

Abstract
Association of seed dormancy with shattering, awn, and black hull and red pericarp colors enhances survival of wild and weedy species, but challenges the use of dormancy genes in breeding varieties resistant to preharvest sprouting. A phenotypic selection and recurrent backcrossing technique was used to introduce dormancy genes from a wild-like weedy rice to a breeding line to determine their effects and linkage with the other traits. Five generations of phenotypic selection alone for low germination extremes simultaneously retained dormancy alleles at five independent QTL, including qSD12 (R2 > 50%), as determined by genome-wide scanning for their main and/or epistatic effects in two BC4F2 populations. Four dormancy loci with moderate to small effects colocated with QTL/genes for one to three of the associated traits. Multilocus response to the selection suggests that these dormancy genes are cumulative in effect, as well as networked by epistases, and that the network may have played a “sheltering” role in maintaining intact adaptive haplotypes during the evolution of weeds. Tight linkage may prevent the dormancy genes from being used in breeding programs. The major effect of qSD12 makes it an ideal target for map-based cloning and the best candidate for imparting resistance to preharvest sprouting.
WEEDS, plants adapted to human disturbances (Harlan and de Wet 1965), compete with crops for environmental resources and occasionally interbreed with conspecific cultivars (Ladizinsky 1985; Langevin et al. 1990). Understanding the genetic basis underlying adaptation is crucial to devising integrated strategies for weed control or to preventing the rise of “superweeds,” that is, weeds with transgenes derived from transgenic crops (Gressel 1999; Basu et al. 2004; Lègére 2005). Conversely, to meet breeding objectives for sustainable agriculture, conspecific weeds, as part of the primary gene pool (Harlan et al. 1973), are valuable to regain some genetic diversity that was eliminated during domestication (Tanksley and McCouch 1997). Weedy rice, including red rice, accompanies cultivated rice worldwide (Oka 1988). Research on weedy rice has been done for its origin, classification, control, and risk assessment related to transgenic cultivars (Suh et al. 1997; Tang and Morishima 1997; Baker et al. 2000; Vaughan et al. 2001; Gealy et al. 2003; Chen et al. 2004), but rarely on the genetics of adaptive traits (Bres-Patry et al. 2001) or for identification of beneficial genes for rice improvement (Ahn et al. 2002).
Seed dormancy optimizes timing of germination for wild and weedy plants and provides resistance to preharvest sprouting (PHS) for cereal crops (Bewley and Black 1982). Both dormancy and PHS are complex traits controlled by many genes (Chang and Tagumpay 1973) or quantitative trait loci (QTL) such as in Arabidopsis (Alonso-Blanco et al. 2003), barley (Oberthur et al. 1995; Li et al. 2003; Prada et al. 2004; Zhang et al. 2005), rice (Lin et al. 1998; Cai and Morishima 2000; Dong et al. 2003; Gu et al. 2004), sorghum (Lijavetzky et al. 2000), and wheat (Anderson et al. 1993; Kato et al. 2001; Mares and Mrva 2001; Groos et al. 2002; Osa et al. 2003; Kulwal et al. 2004). Dormancy alleles at a few QTL have been introduced into the nondormant genetic background to validate their effects or to explore their potential in breeding programs (Han et al. 1999; Gao et al. 2003; Takeuchi et al. 2003). Validated QTL may be cloned to characterize molecular mechanisms directly regulating germination and dormancy (Koornneef et al. 2002).
Utilization of dormancy genes from wild and weedy germplasm to control PHS in cereal crops may be hampered by linkage with some traits that may have an adaptive value under natural conditions but are undesirable for modern cultivars. Dormancy association with red grain color in wheat (Nilsson-Ehle 1914) has prevented the use of these dormancy genes in the development of white grain-colored cultivars with resistance to PHS (Flintham 2000). Dormancy is also associated with seed appendages, or shattering, and black pigmentation in other grass species (Johnson 1935; Oka 1988; Simpson 1992; Khan et al. 1996; Gu et al. 2005a). Some of the associations in wild (Oryza rufipogan) and weedy (O. sativa) rice are explained by QTL clustered on the same chromosomal blocks (Cai and Morishima 2000; Thomson et al. 2003; Gu et al. 2005b). Introduction of the QTL regions into breeding lines, i.e., nondormant pure lines, enables a precise assessment of the linkage strengths, and it is the first step in characterizing the structure of dormancy gene-related linkage disequilibrium and in understanding the evolutionary history of the adaptive haplotypes across grass genomes.
Phenotypic selection with recurrent backcrossing was proposed to isolate genes with a major effect on a quantitative trait (Wright 1952) and is now combined with QTL analysis to simultaneously discover and transfer useful alleles from nondomesticated germplasm into breeding lines or intermediate breeding materials (Tanksley and Nelson 1996). Genetic analysis suggested the presence of major dormancy genes in some weedy rice accessions (Gu et al. 2003). Thus, we used a phenotypic selection technique, without assistance with molecular markers or morphological characteristics, to initiate introduction of dormancy genes from a weedy accession into a breeding line. After completion of dormancy QTL mapping (Gu et al. 2004), the linkage map was employed to determine QTL retained by the phenotypic selection over five generations. Five dormancy QTL regions, including four haplotypes for other adaptive traits, were introduced into the background of a breeding line.
MATERIALS AND METHODS
Plant genotypes and mating scheme:
The breeding scheme (Figure 1) was used to introduce dormancy genes from the weedy accession SS18-2 to the EM93-1 background. A rice seed usually refers to a dispersal unit that consists of the maternal (i.e., hull, pericarp, and testa) and offspring (i.e., endosperm and embryo) tissues (Grist 1986). SS18-2 is a wild-like indica-type weedy rice originally collected from Thailand (Suh et al. 1997; Tang and Morishima 1997) and has a high degree of seed dormancy and shattering, long awns, dark pigmentation on the hull, and red pigmentation on the pericarp/testa. EM93-1 and CO39 are indica-type early and moderate maturation lines, respectively, and do not have dormancy, shattering, awn, and black or red pigmentations on the maternal tissues. Selection started with the F2 CO39/SS18-2, the population with the highest estimate of heritability for dormancy in the experiment (Gu et al. 2003). A strongly dormant, early flowering F2 segregate (i.e., plant 14, ∼30 days earlier than the earlier flowering parent CO39) was selected to cross with EM93-1. Individuals having the lowest germination level in the F2-derived F1 to BC3F1 populations were backcrossed with EM93-1 to develop the next generation (Figure 1). EM93-1 was used to replace CO39 as the recurrent parent in the backcross because this breeding line has the duration to flowering similar to that of the above F2 plant 14 under long day lengths and was used to develop the population to map the dormancy QTL (Gu et al. 2004). Hybridizations were made using ratooning plants from the selected individuals in each generation.
Figure 1.

Breeding scheme for introducing dormancy genes from weedy to cultivated rice. SS18-2, CO39, and EM93-1 are a weedy accession, cultivar, and breeding line, respectively. Plants in parentheses were dormant genotypes selected to develop the next generation.
Similar to SS18-2, the low germination extremes selected in early generations also displayed other weedy traits, although awn length shortened, darkness of the hull lightened, and degree of shattering decreased with each generation. In the BC4F1 generation, plants 44 and 132, which had the traits identical to EM93-1, other than those mentioned above, were selected to develop F2 populations, i.e., BC4F2 (44) and BC4F2 (132) (Figure 1). Plant 132 seeds had the lowest germination rate and red pericarp color, and plant 44 seeds had a moderate level of germination and shattering, relatively long awns, black hull color, and red pericarp color. These BC4F2's were used to determine the effects of retained dormancy QTL and linkage with other weedy characteristics.
Plant cultivation and phenotypic identification:
The populations were grown in the greenhouse from 2000 to 2004. Plants were cultivated in pots (28-cm diameter × 25-cm height), with one plant per pot filled with a mixture of clay soil and SUNSHINE medium (Sun Gro Horticulture Canada, Seba Beach, AB, Canada). Day/night temperatures were set at 29°/21° and the day length was set for 14 hr. Seeds were harvested at 40 days after flowering, which was measured by emergence of the first panicle in the plant. Seeds were cleaned and air dried in the greenhouse for 3 days to ∼12% moisture content. Dried seeds were sealed in plastic containers and stored at −20° to prevent them from after-ripening prior to use.
The degree of dormancy was measured by percentage of germination. Prior to germination, seeds sampled from each plant were after-ripened at room temperature (23°–25°) for different periods of time (1–45 days). Three replications of ∼50 seeds each were placed in 9-cm petri dishes that were lined with a Whatman no. 1 filter paper, wetted with 10 ml deionized water, and incubated at 30° and 100% relative humidity in the dark for 7 days. Germination was evaluated visually by ≥3-mm protrusion of the radicle or coleoptile from the hull. Percentage of germination (x) was transformed by sin−1(x)−0.5 for statistical analysis.
Phenotypes for awn, black hull color, and red pericarp color were identified on the basis of the presence or absence of each characteristic from the F2 to BC4F1 generations, as in previous research (Gu et al. 2003); the presence and absence were scored as 1 and 0, respectively, for correlation analysis. There was no difficulty in distinguishing the red pericarp color genotypes from the white ones on the basis of the appearance of fully matured seeds; thus this trait was also scored as the presence and absence in the two BC4F2's. The other weedy traits in the BC4F2 (44) population were quantified by awn length, intensity of component pigmentations, and shattering rate for QTL analysis (Gu et al. 2005b). Briefly, the awn length was expressed as the mean length averaged over three samples of 50 seeds each. Shattering rate was expressed as the percentage of shattered to total air-dried seed weight. To assess shattering, panicles were cut from the plant and gently shaken for ∼20 sec over a container to collect shattered seeds and then hand threshed to collect nonshattered ones. Pigmentation was expressed as spectral reflectance. Reflectance was measured with a Chroma Meter (Minolta CR310). The Chroma Meter decomposes reflectance spectra of hull color into three (i.e., L, a, and b) dimensions, with low L and high a or b positive values indicating a high intensity of black and red or yellow pigmentations, respectively.
QTL analysis:
The selected plants from the F2 to BC3F1 generations (Figure 1) were genotyped with rice microsatellite (RM) markers flanking the six dormancy QTL (Gu et al. 2004) to track the SS18-2-derived alleles. Genomic DNA was prepared from ∼50 seedlings bulked from individual F2 to BC3F1 plants. BC4F1 plants 44 and 132 were genotyped for 140 RM markers evenly distributed over the framework linkage map (Gu et al. 2004) to scan for chromosome (chr) segments from SS18-2. Genomic DNA for the BC4F1 plants was prepared from the leaves. Additional markers were screened (McCouch et al. 2002) and were used to delimit the chr segments. Two populations of BC4F2 plants were genotyped for all the polymorphic markers retained in the BC4F1 plants. Genomic DNA from individual BC4F2 plants was prepared from young leaves. DNA was extracted, the markers were amplified by polymerase chain reaction (PCR), and the products were resolved using methods previously described (Gu et al. 2004). Markers were positioned using MAPMAKER/EXP 3.0 (Lincoln et al. 1992).
One- or two-way ANOVAs were used to detect QTL segregating in BC4F2 populations. These analyses were based on a linear model in which a phenotypic value was partitioned into the mean, genotypic, and error (including random error and the residual effect unexplained by the genotypic effects) components. The one-way ANOVA was performed for all markers retained on each SS18-2-derived segment. The marker that contributed most to the phenotypic variance as compared with the other markers on the same segment was used to estimate its epistasis with the marker on another SS18-2-derived segment. For two-way ANOVA, the genotypic effect in the model for the above one-way ANOVA was further partitioned into main and interaction effects of individual loci. The threshold for a significant main or epistatic effect was set at the 5% probability level. The contributions (R2) of the main or epistatic effects were calculated as the proportion of component type III sum-of-squares (SS) to the corrected total SS. The software WinQTLCart (Wang et al. 2004) was used to infer the relative order for QTL located in the same chr region.
QTL additive (a) and dominance (d) effects were estimated according to Kearsey and Pooni (1996),
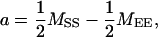 |
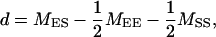 |
where MEE, MSS, and MES are means of the EM93-1-type homozygous, SS18-2-type homozygous, and heterozygous genotypes, respectively, for the marker with the most contribution. Standard errors for the parameters a and d were estimated as
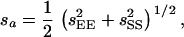 |
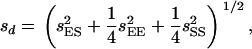 |
where 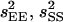 , and
, and 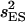 are variances of the means MEE, MSS, and MES, respectively. Significance of a and d estimates was determined by Student's t-test. Epistatic effects were ignored in the above estimation due to limitation of population sizes. The above statistical analyses were implemented using SAS programs (SAS Institute 1999).
are variances of the means MEE, MSS, and MES, respectively. Significance of a and d estimates was determined by Student's t-test. Epistatic effects were ignored in the above estimation due to limitation of population sizes. The above statistical analyses were implemented using SAS programs (SAS Institute 1999).
RESULTS
Responses to phenotypic selection:
Individuals selected from the F2 to BC3F1 populations were dormant genotypes because segregation for germination (0–100%) occurred in the next generation (Figure 2). Segregation also occurred for the traits awn, black hull color, and red pericarp color in all of the above generations. The association between dormancy and red pericarp color was not significant in the F2 plant 14-derived F1 population (Figure 2A), which was similar to that in the F2 CO39/SS18-2 population (Gu et al. 2003), but was significant in the BC1F1 to BC4F1 generations (Figure 2, B–E). There is a dormancy QTL (i.e., qSD7-1) allele linked in coupling with the red pericarp color gene Rc (Gu et al. 2004). Increase in strength of the association with advance of the BC generations (Figure 2, A–E), or with replacement of the CO39 with the EM93-1 genome as a result of backcrossing, confirms the linkage and suggests that the parent CO39 must contain factor(s) elsewhere in the genome affecting expression of this dormancy gene. Phenotypic correlations of dormancy with awn and black hull color also differed by generation (Gu et al. 2003; Figure 2, A–E). The differences might arise from variations in expressivity (i.e., percentage of seeds with an awn or awn length and hull color darkness) of these morphological traits in the populations.
Figure 2.

Distribution of percentage of germination for the five populations (see Figure 1). (A) F1 (EM93-1/F2 no. 14a,b,r) (20 DAR). (B) BC1F1 (EM93-1/F1 no. 38) (10 DAR). (C) BC2F1 (EM93-1/BC1F1 no. 60) (10 DAR). (D) BC3F1 (EM93-1/BC2F1 no. 42) (10 DAR). (E) BC4F1 (EM93-1/BC3F1 no. 81) (7 DAR). The open and solid arrows indicate germination levels for the recipient parent EM93-1 and the plant selected as the parent to develop the next generation, respectively. Superscripts indicate if the selected plant had awn (a), black hull color (b), and/or red pericarp/testa color (r) characteristics. N is the population size, and rr,g, ra,g, and rb,g are correlation coefficients between the characteristics red pericarp color (r), awn (a), and black hull color (b) and the percentage of germination evaluated at the specified days of after-ripening (DAR), respectively; the superscripts indicate the correlations were nonsignificant (ns) or significant at the 0.05 (*), 0.01 (**), and <0.0001 (***) probability levels.
In addition to the qSD7-1 region, selection in early generations also retained dormancy alleles at qSD4, -8, and -12 as indicated by the presence of SS18-2-type alleles at markers flanking the three QTL (Table 1). Plant 14 from the F2 CO39/SS18-2 population was homozygous for CO39-type alleles at two qSD6 flanking markers (Table 1), indicating that the initial selection lost this dormancy allele. Plants selected from the F2 to the BC2F1 generation had the SS18-2-type allele at only one of two markers flanking qSD7-2; we surmise that this dormancy allele could be lost either in the initial selection or in selections before the BC4F1 generation (Table 1).
TABLE 1.
Genotypes of the plants selected in successive generations for markers flanking the putative dormancy QTL
| Genotypes of the plantsb
|
||||||
|---|---|---|---|---|---|---|
| QTLa | Markersa | F2 no. 14 | F2-derived F1 no. 38 | BC1F1 no. 60 | BC2F1 no. 42 | BC3F1 no. 81 |
| qSD4 | RM564A | c/s | e/s | e/s | e/s | e/s |
| RM252 | c/s | e/s | e/s | e/s | e/s | |
| qSD6 | RM549 | c/c | c/e | c/e | e/e | e/e |
| RM527 | c/c* | c/e | e/e | e/e | e/e | |
| qSD7-1 | RM180 | c*/s | e/s | e/s | e/s | e/s |
| RM214 | c/c | c/e | c/e | e/e | e/e | |
| qSD7-2 | RM346 | c/s | e/s | e/s | e/s | e/e |
| RM234 | c/c* | c/e | e/e | e/e | e/e | |
| qSD8 | RM339 | c/s | e/s | e/s | e/s | e/s |
| RM531 | c/s | e/s | e/s | e/s | e/s | |
| qSD12 | RM270 | c*/s | e/s | e/s | e/s | e/s |
| RM235 | c/s | e/s | e/s | e/s | e/s | |
QTL were marked on the basis of the BC1(EM93-1//EM93-1/SS18-2) population (Gu et al. 2004).
Marker alleles for selected plants (Figure 1) are represented by their donor parents CO39 (c), EM93-1 (e), and SS81-2 (s); the asterisk indicates the alleles at this locus are identical between EM93-1 and CO39.
The genome-wide scan detected five SS18-2-derived chr segments for BC4F1 plants 44 and 132 (Figure 3). These plants differed completely in segments on chr 4, 8, and 12, had overlapping segments on chr 7 and 10, and shared a segment on chr 1 as defined by the RM markers in the regions (Figure 3). The length of donor chr segments varying from ∼10 to 40 cM (Figure 3) and the five segments accounted for ∼6.6% of the SS18-2 haploid genome (Gu et al. 2004). The proportion of donor segments is close to the expectation (6.25%) for a BC4 generation. Previous research did not detect dormancy QTL on the chr 1 and 10 segments (Gu et al. 2004); their presence in the BC4F1's suggests they may also harbor dormancy genes or associate with some unidentified factors related to the selection regime.
Figure 3.

Graphic genotypes for the BC4F1 plants 132 (A) and 44 (B). Open or solid bars stand for the EM93-1- or SS18-2-derived chromosomes or chromosomal segments, which were determined by the rice microsatellite (RM) markers at the tick mark positions on the framework linkage map (Gu et al. 2004); the chromosomes not shown were identical to EM93-1. Intermarker distances (centimorgans) and QTL for seed dormancy (qSD), shattering (qSH), awn length (qAL), and hull color (qHC), which were estimated on the basis of the BC4F2 populations, are placed to the left of the segments. Dotted lines indicate a digenic epistasis detected between two QTL.
Dormancy QTL:
The BC4F2 populations differed in distribution patterns for germination (Figure 4). For example, it took the BC4F2 (132) and BC4F2 (44) populations ∼25 and 20 days of after-ripening (DAR), respectively, to display the largest variation in germination and >45 and ∼30 DAR, respectively to reach 80% mean germination (Figure 4). In addition, the BC4F2 (132) and BC4F2 (44) populations displayed bimodal and nearly normal distributions, respectively, when the largest variation occurred. These results suggest that the dormancy alleles differentiated between BC4F1 plants 44 and 132 may also differ in the magnitude of their gene effects.
Figure 4.

Distribution of percentage of germination for the BC4F2 132 (N = 160) (A) and 44 (N = 230) (B) populations. Mean and standard deviation for germination evaluated at 10 (solid circles), 20 or 25 (open columns), and 30 or 45 (open circles) days of after-ripening (DAR) are shown in parentheses.
Dormancy QTL were detected on the chr 1, 7, and 12 segments by their main or epistatic effects from BC4F2 (132). The QTL on chr 7 and 12 coincide with the previously identified qSD7-1 and qSD12, respectively (Gu et al. 2004; Figure 3A). The locus qSD12 accounted for ∼50% of the phenotypic variance and its genetic effect was predominantly additive (Table 2). The locus qSD7-1 accounted for ∼9% of the phenotypic variance and consisted of both gene additive and dominance effects (Table 2). The locus qSD7-1 also was involved in digenic epistases with qSD12 (see Gu et al. 2004) and a locus near marker RM220 on the chr 1 segment; the component two-way interaction effects accounted for 4.6% (P = 0.020, data not shown) and 6.4% (Figure 5A), respectively, of the phenotypic variance. The locus near RM220 was not detected in the primary segregating population (Gu et al. 2004) and is now designated qSD1.
TABLE 2.
Gene effects of dormancy QTL detected at various days of after-ripening (DAR) in the BC4F2 populations
| QTLa | DAR | R2 (%)b | Pb | a ± SEac | d ± SEdc |
|---|---|---|---|---|---|
| BC4F2 (132)d | |||||
| qSD7-1 (RM5672) | 10 | 9.0 | 0.0007 | −0.067 ± 0.033* | −0.124 ± 0.043** |
| 25 | 10.2 | 0.0003 | −0.088 ± 0.040* | −0.144 ± 0.051** | |
| 45 | 8.4 | 0.0012 | −0.070 ± 0.039, NS | −0.142 ± 0.052** | |
| qSD12 (RM270) | 10 | 48.4 | <0.0001 | −0.261 ± 0.021*** | 0.075 ± 0.031* |
| 25 | 53.7 | <0.0001 | −0.319 ± 0.025*** | 0.054 ± 0.034, NS | |
| 45 | 49.9 | <0.0001 | −0.304 ± 0.025*** | 0.060 ± 0.036, NS | |
| BC4F2 (44)d | |||||
| qSD1 (RM220) | 20 | 7.0 | 0.0066 | −0.083 ± 0.025** | −0.013 ± 0.036, NS |
| 30 | 6.9 | 0.0031 | −0.072 ± 0.022** | 0.014 ± 0.029, NS | |
| qSD4 (RM252) | 10 | 10.9 | <0.0001 | −0.076 ± 0.020*** | −0.068 ± 0.028* |
| 20 | 6.4 | 0.0035 | −0.081 ± 0.032* | −0.051 ± 0.044, NS | |
| qSD7-1 (RM5672) | 10 | 18.0 | <0.0001 | −0.114 ± 0.022*** | −0.115 ± 0.027*** |
| 20 | 10.7 | <0.0001 | −0.102 ± 0.025*** | −0.063 ± 0.032* | |
| 30 | 7.0 | 0.0007 | −0.077 ± 0.021*** | −0.028 ± 0.026, NS | |
NS, not significant; *P = 0.05, **P = 0.01, ***P < 0.001.
The codominant markers in parentheses contributed most to the phenotypic variance.
Proportion of phenotypic variance explained by the QTL and the F-test probability computed by one-way ANOVA.
Gene additive (a) and dominance (d) effects and their standard errors.
Standard deviations for arc-sine-transformed germination at 10, 25, and 45 DAR were 0.28, 0.32, and 0.31, respectively, for the BC4F2 (132) and at 10, 20, and 30 DAR were 0.22, 0.25, and 0.19, respectively, for the BC4F2 (44) populations.
Figure 5.

Digenic epistases detected in the BC4F2 (132) qSD1 × qSD7-1 (A) and (44) qSD1 × qSD8 (B) populations. The dormancy loci qSD1, qSD7-1, and qSD8 are represented by their linked markers RM220, RM5672, and RM531, respectively. Marker genotypes are indicated by combinations of alleles from the parents EM93-1 (E) or SS18-2 (S). Lines with open circles (EE), closed circles (SS), and triangles (ES) stand for the three genotypes for RM220. A circle or triangle and vertical bars indicate the mean and standard error for a digenic genotype in arc-sine-transformed percentage of germination (x) evaluated at 20 or 45 days of after-ripening (DAR). R2 and P-values are the proportion of phenotypic variance accounted for by the component epistatic effect and its F-test probability, respectively.
Dormancy QTL were detected on the chr 1, 4, 7, and 8 segments by their main or epistatic effects from BC4F2 (44). The QTL on chr 4, 7, and 8 coincide with the previously identified qSD4, -7-1, and -8, respectively (Gu et al. 2004; Figure 3B). The locus qSD1 in this population had a relatively small main effect (R2 ≈ 7%). It was also involved in digenic epistases with both qSD7-1 and qSD8, where the component two-way interactions accounted for 4.7% (P = 0.047) (pattern similar to Figure 5A) and 9.1% (Figure 5B) of the phenotypic variance, respectively. Intriguingly, the epistases involving qSD1 displayed different patterns. The difference in degree of dormancy between two homozygous genotypes at qSD1 was greater when the dormancy alleles were present at qSD7-1 or absent at qSD8 (Figure 5).
Haplotypes:
The loci qSD7-1 and Rc colocated in the region between markers RM5672 and RM180 on the basis of the two BC4F2 populations (Figure 3, A and B). The Rc locus, which colocates with markers E10534S (Harushima et al. 1998) or aligns with RM5672 (McCouch et al. 2002) in high-resolution maps, was estimated to be 1.5 cM distal from RM5672 on the basis of 390 individuals from the BC4F2 populations. The dominant gene Rc contributed a few percentiles more to phenotypic variance in germination than the marker RM5672 in both BC4F2 populations. The reason that RM5672 was listed in Table 2 as the marker nearest qSD7-1 is because its codominant nature facilitated estimation of gene additive and dominant effects for the locus.
A range of variation in awn length, shattering rate, and intensity of component pigmentations on the hull occurred in the BC4F2 (44) population (Table 3). The distribution patterns for these traits (data not shown), especially the awn length and pigmentations, were similar to those in the primary segregation population (Gu et al. 2005b). Two QTL for awn length were detected on the chr 4 and 8 segments, respectively (Table 4), and their main and epistatic (17.4%, P < 0.0001) effects together accounted for 78.8% of the phenotypic variance. The same segments also harbored QTL for shattering (Table 4). Two QTL for hull color were detected on the chr 1 and 4 segments, respectively; at both loci the SS18-2- and EM93-1-type alleles increased the intensities of black and yellow/red pigmentations, respectively (Table 4). The QTL for awn, shattering, and hull color on chr 4 and 8 were also detected in the primary segregation population (Gu et al. 2005b), but they link more tightly with dormancy QTL in the BC4F2 (44) population as indicated by common nearest markers. The locus for hull color on chr 1 is designated as qHC1, as it was not detected in the primary segregating population (Gu et al. 2005b). The BC4F2 (132) population also segregated for the qHC1 region, but not for hull color as judged by visual examination; we measured intensities of the three-component pigmentations for 140 BC4F2 (132) plants using the same method, but failed to detect a significant effect of qHC1 (R2 < 0.01, P > 0.9) in the subpopulation. Considering its relatively minor effect on black pigmentation (R2 < 5%), we conclude that qHC1 is a modifier to the major locus qHC4 (R2 = 50%, Table 4).
TABLE 3.
Correlation between the hull color, awn length, and shattering traits and the germination evaluated at 20 days of after-ripening in the BC4F2 (44) population
| Traits | Mean | SD | Range | r |
|---|---|---|---|---|
| Awn length (mm) | 3.2 | 3.8 | 0.0–18.8 | −0.2902*** |
| Shattering (%) | 53 | 23 | 0–100 | −0.3816*** |
| Pigmentations on the hulla | ||||
| Black (L-value) | 46.4 | 5.1 | 35.7–56.6 | 0.2953*** |
| Red (a-value) | 2.22 | 0.48 | 1.10–3.82 | 0.3874*** |
| Yellow (b-value) | 8.5 | 3.5 | 2.2–16.2 | 0.2865*** |
,r was significant at P < 0.0001.
Black, red, and yellow pigmentations were quantified by the L, a, and b values, respectively, taken with Chroma Meter (Minolta CR310); a low L and a high a or b value indicates a high intensity of black and red or yellow pigmentations, respectively.
TABLE 4.
QTL for awn length (AL), shattering (SH), and hull color (HC) detected in the BC4F2 (44) population
| QTLa | Nearest marker | R2 (%)b | Pb | a ± SEac | d ± SEdc | Donord |
|---|---|---|---|---|---|---|
| qAL4-2 | RM252 | 32.8 | <0.0001 | 2.64 ± 0.66*** | 0.56 ± 0.71, NS | SS18-2 |
| qAL8 | RM531 | 24.8 | <0.0001 | 1.80 ± 0.24*** | 0.49 ± 0.34, NS | SS18-2 |
| qSH4 | RM252 | 34.9 | <0.0001 | −0.21 ± 0.04*** | −0.10 ± 0.04* | SS18-2 |
| qSH8 | RM531 | 7.5 | 0.0012 | −0.08 ± 0.02** | 0.02 ± 0.04, NS | SS18-2 |
| qHCB1 | RM220 | 4.4 | 0.0160 | 1.42 ± 0.52** | 0.61 ± 0.73, NS | SS18-2 |
| qHCR1 | RM220 | 15.3 | <0.0001 | 0.25 ± 0.04*** | 0.06 ± 0.06, NS | EM93-1 |
| qHCY1 | RM220 | 8.9 | 0.0002 | 1.43 ± 0.33*** | 0.28 ± 0.48, NS | EM93-1 |
| qHCB4 | RM252 | 50.5 | <0.0001 | 4.06 ± 0.49*** | 3.00 ± 0.59*** | SS18-2 |
| qHCY4 | RM252 | 36.6 | <0.0001 | 2.31 ± 0.36*** | 1.78 ± 0.43*** | EM93-1 |
The quantification methods used here improved the correlations of degree of dormancy to the awn, shattering, and hull color traits in the BC4F2 (44) population (Table 3), as compared with the results from early generations (Figure 2). The above advanced BC analysis demonstrated that the correlations arose from the tight linkage among/between QTL on the chr 1, 4, 7, and 8 segments (Figure 3B). The four sets of the SS18-2-derived haplotypes most likely locate within the genetic distance of a few centimorgans because the QTL for different traits on each of the segments shared common nearest markers (Table 4, Figure 5) and identical peak positions as inferred by the WinQTLCart mapping (data not shown).
DISCUSSION
Dormancy gene action over generations of selection:
At least five dormancy QTL, including previously identified qSD4, -7-1, -8, and -12, responded to the single-plant selection in the early generations. Additive effects varied from insignificant (e.g., qSD8) to a magnitude equivalent to 1 unit of standard deviation (i.e., qSD12) in the relatively synchronized genetic backgrounds. This multilocus response was much higher than the theoretical probability (1/32 or 3.1%) for a five-locus heterozygote in a BCF1 population and was even higher than the expectation for segregating loci retained by the selection with recurrent backcrossing technique (Hill 1998). We selected for low germination extremes and determined that most QTL consisted of predominately gene additive effects (Table 2). Thus, we attribute the simultaneous retention of the five dormancy alleles for the most part to the gene cumulative effect postulated by Chang and Tagumpay (1973) for dormancy in rice. In addition, the two- or higher-order interactions in the primary (Gu et al. 2004) and the BC4F2 segregation populations (Figure 5) suggest that all dormancy loci identified from SS18-2 are networked by epistases. It is likely that the phenotypic selection was more favorable for networked genes rather than for a set of random loci. For example, the qSD1 dormancy allele did not display a significant main effect in the BC4F2 (132), yet it was retained during selection. Thus, it must have benefited from its epistasis with qSD7-1, a moderate locus (R2 = 9%) that also interacted with the major QTL qSD12. Similarly, the dormancy allele at qSD8 retained in BC4F1 plant 44 benefited from its epistasis with qSD1, which also interacted with the qSD7-1. Further research will be needed to isolate QTL, which have minor effects or only epistatic effects, from multilocus systems as single Mendelian factors to determine their genetic nature.
Implications of dormancy gene-related haplotypes:
Four haplotypes define the genetic basis for associations of seed dormancy with shattering, awn, black hull color, and red pericarp color traits over generations (Figure 2, Table 3). Although dormancy alleles in the haplotypes had moderate to small effects, their transmission directly affected the fate of groups of alleles, including major genes for other weedy traits in a population. The adaptive significance of dormancy in seed-bearing plants has been limited to the promotion of survival under adverse environmental conditions by distributing germination timing. The other weedy traits also contribute to adaptation in different ways, such as shattering enables weed seeds to escape from harvest, long awn aids seed dispersal, and chemicals underlying hull and pericarp colors aid in seed persistence in the soil. In adverse environments, genotypes with a high level of seed dormancy due to having multiple dormancy genes with epistatic effects can survive longer; these strongly dormant genotypes are those carrying genes for the other weedy traits because of tight linkage in the haplotypes. Our phenotypic selection, as a simulation of natural selection for strongly dormant genotypes, suggests that the multiple-gene system governing dormancy sheltered genes for other important adaptive traits during the evolution of weeds.
Haplotypes are signatures for evolutionary genetics or comparative genomics (Kim and Nielsen 2004). Wild-like weedy rice such as SS18-2 is considered to originate from a natural hybridization between wild Oryza ssp. and cultivars on the basis of morpho-physiological characteristics (Oka 1988; Suh et al. 1997; Tang and Morishima 1997). QTL or QTL clusters for some of the traits we measured have been reported in O. rufipogen accessions (Xiong et al. 1999; Cai and Morishima 2002; Thomson et al. 2003), suggesting that the haplotypes detected in SS18-2 were likely inherited as units from their wild relative, rather than by the hitchhiking effect of allelic mutations (Kim and Nielsen 2004) after differentiation of the wild and weedy species. The origin of cultivar-like weedy rice (Suh et al. 1997; Tang and Morishima 1997), such as red rice, which is distributed in East Asia, America, and Europe where there was no wild rice (O. ssp.), remains uncertain (Vaughan et al. 2001). Examining the haplotypes in cultivar-like weedy rice should be helpful in determining its origin. In addition, the weedy traits studied in this research are also common in other grass species (Harlan et al. 1973). Some genes for shattering and red pigmentation have been used to develop a consensus map for grasses (Devos and Gale 1997). Isolation and characterization of these adaptive haplotypes should enhance our knowledge about evolution of grass genomes and weediness.
There are increasing concerns about the risks of gene flow from transgenic cultivars to conspecific weeds (Oard et al. 2000; Gealy et al. 2003; Chen et al. 2004). Natural hybridization occurs between weeds and cultivars (Harlan et al. 1973; Langevin et al. 1990) and initiates hybridization-differentiation cycles (Ladizinsky 1985; Oka 1988). Once transgenes enter the cycles, the transgenic recombinants have greater opportunity to survive because of seed dormancy. Furthermore, if transgenes integrate into a weedy haplotype region, the recombinants could become superweeds. Gressel (1999) proposed that construction of a transgene (e.g., herbicide-resistant genes) in tandem with a gene for nondormancy or nonshattering would mitigate the risk of transgenic cultivars becoming “volunteer” weeds in the following crop. Theoretically this proposal has merit for managing the incidence of transgenic weeds, but technically many issues must be resolved. For example, there is no information on the molecular structure of genes that directly regulate dormancy because they have not been cloned; there is insufficient information on what dormancy locus region might be the best target for insertion of a transgene as it relates to the effects of genes (Table 2) in natural populations and the effects of genetic background due to epistasis (Figure 5); and, most importantly, there is no information about how the genes for weedy traits evolved to form haplotypes as it relates to the possibility of developing superweeds with an additional trait such as herbicide resistance.
Challenges and promises for the use of dormancy genes:
Domestication and breeding activities have eliminated a substantial degree of seed dormancy from modern cultivars by selection for rapid and uniform germination; thus, PHS has been a worldwide problem in agriculture (Harlan et al. 1973). Linkage drag with undesirable traits like shattering, awn, black hull color, and red pericarp color is a major challenge for the use of weedy rice-derived dormancy genes for improving resistance to PHS. It will be difficult to separate some dormancy alleles from the linked genes as indicated by strong associations over successive generations. Association between red pericarp color and seed dormancy in cereal grains could be due to pleiotrophy or tight linkage of genes (Gfeller and Svejda 1960; Flintham 2000). Fine mapping of the qSD7-1 region or cloning of the dormancy locus will reveal the nature of the association and, therefore, determine if this dormancy allele could be used in breeding. The same approach would also be necessary to determine the usefulness of dormancy genes in other haplotypes. Additional challenges include the variation in gene effects with genetic background (Table 2) and the genotype-by-environmental interaction (Gu et al. 2005b). For example, qSD1 was not detectable in the primary segregating population (Gu et al. 2004), but was detected in the BC4F2 (44) population. It is not unusual that a dormancy allele could offset or even reverse the effect of another dormancy allele on germination due to epistasis (Figure 5B; Gu et al. 2004).
Major dormancy QTL, such as qSD12, are promising candidates for breeding varieties resistant to PHS and convey key genetic information on the regulation of germination and after-ripening. The major effect of qSD12 was detected in the primary segregation population grown in different years (Gu et al. 2005b) and it had no deleterious effects in the BC4F2 (132) population. The large additive effect (Table 2) suggests that gene(s) underlying qSD12 can be incorporated into conventional varieties or parental lines of hybrid rice to markedly improve resistance to PHS. QTL that explained ≥50% of the phenotypic variance in dormancy or dormancy-related traits (e.g., amylase content) have been identified from two- and six-rowed barley varieties (Oberthur et al. 1995; Li et al. 2004; Prada et al. 2004). Fine or comparative mapping suggests that barley QTL may consist of a gene cluster (Han et al. 1999) and may be conserved in rice and wheat (Li et al. 2004). We are developing isogenic lines for qSD12 to clone its underlying gene(s) by taking advantage of the published rice genome sequences.
Acknowledgments
We thank B. Hoffer, T. Nelson, and C. Kimberlin for their technical assistance and anonymous reviewers for their valuable comments. Funding for this work was provided by the U.S. Department of Agriculture-National Research Initiative (0200668).
References
- Ahn, S. N., J. P. Suh, C. S. Oh, S. J. Lee and H. S. Suh, 2002. Development of introgression lines of weedy rice in the background of Tongil-type rice. Rice Genet. Newsl. 19: 14–15. [Google Scholar]
- Alonso-Blanco, C., L. Bentsink, C. J. Hanhart, H. B. E. Vries and M. Koornneef, 2003. Analysis of natural allelic variation at seed dormancy loci of Arabidopsis thaliana. Genetics 164: 711–729. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anderson, J. A., M. E. Sorrells and S. D. Tanksley, 1993. RFLP analysis of genomic regions associated with resistance to pre-harvest sprouting in wheat. Crop Sci. 33: 453–459. [Google Scholar]
- Baker, B., D. V. Chin and M. Mortimer, 2000. Wild and Weedy Rice in Rice Ecosystems in Asia: A Review. Limited Proceedings 2000, No. 2, International Rice Research Institute, Manila, Philippines.
- Basu, C., M. D. Halfhill, T. C. Mueller and C. N. Jr. Stewart, 2004. Weed genomics: new tools to understand weed biology. Trends Plant Sci. 9: 391–398. [DOI] [PubMed] [Google Scholar]
- Bewley, J. D., and M. Black, 1982. Physiological and Biochemistry of Seeds in Relation to Germination, Vol. 2. Springer-Verlag, Berlin, Heidelberg, Germany/New York.
- Bres-Patry, C., M. Lorieu, G. Clement, M. Bangratz and A. Ghesquiere, 2001. Heredity and genetic mapping of domestication-related traits in a temperate japonica weedy rice. Theor. Appl. Genet. 102: 118–126. [Google Scholar]
- Cai, H. W., and H. Morishima, 2000. Genomic regions affecting seed shattering and seed dormancy in rice. Theor. Appl. Genet. 100: 840–846. [Google Scholar]
- Cai, H. W., and H. Morishima, 2002. QTL clusters reflect character associations in wild and cultivated rice. Theor. Appl. Genet. 104: 1217–1228. [DOI] [PubMed] [Google Scholar]
- Chang, T. T., and O. Tagumpay, 1973. Inheritance of grain dormancy in relation to growth duration in 10 rice crosses. SABRAO Newsl. 5: 87–94. [Google Scholar]
- Chen, L. J., D. S. Lee, Z. P. Song, H. S. Suh and B. R. Lu, 2004. Gene flow from cultivated rice (Oryza sativa) to its weedy and wild relatives. Ann. Bot. 93: 1–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Devos, K. M., and M. D. Gale, 1997. Comparative genetics in the grasses. Plant. Mol. Biol. 35: 3–15. [PubMed] [Google Scholar]
- Dong, Y., E. Tsozuki, H. Kamiunten, H. Terao, D. Lin et al., 2003. Identification of quantitative trait loci associated with pre-harvest sprouting resistance in rice (Oryza sativa L.). Field Crops Res. 81: 133–139. [Google Scholar]
- Flintham, J. E., 2000. Different genetic components control coat-imposed and embryo-imposed dormancy in wheat. Seed. Sci. Res. 10: 43–50. [Google Scholar]
- Gao, W., J. A. Clancy, F. Han, D. Prada, A. Kleinhofs et al., 2003. Molecular dissection of a dormancy QTL region near the chromosome 7 (5H) L telomere in barley. Theor. Appl. Genet. 107: 552–559. [DOI] [PubMed] [Google Scholar]
- Gealy, D. R., D. H. Mitten and J. N. Rutger, 2003. Gene flow between red rice (Oryza sativa) and herbicide-resistant rice (O. sativa): Implications for weed management. Weed Technol. 17: 627–645. [Google Scholar]
- Gfeller, F., and F. Svejda, 1960. Inheritance of post-harvest seed dormancy and kernel colour in spring wheat lines. Can. J. Plant Sci. 40: 1–6. [Google Scholar]
- Gressel, J., 1999. Tandem constructs: preventing the rise of superweeds. Trends Biotechnol. 17: 361–366. [DOI] [PubMed] [Google Scholar]
- Grist, D.H., 1986. Rice, Ed. 6. Longman, London/New York.
- Groos, C., G. Gay, M. R. Perretant, L. Gervais, M. Bernard et al., 2002. Study of the relationship between pre-harvest sprouting and grain color by quantitative trait loci analysis in a whitexred grain bread-wheat cross. Theor. Appl. Genet. 104: 39–47. [DOI] [PubMed] [Google Scholar]
- Gu, X.-Y., Z.-X. Chen and M. E. Foley, 2003. Inheritance of seed dormancy in weedy rice. Crop Sci. 43: 835–843. [Google Scholar]
- Gu, X.-Y., S. F. Kianian and M. E. Foley, 2004. Multiple loci and epistases control genetic variation for seed dormancy in weedy rice (Oryza sativa). Genetics 166: 1503–1516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gu, X.-Y., S. F. Kianian and M. E. Foley, 2005. a Seed dormancy imposed by covering tissues interrelates to shattering and seed morphological characteristics in weedy rice. Crop Sci. 45: 948–955. [Google Scholar]
- Gu, X.-Y., S. F. Kianian, G. A. Hareland, B. L. Hoffer and M. E. Foley, 2005. b Genetic analysis of adaptive syndromes interrelated with seed dormancy in weedy rice (Oryza sativa). Theor. Appl. Genet. 110: 1108–1118. [DOI] [PubMed] [Google Scholar]
- Han, F., S. E. Ullrich, J. A. Clancy and I. Romogosa, 1999. Inheritance and fine mapping of a major barley seed dormancy QTL. Plant Sci. 143: 113–118. [Google Scholar]
- Harlan, J. R., and J. M. J. de Wet, 1965. Some thoughts about weeds. Econ. Bot. 19: 16–24. [Google Scholar]
- Harlan, J. R., J. M. J. de Wet and E. G. Price, 1973. Comparative evolution of cereals. Evolution 27: 311–325. [DOI] [PubMed] [Google Scholar]
- Harushima, Y., M. Yano, A. Shomura, M. Sato, T. Shimano et al., 1998. A high-density rice genetic linkage map with 2275 markers using a single F2 population. Genetics 148: 479–494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hill, W. G., 1998. Selection with recurrent backcrossing to develop congenic lines for quantitative trait loci analysis. Genetics 148: 1341–1352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnson, L. P. V., 1935. The inheritance of delayed germination in hybrids of Avena fatua and A. sativa. Can. J. Res. 13: 367–387. [Google Scholar]
- Kato, K., W. Nakamura, T. Tabiki, H. Miura and S. Sawada, 2001. Detection of loci controlling seed dormancy on group 4 chromosomes of wheat and comparative mapping with rice and barley genomes. Theor. Appl. Genet. 102: 980–985. [Google Scholar]
- Kearsey, M. J., and H. S. Pooni, 1996. The Genetical Analysis of Quantitative Traits. Chapman & Hall, London.
- Khan, M., P. B. Cavers, M. Kane and K. Thompson, 1996. Role of the pigmented seed coat of proso millet (Panicum miliaceum L.) in imbibition, germination and seed persistence. Seed Sci. Res. 7: 21–25. [Google Scholar]
- Kim, Y., and R. Nielsen, 2004. Linkage disequilibrium as a signature of selective sweeps. Genetics 167: 1513–1524. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koornneef, M., L. Bentsink and H. Hilhorst, 2002. Seed dormancy and germination. Curr. Opin. Plant Biol. 5: 33–36. [DOI] [PubMed] [Google Scholar]
- Kulwal, P. L., R. Singh, H. S. Balyan and P. K. Gupta, 2004. Genetic basis of pre-harvest sprouting tolerance using single-locus and two-locus QTL analyses in bread wheat. Funct. Integr. Genomics 4: 94–101. [DOI] [PubMed] [Google Scholar]
- Ladizinsky, G., 1985. Founder effect in crop-plant evolution. Econ. Bot. 39: 191–199. [Google Scholar]
- Langevin, S. A., K. Clay and J. B. Grace, 1990. The incidence and effects of hybridization between cultivated rice and its related weed red rice (Oryza sativa L.). Evolution 44: 1000–1008. [DOI] [PubMed] [Google Scholar]
- Lègére, A., 2005. Risks and consequences of gene flow from herbicide-resistant crops: canola (Brassica napus L) as a case study. Pest Manag. Sci. 61: 292–300. [DOI] [PubMed] [Google Scholar]
- Li, C. D., A. Tarr, R. C. M. Lance, S. Harasymow, J. Uhlmann et al., 2003. A major QTL controlling seed dormancy and pre-harvest sprouting/α-amylase in two-rowed barley (Hordeum vulgare L.). Aust. J. Agric. Res. 54: 1303–1313. [Google Scholar]
- Li, C., P. Ni, M. Francki, A. Hunter, Y. Zhang et al., 2004. Genes controlling seed dormancy and pre-harvesting sprouting in a rice-wheat-barley comparison. Funct. Integr. Genomics 4: 84–93. [DOI] [PubMed] [Google Scholar]
- Lijavetzky, D., M. C. Martinez, F. Carrari and H. E. Hopp, 2000. QTL analysis and mapping of pre-harvest sprouting resistance in sorghum. Euphytica 112: 125–135. [Google Scholar]
- Lin, S. Y., T. Sasaki and M. Yano, 1998. Mapping quantitative trait loci controlling seed dormancy and heading date in rice, Oryza sativa L., using backcross inbred lines. Theor. Appl. Genet. 96: 997–1003. [Google Scholar]
- Lincoln, S., M. Daly and E. Lander, 1992. Constructing Genetic Maps With MAPERMAKER/EXP 3.0, Ed. 3. Whitehead Institute, Cambridge, MA.
- Mares, D. J., and K. Mrva, 2001. Mapping quantitative trait loci associated with variation in grain dormancy in Australian wheat. Aust. J. Agric. Res. 52: 1257–1265. [Google Scholar]
- McCouch, S. R., L. Teytelman, Y. B. Xu, K. B. Lobos, K. Clare et al., 2002. Development and mapping of 2240 new SSR markers for rice (Oryza sativa L.). DNA Res. 9: 199–207. [DOI] [PubMed] [Google Scholar]
- Nilsson-Ehle, H., 1914. Zur kenntnis der mit der keimungsphysiologie des weizens in zusammenhang stehenden inneren faktoren. Z. Pflanzenzücht. 2: 153–187. [Google Scholar]
- Oberthur, L., T. K. Blake, W. E. Dyer and S. E. Ullrich, 1995. Genetic analysis of seed dormancy in barley (Hordeum vulgare L.). J. Quant. Trait Loci 1: 5. [Google Scholar]
- Oka, H. I., 1988. Origin of Cultivated Rice. Japan Scientific Society Press, Tokyo.
- Oard, J., M. A. Cohn, S. Linscombe, D. Gealy and K. Gravois, 2000. Field evaluation of seed production, shattering, and dormancy in hybrid populations of transgenic rice (Oryza sativa) and the weed, red rice (Oryza sativa). Plant Sci. 157: 13–22. [DOI] [PubMed] [Google Scholar]
- Osa, M., K. Kato, M. Mori, C. Shindo, A. Torada et al., 2003. Mapping QTLs for seed dormancy and the Vp1 homologue on chromosome 3A in wheat. Theor. Appl. Genet. 106: 1491–1496. [DOI] [PubMed] [Google Scholar]
- Prada, D., S. E. Ullrich, J. L. Molina-Cano, L. Cistue, J. A. Clancy et al., 2004. Genetic control of dormancy in a Triumph/Morex cross in barley. Theor. Appl. Genet. 109: 62–70. [DOI] [PubMed] [Google Scholar]
- SAS Institute, 1999. SAS/STAT User's Guide, Version 8. SAS Institute, Cary, NC.
- Simpson, G. M., 1992. The adaptive significance of awns and hairs in grasses. J. Biol. Educ. 26: 10–11. [Google Scholar]
- Suh, H. S., Y. I. Sato and H. Morishima, 1997. Genetic characterization of weedy rice (Oryza sativa L.) based on morpho-physiology, isozymes and RAPD markers. Theor. Appl. Genet. 94: 316–321. [Google Scholar]
- Takeuchi, Y., S. Y. Lin, T. Sasaki and M. Yano, 2003. Fine linkage mapping enables dissection of closely linked quantitative trait loci for seed dormancy and heading in rice. Theor. Appl. Genet. 107: 1174–1180. [DOI] [PubMed] [Google Scholar]
- Tang, L.-H., and H. Morishima, 1997. Genetic characterization of weedy rices and the inference on their origins. Breed. Sci. 47: 153–160. [Google Scholar]
- Tanksley, S. D., and J. C. Nelson, 1996. Advanced backcross QTL analysis: a method for the simultaneous discovery and transfer of valuable QTLs from unadapted germplasm into elite breeding lines. Theor. Appl. Genet. 92: 191–203. [DOI] [PubMed] [Google Scholar]
- Tanksley, S. D., and S. R. McCouch, 1997. Seed banks and molecular maps: unlocking genetic potential from the wild. Science 277: 1063–1066. [DOI] [PubMed] [Google Scholar]
- Thomson, M. J., T. H. Tai, A. M. McClung, X. H. Lai, M. E. Hinga et al., 2003. Mapping quantitative trait loci for yield, yield components and morphological traits in an advanced backcross population between Oryza rufipogon and the Oryza sativa cultivar Jefferson. Theor. Appl. Genet. 107: 479–493. [DOI] [PubMed] [Google Scholar]
- Vaughan, L. K., B. V. Ottis, A. M. Prazak-Havey, C. Sneller, J. M. Chandler et al., 2001. Is all red rice found in commercial rice really Oryza sativa? Weed Sci. 49: 468–476. [Google Scholar]
- Wang, S., C. J. Basten, P. Gaffney and Z-B. Zeng, 2004. Window QTL Cartographer 2.0 User Manual (http://statgen.ncsu.edu/qtlcart/WQTLCart.html).
- Wright, S., 1952. The genetics of quantitative variability, pp. 5–14 in Quantitative Inheritance, edited by E. C. R. Reeve and C. H. Waddington. Her Majesty's Stationary Office, London.
- Xiong, L. Z., K. D. Liu, X. K. Dai, C. G. Xu and Q. Zhang, 1999. Identification of genetic factors controlling domestication-related traits of rice using an F2 population of a cross between Oryza sativa and O. rufipogon. Theor. Appl. Genet. 98: 243–251. [Google Scholar]
- Zhang, F., G. Chen, Q. Huang, O. Orion, T. Krugman et al., 2005. Genetic basis of barley caryopsis dormancy and seedling desiccation tolerance at the germination stage. Theor. Appl. Genet. 110: 445–453. [DOI] [PubMed] [Google Scholar]