repeat hypothesis. Cytogenet. Cell Genet. 80: 133–137. [DOI] [PubMed] [Google Scholar]
- Marahrens, Y., 1999. X-inactivation by chromosomal pairing events. Genes Dev. 13: 2624–2632. [DOI] [PubMed] [Google Scholar]
- Marahrens, Y., B. Panning, J. Dausman, W. Strauss and R. Jaenisch, 1997. Xist-deficient mice are defective in dosage compensation but not spermatogenesis. Genes Dev. 11: 156–166. [DOI] [PubMed] [Google Scholar]
- Marahrens, Y., J. Loring and R. Jaenisch, 1998. Role of the Xist gene in X chromosome choosing. Cell 92: 657–664. [DOI] [PubMed] [Google Scholar]
- Nesbitt, M. N., and S. M. Gartler, 1970. Replication of the mouse sex chromosomes early in the S period. Cytogenetics 9: 212–221. [DOI] [PubMed] [Google Scholar]
- Penny, G. D., G. F. Kay, S. A. Sheardown, S. Rastan and N. Brockdorff, 1996. Requirement for Xist in X chromosome inactivation. Nature 379: 131–137. [DOI] [PubMed] [Google Scholar]
- Priest, J. H., J. E. Heady and R. E. Priest, 1967. Delayed onset of replication of human X chromosomes. J. Cell Biol. 35: 483–487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Singh, N., F. A. Ebrahimi, A. A. Gimelbrant, A. W. Ensminger, M. R. Tackett et al., 2003. Coordination of the random asynchronous replication of autosomal loci. Nat. Genet. 33: 339–341. [DOI] [PubMed] [Google Scholar]
- Spector, D., and R. Goldman, 1998. In situ hybridization to RNA, pp. 114–117 in Cells: A Laboratory Manual, Vol. 2, edited by D. Spector and R. Goldman. Cold Spring Harbor Laboratory Press, Cold Spring Harbor, NY.
- Tan, B. T., L. Wu and A. J. Berk, 1999. An adenovirus-Epstein-Barr virus hybrid vector that stably transforms cultured cells with high efficiency. J. Virol. 73: 7582–7589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taylor, J. H., 1968. Rates of chain growth and units of replication in DNA of mammalian chromosomes. J. Mol. Biol. 31: 579–594. [DOI] [PubMed] [Google Scholar]
- Taylor, J. H., and P. Miner, 1968. Units of DNA replication in mammalian chromosomes. Cancer Res. 9: 1810–1814. [PubMed] [Google Scholar]
- Tiepolo, L., M. Fraccaro, M. Hulten, J. Lindsten, A. Mannini et al., 1967. Timing of sex chromosome replication in somatic and germ-line cells of the mouse and the rat. Cytogenetics 6: 51–66. [DOI] [PubMed] [Google Scholar]
- Todorovic, V., A. Falaschi and M. Giacca, 1999. Replication origins of mammalian chromosomes: the happy few. Front. Biosci. 4: D859–D868. [DOI] [PubMed] [Google Scholar]
- Waterston, R. H., K. Lindblad-Toh, E. Birney, J. Rogers, J. F. Abril et al., 2002. Initial sequencing and comparative analysis of the mouse genome. Nature 420: 520–562. [DOI] [PubMed] [Google Scholar]