

Abstract
Sequences flanking 73 insertions of the retrotransposon PDR1 have been characterized, together with an additional 270 flanking regions from one side alone, from a diverse collection of Pisum germ plasm. Most of the identified flanking sequences are repetitious DNAs but more than expected (7%) lie within nuclear gene protein-coding regions. The approximate age of 52 of the PDR1 insertions has been determined by measuring sequence divergence among LTR pairs. These data show that PDR1 transpositions occurred within the last 5 MY, with a peak at 1–2.5 MYA. The insertional polymorphism of 68 insertions has been assessed across 47 selected Pisum accessions, representing the diversity of the genus. None of the insertions are fixed, showing that PDR1 insertions can persist in a polymorphic state for millions of years in Pisum. The insertional polymorphism data have been compared with the age estimations to ask what rules control the proliferation of PDR1 insertions in Pisum. Relatively recent insertions (< ∼1.5MYA) tend to be found in small subsets of the Pisum accessions set, “middle-aged” insertions (between ∼1.5 and 2.5 MYA) vary greatly in their occurrence, and older insertions (> ∼2.5 MYA) are mostly found in small subsets of Pisum. Finally, the average age estimate for PDR1 insertions, together with an existing data set for PDR1 retrotransposon SSAP markers, has been used to derive an estimate of the effective population size for Pisum of ∼7.5 × 105.
RETROTRANSPOSONS are mobile genetic elements that transpose into different loci replicatively through reverse transcription of RNA intermediates. Retrotransposons are found in all kingdoms of life and are ubiquitous in the genomes of plants (Flavell et al. 1992a; Voytas et al. 1992; Suoniemi et al. 1998; Noma et al. 1999; Schmidt 1999). Long terminal repeat (LTR) retrotransposons tend to be the dominant retrotransposon class in plants and have been classified into two main groups, the Ty1-copia group and the Ty3-gypsy group, on the basis of conserved sequence features and gene order (Xiong and Eickbush 1990), although more recent findings show this to be an oversimplification (Havecker et al. 2004). Each of the retrotransposon groups typically contains a great variety of different retrotransposons (Konieczny et al. 1991; Flavell et al. 1992b), which are found in widely different numbers of copies per genome, from a few to tens of thousands. Collectively, huge numbers of LTR retrotransposon insertions are found in the genomes of many plant species and can constitute more than half the entire genome in some cases (Sanmiguel et al. 1996; Kumar and Bennetzen 1999).
A variety of PCR-based systems have been developed to detect insertional polymorphism of retrotransposons in plants (Waugh et al. 1997; Ellis et al. 1998; Flavell et al. 1998; Kalendar et al. 1999; Provan et al. 1999; Yu and Wise 2000; Porceddu et al. 2002). Most of these are multiplex approaches, which display the regions flanking individual retrotransposon insertions as bands on gels. Such methods can generate large amounts of data easily and are very useful for determining the genetic diversity of germ plasm (Ellis et al. 1998). In contrast, retrotransposon-based insertion polymorphisms (RBIPs) detect individual insertions by PCR with flanking host sequence primers and a retrotransposon-specific primer (Flavell et al. 1998). RBIP produces less data per experiment than do multiplex approaches but is more accurate for studies of deeper phylogeny in wide germ plasm, because it is a codominant method that uses two simple PCRs to detect both presence and absence of the insertion, whereas multiplex approaches detect only insertion presence and absence is inferred by band absence, which can result from mutation in PCR primer sites.
The field pea (Pisum sativum) has a large genome (∼4.5 × 109 bp), which is relatively stable in size between species (Greilhuber and Ebert 1994; Baranyi et al. 1996). At present, rather little genomic sequence is available for Pisum but the repetitious DNAs of the genus are better understood. A variety of retrotransposons, transposons, and other repetitious DNAs have been characterized for Pisum (Lee et al. 1990; Chavanne et al. 1998; Nouzova et al. 2000; Neumann et al. 2001, 2003; Macas et al. 2003). Pea is a predominantly inbreeding Old World legume crop first cultivated ∼10,000 years ago (Blixt 1972; Zohary 1996; Mithen 2003). Cultivated Pisum retains a wide gene pool, both phenotypically and genotypically, and wild Pisum species extend this diversity still further. Traditionally, one cultivated species, P. sativum, and three wild taxa, P. elatius, P. humile, and P. fulvum, have been recognized. However, a combination of molecular and other approaches has led to the conclusion that only P. fulvum is a truly distinct species, with the others forming a single-species complex (Vershinin et al. 2003). Insertional polymorphism for four Ty1-copia group retrotransposons, a Ty3-gypsy group retrotransposon, and a CACTA transposon have been measured by the multiplex sequence-specific amplification polymorphisms (SSAP) approach in Pisum (Vershinin et al. 2003). These diverse mobile elements all produce roughly similar pictures of the diversity of Pisum. P. fulvum represents a distinct, though diverse clade, and P. abyssinicum forms another, far more compact clade. Finally, P. elatius is the most diverse germ plasm set, with P. humile and P. sativum falling within its boundaries. However, individual SSAP markers from any of these species can frequently be found in another one, suggesting that introgression by outbreeding has played a significant role in the genomic evolution of the genus (Vershinin et al. 2003).
PDR1 was the first Ty1-copia group retrotransposon to be isolated from Pisum (Lee et al. 1990) and remains the best understood. It is one of the smallest and simplest transposition-competent LTR retrotransposons known, with 156-bp LTRs and the typical gag-pr-int-rt-rnaseH gene order of the Ty1-copia group. PDR1 is present across the entire Pisum genus in ∼200 dispersed copies per haploid genome (Lee et al. 1990; Ellis et al. 1998) and >95% of insertions are polymorphic within the genus (Ellis et al. 1998; Vershinin et al. 2003). Linkage mapping has shown it to be broadly distributed within the Pisum genome (Ellis et al. 1998). The purpose of this study was, first, to discover the genomic environment of PDR1 insertions by sequencing the surrounding DNA for a large set of insertions; second, to investigate the distribution of these insertions within the genus Pisum by the RBIP approach; third, to determine the antiquity of the insertions; and finally, to compare the age estimations with the occupancy data to determine what rules control the fates of PDR1 insertions in the Pisum genus.
MATERIALS AND METHODS
Plant material and DNA isolation:
Pisum accessions from the John Innes Pisum Collection were selected on the basis of previous studies (Vershinin et al. 2003) to represent the diversity of the genus. Genomic DNAs were isolated from young leaf tissue using Qiagen (Valencia, CA) DNeasy 96 plant kits following the manufacturer's instructions.
Isolation of genomic sequences flanking PDR1 retrotransposon insertions:
DNAs from a variety of Pisum accessions were digested with TaqI restriction endonuclease, followed by ligation with TaqI adapters (Table 1). SSAP PCRs were then carried out (Ellis et al. 1998), with a PDR1-specific primer (see below) and TaqI adapter primer 9011 (Table 1), to create pools of mixed PCR products, each containing a fragment of a PDR1 LTR, together with its flanking host genomic DNA. These were either cloned directly into bacterial vector (see below) or separated by polyacrylamide gel electrophoresis before isolation and cloning (see below).
TABLE 1.
Oligonucleotides used in this study
| Oligonucleotide no. | Oligonucleotide type | Sequence | Positiona |
|---|---|---|---|
| 9363 | Taq I adaptor (+) strand | GACGATGGATCCTGAG | — |
| 9010 | Taq I adaptor (−) strand | CGCTCAGGATCCAT | — |
| 9011 | Taq adapter PCR primer | GACGATGGATCCTGAGCG | — |
| 9479b | PDR1 LTR primer (+) strand | TAAGGTCCATTAGTCAAAGCCC | 3811–3835 |
| 9124b | PDR1 LTR primer (−) strand | GGGCTTTGACTAATGGACCTC | 67–47 |
| 9975b | PDR1 RNAseH gene primer (+) strand | TCCTGTTCAGCATGACGAGAC | 3561–3581 |
| 15940c | PDR1 polypurine tract primer (+) strandc | ATTCACCAGCTTGAGGGGAG | 3749–3768 |
| 15941 | PDR1 LTR primer (+) strand | GTAATGAGCTCCATTAGTCAAAGC | 3810–3833 |
| 20762 | PDR1 gag gene primer (−) strand | ACAAGAAGCACGATGTGCTAC | 308–288 |
| JIp_101 | PDR1 primer binding site region primer (−) | TACAAGGCGGCTAGGG | 215–200 |
Position on retrotransposon PDR1 sequence (accession no. X66399).
SSAP reactions used either conventional Taq DNA polymerase in conventional buffer (Ellis et al. 1998) or Qiagen Hotstar Taq DNA polymerase in unmodified Qiagen buffer (no extra magnesium or Q buffer) and 0.2 pmol/μl of each primer. Hot-start PCR conditions were 95° for 15 min; then 30× 94°, 60°, 72°, each for 1 min; and then 72° for 7 min. All primers were designed for melting temperatures of 60–65° in 50 mm cation concentration. PCRs amplifying sequences 5′ to the PDR1 insertions (i.e., upstream of the major retrotransposon transcript; 5′ SSAP PCRs) used oligonucleotide 9124 or JIp_101 [Table 1, supplementary Figure S1 (http://www.genetics.org/supplemental/)] and PCRs amplifying sequences 3′ to the insertions (3′ SSAP PCRs) used oligonucleotide 9479, 15940, or nested PCR with primer 15940 followed by 15941. The use of the latter primer pair introduced a SacI cut site into the PDR1 end of the SSAP fragment, which was used with a similarly engineered BamHI site in the TaqI adapter primer (9011; Table 1) to clone fragments directionally into SacI/BamHI double-digested M13mp18 vector DNA. Clones were sequenced by using BigDye v2.0 (PE Biosystems). A total of 131 5′ SSAP sequences and 554 3′ SSAP sequences were obtained, representing 67 unique 5′ sequences and 203 unique 3′ sequences, respectively (415 duplicate sequences were obtained).
Isolation of PDR1 RBIP insertions:
To develop RBIPs, genome sequence data were needed from both sides flanking the insertion (Flavell et al. 1998). Three variant methods were used for this (Figure 1).
Figure 1.

Strategies used for isolating sequences flanking PDR1 retrotransposon insertions. (A) Methods 1 and 2: SSAP reactions, using TaqI restriction enzyme and PDR1-specific primers (PL and PR), are primed outward in both directions from PDR1 insertions, producing multiple different flanking fragment gel bands that are scrutinized for pairs originating from single insertions, by sequencing then comparing their 5-bp TSDs (large triangle flanking the insertion; method 1) and by determining their approximate genetic map locations by SSAP marker analysis in a mapping population (method 2). (B) Method 3: Nested SSAP primer pairs derived from the flanking host DNA and oriented back toward the PDR1 insertion (P1 and P2) are used in SSAP reactions on several highly diverse plant DNAs, including the original sample from which the flanking sequence was obtained. DNAs lacking the insertion produce SSAP bands crossing the insertion site and DNAs containing the insertion produce the original PDR1-host junction sequence. (C) An example of successful use of method 3. Agarose gel electrophoresis of SSAP reaction products from 10 diverse pea DNA samples is shown, using either the P1 primer or nested (P1 → P2) primers. o, occupied site SSAP product; u, unoccupied site SSAP product; *, the original donor (occupied site) accession, JI64.
Method 1: matching target site duplications of 5′- and 3′-flanking sequences:
5′ and 3′ SSAP reactions, oriented outward in both directions from the PDR1 LTR into the flanking host DNA, were carried out with primer oligonucleotides PL (usually 9124) or PR (usually 9479) and Taq adapter oligonucleotide [Table 1, supplementary Figure S1 (http://www.genetics.org/supplemental/)]. The two pools of PCR products were treated with Klenow fragment DNA polymerase (New England Biolabs, Beverly, MA) to generate blunt ends, followed by T4 polynucleotide kinase (New England Biolabs), before cloning into M13mp18 bacteriophage vector linearized with HincII restriction endonuclease (Roche, Indianapolis), and then treated with calf intestinal phosphatase (Roche) to reduce background from insert-lacking clones. Random subclones were sequenced. Sequences derived from 5′ SSAP PCR were then compared with those from 3′ SSAP PCR to identify pairs possessing identical 5-base target site duplications (TSDs) flanking the retrotransposon insertion. Such sequence pairs, representing putative pairs of LTR-host junctions from the same PDR insertion, were then tested by PCR with primer pairs derived from 5′- and 3′-flanking genomic DNA in six highly diverse Pisum accessions. Any pair that generated a new band in one or more of these accessions was tested by sequencing to determine if it represented host genomic sequence unoccupied by the PDR1 insertion (Figure 1). Two RBIPs were obtained by this approach.
Method 2: matching segregation patterns for 5′ and 3′ SSAPs in mapping populations:
Radioactive 5′ and 3′ SSAP reactions (Ellis et al. 1998) were carried out separately using 33P-labeled PDR1-specific primers, 9124 and 15940 [Table 1, supplementary Figure S1 (http://www.genetics.org/supplemental/)], respectively, on DNAs of 20 recombinant inbred lines (RILs) from a mapping population derived from a cross between accessions JI15 and JI399 of the John Innes Pisum Collection. The products were visualized by polyacrylamide gel electrophoresis, followed by autoradiography. Candidate pairs of SSAP bands, which cosegregated in the 20 RILs, represented putative pairs of LTR-host junctions from the same PDR1 insertion. These pairs of bands were extracted from the dried gels (Knox 2005), reamplified, and sequenced using BigDye v 3.0 (ABI, Columbia, MD) to confirm identity of the 5-bp TSD and then the allelic state of the locus (occupied or unoccupied) was investigated in the parents of the mapping population by PCR as for method 1. Four RBIPs were obtained by this approach.
Method 3: SSAP from flanking host sequence:
Genomic sequences flanking PDR1 insertions derived from 3′ SSAP (see method 1) were used to design nested primers (Figure 1, P1 and P2), oriented back toward the PDR1 insertion from the surrounding host genomic sequence. For each insertion a “hot-start” SSAP reaction was carried out with primer P1 in a broad set of germ plasm (typically 10 accessions), using the same PCR conditions as for method 1. The use of a good hot-start PCR was crucial to this approach to minimize nonspecific background signal from other genomic regions. The PCR products were then subjected to a second SSAP PCR with nested primer P2 and the same adapter primer under identical PCR conditions. The PCR products were visualized by agarose gel electrophoresis (Figure 1C). Candidate PCR bands composing putative unoccupied insertion (alleles lacking the retrotransposon insertion) were validated by sequence analysis as above. Sixty-four RBIPs were obtained by this approach.
Isolation and sequence determination of PDR1 LTRs from RBIP insertions:
Matched pairs of complete LTRs from individual PDR1 RBIP insertions were amplified with 5′ and 3′ host genome flanking primers [primers L and R in supplementary Table S1 (http://www.genetics.org/supplemental/)] and PDR1 primer 20762, 9975, or JI_101, respectively [Table 1, supplementary Figure S1 (http://www.genetics.org/supplemental/)], using genomic DNA of the original occupied accession used for RBIP development as template. The PCR products were purified with QIAquick Spin columns (Qiagen), cloned into pGEM-T easy vector (Promega, Madison, WI) following the manufacturer's instructions, and sequenced by BigDye automated sequencing (PE Biosystems). LTRs were sequenced initially on one strand and any polymorphism was confirmed by visual comparison of the sequence trace against the wild-type allele trace. Any remaining ambiguities were resolved by sequencing the complementary strand. The four insertions described in method 2 above were sequenced directly from PCR products as described above.
Bioinformatics analysis:
Homology searches for all the isolated flanking sequences were performed by running BLASTN and TBLASTX programs against the NCBI database (Altschul et al. 1997). BLAST searches were done to a local version of the NCBI nonredundant nucleotide database downloaded in April 2004. Blastall was used to batch BLAST the sequences, using both BLASTN and TBLASTX searches and the results were limited to 30 hits with E-values <0.2. For BLASTN searches a word size of 11 was used and for TBLASTX BLOSUM62 was used for the matrix with a word size of 3. Database hits were visually checked to verify their identities and apparent hits within gene-coding regions were carefully checked to ensure that they did not derive from insertions into non-protein-coding regions or the coding regions of mobile elements.
Estimation of the synonymous nucleotide substitution rate for the Pisum lineage:
To calibrate the molecular clock for the Pisum lineage, divergence times for P. sativum vs. Medicago truncatula, Glycine max, and Acacia mangium of 32, 46, and 54 MYA, respectively, were taken from Wojciechowski (2003). Synonymous nucleotide substitution rates were calculated by comparison of exons of two single-copy nuclear genes, namely GdcH from M. truncatula (EST_BF519088) and P. fulvum JI1010 (AJ938069) and Uni from Acacia (AY229890), Pisum (AF035163), and Medicago (AC139708), using DIVERGE [Wisconsin Package Version 10.0; Genetics Computer Group (GCG), Madison, WI]. Ks-values calculated for Acacia-pea (Uni), Medicago-pea (Uni), and Medicago-pea (Gdch) were 1.0, 0.48, and 0.27. Applying the estimated divergence times for the host species described above, these Ks values correspond to substitution rates of 9.3 × 10−9, 7.5 × 10−9, and 4.2 × 10−9 substitutions/site/year, respectively. The average of these values, 7.0 substitutions/site/year, with standard deviation of 2.6 × 10−9, was used as the rate of synonymous substitutions (r).
Estimations of the antiquities of PDR1 insertions:
For each PDR1 insertion, the two LTR nucleotide sequences were aligned using ClustalW (Thompson et al. 1994) with default options (http://www.ebi.ac.uk/clustalw/index.html). The number of nucleotide substitutions per site was calculated from these alignments using Kimura's two-parameter model (Kimura 1980). Corresponding insertion ages for the PDR1 elements were estimated using the formula T = K/2r, where T is the time of insertion, K is the divergence parameter, and r is the average substitution rate (taken as the Ks value estimated above; Li and Graur 1991).
Estimation of effective population size for Pisum:
Allele frequencies of PDR1 insertions were calculated in a set of 259 SSAP markers scored in 52 Pisum accessions (Vershinin et al. 2003), using an Excel spreadsheet. The same spreadsheet was used to obtain the average heterozygosity value He from the corresponding average homozygosity value (the sum of squares of the allele frequencies) and 4Neν (= M) from He, using Equation 2 in Results.
RESULTS
Isolation of sequences flanking PDR1 insertions:
The overall goal of this study was to gain understanding of the nature, distribution, and antiquity of PDR1 insertions in the Pisum genus. This required flanking genomic sequence information from both sides of numerous PDR1 insertions, together with corresponding sequence information for both LTRs (see below). Only two cloned PDR1 insertions were available at the start of this study (Lee et al. 1990; Flavell et al. 1998) and, surprisingly, database searches failed to add any more (data not shown). Therefore, an efficient way of cloning multiple PDR1 insertions from a wide variety of genetically diverse individuals was needed.
Three different methods for isolating PDR1 insertions were tested in parallel (Figure 1). The first two methods involve first isolating multiple junction regions from both sides of PDR1 insertions, using the SSAP marker approach (Ellis et al. 1998), and then comparing pairs of junction sequences to ascertain which one might derive from the same insertion (Figure 1A). These methods are feasible for PDR1 because of its relatively low copy number (∼200 per genome; Ellis et al. 1998).
Method 1 exploits the fact that PDR1 creates a 5-bp TSD of host sequence upon integration (Lee et al. 1990). Thirty-one different sequences containing host-PDR1 5′ junctions (as defined by the polarity of the PDR1 open reading frame; Lee et al. 1990) were compared with 200 different 3′ junctions from the same plant accession, JI399. Six candidate pairs of junction sequences possessed identical 5-bp duplications. These were tested by PCR in a set of five highly diverse Pisum accessions to search for corresponding loci lacking PDR1 insertions (unoccupied sites). Two putative unoccupied sites were identified and both were validated by sequence analysis, yielding RBIP insertions 399-14-9 and 399-80-46. Later bioinformatics analysis showed that a third sequence pair, which did not produce a putative unoccupied site PCR band in the test set of pea DNAs, derived from a third RBIP insertion, 399-3-6, in a gene coding region.
The second method for RBIP isolation used cosegregation in a genetic mapping population to identify candidate host-PDR1 junction pairs (Figure 1A). For this method, SSAP molecular marker experiments (Ellis et al. 1998) were performed from both ends of the retrotransposon (Figure 1A), using DNAs from 20 individuals of a recombinant inbred mapping population (Ellis et al. 1998). Scores from 96 5′ SSAPs and 130 3′ SSAPs were collated and 47 cosegregating SSAP band pairs were identified, excised from the marker gels, and sequenced. Ten of these pairs were found to have identical 5-bp flanking TSDs and were tested by unoccupied site PCR as above, yielding four confirmed RBIPs, MKRBIP2, MKRBIP3, MKRBIP4, and MKRBIP7.
The third method used for isolating RBIP insertions is based on the genomic walking method (Figure 1 B and C,; Rosenthal and Jones 1990; Siebert et al. 1995). First, host sequences flanking the 3′ ends of PDR1 insertion were used to design pairs of locus-specific nested primers (P1 and P2), oriented back toward the PDR1 insertion. Each primer pair was then used for nested SSAP reactions. In the original plant, which gave rise to the 3′ flank, this regenerated the junction sequence from the PDR1 insertion but a DNA sample lacking the insertion produced a different SSAP band, which could be sequenced to identify the other side of the PDR1 insertion. This method has the advantage that a broad diversity of germ plasm can be used for the initial isolation of flanking sequences, yielding a correspondingly broad variety of insertions from across the Pisum genus.
Initial tests using the genomic walking method on Pisum accession JI1794 were successful, yielding two RBIPs (1794-1 and 1794-2). Subsequently, 973 3′-flanking sequences of PDR1 were obtained from 13 highly diverse Pisum accessions. Cross-comparisons between these sequences showed the presence of large numbers of multiple clonings of the same insertions from different plant samples. The final tally of unique 3′-flanking sequences was 200, 150 of which were long enough to design good nested primers. These 150 primer pairs were subjected to the genomic walking experiment (Figure 1B). Sixty-four nested primer pairs showed SSAP polymorphism in the diverse sample set and sequencing of the polymorphic bands generated confirmed that all were unoccupied alleles. RBIP PCR, using primers derived from both sides of the insertions and PDR1, gave the expected band sizes for unoccupied and occupied sites [an example is shown in supplementary Figure S2 (http://www.genetics.org/supplemental/)]. Fifty-two of the 64 RBIPs gave a single occupied-site band for the original donor-occupied accession and the other 12 RBIPs gave both unoccupied- and occupied-site bands. These accessions are all probably homozygotes as the accessions have been inbred in the John Innes Pisum germ plasm collection for many generations, and the DNA was prepared from single individuals. Therefore, it is likely that, for these 12 RBIPs, the PDR1 elements are inserted into other repetitive sequences. PDR1-1, the first such insertion isolated, is located between two B-type legumin genes (LegJ and LegK) and the unoccupied site is duplicated between the nearby LegL and LegM genes (Turner et al. 1993).
In summary, the three methods for RBIP isolation yielded 73 PDR1 insertions [supplementary Table S1 (http://www.genetics.org/supplemental/)]. Cross-comparison between these revealed two duplicates, giving 71 newly isolated, unique RBIPs together with the 2 already isolated (Lee et al. 1990; Flavell et al. 1998).
Sequence analysis of PDR1 insertion targets:
The studies described above revealed 340 different genomic sequences flanking PDR1 insertion sites in 15 diverse Pisum accessions. To investigate the nature of these sequences, searches against the NCBI genome sequence databases were carried out on 320 of these, omitting those <30 nucleotides. Table 2 summarizes the result of this analysis and the complete information is shown in supplementary Table S2 (http://www.genetics.org/supplemental/).
TABLE 2.
Identities and percentages of targets for PDR1 insertion in the Pisum genome
| DNA sequence type | Total no. | % of total | % of classified sequences |
|---|---|---|---|
| Unclassified (no close homologue) | 204 | 64 | — |
| Ty1-copia retrotransposon | 18 | 6 | 16 |
| Ty3-gypsy retrotransposon | 22 | 7 | 19 |
| LINE retrotransposon | 2 | 1 | 2 |
| DNA transposon | 2 | 1 | 2 |
| Other repetitive DNA | 35 | 11 | 31 |
| Unknown low copy sequence | 10 | 3 | 9 |
| Gene protein-coding sequence | 23 | 7 | 20 |
| Chloroplast | 1 | 0 | 1 |
| Total | 317 | 100 | 100 |
Most of the target sequences (64%) are unknown, with no significant hit in the databases. We believe that this is mainly due to the small sizes of many of these sequences and the incomplete knowledge of the highly diverse repetitious DNAs of Pisum (see discussion). Thirty-nine percent of the identifiable target sequences for PDR1 insertion are themselves transposable elements and a further 31% are unknown repetitive sequences, which are likely to be mainly composed of unidentified mobile elements or their relics. This is unsurprising, because Pisum has a large genome (4.5 × 109 bp haploid) and like other similarly sized plant genomes, including the quite closely related Vicia genus (Hill et al. 2005), is known to be composed predominantly of repetitive DNA (Murray and Thompson 1982). RBIP markers derived from such insertions into repeated sequences should have yielded unoccupied-site PCR products in most or all pea samples but, interestingly, this did not happen in most cases (data not shown). We believe that this is due to the antiquity of these repeats, whose sequences have been eroded by mutation.
Twenty percent of classified target sequences for PDR1 insertion (23 sequences or 7% of the 320 sequences analyzed) are protein-coding regions of genes, none of which derive from transposable elements. Each of these insertions probably generated a null allele for the gene concerned. This is a higher percentage than would be expected, considering the expected “gene space” and overall genome size of Pisum (see discussion).
To determine whether PDR1 shows any nucleotide site specificity for insertion, the 30 nucleotides either side of all 340 unique insertions were searched for characteristic motifs (Figure 2). A weak preference for A or T at bases 1–3 of the 5-base TSD was seen. A stronger preference for A or T bases was seen at nucleotides −8, −7, +7, and +8, relative to the insertion site, with the preferred bases being A, T, A, and T, respectively (Figure 2). As this motif has dyad symmetry around the insertion site, the frequency of AT dinucleotides at these positions was explored. Although AT was the most frequent dinucleotide at both sites (27% in both cases) AA, TT, and TA were found at almost equivalent levels (averaging 21, 18, and 15%, respectively), with the 12 other dinucleotides collectively making up the remaining 19%. Finally, the 72 complete RBIP flanking sequences were scrutinized for dyad symmetry at these base positions. No significant symmetry was apparent (data not shown). Therefore, although there is a consensus AT……. AT motif at ± 7–8 bp surrounding the PDR1 insertion site, there is no evidence that individual insertions occur in regions showing dyad symmetry for these bases.
Figure 2.

Consensus sequence motif surrounding PDR1 insertions in Pisum. The observed averaged A + T/G + C ratios across 340 30-nucleotide sequences flanking PDR1 insertions (solid diamonds) are compared to the same sequence data set randomized (shaded diamonds). Percentages of occurrences for G, A, T, and C between −10 and +10 nucleotides relative to the insertions are shown below, with values >49% in solid boxes, percentages between 40 and 49% in dark shaded boxes, and percentages between 30 and 39% in light shaded boxes. The consensus motif for PDR1 is shown, together with the corresponding consensus for Tos17 (Miyao et al. 2003) and a consolidated consensus for both retrotransposons.
The antiquity of PDR1 insertions:
It is possible to estimate the age of a retrotransposon insertion by looking at the sequence divergence between its LTRs, because these are synthesized from a single LTR RNA template before insertion (Sanmiguel et al. 1998; Bowen and McDonald 2001; Jiang et al. 2002a,b; Ma et al. 2004). Such estimations require knowledge of the neutral nucleotide substitution rate for the corresponding host nuclear genome. Published estimates for the synonymous nucleotide substitution rates of angiosperm nuclear genes vary a lot (between 1.5 and 7.1 × 10−9/site/year; Wolfe et al. 1987; Gaut et al. 1996; Small et al. 1998). Therefore, to estimate a synonymous substitution rate within the legumes, three synonymous substitution values (Ks) and corresponding substitution rates were obtained for the protein-coding regions of two genes, Unifoliata and GDCH, between P. sativum and the related legume genera, Medicago and Acacia (materials and methods). The average of the three synonymous rate values obtained is 7.0 × 10−9 substitutions/site/year (standard deviation of 2.6 × 10−9), in reasonable agreement with the above published estimates.
To estimate the ages of the PDR1 insertions isolated in this study, their LTR pairs were sequenced and K-values (substitutions/site/year; Kimura 1980) were calculated for each pair. Fifty-two pairs of LTRs in total were isolated from the 73 available RBIPs (71 from this study and 2 isolated previously). For the other 21 RBIP markers one or both of the LTRs failed to amplify, presumably because of PCR primer site mutation. The results of this analysis are shown in Figure 3. A broad distribution of K-values is seen, with an average K-value for all LTR pairs at 0.0124 (SD = 0.011), corresponding to an average age of 1.9 ± 0.7 MY. Six of these LTR pairs (11%) have identical LTRs, indicating relatively recent transposition (<1.5 ± 0.5 MYA using the Ks value estimated above); 40 (77%) have K-values between 0 and 0.02 [Figure 3, supplementary Table S3 (http://www.genetics.org/supplemental/)], corresponding to a divergence time ∼1.5 ± 0.5 MYA; and the final 6 LTR pairs have K-values between 0.02 and 0.07, corresponding to estimated divergence times of between 1.5 ± 0.5 MY and 5 ± 1.9 MY. These data suggest that the transpositional activity of PDR1 has varied over time, with a broad peak in the last ∼1–2 MY. Similar conclusions have been reached for LTR retrotransposon insertions in maize and rice (Sanmiguel et al. 1998; Vitte et al. 2004). However, it should be noted that the methods used in this study to isolate PDR1 insertions have relied upon several successive PCRs and it is likely that data are biased against older insertions (see discussion).
Figure 3.

Divergence between LTRs for individual PDR1 insertions. K-values (Kimura 1980) were obtained between the two LTRs of 52 individual PDR1 insertions.
Insertion site polymorphism for PDR1 within the genus Pisum:
To study the distribution of PDR1 RBIP insertions across the Pisum genus a set of 47 highly diverse Pisum accessions, almost identical to a set chosen previously to analyze the evolutionary history of Pisum (Vershinin et al. 2003), was chosen. Sixty-eight of the PDR1 RBIP insertions were scored in the 47 accessions [supplementary Table S4 (http://www.genetics.org/supplemental/)]. To visualize the distribution of the insertions in the Pisum accessions, scores for each PDR1 RBIP insertion were plotted onto a phylogenetic tree previously deduced for these accessions using 892 retrotransposon-based SSAP markers (Vershinin et al. 2003). Representative results from this analysis are shown in Figure 4. The distribution patterns of the PDR1 insertions across the Pisum genus are complex and in general resemble those obtained for individual SSAP markers (Vershinin et al. 2003). The distributions of some insertions, such as 2385x23 and 95x19 (Figure 4, B and C), concur approximately with the overall phylogeny of the tree, previously deduced using 892 retrotransposon-based SSAP markers (Vershinin et al. 2003), but most of the insertions, such as MKRBIP3 and 281x44 (Figure 4, D and E), do not. These data are consistent with our previous conclusion that introgression between diverse Pisum lineages has shaped the present pattern of retrotransposon-associated diversity (Vershinin et al. 2003 and discussion).
Figure 4.

Distribution patterns of individual PDR1 insertions in the Pisum genus. Occupancy scores of 10 PDR1 insertions in a core set of Pisum accessions (Vershinin et al. 2003) are shown. The phylogenetic tree (A) is based on 892 retrotransposon-based SSAP markers and derives from the same reference.
Investigation of the relationship between insertional polymorphism and antiquity of PDR1:
The availability of both insertion polymorphism data and antiquity estimations for PDR1 insertions provides the opportunity to search for relationships between these two parameters. The results of such an analysis for 43 PDR1 insertions for which both data sets are available are shown in Figure 5. Unsurprisingly, relatively recent insertions with identical LTRs tend to be distributed among few accessions within the core germ plasm set. The distributions of two such insertions, 1006nr9 and 3150x11, are shown in Figure 4, F and G. However, insertions of intermediate antiquity show no obvious pattern, with both young and older insertions being either rare or well established in the genus. For example, both the 95x2 and 1006x58 insertions show three polymorphisms between their LTRs, yet the former is found in 25 of the 47 pea accessions and the latter in only 4 accessions (Figure 4, H and I). It thus seems that different PDR1 insertions experience different degrees of success in spreading within the Pisum genus. If the averaged occupancy data for insertions are plotted against the number of polymorphisms between the LTRs a weak pattern emerges (Figure 5), suggesting a tendency for both young and old PDR1 insertions to be less common in the germ plasm set. Intriguingly, 5 of the 6 apparently most ancient insertions studied by us (between 1.5 and 5 MYA) are found in ≤4 accessions of the 47-sample Pisum germ plasm set (the two oldest are shown in Figure 4, J and K). This might imply that PDR1 insertions tend to have a limited life span in the Pisum genus and that these insertions are in the process of slow elimination (see discussion).
Figure 5.

Relationship between antiquity of PDR1 insertions and their distribution in the Pisum genus. Frequencies for 43 PDR1 retrotransposon insertions in the 47-accession Pisum core collection (solid circles) (Vershinin et al. 2003) are plotted against the numbers of combined SNP and INDEL polymorphisms between their LTRs. Average values are shown as large open circles with standard deviations shown as vertical bars. The horizontal bar indicates that this particular frequency value is the average of the five data points with between four and nine polymorphisms.
An estimation of effective population size for Pisum:
The average K-value deduced above can be used to deduce the effective population size of Pisum. According to the neutral theory (Kimura and Crow 1964; Kimura 1983a,b) the expected frequency distribution of allele abundance Φ(x) is determined by the effective population size Ne and the mutation rate ν:
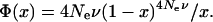 |
(1) |
The 52 RBIP retrotransposon insertion mutations studied here may be considered as neutral alleles but they do not constitute a large enough data set to test reliable fit to the above equation. However, the larger data set of 259 PDR1 SSAP markers in the highly similar Pisum core set used for the generation of the tree shown in Figure 4A (Vershinin et al. 2003) can be used. Figure 6 shows the result of this analysis. The allele frequencies of the occupied sites (horizontal bars) for the 259 markers within this set of accessions (y-axis) are plotted against the proportion of markers with alleles of a given frequency (x-axis). These allele frequency data can be used to derive a value for the average heterozygosity (He), from which 4Neν can be deduced, using the relationship
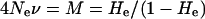 |
(2) |
(Kimura 1983b). The SSAP data give a value for He of 0.615, making 4Neν = 1.60. Using this value in Equation 1 gives the curve in Figure 6, which fits the experimental data nicely if both are scaled to integrate to unity. Substituting ν the average transposition rate obtained from K above for the 52 PDR1 RBIP insertions (1/1.9 × 10−6 = 5.3 × 10−7) gives a value of Ne for Pisum of 7.5 × 105.
Figure 6.

Frequency distribution for PDR1 SSAP markers in a core Pisum set (Vershinin et al. 2003). The proportions of PDR1 markers (y) that have occupied sites (alleles) of frequency x are plotted as solid squares. From the observed distribution of occupied- and unoccupied-site alleles, the frequency distribution as predicted in Equation 1 (Kimura 1983b) is plotted as a line. Note that the five SSAP markers that were fixed in this data set are known to be polymorphic in extended data sets analyzed by Vershinin et al. (2003).
DISCUSSION
The goals of this study were to investigate the nature and antiquity of PDR1 insertions in P. sativum, to gain knowledge of distribution of these insertions across the genus Pisum, and to use these data to investigate the rule(s) controlling the fates of PDR1 insertions in the genus.
The targets for PDR1 insertions:
Three hundred forty distinct sequences flanking PDR1 insertions were obtained in this study, allowing us to deduce a consensus target site sequence for insertion of PDR1, which shows similarities with the specificity for the Tos17 Ty1-copia group retrotransposon of rice (Figure 2; Miyao et al. 2003). Both consensuses show dyad symmetry, both contain strong preferences at positions −3, −2, +2, +3 relative to the TSD (A, T, A, T and G, T, A, C for PDR1 and Tos17, respectively), and both prefer A/T-rich sequence for the middle 3 bases of the TSD. It is premature to deduce a general target site consensus for plant Ty1-copia group retrotransposons on the basis of just two elements, especially since both are found in low copy number and both share the properties of being among the shortest and structurally simplest Ty1-copia group retrotransposons known. It will be interesting to see whether other Ty1-copia group retrotransposons follow the consensus shown in Figure 2. No comparable consensus has yet been reported for the Ty1-copia group retrotransposons of other plants or kingdoms (Le et al. 2000; Kaminker et al. 2002), although TSD preferences are well known for Ty3-gypsy group retrotransposons of Drosophila and plants (Ikenaga and Saigo 1982; Jiang et al. 2002b).
We have also identified the nature of PDR1 insertion sites. Most are other transposons, including retrotransposons and occasionally PDR1 itself [Table 2, supplementary Table S2 (http://www.genetics.org/supplemental/)]. This is unsurprising, as such repetitious DNAs compose a large proportion of pea genomic DNA. More surprisingly, 7% of PDR1 insertion sites are coding regions of nuclear genes that are not derived from transposable elements. This does not take into account PDR1 insertions into introns and gene flanks, because these are difficult to distinguish from nongenic DNA on the basis of short sequence reads, so the actual percentage of insertions within genes is probably higher than this. Assuming that pea has a gene number (∼30,000) and average gene size (∼2 kb) comparable to Arabidopsis and rice (http://www.ostp.gov/NSTC/html/mpgi2001/sequencing.htm), then the gene space of pea is expected to be ∼6 × 107 nucleotides, which represents ∼1.3% of the pea genome. There thus appear to be at least fivefold more PDR1 insertions within genes than expected.
There are two plausible explanations for this discrepancy and these are not mutually exclusive. First, there may be more genes in Pisum than are found in Arabidopsis or rice; for M. truncatula gene number has been estimated at 37,000–46,000 (http://catg.ucdavis.edu/m.truncatula.pdf). Gene duplication may be a factor in this. There clearly is genetic redundancy in pea since the PDR1 insertions into coding regions described here were probably all complete knockouts (these are kilobase-sized inserts). Incidentally, the only PDR1 element to be sequenced entirely (Lee et al. 1990) resides in the close vicinity of a duplicated gene. However, considerable gene duplications are known for both rice and Arabidopsis and it remains unclear whether the level of gene duplication in pea exceeds that seen for these other species. To our knowledge there is no evidence for ancient polyploidy in Pisum.
A second explanation for the higher than expected proportion of PDR1 insertion sites in genes is that PDR1 may have shown a preference for inserting into genic regions. Many cases in the literature of retrotransposons show insertional preferences (e.g., Kim et al. 1998; Presting et al. 1998) and under cell culture conditions both Tos17 in rice (Miyao et al. 2003) and Tnt1 in tobacco (M.-A. Grandbastien, personal communication) insert preferentially in genic regions, presumably as a consequence of an open chromatin configuration. However, in the intact organism every insertion is tested by natural selection and the existence of transposable elements that preferentially target genes would impose a large fitness cost on the host, particularly for diploid, predominantly selfing plants such as pea, as the majority of offspring would be homozygous for the insertions.
A preference for insertion into genes may explain why PDR1 copy number appears to be quite strongly constrained to ∼200 per genome across the genus Pisum (Lee et al. 1990; Ellis et al. 1998), although the exact relationship between PDR1 copy number and fitness would be critical (Brookfield 2005). Nevertheless, the allele frequency data for PDR1 insertions (Figure 6) show good fit with the expected distribution for neutral alleles, suggesting that those insertions that have survived selection do not have large individual fitness effects. Interestingly, in maize the large majority of retrotransposon-induced mutations are caused by elements with copy numbers between ∼2 and 50 (Marillonnet and Wessler 1998), whereas elements, which are present in thousands of copies, cause few or no mutations and generally are found in nested complexes between genes (Sanmiguel et al. 1996; Shirasu et al. 2000; Chantret et al. 2005). The distribution of PDR1 on this scale in Pisum remains to be elucidated.
Ages of PDR1 insertions and their relationship with insertional polymorphism in the genus Pisum:
Our results indicate that PDR1 has been transposing within roughly the last 5 MYA, with a peak at ∼1–2 MYA. We cannot comment on the earlier history of PDR1, because our PCR-based approach for isolating insertions becomes progressively less efficient for isolating older insertions as a result of primer site mutation. Nevertheless, our conclusions are quite similar to corresponding sequence-based data from maize and rice (Sanmiguel et al. 1998; Vitte et al. 2004). Surprisingly, none of the 43 insertions studied here have become fixed in the pea genome during this long period. This may be due to the predominantly selfing character of the species but the distribution of alleles across the Pisum diversity tree (Vershinin et al. 2003) indicates that introgression between highly diverse germ plasm has been an important factor in the evolution of the genus. While this introgression has been sufficient to shuffle many alleles it has apparently not been sufficiently widespread to drive many of these PDR1 elements to fixation. In a larger study using multiplex SSAP markers derived from several LTR retrotransposons including PDR1, ∼2% of SSAP bands were seen to be fixed in a virtually identical set of pea samples (Vershinin et al. 2003). It should be noted that the PDR1 insertions with both LTR sequence and diversity data that have been studied here have successfully produced PCR products in several successive amplifications in diverse germ plasm. It is possible that this experimental approach reduced the detection frequency for older insertions and so effectively excluded ancient, fixed PDR1 elements from this study.
Another possible factor in the distribution of PDR1 insertions is the geography of pea. Wild Pisum is distributed widely across Southern Asia, North Africa, and Southern Europe (Ambrose and Maxted 2000). Ancient insertions that show restricted distributions may represent geographically isolated plant lineages or they may represent the remnants of ancient, more widespread populations that are now in decline. More work is required to clarify this issue.
A final caveat to our insertional polymorphism analysis is the possibility that some of the scores may be inaccurate because many of the PDR1 insertions described in this study are in repetitious DNA. In such cases the unoccupied site is present in multiple copies across the genome. This might lead to inaccuracy in scoring the state of the locus by the production of unoccupied-site PCR products, irrespective of whether the PDR1 insertion is present or not. In an extreme instance, such spurious unoccupied-site amplicons might outcompete the production of the occupied-site product, leading to a misscored sample. In practice, the majority of scores obtained in this study are either occupied or unoccupied [supplementary Figure S4 (http://www.genetics.org/supplemental/)], suggesting that this is not a problem for most of the 64 insertions scored and the exceptions have been largely confined to a small number of insertions that misbehave in multiple accessions. Actually, it is surprising that this potential problem has caused so few difficulties in the scoring of these PDR1 insertions and we suggest that this may be due to sequence decay in the insertion sites, which allows repetitious unoccupied sites to be amplified in effect as pseudo-single-copy sites.
The effective population size of Pisum:
The availability of rate data for retrotransposon insertions has allowed us to reexamine an earlier, larger set of retrotransposon polymorphism data and thereby obtain an estimated value for the effective population size of Pisum. The congruence between the observed allele frequency data and the plot obtained using Equation 1 suggests that this approach is valid. Several assumptions made during the deduction of Equation 1 need to be considered here. Most importantly, these population genetics models assume a random-mating population but Pisum is a predominant inbreeder. Every cross between two different Pisum genotypes would produce effectively a mixed recombinant inbred subpopulation carrying the original parental alleles. For the purposes of analyzing effective population size, such subpopulations approximate to individuals. This consideration suggests that the effective population size is very much smaller than actual population size and dominated by the harmonic mean of the effective population size per lineage per unit time (Kimura 1983a, pp. 40–43).
Effective population size is an important parameter with regard to the domestication of crop plants such as Pisum from their wild progenitors. All of the major food crops fall into this category and the diversity of alleles is important with regard to important traits such as pest resistance and abiotic stress tolerance. The wild gene pool for Pisum, as for the other crop species, is wider than that of the domesticated samples. The effective population size is a useful measure of this diversity and the methods shown here offer a way to measure this parameter in Pisum and, by extrapolation, in other crop plants. The value of <1 million individuals, which we have obtained here, seems quite low for a reasonably common species with such broad geographic distribution.
Acknowledgments
We thank David Martin for help with database searches and Pete Isaac and Alan Schulman for many helpful discussions on all aspects of this work. This work was supported by grants 31502 (TEGERM) and FP6-2002-FOOD-1-506223 (Grain Legumes) from the European Commission under the Frameworks V and VI and by Biotechnology and Biological Sciences Research Council grant 94/BEP17084 (Bioinformatics and E-Science program).
References
- Altschul, S. F., T. L. Madden, A. A. Schaffer, J. Zhang, Z. Zhang et al., 1997. Gapped BLAST and PSI-BLAST: a new generation of protein database search programs. Nucleic Acids Res. 25: 3389–3402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ambrose, M., and N. Maxted, 2000. Peas (Pisum L.), pp. 181–190 in Plant Genetic Resources of Legumes in the Mediterranean, edited by N. Maxted and S. J. Bennett. Kluwer Academic Publishers, The Netherlands.
- Baranyi, M., J. Greilhuber and W. W. Swiecicki, 1996. Genome size in wild Pisum species. Theor. Appl. Genet. 93: 717–721. [DOI] [PubMed] [Google Scholar]
- Blixt, S., 1972. Mutation genetics in Pisum. Agri Hort. Genet. 30: 1–293. [Google Scholar]
- Bowen, N. J., and J. F. McDonald, 2001. Drosophila euchromatic LTR retrotransposons are much younger than the host species in which they reside. Genome Res. 11: 1527–1540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brookfield, J. F. Y., 2005. The ecology of the genome—mobile DNA elements and their hosts. Nat. Rev. Genet. 6: 128–136. [DOI] [PubMed] [Google Scholar]
- Chantret, N., J. Salse, F. Sabot, S. Rahman, A. Bellec et al., 2005. Molecular basis of evolutionary events that shaped the hardness locus in diploid and polyploid wheat species (triticum and aegilops). Plant Cell 17: 1033–1045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chavanne, F., D. X. Zhang, M. F. Liaud and R. Cerff, 1998. Structure and evolution of Cyclops: a novel giant retrotransposon of the Ty3/Gypsy family highly amplified in pea and other legume species. Plant Mol. Biol. 37: 363–375. [DOI] [PubMed] [Google Scholar]
- Ellis, T. H. N., S. J. Poyser, M. R. Knox, A. V. Vershinin and M. J. Ambrose, 1998. Ty1-copia class retrotransposon insertion site polymorphism for linkage and diversity analysis in pea. Mol. Gen. Genet. 260: 9–19. [DOI] [PubMed] [Google Scholar]
- Flavell, A. J., D. B. Smith and A. Kumar, 1992. a Extreme heterogeneity of Ty-copia group retrotransposons in plants. Mol. Gen. Genet. 231: 233–242. [DOI] [PubMed] [Google Scholar]
- Flavell, A. J., E. Dunbar, R. Anderson, S. R. Pearce, R. Hartley et al., 1992. b Ty1-copia group retrotransposons are ubiquitous and heterogeneous in higher plants. Nucleic Acids Res. 20: 3639–3644. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Flavell, A. J., M. R. Knox, S. R. Pearce and T. H. N. Ellis, 1998. Retrotransposon-based insertion polymorphisms (RBIP) for high throughput marker analysis. Plant J. 16: 643–650. [DOI] [PubMed] [Google Scholar]
- Gaut, B. S., B. R. Morton, B. C. McCaig and M. T. Clegg, 1996. Substitution rate comparisons between grasses and palms: synonymous rate differences at the nuclear gene Adh parallel rate differences at the plastid gene rbcL. Proc. Natl. Acad. Sci. USA 93: 10274–10279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Greilhuber, J., and I. Ebert, 1994. Genome size variation in Pisum sativum. Genome 37: 646–655. [DOI] [PubMed] [Google Scholar]
- Havecker, E. R., X. Gao and D. F. Voytas, 2004. The diversity of LTR retrotransposons. Genome Biol. 5: 225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hill, P., D. Burford, D. M. Martin and A. J. Flavell, 2005. Retrotransposon populations of Vicia species with varying genome size. Mol. Genet. Genomics 273: 371–381. [DOI] [PubMed] [Google Scholar]
- Ikenaga, H., and K. Saigo, 1982. Insertion of a movable genetic element, 297, into the T-A-T-A Box for the H3 histone gene in Drosophila melanogaster. Proc. Natl. Acad. Sci. USA 79: 4143–4147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiang, N., Z. Bao, S. Temnykh, Z. Cheng, J. Jiang et al., 2002. a Dasheng: a recently amplified nonautonomous LTR element that is a major component of pericentromeric regions in rice. Genetics 161: 1293–1305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiang, N., I. K. Jordan and S. R. Wessler, 2002. b Dasheng and RIRE2. A nonautonomous long terminal repeat element and its putative autonomous partner in the rice genome. Plant Physiol. 130: 1697–1705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kalendar, R., T. Grob, M. Regina, A. Suoniemi and A. H. Schulman, 1999. IRAP and REMAP: two new retrotransposon-based DNA fingerprinting techniques. Theor. Appl. Genet. 98: 704–711. [Google Scholar]
- Kaminker, J. S., C. M. Bergman, B. Kronmiller, J. Carlson, R. Svirkas et al., 2002. The transposable elements of the Drosophila melanogaster euchromatin: a genomics perspective. Genome Biol. 3: Research0084.1–0084.20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim, J. M., S. Vanguri, J. D. Boeke, A. Gabriel and D. F. Voytas, 1998. Transposable elements and genome organisation: a comprehensive survey of retrotransposons revealed by the complete Saccharomyces cerevisiae genome sequence. Genome Res. 8: 464–478. [DOI] [PubMed] [Google Scholar]
- Kimura, M., 1980. A simple method for estimating evolutionary rates of base substitutions through comparative studies of nucleotide sequences. J. Mol. Evol. 16: 111–120. [DOI] [PubMed] [Google Scholar]
- Kimura, M., 1983. a The Neutral Theory of Molecular Evolution, p. 205. Cambridge University Press, Cambridge, UK.
- Kimura, M., 1983. b Rare variant alleles in the light of the neutral theory. Mol. Biol. Evol. 1: 84–93. [DOI] [PubMed] [Google Scholar]
- Kimura, M., and J. F. Crow, 1964. The number of alleles that can be maintained in a finite population. Genetics 49: 725–738. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Knox, M., 2005. The regional control of chiasmata and recombination frequency in pea (Pisum sativum L.). Ph.D. Thesis, University of East Anglia, Norwich, UK.
- Konieczny, A., D. F. Voytas, M. P. Cummings and F. M. Ausubel, 1991. A superfamily of Arabidopsis thaliana retrotransposons. Genetics 127: 801–809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kumar, A., and J. L. Bennetzen, 1999. Plant retrotransposons. Annu. Rev. Genet. 33: 479–532. [DOI] [PubMed] [Google Scholar]
- Le, Q. H., S. Wright, Z. Yu and T. Bureau, 2000. Transposon diversity in Arabidopsis thaliana. Proc. Natl. Acad. Sci. USA 97: 7376–7381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee, D., T. H. N. Ellis, L. Turner, R. P. Hellens and W. G. Cleary, 1990. A copia-like element in Pisum demonstrates the uses of dispersed repeated in genetic analysis. Plant Mol. Biol. 15: 707–722. [DOI] [PubMed] [Google Scholar]
- Li, W.-H., and D. Graur, 1991. Fundamentals of Molecular Evolution. Sinauer Associates, Sunderland, MA.
- Ma, J., K. M. Devos and J. L. Benetzen, 2004. Analysis of LTR-retrotransposon structures reveals recent and rapid genomic DNA loss in rice. Genome Res. 14: 860–869. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Macas, J., P. Neumann and D. Pozarkova, 2003. Zaba: a novel miniature transposable element present in genomes of legume plants. Mol. Gen. Genomics 269: 624–631. [DOI] [PubMed] [Google Scholar]
- Marillonnet, S., and S. R. Wessler, 1998. Extreme structural heterogeneity among the members of a maize retrotransposon family. Genetics 150: 1245–1256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mithen, S., 2003. After the Ice: A Global Human History 20,000–5,000 BC. Weidenfield & Nicholson, London.
- Miyao, A., T. Katsuyuki, K. Murata, H. Sawaki, S. Takeda et al., 2003. Target site specificity of the Tos17 retrotransposon shows a preference for insertion within genes and against insertion in retrotransposon-rich regions of the genome. Plant Cell 15: 1771–1780. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murray, M. G., and W. F. Thompson, 1982. Repeat sequence interspersion in coding DNA of peas does not reflect that in total DNA. Plant Mol. Biol. 1: 143–153. [DOI] [PubMed] [Google Scholar]
- Neumann, P., M. Nouzova and J. Macas, 2001. Molecular and cytogenetic analysis of repetitive DNA in pea (Pisum sativum L.). Genome 44: 716–728. [PubMed] [Google Scholar]
- Neumann, P., D. Pozarkova and J. Macas, 2003. Highly abundant pea LTR retrotransposon Ogre is constitutively transcribed and partially spliced. Plant Mol. Biol. 53: 399–410. [DOI] [PubMed] [Google Scholar]
- Noma, K., E. Ohtsubo and H. Ohtsubo, 1999. Non-LTR retrotransposons (LINEs) as ubiquitous components of plant genomes. Mol. Gen. Genet. 261: 71–79. [DOI] [PubMed] [Google Scholar]
- Nouzova, M., P. Neumann, A. Navratilova and J. Macas, 2000. Microarray-based survey of repetitive genomic sequences in Vicia spp. Plant Mol. Biol. 45: 229–244. [DOI] [PubMed] [Google Scholar]
- Porceddu, A., E. Albertini, G. Barcaccia, G. Marconi, F. Bertoli et al., 2002. Development of S-SAP markers based on an LTR-like sequence from Medicago sativa L. Mol. Gen. Genet. 267: 107–114. [DOI] [PubMed] [Google Scholar]
- Presting, G. G., L. Malysheva, J. Fuchs and I. Schubert, 1998. A Ty3/gypsy retrotransposon-like sequence localises to the centromeric regions of cereal chromosomes. Plant J. 16: 721–728. [DOI] [PubMed] [Google Scholar]
- Provan, J., W. T. B. Thomas, B. P. Forster and W. Powell, 1999. Copia-SSR: a simple marker technique which can be used on total genomic DNA. Genome 42: 363–366. [Google Scholar]
- Rosenthal, A., and D. S. C. Jones, 1990. Genomic walking and sequencing by oligo-cassette mediated polymerase chain reaction. Nucleic Acids Res. 18: 3095–3096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sanmiguel, P., A. Tikhonov, Y. K. Jin, N. Motchoulskaia, D. Zakharov et al., 1996. Nested retrotransposons in the intergenic regions of the maize genome. Science 274: 765–768. [DOI] [PubMed] [Google Scholar]
- Sanmiguel, P., B. S. Gaut, A. Tikhonov, Y. Nakijama and J. L. Bennetzen, 1998. The paleontology of intergene retrotransposons of maize. Nat. Genet. 20: 43–45. [DOI] [PubMed] [Google Scholar]
- Schmidt, T., 1999. LINEs, SINEs and repetitive DNA: non-LTR retrotransposons in plant genomes. Plant Mol. Biol. 40: 903–910. [DOI] [PubMed] [Google Scholar]
- Shirasu, K., A. H. Schulman, T. Lahaye and P. Schulze-Lefert, 2000. A contiguous 66-kb barley DNA sequence provides evidence for reversible genome expansion. Genome Res. 10: 908–915. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Siebert, P. D., A. Chenchik, D. E. Kellogg, K. A. Lukyanov and S. A. Lukyanov, 1995. An improved PCR method for walking in uncloned genomic DNA. Nucleic Acids Res. 23: 1087–1088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Small, R. L., J. A. Ryburn and J. F. Wendel, 1998. Low levels of nucleotide diversity at homoeologous Adh loci in allotetraploid cotton (Gossypium L.). Mol. Biol. Evol. 16: 491–501. [DOI] [PubMed] [Google Scholar]
- Suoniemi, A., J. Tanskanen and A. H. Schulman, 1998. Gypsy retrotransposons are widespread in the plant kingdom. Plant J. 13: 699–705. [DOI] [PubMed] [Google Scholar]
- Thompson, J. D., D. G. Higgins and T. J. Gibson, 1994. Clustal W: improving the sensitivity of progressive multiple sequence alignment through sequence weighting, position-specific gap penalties and weight matrix choice. Nucleic Acids Res. 22: 4673–4680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Turner, L., R. P. Hellens, D. Lee and T. H. N. Ellis, 1993. Genetic aspects of the organization of legumin genes in pea. Plant Mol. Biol. 22: 101–112. [DOI] [PubMed] [Google Scholar]
- Vershinin, A. V., T. R. Alnutt, M. R. Knox, M. R. Ambrose and T. H. N. Ellis, 2003. Transposable elements reveal the impact of introgression, rather than transposition, in Pisum diversity, evolution and domestication. Mol. Biol. Evol. 20: 2067–2075. [DOI] [PubMed] [Google Scholar]
- Vitte, C., T. Ishii, F. Lamy, D. Brar and O. Panaud, 2004. Genomic paleontology provides evidence for two distinct origins of Asian rice (Oryza sativa L.). Mol. Genet. Genomics 272: 504–511. [DOI] [PubMed] [Google Scholar]
- Voytas, D. F., M. P. Cummings, A. Konieczny, F. M. Ausubel and S. R. Rodermel, 1992. Copia-like retrotransposons are ubiquitous among plants. Proc. Natl. Acad. Sci. USA 89: 7124–7128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Waugh, R., K. McLean, A. J. Flavell, S. R. Pearce, A. Kumar et al., 1997. Genetic distribution of BARE-1 retrotransposable elements in the barley genome revealed by sequence-specific amplification polymorphisms (S-SAP). Mol. Gen. Genet. 253: 687–694. [DOI] [PubMed] [Google Scholar]
- Wolfe, K. H, W.-H. Li and P. M. Sharpe, 1987. Rates of nucleotide substitution vary greatly among plant mitochondrial, chloroplast and nuclear DNAs. Proc. Natl. Acad. Sci. USA 84: 9054–9058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wojciechowski, M. F., 2003. Reconstructing the phylogeny of legumes (Leguminosae): an early 21st century perspective, pp. 5–35 in Advances in Legume Systematics, Part 10, Higher Level Systematics, edited by B. B. Klitgaard and A. Bruneau. Botanic Gardens, Kew, UK.
- Xiong, Y., and T. H. Eickbush, 1990. Origin and evolution of retroelements based upon their reverse transcriptase sequences. EMBO J. 9: 3353–3362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu, G. X., and R. P. Wise, 2000. An anchored AFLP- and retrotransposon-based map of diploid Avena. Genome 43: 736–749. [PubMed] [Google Scholar]
- Zohary, D., 1996. The mode of domestication of the founder crops of near east agriculture, pp. 142–158 in The Origin and Spread of Agriculture and Pastoralism in Eurasia, edited by D. R. Harris. University College London Press, London.